

Abstract
Release of lipopolysaccharide (LPS) endotoxin from Gram negative bacterial membranes triggers macrophages to produce large quantities of cytokines that can lead to septic shock and eventual death. Agents that bind to and neutralize LPS may provide a means to clinically prevent septic shock upon bacterial infection. Previously, we reported the design of antibacterial helix peptide SC4 and β-sheet-forming βpep peptides that neutralize LPS in vitro. We hypothesized that the ability of these and other such peptides to neutralize LPS rested in the common denominator of positively charged amphipathic structure. Here, we describe the design and synthesis of non-peptide, calixarene-based helix/sheet topomimetics that mimic the folded conformations of these peptides in their molecular dimensions, amphipathic surface topology, and compositional properties. From a small library of topomimetics, we identified several compounds that neutralize LPS in the 10−8 M range, making them as effective as bactericidal/permeability increasing (BPI) protein and polymyxin B. In an endotoxemia mouse model, three of the most in vitro effective topomimetics are shown to be at least partially protective against challenges of LPS from different bacterial species. NMR studies provide mechanistic insight by suggesting the site of molecular interaction between topomimetics and the lipid A component of LPS, with binding being mediated by electrostatic and hydrophobic interactions. This research contributes to the development of pharmaceutical agents against endotoxemia and septic shock.
Introduction
A number of diseases result from Gram negative bacterial infection and subsequent release of lipopolysaccharide (LPS) endotoxins from their membranes.1,2 Sepsis and septic shock are systemic complications generally associated with increased levels of LPS in the blood stream. An inflammatory response involving various cell receptors3 (e.gs CD144, the Toll-like receptor 4-MD-2 receptor complex5 and non-CD14 expressing endothelial cells6) and plasma components like cytokines, lipid mediators and reactive oxygen species,7 occurs on exposure to LPS, and this may initiate the cascade to septic shock, organ failure, and ultimately death.8 Standard clinical approaches to this problem are generally aimed at combating the bacterial infection itself via treatment with antibacterial agents, but these themselves may lead to disruption of the very bacterial membranes that release LPS. More recent clinical strategies against sepsis have been focused at targeting specific mediators, primarily cytokines; however, this approach has failed in clinical trials.9 A therapeutic approach that quells LPS stimulation of the inflammatory response at the onset, rather than one that inhibits any individual intermediary mediator or molecular event, may actually be the most effective way to halt the septic shock cascade. In this regard, a therapeutic agent that can bind to and neutralize LPS directly would be highly useful in the clinic. While some bactericidal agents also can neutralize LPS, most are not that active against the endotoxin in vivo. More effective LPS neutralizing agents are clearly needed.
LPS is an integral component of the outer membrane of Gram negative bacteria.10,11 As such, it is composed of hydrophobic, acyl chains at one end, and hydrophilic and negatively charged groups at the other end. Because the chemical structure of LPS is highly variable among species of bacteria,10,12 a generic structure of LPS is illustrated in Figure 1. The lipid A group, which is the most conserved part of LPS from any Gram negative species of bacteria, consists of a poly N- and O-acylated, 4-phosphoglucos-2-amine-[1,6-0]-1-phosphoglucos-2-amine moiety. Bonded to the otherwise free O6 group of one of the glucosamines is an inner core of phosphorylated polysaccharides, followed by an outer core of simpler polysaccharides and an O-specific chain. The outer core and O-specific chain are most highly variable among Gram negative bacterial species, which is one reason why it has been difficult to identify one broad spectrum agent that neutralizes LPS molecules equally well from multiple species of bacteria. The more conserved lipid A moiety is perhaps the best target in this regard.
Figure 1.

LPS structure. A generic structure of lipopolysaccharide (LPS) is shown. The phospholipid “lipid A” moiety is common in LPS from all species of Gram negative bacteria, whereas the polysaccharide moiety is highly variable in LPS from different bacteria.
A number of bactericidal peptides are known to bind to and to neutralize LPS.13 Perhaps the prototypic one is polymyxin B (PmxB), a small cyclic lipopeptide.14 However, due to its high neuro and nephrotoxicity, PmxB is limited to topical application. Other examples of such peptides are the cecropins,15 magainins,16 proline-arginine-rich peptides,17 sapecin,18 tachyplesin,19 the defensins,20,21βpep peptides,22,23 and dodecapeptide SC4.24 More recently, a lactoferrin-based peptide, LF11,25 and an N-acylated form lauryl-LF1126 have been identified. The structures of many of these peptides are known and range from turn/loop to helix to β-sheet. For example, βpep peptides fold as anti-parallel β-sheet sandwiches,27 and tachyplesin,19 as well as anti-bacterial peptide defensins,20,21 form dimeric β-sheets. Even the larger LPS binding protein, bactericidal/permeability increasing (BPI) factor28 forms an amphipathic β-sheet structural motif having a cationic face. On the other hand, the cecropins and magainins are helix-forming peptides,29,30 whereas the sapacins contain both α-helix and β-sheet segments.31 In addition, a number of small, antibiotic peptides based on the structure of PmxB has been designed as short β-hairpins constrained by a disulfide bridge.32
Structural analysis of these peptides can be quite helpful in identifying those molecular features paramount to binding to and neutralizing LPS. Regardless of the molecular structure of a given peptide, the underlying theme for efficient interaction with and binding to LPS is known from many studies to be a net positive charge and high hydrophobicity, usually in the context of an amphipathic structure. Positively charged residues from the peptide presumably promote interaction with negatively charged groups on LPS, i.e. phosphates on the lipid A glucosamines and/or those in the inner core polysaccharide unit, while hydrophobic residues from the peptide interact with acyl chains on lipid A.
Several structural studies of peptides in complex with LPS support this notion and have provided additional insight into the structural origins of peptide-mediated LPS neutralization. In an X-ray crystal of the E. coli iron uptake receptor protein FhuA in complex with an LPS molecule, Ferguson et al33 found a precise spatial arrangement of cationic side chains from a three-stranded antiparallel β-sheet was crucial to bind this LPS. Using NMR spectroscopy, Pristovsek & Kidric34 determined the structure of PmxB in a LPS bound state and concluded that a phenylalanine (F6) side chain and two positively charged, α,γ-diaminobutyric acid groups (Dab 1 and Dab 5) were crucial to binding LPS. From another NMR structural study, Japelj et al25 found that peptide LF11 in the presence of LPS from E. coli serotype 055:B5, folded “in a ‘T-shaped’ arrangement of a hydrophobic core and two clusters of basic residues that match the distance between the two phosphate groups of the lipid A moiety”. All three of these structural studies demonstrate the importance to LPS binding of some specific spatial relationships among both cationic and hydrophobic groups on these peptides.
The present study capitalizes on this recurring theme and uses the NMR structures of βpep peptides22,23 and dodecapeptide SC424 to design a series of non-peptide, calixarene-based compounds that mimic the overall structure of a small unit of helix or β-sheet. This design essentially captures the molecular dimensions and amphipathic surface topology common to all LPS binding peptides. These novel, sheet/helix topomimetics present hydrophobic and positively charged residues in a manner that allows them to effectively bind to and neutralize LPS. We demonstrate here that these topomimetics neutralize LPS in vitro from multiple species of Gram negative bacteria and promote survival of mice challenged with LPS in vivo. The present work contributes to the development of therapeutic agents useful in the clinic against endotoxemia and sepsis.
Results
From the literature, it is evident that molecules that neutralize LPS best have amphipathic character and a net positive charge. With this in mind, we focused our mimetic design around a positively charged, amphipathic structural element with the same molecular dimensions as a few turns of α-helix as in SC424, or as a small stretch of β-sheet from βpep peptides.22,23 These conformational and compositional parameters provide a good working model of a reasonable pharmacophore site for LPS binding. A small segment of β-sheet about 3 residues long on each β-strand or about 2 turns of an α-helix are dimensionally well approximated by the calix[4]arene scaffold, as illustrated in Figure 2 A. Adding various chemical groups (aliphatic hydrophobic groups and hydrophilic amines) onto this calixarene scaffold produces a compound that essentially mimics the molecular dimensions and surface topology of segments of β-sheet or α-helix as in βpep peptides or SC4 described above. From the design perspective, this is why the calixarene scaffold was chosen. From the chemistry prospective, the major criterion was ability and ease of synthesis.
Figure 2.

Molecular design approach. (A) The topological design features influencing our choice of a calixarene scaffold for arraying hydrophobic and hydrophilic substituents, are shown. The folded structures of βpep-25 and SC4 peptides are illustrated to scale with the calix[4]arene scaffold. Chemical structures for calixarene analogs in the topomimetic library are displayed (B–E). These are grouped as (B) tertiary amines, (C) guanidinium, (D) triazole, primary amines, and negatively charged groups, and (E) generic partial cone conformer.
A small library of calixarene-based compounds was then synthesized, whose chemical structures are shown in Figure 2B–E. Representative methods for preparation of several calixarene derivatives are depicted in scheme 1. Most compounds in this library are in the cone conformation (e.g. 2), in which the four aryl units of the calixarene scaffold are all oriented in the same direction. Only two compounds (4 and 18) are partial cone conformers, analogous to compounds 3 and 17, respectively, where one of the four aromatic groups and its substituents are flipped about the methylene bridge (cf. a generic partial cone conformer, Figure 2E). In addition, calix[4]arenes have a broader and a narrower face due to positioning of the methylene bridges between aryl groups. For synthetic reasons, most of our analogs have hydrophobic groups on the broader face, meta to the bridging methylenes. One exception is compound 8, which was synthesized specifically to assess whether switching the phobicity of the two faces affects activity. As will be evident below, we synthesized most of our compounds with hydrophobic groups on the broader face of calixarene because 8 was relatively so ineffective at neutralizing LPS.
Scheme 1.

(A) synthesis of tertiary amine calixarene derivatives 1 and 5. (B) synthesis of guanidine calixarene derivatives 13 and 14 and primary amine calixarene derivative 16. (C) synthesis of triazole linked primary amine calixarene derivatives 17 and 18. Reaction conditions: a) AlCl3, PhOH, toluene, rt; b) ethyl bromoacetate, K2CO3, acetone, reflux; c) N,N-dimethylethylenediamine, toluene, reflux; d) i. NaH, 3-chloro-2-methylpropene, THF, DMF, 80 °C; ii. N,N-dimethylaniline, 200 °C; e) Pd/C, H2, 1atm, EtOAc, rt; f) 4-bromobutyronitrile, K2CO3, acetone, reflux; g) 4-bromobutyronitrile, NaH, DMF, 75 °C; h) NaBH4, CoCl2, MeOH; i) 1,3-bis(tert-butoxycarbonyl)-2-methyl-2-thiopseudourea, HgCl2, Et3N, CH2Cl2; j) TEA, 5% anisole in CH2Cl2, rt; k) NaH, propargyl bromide, THF, DMF, reflux; 1) N3(CH2)2NHBoc, ascorbic acid, NaOAc, CuSO4, t-BuOH, H2O, THF; m) TFA, 5% anisole in CH2Cl2, 0 °C to rt.
