

Abstract
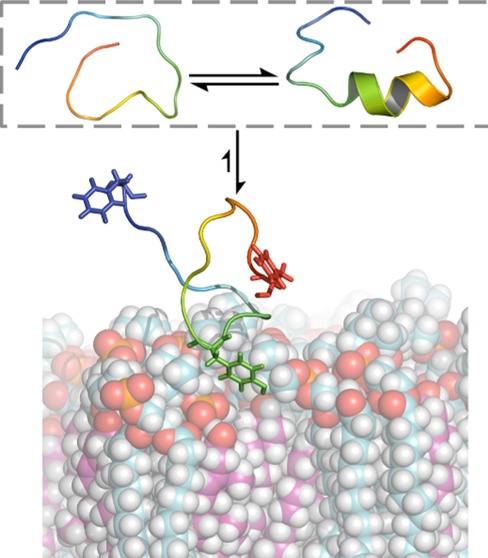
The cholesterol recognition/interaction amino acid consensus (CRAC) motif is a primary structure pattern used to identify regions that may be responsible for preferential cholesterol binding in many proteins. The leukotoxin LtxA, which is produced by a pathogenic bacterium, contains two CRAC seqences, only one of which is responsible for cholesterol binding, and the binding is required for cytotoxicity. The factors, in addition to the CRAC definition, that may be responsible for cholesterol-binding functionality and atomistic interactions between the CRAC region and cholesterol are as yet unknown. This study uses molecular dynamics simulations to identify structural characteristics and specific interactions of the two LtxA CRAC peptides with both pure phospholipid and binary cholesterol–phospholipid bilayers. We have identified changes in the secondary structure of these peptides that occur upon cholesterol binding, which are not seen when it is associated with a cholesterol-devoid membrane, and which show salient coupling of structural disorder and function. Additionally, the central tyrosine residue of the CRAC motif was found to play a significant role in cholesterol binding, though residues outside of the CRAC motif also influence membrane interactions and functionality of the CRAC region.
Introduction
Biological membranes are complex, dynamic systems that are responsible for an array of cellular processes. Interactions between various membrane components, including lipid–lipid, lipid–protein, and protein–protein interactions, all contribute to the structure and function of cell membranes.1,2 The study of membrane-bound proteins is relevant to understanding many native cell processes as well as the cytotoxic effects of some proteins, and much research has been done to elucidate these details;3 however, mechanisms of membrane binding by proteins remain perplexing. The difficulty of experimentally determining structural characteristics of membrane-bound proteins and the small-scale interactions of peptides with membranes makes molecular dynamics simulations a promising technique for the study of membrane–peptide interactions.4 Furthermore, biological membranes have been found to have significant and varied effects on protein structure. While inducing or stabilizing an α-helical structure in some intrinsically disordered proteins (IDPs), such as α-synuclein and islet amyloid polypeptide (IAPP),5−7 near-membrane environments can also cause structural loss of folded proteins; this loss of structure near membranes is associated with the functionality of many proteins, including several protein toxins.8
One area of particular interest in protein–membrane interactions is binding of proteins to membrane-bound cholesterol, which has led to the definition of the cholesterol recognition/interaction amino acid consensus (CRAC) motif.9 Highly conserved among cholesterol-interacting proteins, the CRAC motif takes the form -L/V-X1–5-Y-X1–5-R/K-, where X1–5 may be any sequence of amino acids from one to five residues in length. However, the liberal constraints of the CRAC algorithm overpredict actual cholesterol-binding functionality. For example, a leukotoxin (LtxA) produced by the pathogenic bacterium Aggregatibacter actinomycetemcomitans exhibits binding specific to cholesterol and contains two CRAC sequences, termed CRAC336 (LEEYSKRFKK) and CRAC503 (VDYLKK).10 Of the two CRAC sites, only CRAC336 was shown to be responsible for cholesterol binding of LtxA.10 Furthermore, CRAC336 is highly conserved among the repeats-in-toxin family of proteins, which includes Escherichia coli α-hemolysin and Mannheimia hemolytica leukotoxin, in addition to LtxA. The differences that exist between the two CRAC regions that lead to their differing levels of functionality are unkown but may provide insight into cholesterol binding not only by LtxA but also by other toxins that conserve this functional CRAC region and require cholesterol binding for toxicity.
