

Abstract
Non-selective inhibition of histone deacetylases (HDACs), enzymes that remove acetyl groups from histone core proteins, enhances cognition and NMDAR-dependent long-term potentiation at hippocampal CA3-CA1 synapses. It is not known whether this is a general mechanism by which HDACs modulate plasticity at other hippocampal synapses. Furthermore, it has yet to be tested whether HDAC inhibition can reverse deficits in synaptic plasticity in disease models. Here, we investigated whether inhibition of HDACs, and specifically HDAC3, a class I HDAC isoform known to negatively regulate hippocampus-dependent learning and memory, enhances LTP at medial perforant path-dentate granule cell (MPP-DGC) synapses in wild-type and Fragile X (Fmr1-/y) mice, a model with known LTP deficits at this synapse. The non-selective HDAC inhibitor trichostatin A (TSA) significantly increased the magnitude of LTP at MPP-DGC synapses in wild-type mice, similar to reports at CA3-CA1 synapses. The enhancement of LTP was mimicked by selective HDAC3 inhibition, implicating a role for this isoform in the negative regulation of synaptic plasticity. However, HDAC3 inhibition was completely ineffective in reversing the deficit in LTP at MPP-DGC synapses in slices from Fmr1-/y mice, and in fact, HDAC3 inhibiton was unable to induce any improvement whatsoever. These findings indicate that the enhancing effect of HDAC3 inhibition on LTP in wild-type mice requires FMRP, revealing a novel role for FMRP in hippocampal plasticity.
Keywords: Mental retardation, plasticity, excitatory transmission, hippocampus
1. Introduction
Histone deacetylases (HDACs), enzymes that encourage chromatin condensation by removing acetyl groups from histones, decrease transcription of genes required for memory formation (Kouzarides, 2007). Previous studies demonstrated that non-selective HDAC inhibition enhances NMDAR-dependent long-term potentiation (LTP) at hippocampal CA3-CA1 synapses and hippocampus-dependent learning and memory in wild-type (WT) mice (Levenson et al., 2004; Vecsey et al., 2007; Stefanko et al., 2009). HDAC inhibition also improves learning and memory in an Alzheimer’s disease mouse model and in mice with p25-induced neurodegeneration (Fischer et al., 2007; Kilgore et al., 2010). Although these findings support the therapeutic potential of HDAC inhibitors in treating cognitive deficiencies, their non-specific effects limit clinical use (Tsankova et al., 2006; Bruserud et al., 2007; Renthal et al., 2007), motivating studies to determine which HDAC isoforms can be selectively targeted to improve memory function.
To-date, three Class I HDAC isoforms have been investigated and two have been identified as negative regulators of memory formation. Over-expression of HDAC2, but not HDAC1, impairs LTP at CA3-CA1 synapses associated with deficits in contextual fear conditioning and Morris water maze, while synaptic plasticity and memory are facilitated in HDAC2 knockout mice (Guan et al., 2009). Genetic deletion or selective pharmacological inhibition of HDAC3, the isoform most highly expressed in hippocampus (Broide et al., 2007), enhances object recognition and location memory (McQuown et al., 2011), but whether the behavioral improvements are associated with increased LTP was not explored.
The beneficial effects of HDAC inhibition on synaptic plasticity have been studied primarily at hippocampal CA3-CA1 synapses in WT mice. Therefore, it is not known whether HDAC inhibitors increase LTP at other hippocampal synapses or in animals with known deficits in hippocampal plasticity. In a mouse model of Fragile X Syndrome (Fmr1-/y mice), the most common inherited form of mental retardation (Bakker, 1994), loss of Fragile X Mental Retardation Protein (FMRP) causes a deficit in NMDAR-dependent LTP at medial perforant path-dentate-gyrus granule cell (MPP-DGC) synapses that is accompanied by impaired dentate gyrus associated cognitive tasks (Yun and Trommer, 2011; Eadie et al., 2012; Franklin et al., 2014). The LTP deficit can be reversed by acute application of glycine or D-serine (Bostrom et al., 2013) in brain slice recordings or by selective inhibition of glycogen synthase kinase-3 (GSK3) (Franklin et al., 2014). Acute GSK3 inhibition in vivo also reverses dentate gyrus specific behavioral deficits (Franklin et al., 2014), mechanistically linking impaired LTP and behavior. Here, we sought to determine whether HDAC3 inhibition increases the LTP magnitude at MPP-DGC synapses in WT mice, and whether it also reverses the LTP deficit at these synapses in Frm1-/y mice, with hopes of identifying a novel therapeutic target to treat cognitive impairment in Fragile X Syndrome (FXS).