Moreover, calix[4]arenes can exist in four topological isomers (4-up, 3-up/1-down, and two 2-up/2-down) that can interconvert, in principle, by rotation of the oxygenated “head” of each ring through the core.35 However, calix[4]arene derivatives bearing groups larger than ethoxy on the lower, more narrow rim (which includes all those studied here) are essentially inert with respect to this topological change on the laboratory/pharmacology time scale. Even for derivatives unsubstituted para to the oxygenated carbon, rotation of the larger “tail” of the ring through the core is too slow to be of consequence.36 In this regard, those calixarene compounds that we synthesized to be amphipathic, remain so and do not interconvert.
Initially, we screened our topomimetic library in vitro for the ability of compounds to neutralize E. coli 055:B5 LPS at 10 μM. Full dose response curves were then generated for those compounds that neutralized LPS by 50% or greater. The same approach was used for other species of LPS tested. We found several members from our library to be effective at binding to and neutralizing LPS from various species of bacteria. A few dose response curves for some of these analogs are shown in Figure 3, and IC50 values for all analogs determined by sigmoidal curve fitting of dose responses are listed in Table 1.
Figure 3.
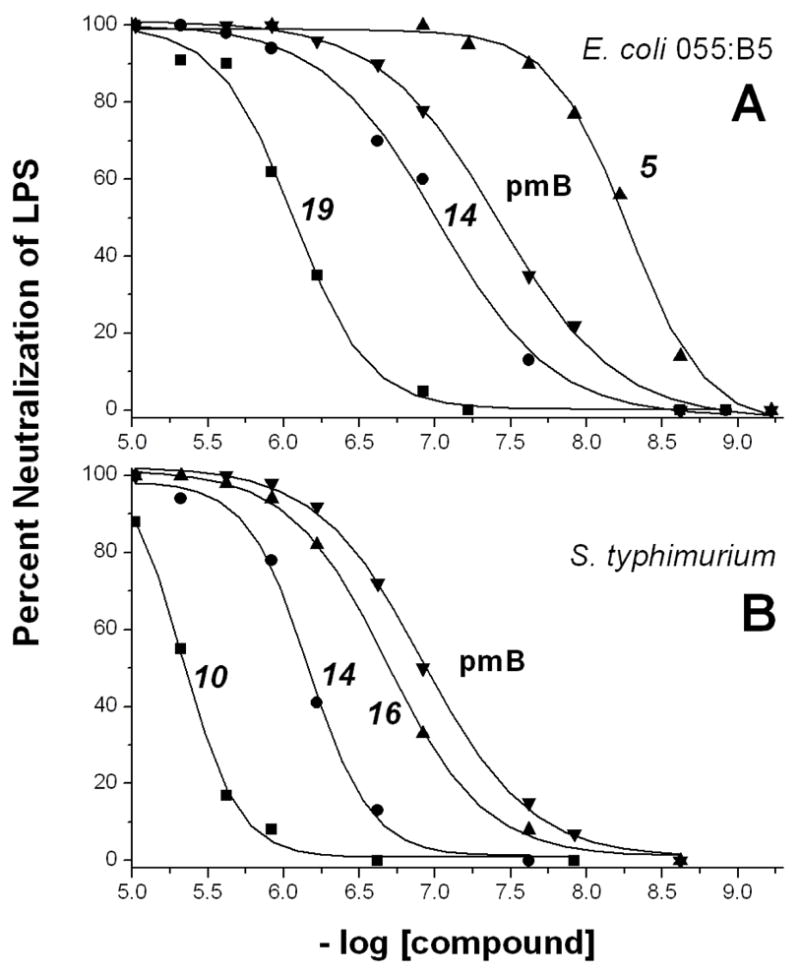
Examples of dose response curves for endotoxin neutralization. Several dose response curves for LPS neutralization are shown for some of the sheet/helix mimetics listed in Table 1. Symbols indicate actual data points, and lines are the best fit of a sigmoidal function to the data points. For all compounds listed in Table 1, IC50 values have been derived from dose response curves fitted similarly.
Table 1.
IC50 values (μM) of the calixarene derivatives for LPS binding.a
| Compound | E. coli 055:B5 | E. coli 0111:B4 | Pseudomonas aeruginosa | Klebsiella pneumoniae | Salmonella typhimurium | Serratia marcascens |
|---|---|---|---|---|---|---|
|
Tertiary Amine Derivatives
| ||||||
| 1 | 3.4 | >5 | ND | 1.5 | ND | >5 |
| 2 | >5 | 1.4 | >5 | 1.5 | 0.6 | 3.4 |
| 3 | 4.4 | >5 | ND | 0.08 | 4.1 | ND |
| 4 | 0.05 | 2.7 | ND | 0.4 | 0.8 | ND |
| 5 | 0.006 | 3.1 | 4.2 | 1.0 | 0.4 | ND |
| 6 | 3.6 | 4.7 | >5 | ND | >5 | ND |
| 7 | 3.8 | 3.7 | >5 | 2.1 | 3.1 | ND |
| 8 | >5 | >5 | ND | 2.4 | 3.2 | ND |
| 9 | >5 | >5 | ND | ND | >5 | ND |
|
| ||||||
|
Guanidine Derivatives
| ||||||
| 10 | >5 | ND | ND | ND | 4.4 | ND |
| 11 | 4.1 | 3.9 | ND | ND | >5 | ND |
| 12 | 4.1 | >5 | ND | ND | 2.6 | ND |
| 13 | 0.04 | 0.7 | 1.5 | 1.0 | 0.6 | ND |
| 14 | 0.1 | 0.4 | 0.8 | 0.5 | 0.6 | 3.2 |
|
| ||||||
|
Triazole Derivative
| ||||||
| 15 | ND | ND | ND | ND | ND | ND |
|
| ||||||
|
Primary Amine Derivatives
| ||||||
| 16 | 1.3 | 0.1 | 0.5 | 0.5 | 0.2 | 4.5 |
| 17 | 0.05 | 2.2 | 2.6 | 1.0 | 1.1 | >5 |
| 18 | 0.6 | 1.5 | 1.0 | 0.9 | ND | ND |
| 19 | 0.9 | 1.6 | 0.8 | 0.3 | 0.6 | 1.5 |
| 20 | >5 | ND | ND | ND | >5 | ND |
|
| ||||||
|
Negatively Charged Derivatives
| ||||||
| 21 | >5 | >5 | ND | ND | ND | ND |
| 22 | >5 | ND | ND | ND | ND | ND |
| 23 | 5 | 5 | ND | 2.1 | ND | 4.2 |
|
| ||||||
|
Peptides
| ||||||
| SC4 | 2 | >5 | 2 | >5 | 4 | >5 |
|
| ||||||
| Bpep-25 | 2.5 | 2 | 1.2 | 2 | 2.5 | 4.1 |
|
| ||||||
| PmxB | 0.03 | 0.03 | 0.003 | 0.01 | 0.1 | ND |
ND = no detectable activity at 5 × 10−6 M
>5 = minimal activity at 5 × 10−6 M; no IC50 determined.
errors are estimated to be ± 15% of the value indicated in the table.
Values in sub-micro molar range are shown in bold.
Although a number of these compounds have IC50 values in the single digit micromolar range, some fall in the sub-micromolar range, and a few are active in the 5 to 50 nanomolar range. The best LPS neutralizing compounds are 5, 13, 14, 16, 17, and 19. IC50 values in the 5 nM to 50 nM range are exceptional, not only because this level of activity is better than that for peptides SC4 and βpep-25, but because it is, in some instances, on par with the LPS binding protein bactericidal/permeability increasing (BPI) factor and polymyxin B (see Table 1).
To demonstrate in vivo efficacy of these agents, one compound from each derivative subset (primary amine, tertiary amine, and guanidinium group, see Table 1 and Figure 2) was selected to treat LPS-challenged mice. Selection was based on which compounds were most active in vitro against LPS from E. coli serotype 055:B5, namely 5, 13, and 17. For this initial in vivo study, mice were administered a lethal dose of E. coli 055:B5 LPS (600 μg) and treated with each compound at doses of 5 mg/kg and 50 mg/kg. Although the 5 mg/kg dose was relatively ineffective, the 50 mg/kg dose of 5 and 13 demonstrated protection from LPS challenge, with 60% and 40% survival, respectively, compared to 0% for controls (Figure 4A). This encouraging result prompted us to test these compounds against LPS derived from E. coli strain 0111:B4, where in vitro activities were significantly less (see Table 1). Once again, mice were administered a lethal dose of E. coli 0111:B4 LPS (500 μg), and treated (50 mg/kg) with each of these three compounds. In this study, mice treated with 13 and 17 had a 100% and 25% survival, respectively, compared to 0% for controls (Figure 4B). Lastly, we tested these compounds in mice challenged with a lethal dose of LPS derived from Salmonella (600 μg). In this case, all three compounds were effective, with 17 showing 100% survival, and 5 and 13 showing 80% and 60% survival, respectively, compared to 0% for controls (Figure 4C). Compound 13 was the best overall. Moreover, we observed no toxic side effects from these calixarene compounds, as control mice administered only calixarene compounds all survived, displayed normal behavior, and gained weight normally.
Figure 4.

Helix/sheet topomimetics protect mice from LPS endotoxin. Three helix/sheet topomimetics (5, 13 and 17) were used in mouse endotoxemia models to assess in vivo efficacy. In three separate studies, these compounds were tested against LPS derived from E. coli serotype 0111 :B4, E. coli serotype 055:B5 LPS, and Salmonella. The compounds (in a final concentration of 2 % DMSO v/v), were first mixed individually with LPS, and incubated for 30 minutes prior to i.p. injection into C57/BL6 mice (n = 4–8/group). The control mice were treated with DMSO (2% v/v) alone. Each mouse received a lethal dose of LPS, with or without one of the compounds. (A) Survival of mice after being challenged with 600 μ1 LPS form E. coli serotype 055:B5 with or without one of the compounds. The survival percentage of treatment with compound 5 and 13 are significantly significant (p = 0.03 and 0.006 respectively). (B) Survival of mice after being challenged with 500 μl LPS form E. coli serotype 0111:B4 with or without one of the compounds. The survival percentage of treatment with compound 13 is significantly increased (p = 1.3 10−5). (C) Survival of mice after being challenged with 600 μ1 LPS form Salmonella with or without one of the compounds. The survival percentage of treatment with compound 17, 5 and 13 are significantly increased (p = 9×l0−8, 0.008 and 0.002 respectively). In all panels, symbols are defined as: control (□), 5 (•), 17 (π), 13(θ).