We have performed simulations of the peptides corresponding to the CRAC336 and CRAC503 sequences (acetyl-FDRARMLEEYSKRFKKFGY-NH2 and acetyl-QSGKAYVDYLKKGEELA-NH2, respectively) used in the experiments by Brown et al.10 By comparing simulations carried out both in solution and near membrane bilayers, we are able to identify differences in the interactions of the two peptides with a lipid bilayer in the presence and absence of cholesterol. Consistent with former experiments, CRAC336, but not CRAC503, showed strong membrane affinity specific to the cholesterol-bearing bilayer. Additionally, significant loss of the peptide’s secondary structure was also correlatively associated with its interaction with this membrane.
Computational Methods
Simulation of each of the two peptides in solution was first performed using Gromacs 4.5.611 with the Amber03d force field12 and the TIP3P water model.13 Each peptide was solvated in a truncated octahedron box with 3.7 nm between the nearest faces, and ions were added to neutralize charge. Replica-exchange molecular dynamics (REMD)14 was performed using an NVT ensemble with 24 replicas from 300 to 499 K. Temperatures for each replica were 300, 304, 308, 313, 318, 324, 330, 337, 344, 352, 360, 369, 378, 388, 398, 409, 420, 431, 444, 455, 466, 477, 488, and 499 K. Electrostatic interactions were calculated using the particle mesh Ewald method15 with a real space cutoff of 0.9 nm. For van der Waals interactions, a 1.2 nm cutoff was used. The simulation was initiated from a random-coil-like conformation. Each replica was simulated for 151 ns, with a time step of 2 fs, and exchange attempted every 1000 steps. The first 51 ns were discarded as equilibration. Though frames that allowed the peptide to interact with its periodic image were not significant (less than 4% of frames for CRAC503 with a minimum distance between periodic images of less than 0.6 nm), they were discarded from the analysis.16,17
Both peptides were also simulated near a pure DMPC bilayer composed of 64 lipid molecules (32 in each leaflet) and a 40% cholesterol bilayer composed of 128 molecules (26 cholesterol and 38 DMPC molecules in each leaflet), both of which were obtained from the Slipids Web site (http://people.su.se/∼jjm/). The starting configurations for these simulations were taken from the equilibrium trajectory of the solution simulations. To enhance conformational sampling of the peptide, these simulations utilized replica exchange with solute tempering (REST)18−20 using Gromacs (4.6.5)11 patched with the PLUMED plugin (version 2.0.2).21 The Amber03d force field and the TIP3P water model were used for protein and solvent parameters, and the Slipids force field was used for the lipid parameters.
The Amber03d force field12 has been found to accurately balance helix and coil populations and to fold both α-helix and β-sheet structures,22,23 and the Slipids force field with TIP3P water has been validated for the simulation of lipids, including binary bilayers.24−26 Force-field accuracy continues to be an ongoing endeavor, and agreement between our simulation and experimental results suggests that our force-field parameters are appropriate. However, further work comparing results from different force fields is needed in the future.