2. Materials and Methods
2.1 Animals
Adult male wild-type (WT) C57Bl/6 mice and Fmr1-/y mice (two-six months of age) on a pure C57Bl/6 background were used in all experiments. Male (Fmr1 -/y) and female (Fmr1 -/x) mice were mated to generate Fmr1-/y and WT littermates. Mice were housed on a 12h light/dark cycle in temperature controlled rooms. The care and use of all mice followed an approved protocol by the University of Alabama at Birmingham Institutional Animal Care and Use Committee and in accordance National Institutes of Health guidelines.
2.2 Hippocampal slice preparation
Mice were decapitated following isoflurane anesthesia and brains were rapidly removed and placed in modified ice-cold artificial cerebrospinal fluid (aCSF) [in mM: 85 NaCl, 2.5 KCl, 4 MgSO4, 0.5 CaCl2, 1.25 NaH2PO4, 25 NaHCO3, 25 glucose and 75 sucrose saturated in 95% O2 and 5% CO2]. Coronal slices (400uM) of dorsal hippocampus (defined as Bregma −1.46mm to −2.46; Franklin and Paxinos atlas) were prepared using a vibratome (Vibratome 1000 Plus; St. Louis MO) then immediately transferred into a holding chamber containing standard aCSF [in mM: 124 NaCl, 3 KCl, 1 MgSO4, 2 CaCl2, 1.25 NaH2PO4, 26 NaHCO3, 15 glucose saturated in 95% O2 and 5% CO2] where the slices remained until experimentation or drug treatment. In experiments where pre-incubation of drug was required, slices were stored in standard aCSF for 30 min following slicing then transferred to a holding chamber that contained standard aCSF with either DMSO or RGFP966. All slices, regardless of treatment, rested at least 1 hr prior to recording.
2.3 Electrophysiology
Brain slice electrophysiology experiments were performed as previously reported (Franklin et al., 2014). Briefly, for extracellular field dendritic field potential (fEPSP) recordings, slices were transferred to a submersion chamber and perfused with standard aCSF at 26–28° F. Baseline fEPSPs were generated by stimulating the medial perforant path input onto dentate granule cell synapses (0.1 Hz, 200uS) (MPP-DGC). Correct electrode placement was confirmed visually and by the presence of paired-pulse depression characteristic of MPP-DGC synapses (McNaughton 1980, Colino and Malenka 1993), through the duration of the experiment. LTP was induced using high-frequency stimulation (HFS, 100 Hz, 1 s duration × 4, 60s interval). All recordings were performed in the presence of the GABAAR antagonist picrotoxin (100mM) so that fEPSPs could be measured in isolation, as previously reported (Franklin et al., 2014).
2.4 Reagents
Trichostatin A (TSA) (Tocris Bioscience; Ellisville, MO) was dissolved in DMSO and used at a final concentration of 1.65uM (Levenson et al., 2004). RGFP966 (generous gift from Repligen Corp) is an HDAC3 inhibitor with an IC50 of 0.8uM and is specific to HDAC3 up to 15uM (Malvaez et al., 2013). It was dissolved in DMSO and used at a final concentration of 10 uM in all experiments.
2.5 Statistics
Data are expressed as mean ± SEM. Significant differences were determined using Student’s t tests at P< 0.05. n represents animal number. For each experiment, at least 6 animals were used. Data were included only when both control and drug experiments were successfully completed from an individual animal, except when LTP was compared between WT and Fmr1-/y mice. In this case all control experiments were included (Figure 3A). Experiments were discarded from the data set if there was >10% change in fEPSP slope during baseline recording.
Figure 3.