For insight into the molecular mechanism of LPS neutralization by our topomimetics, we performed NMR titration experiments on lipid A using two of them, 5 and 13. From an NMR perspective, lipid A is cleaner to work with than LPS, and, moreover, lipid A is considered to be the toxic part of LPS that is key to binding of LPS to cell receptors on macrophages.3 By acquiring TOCSY spectra as a function of the lipid A:compound molar ratio, we found that specific resonances from lipid A and from 5 or 13, were chemically shifted during the titration. Figure 5A exemplifies changes to TOCSY spectra for some resonances of lipid A before and after titration with 13. Actual chemical shift changes (Δδ) for some resonances of lipid A, and of 5 and 13, are plotted in Figures 5B–5E. Although results are similar with either compound, chemical shift changes were generally greater for the tertiary amine compound 5, suggesting somewhat stronger binding against this species of lipid A from E. coli. However, because lipid A resonances are merely shifted during the titration using either compound, interactions must be occurring within the fast exchange limit, indicating that the equilibrium dissociation binding constant, Kd, is in the μM or greater range.37 The normalized chemical shift change averaged over all resonances for 5 and 13 (Figure 5F) also suggests a binding stoichiometry of 2:1 (lipid A:compound). Nevertheless, we can not conclude definitively that this is the actual binding stoichiometry, as it could as well be 1:1, because interaction events occur in the fast exchange regime on the NMR chemical shift timescale. NMR gradient diffusion measurements were also performed (data not shown) and demonstrate that molecular sizes change very little during the titration (also consistent with fast exchange), indicating that large complexes among lipid A molecules or between/among lipid A and 5 or 13, are not being formed to any appreciable degree.
Figure 5.
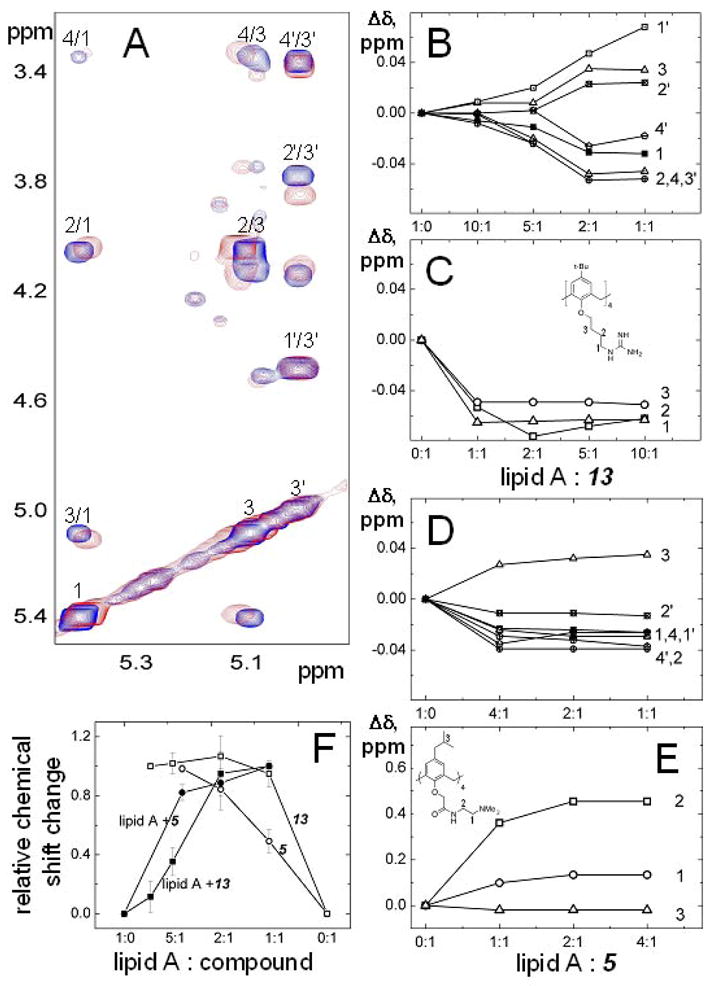
TOCSY spectra and chemical shift changes upon titration of 5 and 13 with lipid A. Spectra were collected on Inova 800 MHz NMR-spectrometer at 25 °C. Lipid A was dissolved in chloroform/methanol/water 74/23/3 mixture at the concentration of 1 mg/ml. (A) Expanded regions of two-dimensional proton TOCSY spectra are displayed (overlaid) in the absence (blue) and presence (red) of compound 13 at 1:2 lipid A:compound molar ratio. Assignments for anomeric protons are indicated. (B–E) Chemical shift changes for some proton resonances of lipid A and compounds 13 and 5 are shown as a function of the lipid A:compound molar ratio. These changes were calculated by subtraction of the chemical shift of a given proton resonance from pure lipid A from the chemical shift of the same proton resonance from lipid A to which the calixarene compound was added. Resonances for protons 1 to 4 (blue) and 1′ to 4′ (red) are shown using different colors for clarity. (F) Relative chemical shift changes are illustrated for resonances from lipid A, and compounds 13 and 5, induced by addition of compounds to lipid A at different ratios. Each point was obtained by dividing the chemical shift change shown in panels B–E by the chemical shift change between pure lipid A and after addition of compound at the ratio 1:1. The arithmetic averages of relative chemical shifts are plotted. Error bars represent the standard deviation of chemical shifts calculated for different proton resonances.
Figure 6A indicates chemical groups on lipid A that are most affected in terms of chemical shift changes via interaction with compounds 5 or 13. These tend to be located near the two phosphorylated glucosamine rings and at the α- and β-carbons of the acyl chains. The proximity of these groups is better appreciated in the topologic space filling model of the X-ray structure of lipid A38; pdb code 1qff) (Figure 6A right), where the most chemically shifted groups are color coded. The two phosphates are positioned at the upper right and left regions of the lipid A structure as labeled. This picture suggests that one region in particular forms the binding site for our topomimetic compounds. The dimensions of this site, vis a vis that of our calixarenes, suggest that, if two-point electrostatic binding (between phosphate and ammonium ions) is operative, then only one molecule of 5 or 13 could bind lipid A at any given time, supporting the idea of a 1:1 binding stoichiometry.
Figure 6.

Structures of lipid A and compounds 5 and 13 highlighting probable binding sites. The chemical structure (A left) and calculated Connoly surface (A right) of hexaacyl lipid A from E. coli are shown. In panel A left, proton resonances affected by the binding of compounds 5 and 13 are labeled using solid circles. Large red circles correspond to the largest chemical shift changes (>0.03 ppm), whereas orange and yellow circles indicate intermediate (0.02–0.03 ppm) and small (<0.02 ppm) chemical shift changes. The same color codes are used in A right to indicate the probably site of interaction with 5 and 13. (B) Energy minimized structures of compounds 5 and 13 are displayed. Chemical groups whose resonances are most affected by interaction with lipid A are color coded as described above.
The groups that are most affected in our topomimetics are indicated in the modeled structures of 5 and 13 (Figure 6B). While some minor changes are observed within the calixarene scaffold itself and the hydrophobic face, the most significant changes are found within the linker from the scaffold to the positively charged moiety, either the guanidinium group or the tertiary amine. Although clear proof for the specifics of the interaction is lacking, it seems reasonable to propose that the positively charged groups on 5 or 13 interact with the negatively charged phosphate groups on lipid A. This would then tend to position the hydrophobic face of the topomimetics towards the acyl chains of lipid A. Although in this case one might have expected greater chemical shift changes (i.e. stronger interactions) from hydrophobic groups due to their potential mutual interactions, stronger interactions between hydrophobic groups from lipid A and the topomimetics were probably attenuated by the presence of lower dielectric solvent conditions, i.e. chloroform/methanol or DMSO, that were necessary for the NMR studies. In aqueous media, it is more probable that such hydrophobic interactions between lipid A and the calixarene compounds would occur. Moreover, binding of these compounds to an intact lipopolysaccharide (LPS) molecule may be different from that observed here with lipid A, the conserved part of the LPS molecule.
Discussion
Our helix/sheet topomimetic design has led to the discovery of novel calixarene-based compounds with the ability to bind to and neutralize bacterial LPS endotoxin in vitro and in vivo. Of particular note, we have demonstrated that three of our topomimetic compounds (5,13, 17) can significantly improve survival of mice directly challenged with LPS endotoxin from different bacterial sources, with 13 being the best overall. In addition, our NMR studies have provided mechanistic and structural insight as to where on the lipid A moiety of LPS our calixarene compounds are binding.
Although the concept of designing protein surface mimetics (topomimetics) has been around for many years,39 it has usually been based on the use of peptide mimetics (or peptidomimetics). These are typically small, linear (or sometimes cyclized) peptides derived from larger proteins and often have unnatural amino acids or amino acid replacements incorporated into the peptide backbone to induce secondary structure and/or stability. Examples include mimetics of α-helices,40–42 β-strands,43 β-sheets,44,45 β-turns,46–48 and loops.49–51 Calixarene has also been used in one study as a scaffold to present and constrain small looped peptides that bind platelet-derived growth factor,52 but not in the context of fully non-peptidic compounds, as reported here. In fact, little has been done to design non-peptide topomimetic compounds that mimic a portion of the surface of a folded protein or peptide. We know of only a few examples.41,53 One uses an oligosaccharide53 and the other a linear aromatic template41 to arrange chemical substituents with similar distances and orientations as those found in a helical peptide.
To our knowledge, the results we present here represent the first successful attempt to design a non-peptidic protein surface topomimetic that mimics the general topology and molecular dimensions of segment of an α-helix or B-sheet to neutralize LPS. Nevertheless, although compounds in our small library are both amphipathic and have similar overall molecular dimensions as a segment of β-sheet-folded βpep peptides or helix-forming SC4, none matches exactly the surface topology of either peptide. For example, the key side chains in βpep peptides are mixed and heterogeneous within the context of the amphipathic surface. Our helix/sheet topomimetics, on the other hand, mostly display the same chemical substituents on each respective surface of the calixarene scaffold. The primary reason for this lies in the practicality of chemical synthesis, in that it would be difficult to synthesize calixarene with four different substituents on each side of the scaffold. This limitation aside, we apparently have captured in these helix/sheet topomimetics elements responsible for the LPS neutralizing activity of our peptides and, in fact, have serendipitously produced compounds with greater activity overall. This may be due in part to the smaller negative conformational entropy change that occurs upon binding their target LPS molecule because calixarene-based compounds are not as internally flexible as short linear peptides.
Analysis of LPS binding data for compounds in our library reveals some insightful structure-activity relationships. Although the activity of a given compound depends on the bacterial source of LPS, some generalizations may be made, (a) The presence of alkyl groups on the hydrophobic face of the calixarene scaffold is essential. For example, compounds 1 and 9 with only hydrogens on the hydrophobic face have minimal, if any, activity, whereas addition of tert-butyl (2) or propyl (3) group increases activity significantly. Even though LPS binding activity is somewhat further improved by the presence of iso-butyl groups (5), variations in activity from one alkyl group to another is not that dramatic. One exception is LPS from E. coli 055:B5 where the activity of 5 is 6 nM compared to, for example, >5 μM for 2 (tert-butyl groups) or 4.4 μM for 3 (propyl groups), (b) The presence of unsaturated hydrocarbon groups, as in 6, decreases activity, (c) Activity is greater when aliphatic hydrophobic groups are displayed on the broader face of the calix[4]arene scaffold. This is evident by comparing activities of homologs 5 and 8, which display the same hydrophilic and hydrophobic groups, but on opposite sides on the calixarene scaffold. Compound 8 has its hydrophobic groups on the narrower face of the scaffold and is considerably less active than 5 overall, (d) The presence of four positively charged groups (13, 14 and 19) is better than two (10, 12 and 20). (e) Selection of positively charged groups is important. If we compare compounds with t-butyl groups on their hydrophobic faces, the presence of primary amines (16 or 19) and guanidinium (13 or 14) groups promotes much better broad spectrum activity than the presence of tertiary amines (2) or triazole groups (15).
