

Abstract
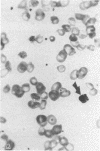
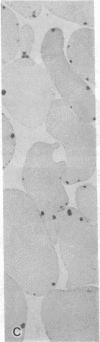
Hemoglobin Mississippi (HbMS: beta 44ser----cys) has anomalous properties that include disulfide linkages with normal beta-, delta-, gamma-, and alpha-chains, and the formation of high molecular weight multimers. While heterozygotes for HbMS are clinically and hematologically normal and carriers of the beta +-thalassemia gene in our family had mild microcytic anemia, the proband with HbMS-beta +-thalassemia had a hemoglobin level of 7 g/dl, mean corpuscular volume (MCV) of 68 fl, reticulocytes of 2-6%, HbF of 18%, marked anisocytosis and poikilocytosis, and splenomegaly, all features of thalassemia intermedia. With oxidant stress, her erythrocytes developed multiple dispersed Heinz bodies, but HbMS was only mildly unstable. HbMS was susceptible to proteolytic degradation in the presence of ATP. The unexpectedly severe clinical findings in HbMS-beta +-thalassemia may result from the proteolytic digestion of HbMS, as well as the excessive alpha-chains characteristic of beta +-thalassemia, which combined provide the increment of cellular damage that results in the phenotype of thalassemia intermedia.
Full text
PDF







Images in this article






Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Adams J. G., 3rd, Boxer L. A., Baehner R. L., Forget B. G., Tsistrakis G. A., Steinberg M. H. Hemoglobin Indianapolis (beta 112[G14] arginine). An unstable beta-chain variant producing the phenotype of severe beta-thalassemia. J Clin Invest. 1979 May;63(5):931–938. doi: 10.1172/JCI109393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams J. G., 3rd, Steinberg M. H., Newman M. V., Morrison W. T., Benz E. J., Jr, Iyer R. beta-Thalassemia present in cis to a new beta-chain structural variant, Hb Vicksburg [beta 75 (E19)Leu leads to 0]. Proc Natl Acad Sci U S A. 1981 Jan;78(1):469–473. doi: 10.1073/pnas.78.1.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonarakis S. E., Irkin S. H., Cheng T. C., Scott A. F., Sexton J. P., Trusko S. P., Charache S., Kazazian H. H., Jr beta-Thalassemia in American Blacks: novel mutations in the "TATA" box and an acceptor splice site. Proc Natl Acad Sci U S A. 1984 Feb;81(4):1154–1158. doi: 10.1073/pnas.81.4.1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BAGLIONI C. ABNORMAL HUMAN HEMOGLOBINS. X. A STUDY OF HEMOGLOBIN LEPORE BOSTON. Biochim Biophys Acta. 1965 Jan 4;97:37–46. doi: 10.1016/0304-4165(65)90267-9. [DOI] [PubMed] [Google Scholar]
- BAGLIONI C. The fusion of two peptide chains in hemoglobin Lepore and its interpretation as a genetic deletion. Proc Natl Acad Sci U S A. 1962 Nov 15;48:1880–1886. doi: 10.1073/pnas.48.11.1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BETKE K., MARTI H. R., SCHLICHT I. Estimation of small percentages of foetal haemoglobin. Nature. 1959 Dec 12;184(Suppl 24):1877–1878. doi: 10.1038/1841877a0. [DOI] [PubMed] [Google Scholar]
- Baird M., Schreiner H., Driscoll C., Bank A. Localization of the site of recombination in formation of the Lepore Boston globin gene. J Clin Invest. 1981 Aug;68(2):560–564. doi: 10.1172/JCI110289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballas S. K., Burka E. R., Gill F. M. Abnormal red cell membrane proteolytic activity in severe heterozygous beta-thalassemia. J Lab Clin Med. 1982 Feb;99(2):263–274. [PubMed] [Google Scholar]
- Bank A., O'Donnell J. V. Intracellular loss of free alpha chains in beta thalassemia. Nature. 1969 Apr 19;222(5190):295–296. doi: 10.1038/222295a0. [DOI] [PubMed] [Google Scholar]