Electrostatics were calculated using the particle mesh Ewald method with a real space cutoff of 1.0 nm. For van der Waals interactions, a 1.0 nm cutoff was also used. Eight replicas were used to scale the protein energy with effective temperatures of 300, 331.227, 365.704, 403.77, 445.798, 492.201, 543.434, and 600 K as described previously.20 Each replica was simulated for 100 ns, with a time step of 2 fs, and exchange attempted every 500 steps. Though sufficient for the CRAC503 peptide, eight replicas were found to limit the exchange of the lowest-temperature replicas in the case of the CRAC336 peptide. As a result, the final configurations of this 100 ns run were used to populate 12 replicas to scale the protein energy with effective temperatures of 300, 319.512, 340.294, 362.427, 385.999, 411.105, 437.844, 466.322, 496.652, 528.955, 563.359, and 600 K. Each of the 12 replicas was simulated for an additional 100 ns, again with a time step of 2 fs, and exchange attempted every 500 steps. Percolation of replicas demonstrates movement of replicas from lowest to highest temperature nodes and is shown for the CRAC336 peptide with the 40% cholesterol membrane system in the Supporting Information. Semi-isotropic pressure coupling was maintained with the Parrinello–Rahman barostat27 using a coupling time of 5 ps. In each case, the first 5 ns of the trajectory of interest (300 K) were discarded as equilibration time. Equilibration times were determined from cumulative averaging of secondary structure properties; these cumulative averages of secondary structure propensities for the CRAC336 DMPC membrane system are available in the Supporting Information.
The CRAC336 Y19A mutant peptide was simulated near the 40% cholesterol membrane for 50 ns using eight replicas and discarding 5 ns of equilibration time as described above. The CRAC336 Y10A mutant required greater equilibration time and was simulated for 150 ns, using the last 100 ns for analysis, and using 12 replicas.
Results and Discussion
Both CRAC336 and CRAC503 were simulated in three environments: in solution, near a 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) bilayer, and near a bilayer composed of 60 mol % DMPC and 40 mol % cholesterol (DMPC/CHO). When comparing LtxA binding to membranes with 0, 20, 40, and 60% cholesterol, Brown et al. found that the protein had the greatest affinity for membranes composed of 40% cholesterol.10 In the same study, it was found that LtxA’s affinity for the 40% cholesterol membrane was not attributable merely to its raft-like nature because cholesterol binding of LtxA to a non-raft-like bilayer composed of 60 mol % 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) and 40 mol % cholesterol (60:40 DOPC/CHO) was not significantly different from its binding to the raft-like DMPC/CHO bilayer.
The extent to which each of the CRAC peptides interacted with the membrane in our simulations varied between both the membrane environments and the peptides. To compare each peptide’s propensity to remain close to or even penetrate the membrane surface, the time-averaged density profile along the membrane normal for each case is plotted in Figure 1. Near the pure DMPC bilayer, both CRAC336 and CRAC503 show sampling of space both at the membrane interface and throughout the aqueous region. However, the density profile of CRAC336 near the 40% cholesterol bilayer shows strong localization of the peptide close to the bilayer surface. Though CRAC503 shows some increase in near-membrane density near the 40% cholesterol membrane, this peptide continues to show significant sampling of space throughout the aqueous region as well.
Figure 1.

Density profiles of each CRAC peptide/membrane system along the membrane normal. CRAC336 and CRAC503 systems are in the left and right columns, respectively, and density profiles near the 60:40 DMPC/CHO and pure DMPC bilayer are represented in the top and bottom rows, respectively. Peptide density is shown in red; cholesterol density is magenta, and DMPC densities are broken into hydrocarbon, carbonyl, glycerol, phosphate, and choline groups (see legend for color definitions).
To identify whether the inherent structural characteristics of the two peptides are responsible for their cholesterol interaction, secondary structure assignments were determined for each 300 K solution trajectory using the DSSP algorithm.28 Averaging the number of residues assigned to each secondary structure over the trajectory yields similar results for the two peptides, with both sharing comparable tendencies toward each secondary structure assignment (Figure 2A, black). Secondary structure assignments per residue are plotted in Figure 2B, again showing similar results for both peptides in solution. Both peptides share an overall high coil, low-to-moderate β-sheet propensity as well as a helical propensity over their central residues. CRAC336 does show greater β-sheet and lower α-helix propensity than CRAC503, and it is possible that these structural differences may contribute to the cholesterol-binding activity of the peptides. However, greater structural differences are seen in the near-membrane environments.
Figure 2.