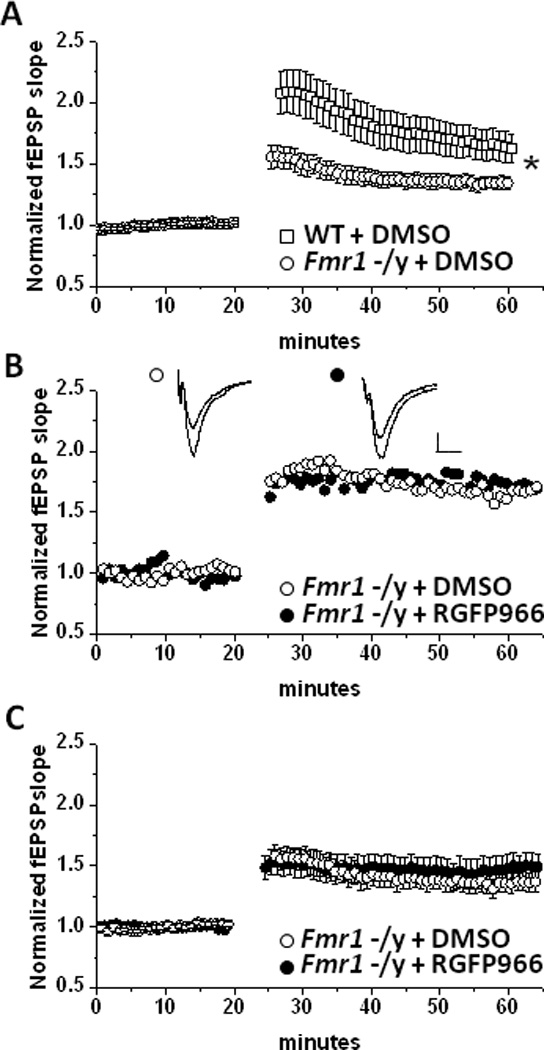
Selective HDAC3 inhibition does not enhance LTP at MPP-DGC Synapses in Frm1-/y mice. (A) Summary plot of the average LTP magnitude induced by HFS at MPP-DGC synapses in slices from WT (n=15) or Fmr1 -/y (n=12). (B) Single representative experiment and (C) summary plot of the average LTP magnitude induced by HFS at MPP-DGC synapses in slices from Fmr1-/y mice treated with DMSO (n=6) or RGFP966 (n=6). n represents animal number. Scale bar 0.2mV/100u.
3. Results
3.1 TSA LTP results
We sought to determine whether HDAC inhibition increases the LTP magnitude at MPP-DGC synapses similarly to what has been previously reported at CA3-CA1 synapses in acute slices from adult WT mice (Levenson et al., 2004; Guan et al., 2009). Extracellular dendritic fEPSPs were recorded at MPP-DGC synapses in acute slices from adult WT mice. LTP was induced using high frequency stimulation in the presence or absence of TSA, a non-selective HDAC inhibitor. Slices were continuously perfused with either DMSO (vehicle) or TSA for 1 hr prior to delivering HFS to induce LTP. We found that in the presence of TSA the magnitude of LTP at MPP-DGC synapses is significantly increased compared to DMSO alone (DMSO: 136±2% of baseline fEPSP slope vs TSA: 162±5 % of baseline fEPSP slope; p=.01) (Figure 1A–1B).
Figure 1.
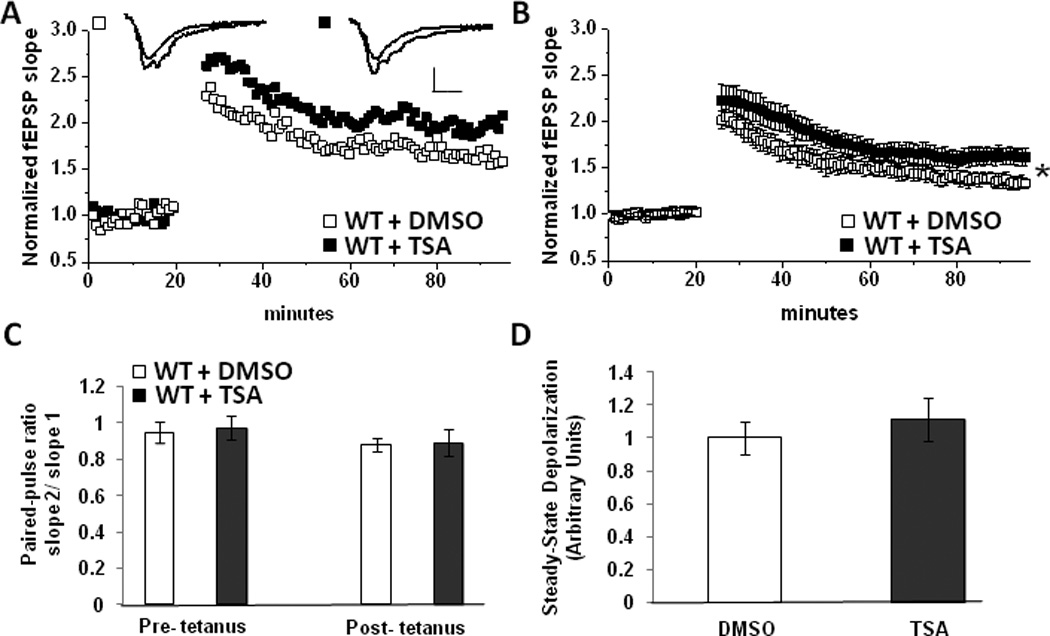
Non-Selective HDAC inhibition enhances LTP at MPP-DGC Synapses. (A) Single representative experiment and (B) summary plot of the average LTP magnitude at MPP-DGC synapses in WT slices treated with DMSO (n=7) or TSA (n=7) n represents animal number. Scale bar 0.2mV/100uS (C) Average PPR in DMSO and TSA treated groups (from experiments in B) are not significantly different. (D) No difference in depolarization during HFS between DMSO and TSA treated groups (from experiments in B). (*p<0.05 Student’s t test)
3.2 TSA paired-pulse and steady state depolarization analysis
We next assessed whether the enhanced LTP magnitude in TSA is associated with an increase in presynaptic neurotransmitter release. To this end, we measured the paired-pulse ratio (PPR), an indirect measure of presynaptic release probability, in DMSO and TSA treated slices pre- and post-tetanus and found significant differences between groups (pre-HFS, DMSO: 0.95± .05 vs TSA: 0.97±.06; p=.38; post-HFS, DMSO: 0.87± .03 vs TSA: 0.89±.067; p=.44) (Figure 1C). This suggests that an increase in neurotransmitter release is likely not responsible for the enhanced LTP magnitude. We also found no difference in the magnitude of the depolarization during HFS when comparing DMSO and TSA groups (arbitrary units, DMSO: 1.0± .09 vs TSA: 1.11±.13; p=.26) (Figure 1D). Taken together these results suggest a likely post-synaptic mechanism for the enhanced magnitude of LTP following HDAC inhibition.