- Boches F. S., Goldberg A. L. Role for the adenosine triphosphate-dependent proteolytic pathway in reticulocyte maturation. Science. 1982 Feb 19;215(4535):978–980. doi: 10.1126/science.7156977. [DOI] [PubMed] [Google Scholar]
- Botbol V., Scornik O. A. Degradation of abnormal proteins in intact mouse reticulocytes: accumulation of intermediates in the presence of bestatin. Proc Natl Acad Sci U S A. 1979 Feb;76(2):710–713. doi: 10.1073/pnas.76.2.710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalevelakis G., Clegg J. B., Weatherall D. J. Imbalanced globin chain synthesis in heterozygous beta-thalassemic bone marrow. Proc Natl Acad Sci U S A. 1975 Oct;72(10):3853–3857. doi: 10.1073/pnas.72.10.3853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clegg J. B., Naughton M. A., Weatherball D. J. Abnormal human haemoglobins. Separation and characterization of the alpha and beta chains by chromatography, and the determination of two new variants, hb Chesapeak and hb J (Bangkok). J Mol Biol. 1966 Aug;19(1):91–108. doi: 10.1016/s0022-2836(66)80052-9. [DOI] [PubMed] [Google Scholar]
- Clegg J. B., Weatherall D. J., Milner P. F. Haemoglobin Constant Spring--a chain termination mutant? Nature. 1971 Dec 10;234(5328):337–340. doi: 10.1038/234337a0. [DOI] [PubMed] [Google Scholar]
- Etlinger J. D., Goldberg A. L. A soluble ATP-dependent proteolytic system responsible for the degradation of abnormal proteins in reticulocytes. Proc Natl Acad Sci U S A. 1977 Jan;74(1):54–58. doi: 10.1073/pnas.74.1.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagan J. M., Waxman L., Goldberg A. L. Red blood cells contain a pathway for the degradation of oxidant-damaged hemoglobin that does not require ATP or ubiquitin. J Biol Chem. 1986 May 5;261(13):5705–5713. [PubMed] [Google Scholar]
- Fessas P., Loukopoulos D., Loutradi-Anagnostou A., Komis G. 'Silent' beta-thalassaemia caused by a 'silent' beta-chain mutant: the pathogenesis of a syndrome of thalassaemia intermedia. Br J Haematol. 1982 Aug;51(4):577–583. doi: 10.1111/j.1365-2141.1982.tb02821.x. [DOI] [PubMed] [Google Scholar]
- Flavell R. A., Kooter J. M., De Boer E., Little P. F., Williamson R. Analysis of the beta-delta-globin gene loci in normal and Hb Lepore DNA: direct determination of gene linkage and intergene distance. Cell. 1978 Sep;15(1):25–41. doi: 10.1016/0092-8674(78)90080-6. [DOI] [PubMed] [Google Scholar]
- Goldberg A. L., Dice J. F. Intracellular protein degradation in mammalian and bacterial cells. Annu Rev Biochem. 1974;43(0):835–869. doi: 10.1146/annurev.bi.43.070174.004155. [DOI] [PubMed] [Google Scholar]
- Goossens M., Lee K. Y., Liebhaber S. A., Kan Y. W. Globin structural mutant alpha 125Leu leads to Pro is a novel cause of alpha-thalassaemia. Nature. 1982 Apr 29;296(5860):864–865. doi: 10.1038/296864a0. [DOI] [PubMed] [Google Scholar]
- Hanash S. M., Rucknagel D. L. Proteolytic activity in erythrocyte precursors. Proc Natl Acad Sci U S A. 1978 Jul;75(7):3427–3431. doi: 10.1073/pnas.75.7.3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershko A., Ciechanover A., Rose I. A. Resolution of the ATP-dependent proteolytic system from reticulocytes: a component that interacts with ATP. Proc Natl Acad Sci U S A. 1979 Jul;76(7):3107–3110. doi: 10.1073/pnas.76.7.3107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honig G. R., Shamsuddin M., Zaizov R., Steinherz M., Solar I., Kirschmann C. Hemoglobin Petah Tikva (alpha 110 ala replaced by asp): a new unstable variant with alpha-thalassemia-like expression. Blood. 1981 Apr;57(4):705–711. [PubMed] [Google Scholar]