Clearly, some of our compounds have specific attributes that make them more effective against LPS from a particular species of bacteria. For example, calixarenes displaying guanidinium groups promote better activity than those with primary amines against both species of E. coli LPS that we tested. This observation is consistent with structural studies performed on peptide LF11 in complex with LPS from E. coli serotype 055:B5. Japelj et al25 reported that two of the arginines in peptide LF11 are positioned close to the two phosphate groups of the lipid A moiety. The distance of about 13 Å separating these two phosphate groups nearly matches that between these two guanidinium groups in the complex. Nevertheless, other arginine and lysine groups in peptide LF11 could also interact with the lipid A phosphate groups. Moreover, these authors mentioned that one of the arginine guanidinium groups may hydrogen bond to one of the sugar units in LPS. The guanidinium group, rather than the ammonium group, forms an inherently stronger electrostatic interaction with the phosphate group compared to the primary amine in lysines.54 Riordan et al55 suggest that arginyl residues play a unique role in anion recognition. A large diffuse cation like arginine is well suited to interact with large bio-anions like phosphates and sulfates. In modulating interactions between glycosaminoglycans and proteins, arginines are also generally preferred over lysines, with histidines being a distant third.56 The trailing effect from histidine parallels our finding that while the guanidinium (arginine) and primary amine (lysine) groups can effectively neutralize LPS, the triazole group (a histidine surrogate) makes the compound relatively ineffective. Nevertheless, compound 17 with its primary amine (a hydrogen bonding donor like the guanidinium group) linked through a triazole group, displays essentially the same activity against E. coli LPS, as does guanidinium derivative 13. In contrast, our NMR studies suggest that tertiary amine derivative 5 interacts more strongly with E. coli lipid A than does guanidinium derivative 13, which in fact parallels LPS neutralizing activity differences of these two compounds against E. coli 055:B5 LPS (see Table 1).
Against LPS from other Gram negative bacteria, our topmimetics demonstrate no great overall preference for guanidinium over ammonium groups. The apparent overall preference of E. coli LPS for guanidinium is most likely due to structural differences in LPS among different geni of bacteria. Some topomimetic compounds in the library do demonstrate better activities against strains of bacteria other than E. coli. For example, primary amine analogs 16 and 19 have IC50 values in the sub-micrommolar range against LPS from Pseudomonas, Salmonella, and Klebsiella. Furthermore, the topological difference between the cone conformer and the partial cone conformer affects LPS binding activity apparently selectively, as evidenced by the high selectivity of partial cone conformer 3 for Klebsiella LPS with an IC50 value of 80 nanomolar. Information of this nature may be used to design compounds with specificity against LPS from a subset of bacteria.
In conclusion, we demonstrated here the proof of principle that a relatively small, non-peptidic calixarene-based compound can be designed and synthesized to mimic the molecular dimensions and amphipathic surface topology of LPS binding β-sheet and helical peptides. These novel sheet/helix topomimetics present hydrophobic and positively charged residues in a manner that allows some of them to effectively bind to and neutralize LPS endotoxin in vitro, as well as in vivo. These agents may have utility in the clinic by providing a new means to combat LPS-induced endotoxemia and septic shock.
Experimental Section
Peptide preparation
Peptides were synthesized using a Milligen/Biosearch 9600 peptide solid-phase synthesizer using fluorenylmethoxycarbonyl chemistry. Lyophilized crude peptides were purified by preparative reversed-phase HPLC on a C18 column with an elution gradient of 0–60% acetonitrile with 0.1% trifluoroacetic acid in water. Purity and composition of the peptides were verified by HPLC (Beckman Model 6300), amino acid analysis and mass spectrometry.
Chemical synthesis of calixarene derivatives
Calix[4]arene 25,57 tetraethyl calix[4]arenetetraacetate 26,58 and calix[4]arenetetraamide 159 were prepared according to procedures in the literature.
4-Methallyl calix[4]arene 27
To a solution of calix[4]arene (212.2 mg, 0.5 mmol) in THF (5.0 mL) and DMF (0.5 mL) was added NaH (200.0 mg, 8.2 mmol). The reaction mixture was refluxed for 1 hour and methylallylchloride (2.2 mL, 25 mmol) was added. Continuously heated for 5 hours, the reaction mixture was cooled and filtered through a pad of Celite. The filtrate was diluted with EtOAc and washed with brine. Organic phase was separated, dried (Na2SO4) and concentrated. The residue was purified by flash chromatography (1% EtOAc in hexanes) gave O-methylallyl calix[4]arene (320.3 mg, 100%) as colorless oil.
O-Methallyl calix[4]arene 6 (44.5 mg, 0.069 mmol) was dissolved in neat N,N-dimethylaniline (1 mL) and stirred at 210 °C for 2 hours. The reaction mixture was cooled and poured into ice-water (10.0 mL) with concentrated HCl (10.0 mL). The mixture was stirred for 10 min and extracted by CHCl3. The organic phase was separated, dried (Na2SO4), and concentrated. Crystallization from ethanol provided 4-methallyl calix[4]arene 8 (27.6 mg) as white fine crystals. The mother liquor was concentrated and crystallized to give more desired product (2.4 mg) to provide 67% total yield. 1H NMR (500 MHz, CDCl3) δ 10.19 (s, 4H), 6.88 (s, 8H), 4.77 (br s, 4H), 4.68 (br s, 4H), 4.21 (br d, J = 12.5 Hz, 4H), 3.45 (br d, J = 12.5 Hz, 4H), 3.10 (s, 8H), 1.63 (s, 12H); 13C NMR (125 MHz) δ 147.3 (4C), 145.4 (4C), 133.3 (8C), 129.5 (8C), 128.2 (4C), 112.0 (4C), 44.0 (4C), 32.0 (4C), 22.3 (4C); HRMS (ESI) m/z calcd for C44H48Na1O4 (M+Na)+ 663.3450, found 663.3446.
Tetra-ester 28
4-Methallyl calix[4]arene 27 (96.1 mg, 0.15 mmol) and K2CO3 (165.8 mg, 1.2 mmol)were refluxed in acetone (3.0 mL) for one hour and then ethyl bromoacetate (0.13 mL, 1.2 mmol). This reaction mixture was refluxed for 24 hours. After cooling, the reaction mixture was filtered through a pad of Celite and rinsed with CH2Cl2. The filtrate was concentrated under vacuo to give the crude product, which was subjected to hydrogenation with Pd on activated carbon (10%, 70 mg) in EtOAc (3 mL). Purification of the crude reduced product with flash chromatography (15% EtOAc in hexanes) provided oil-like tetra-ester 28 (148.0 mg, 100%) in the cone conformation. 1H NMR (500 MHz, CDCl3) δ 6.47 (s, 8H), 4.80 (d, J = 13.2 Hz, 4H), 4.75 (s, 8H), 4.20 (q, J = 7.4 Hz, 8H), 3.14 (d, J = 13.2 Hz, 4H), 2.11 (d, j= 7.4 Hz, 8H), 1.59 (m, 4H), 1.28 (t, J = 7.4 Hz, 12H), 0.75 (d, J = 6.1 Hz, 24H); 13C NMR (75 MHz, CDCl3) δ 170.7 (4C), 153.7 (4C), 135.8 (4C), 134.0 (8C), 129.4 (8C), 71.5 (4C), 60.5 (4C), 44.9 (4C), 31.6 (4C), 30.4 (4C), 22.4 (8C), 14.4 (4C); HRMS (ESI) m/z calcd for C60H80Na1O12 (M+Na)+ 1015.5547, found 1015.5575.
Tetra-amide 5
Tetra-ester 28 (146.8 mg, 0.15 mmol) in toluene (1.5 mL) and methanol (1.5 mL) was treated with N, N-dimethylethylenediamine (0.32 mL, 2.96 mmol) and stirred in a sealed tube at 80 °C for 24 hours. The volatile components were removed under vacuo (~ 60 °C bath temperature) to give a light brown sticky solid. The solid was dissolved in a minimum amount of CH2Cl2 and tetra-amide 5 (103.6 mg, 60%) was obtained as a light yellow solid by the sequence of dropwise addition of ether (~10:1 Et2O : CH2Cl2 final ratio), centrifugation, decantation, and drying. 1H NMR (500 MHz, CDCl3) δ 7.74 (br t, J = 5.8 Hz, 4H), 6.39 (s, 8H), 4.48 (s, 8H), 4.44 (d, J= 13.5 Hz, 4H), 3.46 (dt, J= 6.2, 5.8 Hz, 8H), 3.16 (d, J= 13.5 Hz, 4H), 2.50 (t, J = 6.2 Hz, 8H), 2.25 (s, 24 H), 2.10 (d, J = 7.0 Hz, 8H), 1.65 (m, 4H), 0.80 (d, J = 6.2 Hz, 24H); 13C NMR (125 MHz, CDCl3) δ 170.0 (4C), 153.8 (4C), 136.2 (4C), 133.4 (8C), 129.7 (8C), 74.5 (4C), 58.2 (4C), 45.4 (8C), 45.0 (4C), 37.1 (4C), 31.3 (4C), 30.5 (4C), 22.4 (8C); HRMS (ESI) m/z calcd for C68H105N8O8 (M+H)+ 1161.8055, found 1161.8091.
5,11,17,23-Tetra-tert-butyl-25,27-bis(3-cyanopropyloxy)-26,28-dihydroxy calix[4]arene (29)
According to the literature,60 4-tert-butylcalixarene 24 (1.3 g, 2.0 mmol) in acetone (20 mL) was treated with K2CO3 (1.6 g, 12.0 mmol). The reaction mixture was refluxed for 1 hour followed by addition of 4-bromo-butyronitrile (1.6 mL, 16.0 mmol). After 16 hours the reaction mixture was cooled and filtered through Celite. The filtrate was washed with brine, dried (Na2SO4), and concentrated. Crystallization from ethanol gave dinitrile 29 (1.3 g) as a white crystal. More desired product (158.6 mg, 91% in total) was collected from the mother liquor by flash chromatography (20% EtOAc in hexanes). 1H NMR (500 MHz, CDCl3) δ 7.44 (s, 2H), 7.06 (s, 4H), 6.86 (s, 4H), 4.17 (d, J = 13.2 Hz, 4H), 4.09 (t, J = 5.5 Hz, 4H), 3.38 (d, J = 13.2 Hz, 4H), 3.05 (t, J = 7.0 Hz, 4H), 2.34 (tt, J = 7.0, 5.5 Hz, 4H), 1.28 (s, 18H), 1.00 (s, 18H); HRMS (ESI) m/z calcd for C52H66N2Na1O4 (M+Na)+ 805.4920, found 805.4908.