3.3 RGFP966 LTP, paired-pulse, and steady state depolarization results and analysis
Given that selective pharmacological inhibition of HDAC3 improves object recognition and location memory (McQuown et al., 2011), behaviors requiring the dentate gyrus (Goodrich-Hunsaker et al., 2008), we next tested whether selective HDAC3 inhibition would increase the LTP magnitude at MPP-DGC synapses, mimicking the effect of the non-selective HDAC inhibitor TSA. To accomplish this, slices were incubated for at least 3 hr in DMSO (vehicle) or 10 uM RGFP966 prior to inducing LTP with HFS. We found that RGFP966 robustly enhanced the LTP magnitude at MPP-DGC synapses (DMSO: 152±23% of baseline fEPSP slope vs RGFP966: 216±91% of baseline fEPSP slope; p=.005) (Figure 2A–2B). However, in contrast to TSA where significant potentiation does not occur until 45 min post-tetanus (DMSO: 142±33% of baseline fEPSP slope vs TSA: 164±7 % of baseline fEPSP slope; p=.03) (Figure 1B), with selective HDAC3 inhibition, significant potentiation occurred as soon as 10 min post-tetanus (DMSO: 176±35% of baseline fEPSP slope vs RGFP966: 244±181% of baseline fEPSP slope; p=.046). Similar to TSA, there were no differences in PPR (pre-HFS, DMSO: 0.95± .004 vs RGFP966: 0.98±.03; p=.30; post-HFS, DMSO: 0.93± .02 vs RGFP: 1.12±.44; p=.19) (Figure 2C) or depolarization during HFS between the DMSO and RGFP966 groups (in arbitrary units, DMSO: 1.0±.13 vs TSA: 0.8±.15; p=.29) (Figure 2D).
Figure 2.
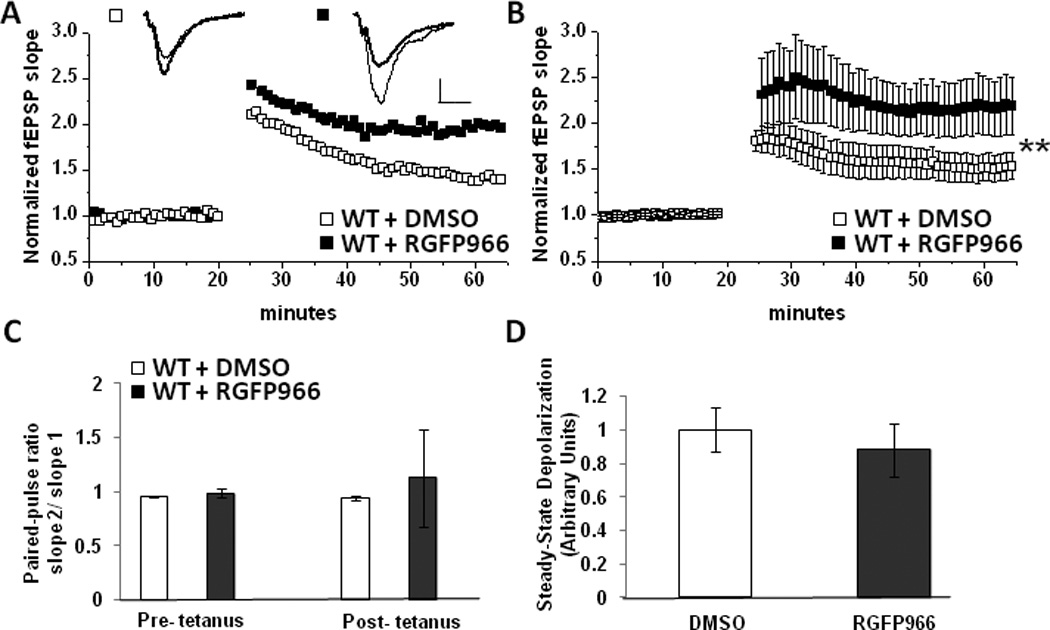
Selective HDAC3 inhibition enhances LTP at MPP-DGC Synapses. (A) Single representative experiment and (B) summary plot of the average LTP magnitude at MPP-DGC synapses in WT slices treated with DMSO (n=10) or RGFP966 (n=10). n represents animal number. Scale bar 0.2mV/100uS (C) Average PPR in DMSO and TSA treated groups (from experiments in B) are not significantly different. (D) No difference in depolarization during HFS between DMSO and TSA treated groups (from experiments in B). (**p<0.01 Student’s t test)
3.4 RGFP966 LTP results in Fmr1 -/y mice
Because of the robust effects of HDAC3 inhibition on the LTP magnitude at MPP-DGC synapses in WT mice, we next sought to determine whether the inhibitor would rescue deficits in LTP at MPP-DGC synapses in Fmr1-/y mice. First, we confirmed that the magnitude of LTP at MPP-DGC synapses is impaired in slices from Fmr1-/y mice (WT: 163±21% of baseline fEPSP slope vs Fmr1-/y: 135±3% of baseline fEPSPs slope; p=0.03) (Figure 3A), consistent with our previously published findings (Franklin et al., 2014). Slices from a subset of the Fmr1- /y mice (n=8/12 mice) recorded in the data set shown in Figure 3A were incubated for at least 3 hrs in either DMSO or RGFP966, consistent with the protocol used in experiments with slices from WT mice, and were recorded interleaved. Surprisingly, HDAC3 inhibition with RGFP966 was completely ineffective at increasing the LTP magnitude at MPP-DGC synapses in Fmr1-/y mice (DMSO: 135±3% of baseline fEPSP slope vs RGFP966: 146±7% of baseline fEPSPs slope; p=.24) (Figure 3B,C).