Secondary structure comparison of CRAC336 (left) and CRAC503 (right) in solution (black), near a DMPC/CHO (red) membrane, and near a pure DMPC membrane (green). (A) Average number of residues assigned to coil, β-sheet, β-bridge, bend, turn, α-helix, and 310-helix over each trajectory. Not shown, π-helix averaged less than 0.1% of residues in each case. Error bars represent statistical uncertainty based on block averaging. (B) Average fraction of time spent in coil (including random coil, bend, and turn), helix (α-, π-, and 310-helix), and β-sheet (β-sheet and β-bridge) conformations per residue.
To determine whether membrane interactions induce structural changes in either of the two CRAC peptides, DSSP secondary structure assignments were determined for the ensemble of each peptide near both the DMPC/CHO and the pure DMPC bilayers (Figure 2, red and green, respectively). Despite a quantitative change in β-sheet propensity, the qualitative structural characteristics of CRAC503 near both bilayers remain similar to those in aqueous solution, which could be expected based on the relatively weak interaction between the peptide and the bilayers. In the secondary structure of all three environments, the average number of residues shows some β-sheet propensity in addition to a more considerable helical propensity. However, we observe significant changes in secondary structure of CRAC336 near the 40% cholesterol membrane compared to its secondary structure in solution. Near the cholesterol-bearing membrane, the average number of total residues in α- and 310-helical structures and in summed β-sheet and β-bridge structures drops to less than 20 and 10%, respectively, of their original values in solution. The secondary structure of this peptide per residue reveals diminished helicity and β-sheet propensity in favor of increased coil conformation across the entire sequence. This effect is not attributable merely to the presence of a membrane because CRAC336 maintains both helical and β-sheet structures near the pure DMPC membrane. In contrast to this marked difference in secondary structure propensities near the cholesterol-bearing membrane, CRAC336 and CRAC503 share similar structural characteristics near the pure DMPC membrane. These results provide a striking example of the functionality of disorder. Of the two CRAC peptides in both bilayer environments, only CRAC336 near the 40% cholesterol membrane shows a strong singular affinity for the peptide to remain close to the bilayer interface, and the cholesterol-binding functionality of this peptide is coupled to its secondary structure loss.
By the definition of a CRAC motif (-L/V-X1–5-Y-X1–5-R/K-), the sole residue that is always required is the central tyrosine, and CRAC336 near the 40% cholesterol bilayer shows insertion of this residue at the membrane interface during our equilibrium trajectory. Initial interactions between the CRAC336 peptide and the 40% cholesterol bilayer show insertion of not only this central Tyr but also a C-terminal Tyr at the bilayer interface. Near-membrane structures of the CRAC336 peptide near the pure DMPC bilayer frequently display insertion of the N-terminal Phe or C-terminal Tyr; there is no preference for interaction between the central Tyr residue and the membrane (Figure 3). To explore the role of both tyrosine residues in the interation of CRAC336 with the 40% cholesterol membrane, two tyrosine-to-alanine single point mutants, CRAC336 Y10A and CRAC336 Y19A, were also simulated near the 40% cholesterol membrane. The Y10A and Y19A mutants have an average of 2.8 ± 0.7 and 3.8 ± 0.6 residues, respectively, in the summed helical secondary structure versus an average of 0.22 ± 0.09 for the wild-type (WT) CRAC336 peptide, indicating that unlike the WT peptide, neither mutant loses its α-helical propensity near the 40% cholesterol bilayer. Furthermore, density profiles of each system show that the CRAC336 single point mutants sample structures both near the membrane and in the aqueous solution region (Figure 4).
Figure 3.