- Klemes Y., Etlinger J. D., Goldberg A. L. Properties of abnormal proteins degraded rapidly in reticulocytes. Intracellular aggregation of the globin molecules prior to hydrolysis. J Biol Chem. 1981 Aug 25;256(16):8436–8444. [PubMed] [Google Scholar]
- Lessin L. S., Jensen W. N., Klug P. Ultrastructure of the normal and hemoglobinopathic red blood cell membrane. Freeze-etching and stereoscan electron microscopic studies. Arch Intern Med. 1972 Feb;129(2):306–319. [PubMed] [Google Scholar]
- Liebhaber S. A., Kan Y. W. alpha-Thalassemia caused by an unstable alpha-globin mutant. J Clin Invest. 1983 Mar;71(3):461–466. doi: 10.1172/JCI110790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nienhuis A. W., Anagnou N. P., Ley T. J. Advances in thalassemia research. Blood. 1984 Apr;63(4):738–758. [PubMed] [Google Scholar]
- Orkin S. H., Kazazian H. H., Jr, Antonarakis S. E., Ostrer H., Goff S. C., Sexton J. P. Abnormal RNA processing due to the exon mutation of beta E-globin gene. Nature. 1982 Dec 23;300(5894):768–769. doi: 10.1038/300768a0. [DOI] [PubMed] [Google Scholar]
- Polliack A., Rachmilewitz E. A. Ultrastructural studies in -thalassaemia major. Br J Haematol. 1973 Mar;24(3):319–326. doi: 10.1111/j.1365-2141.1973.tb01656.x. [DOI] [PubMed] [Google Scholar]
- Rice R. H., Means G. E. Radioactive labeling of proteins in vitro. J Biol Chem. 1971 Feb 10;246(3):831–832. [PubMed] [Google Scholar]
- Rieder R. F. Human hemoglobin stability and instability: molecular mechanisms and some clinical correlations. Semin Hematol. 1974 Oct;11(4):423–440. [PubMed] [Google Scholar]
- Rieder R. F., Ibrahim A., Etlinger J. D. A soluble adenosine triphosphate-dependent proteolytic system in human peripheral red blood cells. Blood. 1986 May;67(5):1293–1297. [PubMed] [Google Scholar]
- Sancar G. B., Cedeno M. M., Rieder R. F. Rapid destruction of newly synthesized excess beta-globin chains in HbH disease. Blood. 1981 May;57(5):967–971. [PubMed] [Google Scholar]
- Sanguansermsri T., Matragoon S., Changloah L., Flatz G. Hemoglobin Suan-Dok (alpha 2 109 (G16) Leu replaced by Arg beta 2): an unstable variant associated with alpha-thalassemia. Hemoglobin. 1979;3(2-3):161–174. doi: 10.3109/03630267908998911. [DOI] [PubMed] [Google Scholar]
- Shaeffer J. R. Turnover of excess hemoglobin alpha chains in beta-thalassemic cells is ATP-dependent. J Biol Chem. 1983 Nov 10;258(21):13172–13177. [PubMed] [Google Scholar]
- Smith C. M., 2nd, Hedlund B., Cich J. A., Tukey D. P., Olson M., Steinberg M. H., Adams J. G., 3rd Hemoglobin North Shore: a variant hemoglobin associated with the phenotype of beta-thalassemia. Blood. 1983 Feb;61(2):378–383. [PubMed] [Google Scholar]
- Speiser S., Etlinger J. D. Loss of ATP-dependent proteolysis with maturation of reticulocytes and erythrocytes. J Biol Chem. 1982 Dec 10;257(23):14122–14127. [PubMed] [Google Scholar]
- Steinberg M. H., Adams J. G. Thalassemic hemoglobinopathies. Am J Pathol. 1983 Dec;113(3):396–409. [PMC free article] [PubMed] [Google Scholar]
- Testa U., Hinard N., Beuzard Y., Tsapis A., Galacteros F., Thomopoulos P., Rosa J. Excess alpha chains are lost from beta-thalassemic reticulocytes by proteolysis. J Lab Clin Med. 1981 Sep;98(3):352–363. [PubMed] [Google Scholar]
- Traeger J., Winichagoon P., Wood W. G. Instability of beta E-messenger RNA during erythroid cell maturation in hemoglobin E homozygotes. J Clin Invest. 1982 Apr;69(4):1050–1053. doi: 10.1172/JCI110510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vettore L., De Matteis M. C., Di Iorio E. E., Winterhalter K. H. Erythrocytic proteases: preferential degradation of alpha hemoglobin chains. Acta Haematol. 1983;70(1):35–42. doi: 10.1159/000206686. [DOI] [PubMed] [Google Scholar]