5,11,17,23-Tetra-tert-butyl-25,26,27,28-tetrakis(3-cyanopropyloxy) calix[4]arene (30)
Dinitrile (1.4 g, 1.8 mmol) in DMF (20 mL) was treated with NaH (432 mg, 18.0 mmol) at room temperature for 1 hour followed with addition of 4-bromo-butyronitrile (9.0 mL, 90.0 mmol). The reaction mixture was stirred at 75 °C for 20 hours, and then was partitioned between CH2Cl2 and NH4Cl sat. aqueous solution. The organic layer was washed with NH4Cl sat. aqueous solution (3×100 mL), dried (Na2SO4), and concentrated. After removal of remained 4-bromo-butyronitrile at 68 °C under vacuo, the residue was purified by flash chromatography (35% EtOAc in hexanes) to give the tetra-nitrile 30 (1.3 g, 81%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 6.80 (s, 8H), 4.26 (d, J = 12.3 Hz, 4H), 4.02 (t, J = 7.5 Hz, 8H), 3.22 (d, J = 12.3 Hz, 4H), 2.62 (t, J = 7.5 Hz, 8H), 2.28 (tt, J = 7.5, 7.5 Hz, 8H), 1.08 (s, 36H); 13C NMR (75 MHz, CDCl3) δ 152.6 (4C), 145.6 (4C), 133.4 (8C), 125.6 (8C), 119.6 (4C), 73.1 (4C), 34.1 (4C), 31.5 (12C), 31.2 (4C), 26.0 (4C), 14.4 (4C); HRMS (ESI) m/z calcd for C60H76N4Na1O4 (M+Na)+939.5764, found 939.5776.
Tetra-amine 16
According to the literature,60 NaBH4 (227.0 mg, 6.0 mmol) was added batchwise to the solution of tetranitrile (137.5 mg, 0.15 mmol) and CoCl2 (155.8 mg, 1.2 mmol) in methanol (10 mL). After being stirred at room temperature for 26 hours, the reaction mixture was diluted with CH2Cl2. 3N HCl (~30 mL) was added and vigorously stirred until the black precipitate was completely dissolved. The aqueous layer was adjusted to pH = 10 with concentrated NH4OH, and then extracted with CH2Cl2. The combined CH2Cl2 phase was washed with brine, dried (Na2SO4), and concentrated to provide the known crude tetra-amine (140.0 mg, 100%) as an off-white solid.60 Without further purification, this solid was brought to next step. 1H NMR (500 MHz, CDCl3) δ 6.77 (s, 8H), 4.37 (d, J = 12.5 Hz, 4H), 3.87 (t, J = 7.7 Hz, 8H), 3.12 (d, J = 12.5 Hz, 4H), 2.79 (t, J = 7.4 Hz, 8H), 2.02 (tt, J ≈ 7.5, 7.5 Hz, 8H), 1.55 (tt, J = 7.5, 7.5 Hz, 8H), 1.07 (s, 36 H).
Boc protected tetra-guanidine 31 and Boc protected tri-guanidine-mono-amine 32
To a solution of tetra-amine 16 (140.0 mg, 0.15 mmol) was added in sequence 1,3-bis(tert-butoxycarbonyl)-2-methyl-2-thiopseudourea (191.6 mg, 0.66 mmol), HgCl2 (179.2 mg, 0.66 mmol), and Et3N (0.26 mL, 2.0 mmol). After being stirred for 15 hours the reaction mixture was filtered through Celite, and the filtrate was partitioned between CH2Cl2 and NaHCO3 aqueous solution. The organic phase was dried (Na2SO4) and concentrated. Purification with MPLC (15% EtOAc in hexanes) provided Boc protected tetra-guanidine 31 (101.9 mg, 36%) and Boc protected tri-guanidine-mono-amine 32 (35.4 mg, 13%). For 31: 1H NMR (500 MHz, CDCl3) δ 11.51 (s, 4H), 8.38 (t, J = 5.0 Hz, 4H), 6.75 (s, 8H), 4.33 (d, J = 12.6 Hz, 4H), 3.90 (t, J = 7.6 Hz, 8H), 3.48 (td, J = 7.3, 5.0 Hz, 8H), 3.12 (d, J = 12.6 Hz, 4H), 2.00 (m, 8H), 1.68 (m, 8H), 1.48+ (s, 36H), 1.48− (s, 36H), 1.07 (s, 36H); 13C NMR (75 MHz, CDCl3) δ 163.8 (4C), 156.3 (4C), 153.5 (4C), 153.4 (4C), 144.6 (4C), 133.9 (8C), 125.1 (8C), 83.0 (4C), 79.2 (4C), 74.6 (4C), 41.1 (4C), 34.0 (4C), 31.6 (12C), 31.4 (4C), 28.5 (12C), 28.3 (12C), 27.7 (4C), 25.9(4C); HRMS (ESI) m/z calcd for C104H165N12NaO20 (M+H+Na)2+ 962.6080, found 962.6090. For 32: 1H NMR (500 MHz, CDCl3) δ 11.52 (s, 3H), 8.39 (m, 3H), 6.79 (s, 4H), 6.74 (s, 4H), 5.25 (m, 1H), 4.34 (d, J = 12.5 Hz, 2H), 4.33 (d, J = 12.5 Hz, 2H), 3.88 (m, 8H), 3.48 (m. 6H), 3.21 (m, 2H), 3.12+ (d, J = 12.5 Hz, 2H), 3.12− (d, J = 12.5 Hz, 2H), 2.01 (m, 8H), 1.69 (m, 8H), 1.48 (s, 54 H), 1.41 (s, 9H), 1.09 (s, 18H), 1.05 (s, 18H); 13C NMR (75 MHz, CDCl3) 163.8 (3C), 156.3 (4C), 153.7 (1C), 153.4+(4C), 153.4− (2C), 144.6+ (1C), 144.6 (1C), 144.5 (2C), 134.0 (4C), 133.8 (2C), 133.7 (2C), 125.1 (8C), 83.1 (4C), 79.3 (3C), 74.7 (4C), 41.2 (1C), 41.1 (3C), 34.0+ (2C), 34.0−(2C), 31.7 (6C), 31.6 (6C), 31.5 (2C), 31.4 (2C), 28.6 (3C), 28.5 (9C), 28.3 (9C), 27.8 (4C), 26.0 (3C), 25.9 (1C); HRMS (ESI) m/z calcd for C98H154N10O18Na2 (M+2Na)2+ 902.5619, found 902.5723.
5,11,17,23-Tetra-tert-butyl-25,26,27,28-tetrakis(4-guanidinobutoxy) calix[4]arene trifloroacetic acid salt (13)
Tetra-guanidine 31 (101.9 mg, 0.05 mmol) was dissolved in a solution of CH2Cl2 with 40% TFA and 5% anisole (1.0 mL) and the mixture was stirred at room temperature for 3 hours. The volatile components were removed under vacuo. The residue was partitioned between CH2Cl2 and water and the aqueous phase was adjusted to pH=8 with NaHCO3 aqueous solution. The organic phase was separated, dried (Na2SO4), and concentrated to give the calixarene 13-TFA salt (104.5 mg, 98% assuming an octatrifluoroacetate salt) as an off-white solid. 1HNMR (500 MHz, CD3OD) δ 6.81 (s, 8H), 4.42 (d, J = 12.8 Hz, 4H), 3.96 (t, J = 7.2 Hz, 8H), 3.29 (m, 8H), 3.15 (d, J = 12.8 Hz, 4H), 2.08 (m, 8H), 1.79 (m, 8H), 1.08 (s, 36H); HRMS (ESI) m/z calcd for C64H102N12O4 (M+2H)2+551.4073, found 551.4107.
5,11,17,23-Tetra-tert-butyl-25,26,27-tris(4-guanidinobutyroxy)-28-(4-aminobutyroxy) calix[4]arene trifloroacetic acid salt (14)
According to the procedure described for tetra-guanidine 13, the Boc protected mono-amino-tri-guanidine 32 (34.5 mg, 0.02 mmol) in 5% anisole CH2Cl2 solution (0.6 mL) was treated TFA (0.4 mL) at room temperature. Standard workup and purification gave rise to the calixarene 14 TFA salt (30.4 mg, 82% assuming an heptatrifluoroacetate salt). 1H NMR (500 MHz, CD3OD) δ 6.97 (s, 4H), 6.67 (s, 4H), 4.42+ (d, J = 12.4 Hz, 2H), 4.42− (d, J = 12.4 Hz, 2H), 4.07 (t, J = 8.0 Hz, 2H), 3.91 (m, 6H), 3.28 (m, 8H), 3.15 (d, J = 12.4 Hz, 2H), 3.13 (d, J = 12.4 Hz, 2H), 2.17 (m, 2H), 2.02 (m, 6H), 1.77 (m, 6H), 1.66 (m, 2H), 1.20+ (s, 9H), 1.20− (s, 9H), 0.97 (s, 18H); 13C NMR (75 MHz, CDCl3 and CD3OD) 157.4 (3C), 153.5 (2C), 153.3 (1C), 153.2 (1C), 144.6+(3C), 144.6− (1C), 133.7+ (2C), 133.7− (2C), 133.4 (4C), 125.1 (4C), 125.0 (4C), 74.6 (4C), 41.8 (1C), 41.5 (3C), 33.8+ (2C), 33.8+ (2C), 31.4+ (6C), 31.4− (6C),31.2 (4C), 27.8+(2C), 27.8− (2C), 25.9 (4C); HRMS (ESI) m/z calcd for C63H100N10O4 (M+2H)2+530.3964, found 530.4001.
O-Propargyl- 4-tert-butyl calix[4]arene 33 and 34
A suspension of 4-tert-butyl calix[4]arene (649.0 mg, 1.0 mmol) in THF and DMF (15 mL, 10:1) was treated with NaH (192.0 mg, 8.0 mmol) and propargyl bromide (80% in toluene, 2.23 mL, 20 mmol). The reaction mixture was refluxed for 18 hours and cooled to room temperature. After filtration through Celite, the filtrate was diluted with CH2Cl2, washed with brine, dried (Na2SO4) and concentrated. The residue was first purified by flash chromatography (3% EtOAc in hexanes) to give cone conformer 33 (255.9 mg). The components with cone conformer 33 and partial cone analog 34 as a mixture was further purified by MPLC to give more compound 33 (146.3 mg, 50% in total) and compound 34 (297.3 mg, 37%). For 33, 1H NMR (300 MHz, CDCl3) 8 6.79 (s, 8H), 4.80 (d, J = 2.3 Hz, 8H), 4.60 (d, J = 12.9 Hz, 4H), 3.16 (d, J = 12.9 Hz, 4H), 2.47 (t, J = 2.3 Hz, 4H), 1.07 (s, 36H); 13C NMR (75 MHz) δ 152.6 (4C), 145.7 (4C), 134.5 (8C), 125.2 (8C), 81.4 (4C), 74.5 (4C), 61.2 (4C), 34.1 (4C), 32.6 (4C), 31.6 (12C); HRMS (ESI) m/z calcd for C56H64Na1O4 (M+Na)+ 823.4702, found 823.4725. For 34, 1H NMR (300 MHz, CDCl3) δ 7.43 (s, 2H), 7.06 (s, 2H), 6.99 (d, J = 2.5 Hz, 2H), 6.52 (d, J = 2.5 Hz, 2H), 4.48 (dd, J = 15.3, 2.5 Hz, 2H), 4.44 (dd, J = 15.3, 2.5 Hz, 2H), 4.35 (d, J = 2.5 Hz, 2H), 4.31 (d, J = 13.0 Hz, 2H), 4.24 (d, J = 2.5 Hz, 2H), 3.85 (d, J = 14.0 Hz, 2H), 3.73 (d, J = 14.0 Hz, 2H), 3.08 (d, J = 13.0 Hz, 2H), 2.50 (t, J = 2.3, 2H), 2.44 (t, J = 2.5 Hz, 1H), 2.24 (t, J = 2.5 Hz, 1H), 1.45 (s, 9H), 1.33 (s, 9H), 1.04 (s, 18H); 13C NMR (75 MHz) δ 154.3 (1C), 153.3 (2C), 151.8 (1C), 146.1 (1C), 145.2 (2C), 144.3 (1C), 136.5 (2C), 133.1 (2C), 132.5 (2C), 132.1 (2C), 128.8 (2C), 126.3 (2C), 125.7 (2C), 125.6 (2C), 82.2 (1C), 81.2 (2C), 80.9 (1C), 74.7 (2C), 74.5 (1C), 73.9 (1C), 61.0 (2C), 59.3 (1C), 58.8 (1C), 37.9 (2C), 34.3 (2C), 34.0 (2C), 32.6 (2C), 32.0 (3C), 31.8 (3C), 31.6 (6C).