4. Discussion
Here we report that inhibition of class I HDACs, and more specifically, that inhibition of HDAC3, enhances the LTP magnitude at MPP-DGC synapses in adult WT mice. This finding confirms that modulation of LTP by HDACs is not selective for CA3-CA1 synapses, but also occurs at least at one other synapse in the hippocampal formation. Moreover, this finding raises the question as to whether the increase in LTP magnitude following HDAC inhibition is a general mechanism impacting many, if not all, excitatory synapses throughout the CNS. These findings are also consistent with the interpretation that the increase in LTP magnitude at MPP-DGC synapses we observed following HDAC3 inhibition could be mechanistically linked to the previously reported improved object recognition and location memory in WT mice following pharmacological HDAC3 inhibition, because these behaviors require the dentate gyrus (Goodrich-Hunsaker et al., 2008).
While our finding that HDAC3 inhibition is unable to enhance plasticity in Fmr1-/y mice suggests HDAC inhibitors may not be useful treatment options in Fragile X Syndrome, these results are exciting in that they provide unique insight into a potential mechanism for enhanced plasticity at excitatory synapses in WT mice. Previous studies have demonstrated that the cAMP response element binding protein (CREB) is required for the enhancement of LTP following HDAC inhibition (Vecsey et al., 2007). Interestingly, cAMP production is significantly decreased in platelets from FXS patients (Berry-Kravis and Sklena, 1993) and induced cAMP levels are reduced in brain of Fmr1 y/- mice, human neural cells from FXS patients and in the dmfr1 mutant fly (Kelley et al., 2007), a model of FXS which results in impaired CREB output (Dockendorff et al., 2002). Taken together, this suggests that HDAC inhibition might fail to enhance LTP in Fmr1 -/y mice due to impaired cAMP/CREB function. Additionally, FMRP is an RNA–binding protein known to suppress translation of proteins, many of which are involved in synaptic plasticity (Ashley et al., 1993; Siomi et al., 1993; Feng et al., 1997; Brown et al., 2001; Laggerbauer et al., 2001; Todd et al., 2003; Zalfa et al., 2003; Khandjian et al., 2004; Lu et al., 2004; Weiler et al., 2004). Interestingly, the Fmr1 promoter region contains binding sites for CREB as well as Nrf2 (Smith et al., 2006), a transcription factor activated following HDAC inhibition (Wang et al., 2012). Therefore it is possible that HDAC inhibition modulates LTP by increasing FMRP transcription through CREB and Nrf2 activation. Subsequently, increased FMRP could contribute to enhanced LTP by preventing translation of proteins that normally constrain LTP expression. When FMRP is absent, the brake on translation of “LTP suppressing” proteins is removed and LTP is no longer enhanced.