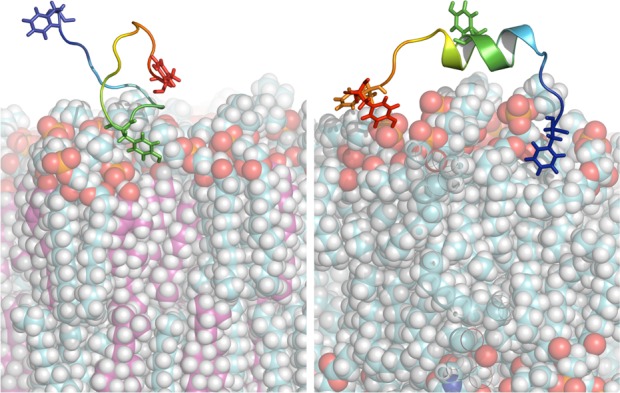
Snapshots from the trajectories of CRAC336 near a DMPC/CHO (left) and pure DMPC bilayer (right). Near the DMPC/CHO membrane, a characteristic interaction takes place between Tyr336 (green) and the membrane. Near the pure DMPC membrane, other residues, including Phe327 (blue), Phe343 (orange), and Tyr345 (red), interact with the membrane more frequently than Tyr336, and helical structures continue to be present during the interaction of the peptide with the membrane. DMPC carbons are cyan; cholesterol carbons are magenta.
Figure 4.
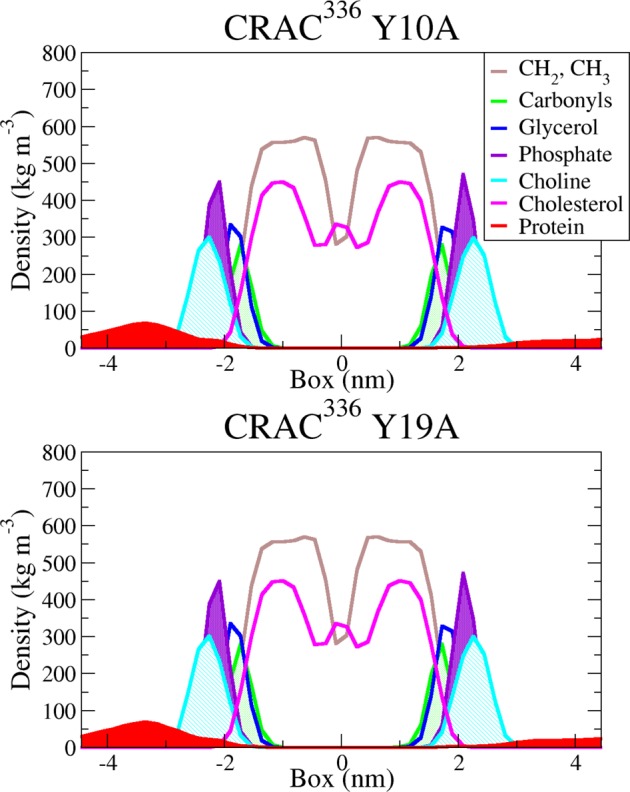
Density profiles along the membrane normal of CRAC336 Y10A and Y19A near the DMPC/CHO bilayer. Peptide density is shown in red; cholesterol density is magenta, and DMPC densities are broken into hydrocarbon, carbonyl, glycerol, phosphate, and choline groups (see legend for color definitions).
Together with the observed interactions between the central Tyr and the cholesterol-bearing membrane and between the terminal Tyr and the pure DMPC membrane, the results of the Y10A and Y19A mutation simulations described above suggest that the central Tyr of the CRAC motif plays a significant role in cholesterol binding. However, the terminal Tyr of CRAC336 may function in stabilizing near-membrane structures, thereby allowing the central Tyr to interact with the membrane. Additionally, the lack of interaction between the central Tyr and the pure DMPC membrane further suggests that the characteristic interaction of the peptide near the 40% cholesterol membrane is cholesterol-specific.