1,2,3-Triazole derivative of calix[4]arene 35
Follow the literature,61 water (0.4 mL) was added to the solution of alkyne 33 (80.1 mg, 0.1 mmol) and N-Boc-2-azido-ethylamine (149.0 mg, 0.8 mmol) in tBuOH (0.4 mL) and THF (0.2 mL). The reaction mixture turned cloudy, and then ascorbic acid (7.0 mg, 0.04 mmol), NaOAc (6.6 mg, 0.08 mmol), and CuSO4-5H2O (5.0 mg, 0.02 mmol) was added to the suspension. The reaction mixture was stirred at room temperature for 36 hrs. NH4Cl aqueous solution (3 mL) was added to stop the reaction. After stirred for 5 min, the reaction mixture was extracted by CH2Cl2. The combined organic phase was dried (Na2SO4) and concentrated. The residue was purified by flash chromatography (4% MeOH in CH2Cl2) to give the tetra-triazole 35 (75.7 mg, 50%) as a light yellow solid. 1H NMR (300 MHz, CDCl3, 55 °C) δ 7.81 (br s, 4H), 6.77 (s, 8H), 5.68 (br s, 4H), 5.00 (br s, 8H), 4.45 (br m, 8H), 4.32 (d, J = 12.8 Hz, 4H), 3.55 (br m, 8H), 3.09 (d, J = 12.8 Hz, 4H), 1.42 (s, 36H), 1.08 (br s, 36H); 13C NMR (75 MHz, CDCl3) δ 156.3 (4C), 152.6 (4C), 145.4 (4C), 144.7 (4C), 134.0 (8C), 125.3 (8C), 124.8 (4C), 79.7 (4C), 67.2 (4C), 49.9 (4C), 40.9 (4C), 34.0 (4C), 31.5 (16C), 28.6 (12C); HRMS (ESI) m/z calcd for C84H120N16Na2O12 (M+2Na)2+ 795.4534, found 795.4567.
1,2,3-Triazole derivative of calix[4]arene 36
According to the procedure described for tetra-triazole 35, alkyne 34 (58.6 mg, 0.07 mmol) and N-Boc-2-azido-ethylamine (114.3 mg, 0.58 mmol) in tBuOH (0.3 mL), THF (0.2 mL), and water (0.4 mL) were treated with ascorbic acid (5.1 mg, 0.03 mmol), NaOAc (4.8 mg, 0.06 mmol), and CuSO4·5H2O (3.6 mg, 0.01 mmol). The reaction mixture was stirred for 36 hrs. Standard workup and purification as described for tetra-triazole 35 gave the tetra-triazole 36 (80.0 mg, 88%) in partial cone conformation as a light yellow solid. 1H NMR (300 MHz, CD3Cl) 8.04 (br s, 2H), 7.49 (br s, 2H), 7.13 (s, 2H), 6.92 (s, 2H), 6.86 (d, J = 2.0 Hz, 2H), 6.42 (d, J = 2.0 Hz, 2H), 5.19 (br s, 2H), 4.99+ (d, J = 11.0 Hz, 2H), 4.99− (br s, 1H), 4.93 (br s, 1H), 4.88 (s, 2H), 4.74 (d, J = 11.0 Hz, 2H), 4.73 (s, 2H), 4.44 (m, 6H), 4.35 (br m, 2H), 4.09 (d, J = 13.2 Hz, 2H), 3.79 (d, J = 13.8 Hz, 2H), 3.72 (d, J = 13.8 Hz, 2H), 3.67 (br m, 2H), 3.59 (m, 4H), 3.35 (br m, 2H), 2.95 (d, J = 13.2 Hz, 2H), 1.45+ (s, 9H), 1.45− (s, 9H), 1.43 (s, 18H), 1.25 (s, 9H), 0.95 (s, 9H), 0.92 (s, 18H); 13C NMR (75 MHz) δ 156.1 (1C), 156.0 (3C), 154.3 (1C), 153.4 (2C), 151.0 (1C), 145.3 (1C), 144.7 (3C), 144.6 (2C), 144.2 (1C), 143.4 (1C), 136.6 (2C), 133.0 (2C), 132.2 (2C), 132.1 (2C), 128.6 (2C), 126.2 (2C), 125.3 (3C), 125.0 (1C), 124.8 (2C), 124.2 (2C), 80.2 (1C), 80.0 (2C), 79.9 (1C), 67.0 (2C), 65.1 (1C), 62.3 (1C), 50.2 (3C), 49.9 (1C), 41.1 (1C), 40.6 (2C), 40.5 (1C), 37.4 (2C), 34.2 (1C), 33.8 (2C), 33.7 (1C), 32.2 (2C), 31.7 (3C), 31.4 (6C), 31.3 (3C), 28.6 (3C), 28.5 (9C); HRMS (ESI) m/z calcd for C84H120N16Na2O12 (M+2Na)2+ 795,4534, found 795.4547.
Aminoethyl triazole derivative of calix[4]arene 17
Tetra-triazole 35 (30.9 mg, 0.02 mmol) was dissolved in a solution of CH2Cl2 with 5% anisole (0.6 mL), and then cooled to 0 °C. TFA (0.4 mL) was added dropwise and the reaction mixture was allowed warm to room temperature. After 3 hours, the reaction mixture was concentrated under vacuo. The residue was triturated in ether to give aminoethyl triazole 17·TFA salt (24.8 mg, 77% assuming an tetratrifluoroacetate) as a white solid. 1H NMR (500 MHz, CD3OD) 8.08 (s, 4H), 6.83 (s, 8H), 5.05 (s, 8H), 4.81 (t, J = 6.0 Hz, 8H), 4.15 (d, J = 12.7 Hz, 4H), 3.58 (t, J = 6.0 Hz, 8H), 2.97 (d, J = 12.7 Hz, 4H), 1.09 (s, 36H); 13C NMR (75 MHz) 8 153.7 (4C), 146.7 (4C), 146.3 (4C), 135.4 (8C), 127.0 (4C), 126.6 (8C), 67.8 (4C), 48.6 (4C), 40.5 (4C), 35.0 (4C), 32.7 (4C), 32.1 (12C); HRMS (ESI) m/z calcd for C64H89N16O4 (M+H)+ 1145.7247, found 1145.7275.
Aminoethyl triazole derivative of calix[4]arene 18
According to the procedure described for aminoethyl triazole 17, tetra-triazole 36 (50.8 mg, 0.03 mmol) in a solution of CH2Cl2 with 5% anisole (0.6 mL) was treated with TFA (0.4 mL). After 3 hour, the reaction mixture was concentrated under vacuo. The residue was triturated in ether to give aminoethyl triazole 18-TFA salt (55.8 mg, 100% assuming an tetratrifluoroacetate) in partial cone conformation as a white solid. 1H NMR (300 MHz, CD3OD) 8.28 (s, 2H), 8.22 (s, 1H), 8.16 (s, 1H), 7.18 (s, 2H), 6.95 (s, 2H), 6.94 (d, J = 2.4 Hz, 2H), 6.42 (d, J = 2.4 Hz, 2H), 4.93 (d, J = 11.6 Hz, 2H), 4.90 (s, 4H), 4.84 (d, J = 11.6 Hz, 2H), 4.74 (m, 8H), 3.96 (d, J = 12.9 Hz, 2H), 3.81 (br s, 4H), 3.54 (m, 8H), 2.85 (d, J = 12.9 Hz, 2H), 1.27 (s, 9H), 1.01 (s, 9H), 0.91 (s, 18H); 13C NMR (75 MHz) 8 156.0 (1C), 154.7 (2C), 152.3 (1C), 146.6 (1C), 146.1 (2C), 146.0 (2C), 145.8 (2C),144.5 (1C), 137.7 (2C), 134.3 (2C), 133.4 (4C), 129.8 (2C), 127.8 (2C), 127.5 (1C), 127.2 (2C), 127.1 (1C), 126.8 (2C), 126.4 (2C), 67.5 (2C), 65.9(1C), 63.5 (1C), 48.9 (1C), 48.4 (2C), 48.2 (1C), 40.4 (4C), 38.0 (2C), 35.0 (1C), 34.8+ (2C), 34.8- (1C), 33.2 (2C), 32.2 (3C), 32.1 (6C), 32.0 (3C); HRMS (ESI) m/z calcd for C64H90N16O4 (M+2H)2+ 573.3660, found 573.3675. A small library of calix[4]arene derivatives was synthesized using methods discussed in Supplemental Material. Chemical structures of all calixarene analogs in the library are presented in the Results section.
Limulus amoebocyte lysate assay for LPS neutralization
The ability of synthetic peptides and calixarene-based compounds to neutralize endotoxin was detected using the chromogenic QCL-1000 kit from BioWhittaker, Inc. (Walkersville, MD), and as described in their protocol and by Mayo et al.24 This method is quantitative for Gram negative bacterial endotoxin (LPS, lipopolysaccharide). In this Limulus amebocyte lysate (LAL) assay, compounds that are active, inhibit the LPS-mediated activation of a proenzyme,62 whose active form would release p-nitroaniline (pNA) from a colorless synthetic substrate (Ac-Ile-Glu-Ala-Arg-pNA), producing a yellow color (pNA) whose absorbtion is monitored spectrophotometrically at 405–410 nm. The initial rate of enzyme activation is proportional to the concentration of endotoxin present.
Variants of LPS from six Gram negative bacteria were used: E. coli serotypes 0111:B4 (Combrex, Walkersville, MD) and 055:B5 (Sigma, St. Louis, MO); Klebsiella pneumoniae (List Biologies, San Jose, CA); Pseudomonas aeruginosa (List Biologies, San Jose, CA); Salmonella typhimurium (List Biologies, San Jose, CA), and Serratia marcascens (List Biologies, San Jose, CA). The concentration of compound required to neutralize a given LPS and therefore to inhibit the Limulus amoebocyte lysate driven by 0.04 units of any given LPS was determined by dose response curves fit by using a standard sigmoidal function to determined the IC50 value for each topomimetic compound. The 0.04 units corresponds to 0.01 ng LPS from E. coli serotype 055:B5, 0.01 ng LPS from E. coli serotype 0111:B4, 0.003 ng LPS from K. pneumoniae, 0.01 ng LPS from P. aeruginosa, 0.03 ng LPS from S. typhimurium, and 0.03 ng LPS from S. marcascens.