An interesting observation is that selective HDAC3 inhibition induces significant potentiation as early as 10 min post-tetanus, while significant potentiation does not occur until 45 min post-tetanus using the non-selective HDAC inhibitor TSA. One possible explanation for the difference in timing is that some HDAC isoforms may act to positively regulate LTP, such as HDAC4 (Kim et al., 2012). Perhaps the delayed onset of the increase in LTP magnitude after TSA is the net result of inhibition of both negative and positive regulators of early plasticity. However, when RGFP966 is applied, only HDAC3 is inhibited, therefore there are no opposing effects from other HDACs and LTP is robustly enhanced immediately after induction.
In summary, our data demonstrate that HDACs, specifically HDAC3, are negative regulators of synaptic plasticity at synapses in dentate gyrus. While selective inhibition of HDAC3 is not beneficial for LTP deficits in a mouse model of Fragile X Syndrome due to the requirement of FMRP to enhance plasticity, our data do not rule out the possibility of using HDAC3 as a therapeutic target in other cognitive disorders where MPP-DGC synapses are affected, like the Ts65Dn model of Down’s syndrome (Kleschevnikov et al., 2004). Furthermore, the question of whether enhanced LTP is an absolute requirement for reversal of cognitive impairments in Fmr1-/y mice remains. To date only CREB (Vecsey et al., 2007) and NR4A (Bridi and Abel, 2013) have been identified as required molecules for the enhancing effects of HDAC inhibition on LTP. We have discovered a crucial role for FMRP in HDAC3 modulation of LTP however future studies are necessary to dissect the precise mechanism by which HDAC3 and FMRP interact.
Acknowledgements
This work was supported by awards from the NIMH (F31MH097362), NINDS (T32 NS061788), University of Alabama at Birmingham Civitan Emerging Scholars Fellowship, and the Howard Hughes Medical Institute by a grant awarded to the University of Alabama at Birmingham. We thank Dr. J. David Sweatt for helpful comments on the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributions:
Aimee V. Franklin designed research, performed research, analyzed data, and wrote the paper.
James R. Rusche designed research.
Lori L. McMahon designed research and wrote the paper.
Conflict of Interest: J.R.R. is employed by Repligen Corp. and provided RGFP966 free of charge. No financial support was provided by Repligen Corp. to carry out the proposed research. A.V.F. and L.L.M declare no competing financial interests
References
- Ashley CT, Jr, Wilkinson KD, Reines D, Warren ST. FMR1 protein: conserved RNP family domains and selective RNA binding. Science. 1993;262:563–566. doi: 10.1126/science.7692601. [DOI] [PubMed] [Google Scholar]
- Bakker DB. Fmr1 knockout mice: a model to study fragile X mental retardation. The Dutch-Belgian Fragile X Consortium. Cell. 1994;78:23–33. [PubMed] [Google Scholar]
- Berry-Kravis E, Sklena P. Demonstration of abnormal cyclic AMP production in platelets from patients with fragile X syndrome. Am J Med Genet. 1993;45:81–87. doi: 10.1002/ajmg.1320450120. [DOI] [PubMed] [Google Scholar]
- Bostrom CA, Majaess NM, Morch K, White E, Eadie BD, Christie BR. Rescue of NMDAR-Dependent Synaptic Plasticity in Fmr1 Knock-Out Mice. Cerebral cortex. 2013 doi: 10.1093/cercor/bht237. [DOI] [PubMed] [Google Scholar]
- Bridi MS, Abel T. The NR4A orphan nuclear receptors mediate transcription-dependent hippocampal synaptic plasticity. Neurobiol Learn Mem. 2013;105:151–158. doi: 10.1016/j.nlm.2013.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broide RS, Redwine JM, Aftahi N, Young W, Bloom FE, Winrow CJ. Distribution of histone deacetylases 1–11 in the rat brain. J Mol Neurosci. 2007;31:47–58. doi: 10.1007/BF02686117. [DOI] [PubMed] [Google Scholar]
- Brown V, Jin P, Ceman S, Darnell JC, O'Donnell WT, Tenenbaum SA, Jin X, Feng Y, Wilkinson KD, Keene JD, Darnell RB, Warren ST. Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell. 2001;107:477–487. doi: 10.1016/s0092-8674(01)00568-2. [DOI] [PubMed] [Google Scholar]