Conclusions
In conclusion, we have used replica-exchange molecular dynamics simulations to compare the effects of lipid bilayer environment on two peptides. Consistent with a previous experimental study,10 our results show a decided preference for the membrane only in the case of CRAC336 near a 40% cholesterol, 60% DMPC bilayer. This preference is not seen for the same peptide near a pure DMPC bilayer or for CRAC503 near either bilayer. Furthermore, our work adds to ongoing experimental work in two key ways: by identifying the role of disorder in cholesterol interactions and by examining the role of the central Tyr in the CRAC motif. As the CRAC336 peptide moves from solution to interact with the 40% cholesterol bilayer, the peptide loses its helical propensity and adopts a random coil conformation. The significance of disorder in protein behavior is becoming more clear; numerous studies have contributed examples of proteins that are disordered in their bound state.29 Our work here highlights the role of disorder in protein–membrane interactions. A single point mutation of the central Tyr, converting it to Ala, resulted in loss of the characteristic behavior of CRAC336 near the 40% cholesterol bilayer, and though the central Tyr of the WT CRAC336 peptide interacts significantly with the cholesterol-bearing bilayer, the same residue does not show the same high propensity for interaction with the cholesterol-devoid bilayer. Furthermore, the context dependence of the functionality of the CRAC domain is demonstrated through a single-point mutation of the CRAC336 peptide outside of the CRAC motif.
Acknowledgments
We thank Giovanni Bussi for providing the necessary scripts to perform Hamiltonian-exchange simulations with Gromacs. This work was supported by National Institutes of Health Grant R00DE022795 (A.C.B.). Use of the high-performance computing capabilities of the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation Grant TG-MCB-120014, is also gratefully acknowledged.
Supporting Information Available
Percolation of replicas of CRAC336 near the 40% bilayer system and cumulative averaging of secondary structure characteristics of CRAC336 near the pure DMPC bilayer system, which were used to determine equilibration time. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Singer S. J.; Nicolson G. L. The Fluid Mosaic Model of the Structure of Cell Membranes. Science 1972, 175, 720–731. [DOI] [PubMed] [Google Scholar]
- Engelman D. M. Membranes are More Mosaic than Fluid. Nature 2005, 438, 578–580. [DOI] [PubMed] [Google Scholar]
- Cho W.; Stahelin R. V. Membrane–Protein Interactions in Cell Signaling and Membrane Trafficking. Annu. Rev. Biophys. Biomol. Struct. 2005, 34, 119–151. [DOI] [PubMed] [Google Scholar]
- Dror R. O.; Jensen M. Ø.; Borhani D. W.; Shaw D. E. Exploring Atomic Resolution Physiology on a Femtosecond to Millisecond Timescale Using Molecular Dynamics Simulations. J. Gen. Physiol. 2010, 135, 555–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyson H. J.; Wright P. E. Coupling of Folding and Binding for Unstructured Proteins. Curr. Opin. Struct. Biol. 2002, 12, 54–60. [DOI] [PubMed] [Google Scholar]
- Uversky V. N. Intrinsically Disordered Proteins and Their Environment: Effects of Strong Denaturants, Temperature, pH, Counter Ions, Membranes, Binding Partners, Osmolytes, and Macromolecular Crowding. Protein J. 2009, 28, 305–325. [DOI] [PubMed] [Google Scholar]
- Morriss-Andrews A.; Shea J.-E. Simulations of Protein Aggregation: Insights from Atomistic and Coarse-Grained Models. J. Phys. Chem. Lett. 2014, 5, 1899–1908. [DOI] [PubMed] [Google Scholar]
- Jakob U.; Kriwacki R.; Uversky V. N. Conditionally and Transiently Disordered Proteins: Awakening Cryptic Disorder To Regulate Protein Function. Chem. Rev. 2014, 114, 6779–6805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.; Papadopoulos V. Peripheral-Type Benzodiazepine Receptor Function in Cholesterol Transport. Identification of a Putative Cholesterol Recognition/Interaction Amino Acid Sequence and Consensus Pattern 1. Endocrinology 1998, 139, 4991–4997. [DOI] [PubMed] [Google Scholar]