Endotoxemia studies in mice
C57 male black mice were injected i.p. with a solution that contained a lethal dose of LPS [600 μg of LPS from E. coli 055:B5 and Salmonella, and 500 μg of LPS from E. coli 011:B4] and 1.25 mg of the topomimetic compound (a dose of 50 mg/kg). Control groups of mice were administered either only the bacteria, or only the topomimetic compounds. Mice were provided food and water as usual ad libitum, and monitored for several days. Experimental protocol for these animal studies was approved by the University of Minnesota Research Animal Resources Ethical Committee. Data are plotted as the number of surviving mice versus time in hours. Statistical analysis was performed on the average amount of survival time per group with a maximum of 120 hours (surviving mice) by using the Student’s t-test.
NMR Spectroscopy
Hexaacyl Lipid A from E. coli was purchased from Sigma, Inc. Deuterated solvents were purchased from Cambridge Isotope Laboratory and used for the NMR experiments. 1 mg of lipid A was dissolved in 600 μl of chloroform/methanol/water (74/23/3) solvent system and sonicated for 20 minutes. Freeze-dried calixarene-based compounds 5 and 13 were dissolved in 12 mu;l of the same solvent system and then added to the lipid A solution at different molar ratios as indicated in the text. 2D-homonuclear TOCSY (mixing time of 60 ms) spectra with WATERGATE for water suppression were acquired at 25 °C on a Varian UNITY Plus-800 NMR spectrometer. 2048 complex data points along the t2 time dimension and 256 increments along the tl time dimension were collected using a spectral width of 9000 Hz. Data were processed using the program NMRPipe63 and a Gaussian window function.
Gradient diffusion measurements were performed on a Varian UNITY Plus-600 NMR spectrometer. The maximum magnitude of the gradient, g, was calibrated using a deuterated water standard supplied by the vendor. Measurements were performed using a double-stimulated echo pulse sequence to suppress convection effects.64 The diffusion coefficient, D, was estimated from the slope of the diffusion attenuation of spin-echo as described previously.65
Supplementary Material
Full spectroscopic characterization data of calixarene derivatives 2– 4, 6–12, 15, 19–23. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
We are grateful to Denisha Walek of the Microchemical Facility for expertise in the synthesis of peptides. This work was supported by research grants to KHM from the National Institute of Allergy and Infectious Diseases through the Greater Lakes Research Center of Excellence (GLRCE) for Biodefense and Emerging Infectious Diseases (U54 AI057153) and the National Cancer Institute (RO1 CA096090).
Footnotes
Abbreviations: BPI, bactericidal/permeability increasing protein; LALF, Limulus anti lipopolysaccharide factor; LPS, lipopolysaccharide; NMR, nuclear magnetic resonance spectroscopy; NOE, nuclear Overhauser effect; rf, radio frequency; FID, free induction decay; HPLC, high performance liquid chromatography; ip, interperitoneal.
References
- 1.Carcillo JA, Cunnion RE. Septic shock. Crit Care Clin. 1997;13:553–574. doi: 10.1016/s0749-0704(05)70328-6. [DOI] [PubMed] [Google Scholar]
- 2.Gagliardi AR, Hennig B, Collins DC. Antiestrogens inhibit endothelial cell growth stimulated by angiogenic growth factors. Anticancer Res. 1996;16:1101–1106. [PubMed] [Google Scholar]
- 3.Aderem A, Ulevitch RJ. Toll-like receptors in the induction of the innate immune response. Nature. 2000;406:782–787. doi: 10.1038/35021228. [DOI] [PubMed] [Google Scholar]
- 4.Ulevitch RJ. Recognition of bacterial endotoxins by receptor-dependent mechanisms. Adv Immunol. 1993;55:267–289. doi: 10.1016/s0065-2776(08)60502-7. [DOI] [PubMed] [Google Scholar]
- 5.Zughaier SM, Zimmer SM, Datta A, Carlson RW, Stephens DS. Differential induction of the toll-like receptor 4-MyD88-dependent and -independent signaling pathways by endotoxins. Infect Immun. 2005;73:2940–2950. doi: 10.1128/IAI.73.5.2940-2950.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bannerman DD, Goldblum SE. Direct effects of endotoxin on the endothelium: barrier function and injury. Lab Invest. 1999;79:1181–1199. [PubMed] [Google Scholar]
- 7.Cohen J. The immunopathogenesis of sepsis. Nature. 2002;420:885–891. doi: 10.1038/nature01326. [DOI] [PubMed] [Google Scholar]
- 8.Abad MC, Ami RK, Grella DK, Castellino FJ, Tulinsky A, Geiger JH. The X-ray crystallographic structure of the angiogenesis inhibitor angiostatin. J Mol Biol. 2002;318:1009–1017. doi: 10.1016/S0022-2836(02)00211-5. [DOI] [PubMed] [Google Scholar]
- 9.Remick DG, Ward PA. Evaluation of endotoxin models for the study of sepsis. Shock. 2005;24(Suppl 1):7–11. doi: 10.1097/01.shk.0000191384.34066.85. [DOI] [PubMed] [Google Scholar]
- 10.Rietschel ET, Brade H, Holst O, Brade L, Muller-Loennies S, Mamat U, Zahringer U, Beckmann F, Seydel U, Brandenburg K, Ulmer AJ, Mattern T, Heine H, Schletter J, Loppnow H, Schonbeck U, Had HD, Hauschildt S, Schade UF, Di Padova F, Kusumoto S, Schumann RR. Bacterial endotoxin: Chemical constitution, biological recognition, host response, and immunological detoxification. Curr Top Microbiol Immunol. 1996;216:39–81. doi: 10.1007/978-3-642-80186-0_3. [DOI] [PubMed] [Google Scholar]
- 11.Rehn M, Veikkola T, Kukk-Valdre E, Nakamura H, Hmonen M, Lombardo C, Pihlajaniemi T, Alitalo K, Vuori K. Interaction of endostatin with integrins implicated in angiogenesis. ProcNatlAcadSci USA. 2001;98:1024–1029. doi: 10.1073/pnas.031564998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ulevitch RJ, Tobias PS. Receptor-dependent mechanisms of cell stimulation by bacterial endotoxin. Annual Review of Immunology. 1995;13:437–457. doi: 10.1146/annurev.iy.13.040195.002253. [DOI] [PubMed] [Google Scholar]
- 13.Andreu D, Rivas L. Animal antimicrobial peptides: an overview. Biopolymers. 1998;47:415–433. doi: 10.1002/(SICI)1097-0282(1998)47:6<415::AID-BIP2>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 14.Rifkind D. Prevention by polymyxin B of endotoxin lethality in mice. J Bacteriol. 1967;93:1463–1464. doi: 10.1128/jb.93.4.1463-1464.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee JY, Boman A, Sun CX, Andersson M, Jornvall H, Mutt V, Boman HG. Antibacterial peptides from pig intestine: isolation of a mammalian cecropin. Proc Natl Acad Sci USA. 1989;86:9159–9162. doi: 10.1073/pnas.86.23.9159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zasloff M, Martin B, Chen HC. Antimicrobial activity of synthetic magainin peptides and several analogues. Proc Natl Acad Sci USA. 1988;85:910–913. doi: 10.1073/pnas.85.3.910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Agerberth B, Lee JY, Bergman T, Carlquist M, Boman HG, Mutt V, Jornvall H. Amino acid sequence of PR-39. Isolation from pig intestine of a new member of the family of proline-arginine-rich antibacterial peptides. Eur J Biochemistry. 1991;202:849–854. doi: 10.1111/j.1432-1033.1991.tb16442.x. [DOI] [PubMed] [Google Scholar]
- 18.Matsuyama K, Natori S. Purification of three antibacterial proteins from the culture medium of NIH-Sape-4, an embryonic cell line of Sarcophaga peregrina. J Biol Chem. 1988;263:17112–17116. [PubMed] [Google Scholar]
- 19.Kawano K, Yoneya T, Miyata T, Yoshikawa K, Tokunaga F, Terada Y, Iwanaga S. Antimicrobial pep tide, tachyplesin I, isolated from hemocytes of the horseshoe crab (Tachypleus tridentatus). NMR determination of the beta-sheet structure. J Biol Chem. 1990;265:15365–15367. [PubMed] [Google Scholar]
- 20.Selsted ME, Harwig SS. Determination of the disulfide array in the human defensin HNP-2. A covalently cyclized peptide. J Biol Chem. 1989;264:4003–4007. [PubMed] [Google Scholar]
- 21.Lehrer RI, Ganz T, Selsted ME. Defensins: endogenous antibiotic peptides of animal cells. Cell. 1991;64:229–230. doi: 10.1016/0092-8674(91)90632-9. [DOI] [PubMed] [Google Scholar]
- 22.Mayo KH, Ilyina E, Park H. A recipe for designing water-soluble, beta-sheet-forming peptides. Protein Sci. 1996;5:1301–1315. doi: 10.1002/pro.5560050709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mayo KH, Haseman JR, Ilyina E, Gray B. Designed beta-sheet-forming peptide 33mers with potent human bactericidal/permeability increasing protein-like bactericidal and endotoxin neutralizing activities. Biochem Biophys Acta. 1998;1425:81–92. doi: 10.1016/s0304-4165(98)00053-1. [DOI] [PubMed] [Google Scholar]
- 24.Mayo KH, Haseman J, Young HC, Mayo JW. Structure-function relationships in novel peptide dodecamerswith broad-spectrum bactericidal and endotoxin-neutralizing activities. Biochem J. 2000;349(Pt 3):717–728. doi: 10.1042/bj3490717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Japelj B, Pristovsek P, Majerle A, Jerala R. Structural origin of endotoxin neutralization and antimicrobial activity of a lactoferrin-based peptide. J Biol Chem. 2005;280:16955–16961. doi: 10.1074/jbc.M500266200. [DOI] [PubMed] [Google Scholar]
- 26.Andra J, Lohner K, Blondelle SE, Jerala R, Moriyon I, Koch MH, Garidel P, Brandenburg K. Enhancement of endotoxin neutralization by coupling of a C12-alkyl chain to a lactoferricin-derived peptide. Biochem J. 2005;385:135–143. doi: 10.1042/BJ20041270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ilyina E, Roongta V, Mayo KH. NMR structure of a de novo designed, peptide 33mer with two distinct, compact beta-sheet folds. Biochemistry. 1997;36:5245–5250. doi: 10.1021/bi963064o. [DOI] [PubMed] [Google Scholar]