- Bruserud O, Stapnes C, Ersvaer E, Gjertsen BT, Ryningen A. Histone deacetylase inhibitors in cancer treatment: a review of the clinical toxicity and the modulation of gene expression in cancer cell. Curr Pharm Biotechnol. 2007;8:388–400. doi: 10.2174/138920107783018417. [DOI] [PubMed] [Google Scholar]
- Dockendorff TC, Su HS, McBride SM, Yang Z, Choi CH, Siwicki KK, Sehgal A, Jongens TA. Drosophila lacking dfmr1 activity show defects in circadian output and fail to maintain courtship interest. Neuron. 2002;34:973–984. doi: 10.1016/s0896-6273(02)00724-9. [DOI] [PubMed] [Google Scholar]
- Eadie BD, Cushman J, Kannangara TS, Fanselow MS, Christie BR. NMDA receptor hypofunction in the dentate gyrus and impaired context discrimination in adult Fmr1 knockout mice. Hippocampus. 2012;22:241–254. doi: 10.1002/hipo.20890. [DOI] [PubMed] [Google Scholar]
- Feng Y, Absher D, Eberhart DE, Brown V, Malter HE, Warren ST. FMRP associates with polyribosomes as an mRNP, and the I304N mutation of severe fragile X syndrome abolishes this association. Mol Cell. 1997;1:109–118. doi: 10.1016/s1097-2765(00)80012-x. [DOI] [PubMed] [Google Scholar]
- Fischer A, Sananbenesi F, Wang X, Dobbin M, Tsai LH. Recovery of learning and memory is associated with chromatin remodelling. Nature. 2007;447:178–182. doi: 10.1038/nature05772. [DOI] [PubMed] [Google Scholar]
- Franklin AV, King MK, Palomo V, Martinez A, McMahon LL, Jope RS. Glycogen Synthase Kinase-3 Inhibitors Reverse Deficits in Long-term Potentiation and Cognition in Fragile X Mice. Biol Psychiatry. 2014;75:198–206. doi: 10.1016/j.biopsych.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodrich-Hunsaker NJ, Hunsaker MR, Kesner RP. The interactions and dissociations of the dorsal hippocampus subregions: how the dentate gyrus, CA3, and CA1 process spatial information. Behavioral neuroscience. 2008;122:16–26. doi: 10.1037/0735-7044.122.1.16. [DOI] [PubMed] [Google Scholar]
- Guan JS, Haggarty SJ, Giacometti E, Dannenberg JH, Joseph N, Gao J, Nieland TJ, Zhou Y, Wang X, Mazitschek R, Bradner JE, DePinho RA, Jaenisch R, Tsai LH. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature. 2009;459:55–60. doi: 10.1038/nature07925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley DJ, Davidson RJ, Elliott JL, Lahvis GP, Yin JC, Bhattacharyya A. The cyclic AMP cascade is altered in the fragile X nervous system. PLoS One. 2007;2:e931. doi: 10.1371/journal.pone.0000931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khandjian EW, Huot ME, Tremblay S, Davidovic L, Mazroui R, Bardoni B. Biochemical evidence for the association of fragile X mental retardation protein with brain polyribosomal ribonucleoparticles. Proc Natl Acad Sci U S A. 2004;101:13357–13362. doi: 10.1073/pnas.0405398101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilgore M, Miller CA, Fass DM, Hennig KM, Haggarty SJ, Sweatt JD, Rumbaugh G. Inhibitors of class 1 histone deacetylases reverse contextual memory deficits in a mouse model of Alzheimer's disease. Neuropsychopharmacology. 2010;35:870–880. doi: 10.1038/npp.2009.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MS, Akhtar MW, Adachi M, Mahgoub M, Bassel-Duby R, Kavalali ET, Olson EN, Monteggia LM. An essential role for histone deacetylase 4 in synaptic plasticity and memory formation. J Neurosci. 2012;32:10879–10886. doi: 10.1523/JNEUROSCI.2089-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleschevnikov AM, Belichenko PV, Villar AJ, Epstein CJ, Malenka RC, Mobley WC. Hippocampal long-term potentiation suppressed by increased inhibition in the Ts65Dn mouse, a genetic model of Down syndrome. J Neurosci. 2004;24:8153–8160. doi: 10.1523/JNEUROSCI.1766-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Laggerbauer B, Ostareck D, Keidel EM, Ostareck-Lederer A, Fischer U. Evidence that fragile X mental retardation protein is a negative regulator of translation. Human molecular genetics. 2001;10:329–338. doi: 10.1093/hmg/10.4.329. [DOI] [PubMed] [Google Scholar]
- Levenson JM, O'Riordan KJ, Brown KD, Trinh MA, Molfese DL, Sweatt JD. Regulation of histone acetylation during memory formation in the hippocampus. J Biol Chem. 2004;279:40545–40559. doi: 10.1074/jbc.M402229200. [DOI] [PubMed] [Google Scholar]