- Brown A. C.; Balashova N. V.; Epand R. M.; Epand R. F.; Bragin A.; Kachlany S. C.; Walters M. J.; Du Y.; Boesze-Battaglia K.; Lally E. T. Aggregatibacter actinomycetemcomitans Leukotoxin Utilizes a Cholesterol Recognition/Amino Acid Consensus Site for Membrane Association. J. Biol. Chem. 2013, 288, 23607–23621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess B.; Kutzner C.; Van Der Spoel D.; Lindahl E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [DOI] [PubMed] [Google Scholar]
- Best R. B.; Hummer G. Optimized Molecular Dynamics Force Fields Applied to the Helix–Coil Transition of Polypeptides. J. Phys. Chem. B 2009, 113, 9004–9015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar]
- Sugita Y.; Okamoto Y. Replica-Exchange Molecular Dynamics Method for Protein Folding. Chem. Phys. Lett. 1999, 314, 141–151. [Google Scholar]
- Essmann U.; Perera L.; Berkowitz M. L.; Darden T.; Lee H.; Pedersen L. G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar]
- Knott M.; Best R. B. A Preformed Binding Interface in the Unbound Ensemble of an Intrinsically Disordered Protein: Evidence from Molecular Simulations. PLoS Comput. Biol. 2012, 8, e1002605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ensign D. L.; Kasson P. M.; Pande V. S. Heterogeneity Even at the Speed Limit of Folding: Large-Scale Molecular Dynamics Study of a Fast-Folding Variant of the Villin Headpiece. J. Mol. Biol. 2007, 374, 806–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P.; Kim B.; Friesner R. A.; Berne B. Replica Exchange with Solute Tempering: A Method for Sampling Biological Systems in Explicit Water. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 13749–13754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Friesner R. A.; Berne B. Replica Exchange with Solute Scaling: A More Efficient Version of Replica Exchange with Solute Tempering (REST2). J. Phys. Chem. B 2011, 115, 9431–9438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussi G. Hamiltonian Replica Exchange in GROMACS: A Flexible Implementation. Mol. Phys. 2014, 112, 379–384. [Google Scholar]
- Bonomi M.; Branduardi D.; Bussi G.; Camilloni C.; Provasi D.; Raiteri P.; Donadio D.; Marinelli F.; Pietrucci F.; Broglia R. A.; et al. PLUMED: A Portable Plugin for Free-Energy Calculations with Molecular Dynamics. Comput. Phys. Commun. 2009, 180, 1961–1972. [Google Scholar]
- Best R. B.; Mittal J. Balance Between α and β Structures in Ab Initio Protein Folding. J. Phys. Chem. B 2010, 114, 8790–8798. [DOI] [PubMed] [Google Scholar]
- Mittal J.; Best R. B. Tackling Force-Field Bias in Protein Folding Simulations: Folding of Villin HP35 and Pin WW Domains in Explicit Water. Biophys. J. 2010, 99, L26–L28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jämbeck J. P.; Lyubartsev A. P. Derivation and Systematic Validation of a Refined All-Atom Force Field for Phosphatidylcholine Lipids. J. Phys. Chem. B 2012, 116, 3164–3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jämbeck J. P.; Lyubartsev A. P. An Extension and Further Validation of an All-Atomistic Force Field for Biological Membranes. J. Chem. Theory Comput. 2012, 8, 2938–2948. [DOI] [PubMed] [Google Scholar]
- Jämbeck J. P.; Lyubartsev A. P. Another Piece of the Membrane Puzzle: Extending Slipids Further. J. Chem. Theory Comput. 2013, 9, 774–784. [DOI] [PubMed] [Google Scholar]
- Parrinello M.; Rahman A. Polymorphic Transitions in Single Crystals: A New Molecular Dynamics Method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar]
- Kabsch W.; Sander C. Dictionary of Protein Secondary Structure: Pattern Recognition of Hydrogen-Bonded and Geometrical Features. Biopolymers 1983, 22, 2577–2637. [DOI] [PubMed] [Google Scholar]
- van der Lee R.; Buljan M.; Lang B.; Weatheritt R. J.; Daughdrill G. W.; Dunker A. K.; Fuxreiter M.; Gough J.; Gsponer J.; Jones D. T.; et al. Classification of Intrinsically Disordered Regions and Proteins. Chem. Rev. 2014, 114, 6589–6631. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.