- 28.Beamer LJ, Carroll SF, Eisenberg D. Crystal structure of human BPI and two bound phospholipids at 2.4 angstrom resolution. Science. 1997;276:1861–1864. doi: 10.1126/science.276.5320.1861. [DOI] [PubMed] [Google Scholar]
- 29.Holak TA, Engstrom A, Kraulis PJ, Lindeberg G, Bennich H, Jones TA, Gronenborn AM, Clore GM. The solution conformation of the antibacterial peptide cecropin A: a nuclear magnetic resonance and dynamical simulated annealing study. Biochemistry. 1988;27:7620–7629. doi: 10.1021/bi00420a008. [DOI] [PubMed] [Google Scholar]
- 30.Marion D, Zasloff M, Bax A. A two-dimensional NMR study of the antimicrobial peptide magainin 2. FEBS Letters. 1988;227:21–26. doi: 10.1016/0014-5793(88)81405-4. [DOI] [PubMed] [Google Scholar]
- 31.Hanzawa H, Shimada I, Kuzuhara T, Komano H, Kohda D, Inagaki F, Natori S, Arata Y. 1H nuclear magnetic resonance study of the solution conformation of an antibacterial protein, sapecin. FEBS Lett. 1990;269:413–420. doi: 10.1016/0014-5793(90)81206-4. [DOI] [PubMed] [Google Scholar]
- 32.Rustici A, Velucchi M, Faggioni R, Sironi M, Ghezzi P, Quataert S, Green B, Porro M. Molecular mapping and detoxification of the lipid A binding site by synthetic peptides. Science. 1993;259:361–365. doi: 10.1126/science.8420003. [DOI] [PubMed] [Google Scholar]
- 33.Ferguson AD, Hofmann E, Coulton JW, Diederichs K, Welte W. Siderophore-mediated iron transport: crystal structure of FhuA with bound lipopolysaccharide. Science. 1998;282:2215–2220. doi: 10.1126/science.282.5397.2215. [DOI] [PubMed] [Google Scholar]
- 34.Pristovsek P, Kidric J. Solution structure of polymyxins B and E and effect of binding to lipopolysaccharide: an NMR and molecular modeling study. J Med Chem. 1999;42:4604–4613. doi: 10.1021/jm991031b. [DOI] [PubMed] [Google Scholar]
- 35.Gutsche CD, Bauer LJ. Calixarenes. 13 The conformational properties of calix[4]arenes, calix[6]arenes, calix[8]arenes, and oxacalixarenes. J Am Chem Soc. 1985;107:6052–6059. [Google Scholar]
- 36.Gutsche CD, Dhawan B, Levine JA, No KH, Bauer LJ. Calixarenes. 9 Conformational isomers of the ethers and esters of calix[4]arenes. Tetrahedron. 1983;39:406–426. [Google Scholar]
- 37.Cavanagh J, Fairbrother WJ, Palmer NJ, Skelton NJ. Protein NMR spectroscopy: principles and practice. Academic Press; San Diego: 1996. p. 587. [Google Scholar]
- 38.Ferguson AD, Welte W, Hofmann E, Lindner B, Hoist O, Coulton JW, Diederichs K. A conserved structural motif for lipopolysaccharide recognition by procaryotic and eucaryotic proteins. Structure. 2000;8:585–592. doi: 10.1016/s0969-2126(00)00143-x. [DOI] [PubMed] [Google Scholar]
- 39.Fairlie DP, West ML, Wong AK. Towards protein surface mimetics. CurrMed Chem. 1998;5:29–62. [PubMed] [Google Scholar]
- 40.Yin H, Hamilton AD. Terephthalamide derivatives as mimetics of the helical region of Bak peptide target Bcl-xL protein. Bioorg Med Chem Lett. 2004;14:1375–1379. doi: 10.1016/j.bmcl.2003.09.096. [DOI] [PubMed] [Google Scholar]
- 41.Yin H, Lee GI, Sedey KA, Rodriguez JM, Wang HG, Sebti SM, Hamilton AD. Terephthalamide derivatives as mimetics of helical peptides: disruption of the Bcl-x(L)/Bak interaction. J Am Chem Soc. 2005;127:5463–5468. doi: 10.1021/ja0446404. [DOI] [PubMed] [Google Scholar]
- 42.Kelso MJ, Beyer RL, Hoang HN, Lakdawala AS, Snyder JP, Oliver WV, Robertson TA, Appleton TG, Fairlie DP. Alpha-turn mimetics: short peptide alpha-helices composed of cyclic metallopentapeptide modules. J Am Chem Soc. 2004;126:4828–4842. doi: 10.1021/ja037980i. [DOI] [PubMed] [Google Scholar]
- 43.Loughlin WA, Tyndall JD, Glenn MP, Fairlie DP. Beta-strand mimetics. Chem Rev. 2004;704:6085–6117. doi: 10.1021/cr040648k. [DOI] [PubMed] [Google Scholar]
- 44.Schneider JP, Kelly JW. Templates that induce alpha-helical, beta-sheet, and loop conformations. Chem Rev. 1995;95:2169–2187. [Google Scholar]
- 45.Nowick JS, Pairish M, Lee IQ, Holmes DL, Ziller JW. An extended beta-strand mimic for a larger artificial beta-sheet. J Am Chem Soc. 1997;119:5413–5424. [Google Scholar]
- 46.Eguchi M, Kahn M. Design, synthesis, and application of peptide secondary structure mimetics. Mini Rev Med Chem. 2002;2:447–462. doi: 10.2174/1389557023405783. [DOI] [PubMed] [Google Scholar]
- 47.Kee KS, Jois SD. Design of beta-turn based therapeutic agents. Curr Pharm Des. 2003;9:1209–1224. doi: 10.2174/1381612033454900. [DOI] [PubMed] [Google Scholar]
- 48.Perkins JJ, Duong LT, Fernandez-Metzler C, Hartman GD, Kimmel DB, Leu CT, Lynch JJ, Prueksaritanont T, Rodan GA, Rodan SB, Duggan ME, Meissner RS. Non-peptide alpha(v)beta(3) antagonists: identification of potent, chain-shortened RGD mimetics that incorporate a central pyrrolidinone constraint. Bioorg Med Chem Lett. 2003;13:4285–4288. doi: 10.1016/j.bmcl.2003.09.055. [DOI] [PubMed] [Google Scholar]
- 49.Sheppard GS, Kawai M, Craig RA, Davidson DJ, Majest SM, Bell RL, Henkin J. Lysyl 4-aminobenzoic acid derivatives as potent small molecule mimetics of plasminogen kringle 5. Bioorg Med Chem Lett. 2004;14:965–966. doi: 10.1016/j.bmcl.2003.12.034. [DOI] [PubMed] [Google Scholar]
- 50.Hasegawa A, Cheng X, Kajino K, Berezov A, Murata K, Nakayama T, Yagita H, Murali R, Greene MI. Fas-disabling small exocyclic peptide mimetics limit apoptosis by an unexpected mechanism. Proc Natl Acad Sci USA. 2004;101:6599–6604. doi: 10.1073/pnas.0401597101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Alexopoulos K, Fatseas P, Melissari E, Vlahakos D, Roumelioti P, Mavromoustakos T, Mihailescu S, Paredes-Carbajal MC, Mascher D, Matsoukas J. Design and synthesis of novel biologically active thrombin receptor non-peptide mimetics based on the pharmacophoric cluster Phe/Arg/NH2 of the Ser42-Phe-Leu-Leu-Arg46 motif sequence: platelet aggregation and relaxant activities. J Med Chem. 2004;47:3338–3352. doi: 10.1021/jm031080v. [DOI] [PubMed] [Google Scholar]
- 52.Blaskovich MA, Lin Q, Delarue FL, Sun J, Park HS, Coppola D, Hamilton AD, Sebti SM. Design of GFB-111, a platelet-derived growth factor binding molecule with antiangiogenic and anticancer activity against human tumors in mice. Nat Biotechnol. 2000;18:1065–1070. doi: 10.1038/80257. [DOI] [PubMed] [Google Scholar]
- 53.Xuereb H, Maletic M, Pelczer I, Gildersleeve J, Kahne D. Design of an Oligosaccharide Scaffold That Binds in the Minor Groove of DNA. J Am Chem Soc. 2000;122:1883–1890. [Google Scholar]
- 54.Mayo KH, Ilyina E, Roongta V, Dundas M, Joseph J, Lai CK, Maione T, Daly TJ. Heparin binding to platelet factor-4. An NMR and site-directed mutagenesis study: arginine residues are crucial for binding. Biochem J. 1995;312(Pt 2):357–365. doi: 10.1042/bj3120357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Riordan JF, McElvany KD, Borders CL., Jr Arginyl residues: anion recognition sites in enzymes. Science. 1977;195:884–886. doi: 10.1126/science.190679. [DOI] [PubMed] [Google Scholar]
- 56.Mikhailov D, Young HC, Linhardt RJ, Mayo KH. Heparin dodecasaccharide binding to platelet factor-4 and growth-related protein-alpha. Induction of a partially folded state and implications for heparin-induced thrombocytopenia. J Biol Chem. 1999;274:25317–25329. doi: 10.1074/jbc.274.36.25317. [DOI] [PubMed] [Google Scholar]
- 57.Gutsche CD, Levine JA. Calixarenes. 6 Synthesis of a functionalizable calix[4]arene in a conformationally rigid cone conformation. J Am Chem Soc. 1982;104:2652–2653. [Google Scholar]
- 58.Arnaud-Neu F, Collins EM, Deasy M, Ferguson G, Harris SJ, Kaitner B, Lough AJ, McKervey MA, Marques E, Ruhl BL, Schwing-Weill MJ, Seward EM. Synthesis, X-ray crystal structures, and cation-binding properties of alkyl calixaryl esters and ketones, a new family of macrocyclic molecular receptors. J Am Chem Soc. 1989;111:8681–8691. [Google Scholar]
- 59.Bryant JLH, Yordanov AT, Linnoila JJ, Brechbiel MW, Frank JA. First noncovalently bound calix[4]arene - Gdffl - albumine complex. Angew Chem Int Ed Engl. 2000;39:1641–1643. doi: 10.1002/(sici)1521-3773(20000502)39:9<1641::aid-anie1641>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 60.Scheerder JJAM, Fochi M, Engbersen JFJ, Reinhoudt DN. Urea-derivatized p-tert-butylcalix[4]arenes: neutral ligands for selective anion complexation. J Org Chem. 1994;59:7815–7820. [Google Scholar]
- 61.Wu P, Feldman AK, Nugent AK, Hawker CJ, Scheel A, Voit B, Pyun J, Frechet JM, Sharpless KB, Fokin VV. Efficiency and fidelity in a click-chemistry route to triazole dendrimers by the copper(i)-catalyzed ligation of azides and alkynes. Angew Chem Int Ed Engl. 2004;43:3928–3932. doi: 10.1002/anie.200454078. [DOI] [PubMed] [Google Scholar]
- 62.Young NS, Levin J, Prendergast RA. An invertebrate coagulation system activated by endotoxin: evidence for enzymatic mediation. J Clin Invest. 1972;51:1790–1797. doi: 10.1172/JCI106980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 64.Jerschow A, Muller N. Suppression of convection artifacts in stimulated-echo diffusion experiments. Double-stimulated-echo experiments. J Magn Reson. 2000;142:323–325. [Google Scholar]
- 65.Nesmelova IV, Sham Y, Dudek AZ, van Eijk LI, Wu G, Slungaard A, Mortari E, Griffioen AW, Mayo KH. Platelet factor 4 and interleukin-8 CXC chemokine heterodimer formation modulates function at the quaternary structural level. J Biol Chem. 2005;280:4948–4958. doi: 10.1074/jbc.M405364200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Full spectroscopic characterization data of calixarene derivatives 2– 4, 6–12, 15, 19–23. This material is available free of charge via the Internet at http://pubs.acs.org.