- Lu R, Wang H, Liang Z, Ku L, O'Donnell WT, Li W, Warren ST, Feng Y. The fragile X protein controls microtubule-associated protein 1B translation and microtubule stability in brain neuron development. Proc Natl Acad Sci U S A. 2004;101:15201–15206. doi: 10.1073/pnas.0404995101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malvaez M, McQuown SC, Rogge GA, Astarabadi M, Jacques V, Carreiro S, Rusche JR, Wood MA. HDAC3-selective inhibitor enhances extinction of cocaine-seeking behavior in a persistent manner. Proc Natl Acad Sci U S A. 2013;110:2647–2652. doi: 10.1073/pnas.1213364110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuown SC, Barrett RM, Matheos DP, Post RJ, Rogge GA, Alenghat T, Mullican SE, Jones S, Rusche JR, Lazar MA, Wood MA. HDAC3 is a critical negative regulator of long-term memory formation. J Neurosci. 2011;31:764–774. doi: 10.1523/JNEUROSCI.5052-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renthal W, Maze I, Krishnan V, Covington HE, 3rd, Xiao G, Kumar A, Russo SJ, Graham A, Tsankova N, Kippin TE, Kerstetter KA, Neve RL, Haggarty SJ, McKinsey TA, Bassel-Duby R, Olson EN, Nestler EJ. Histone deacetylase 5 epigenetically controls behavioral adaptations to chronic emotional stimuli. Neuron. 2007;56:517–529. doi: 10.1016/j.neuron.2007.09.032. [DOI] [PubMed] [Google Scholar]
- Siomi H, Siomi MC, Nussbaum RL, Dreyfuss G. The protein product of the fragile X gene, FMR1, has characteristics of an RNA-binding protein. Cell. 1993;74:291–298. doi: 10.1016/0092-8674(93)90420-u. [DOI] [PubMed] [Google Scholar]
- Smith KT, Nicholls RD, Reines D. The gene encoding the fragile X RNA-binding protein is controlled by nuclear respiratory factor 2 and the CREB family of transcription factors. Nucleic Acids Res. 2006;34:1205–1215. doi: 10.1093/nar/gkj521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanko DP, Barrett RM, Ly AR, Reolon GK, Wood MA. Modulation of long-term memory for object recognition via HDAC inhibition. Proc Natl Acad Sci U S A. 2009;106:9447–9452. doi: 10.1073/pnas.0903964106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd PK, Mack KJ, Malter JS. The fragile X mental retardation protein is required for type-I metabotropic glutamate receptor-dependent translation of PSD-95. Proc Natl Acad Sci U S A. 2003;100:14374–14378. doi: 10.1073/pnas.2336265100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsankova NM, Berton O, Renthal W, Kumar A, Neve RL, Nestler EJ. Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat Neurosci. 2006;9:519–525. doi: 10.1038/nn1659. [DOI] [PubMed] [Google Scholar]
- Vecsey CG, Hawk JD, Lattal KM, Stein JM, Fabian SA, Attner MA, Cabrera SM, McDonough CB, Brindle PK, Abel T, Wood MA. Histone deacetylase inhibitors enhance memory and synaptic plasticity via CREB:CBP-dependent transcriptional activation. J Neurosci. 2007;27:6128–6140. doi: 10.1523/JNEUROSCI.0296-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Zhu X, Kim Y, Li J, Huang S, Saleem S, Li RC, Xu Y, Dore S, Cao W. Histone deacetylase inhibition activates transcription factor Nrf2 and protects against cerebral ischemic damage. Free Radic Biol Med. 2012;52:928–936. doi: 10.1016/j.freeradbiomed.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiler IJ, Spangler CC, Klintsova AY, Grossman AW, Kim SH, Bertaina-Anglade V, Khaliq H, de Vries FE, Lambers FA, Hatia F, Base CK, Greenough WT. Fragile X mental retardation protein is necessary for neurotransmitter-activated protein translation at synapses. Proc Natl Acad Sci U S A. 2004;101:17504–17509. doi: 10.1073/pnas.0407533101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun SH, Trommer BL. Fragile X mice: reduced long-term potentiation and N-Methyl-DAspartate receptor-mediated neurotransmission in dentate gyrus. J Neurosci Res. 2011;89:176–182. doi: 10.1002/jnr.22546. [DOI] [PubMed] [Google Scholar]
- Zalfa F, Giorgi M, Primerano B, Moro A, Di Penta A, Reis S, Oostra B, Bagni C. The fragile X syndrome protein FMRP associates with BC1 RNA and regulates the translation of specific mRNAs at synapses. Cell. 2003;112:317–327. doi: 10.1016/s0092-8674(03)00079-5. [DOI] [PubMed] [Google Scholar]