

Abstract
• Background and Aims Native horseradish (Armoracia rusticana) peroxidase, HRP (EC 1.11.1.7), isoenzyme C is synthesized with N‐terminal and C‐terminal peptide extensions, believed to be associated with protein targeting. This study aimed to explore the specific functions of these extensions, and to generate transgenic plants with expression patterns suitable for exploring the role of peroxidase in plant development and defence.
• Methods Transgenic Nicotiana tabacum (tobacco) plants expressing different versions of a synthetic horseradish peroxidase, HRP, isoenzyme C gene were constructed. The gene was engineered to include additional sequences coding for either the natural N‐terminal or the C‐terminal extension or both. These constructs were placed under the control of a constitutive promoter (CaMV‐35S) or the tobacco RUBISCO‐SSU light inducible promoter (SSU) and introduced into tobacco using Agrobacterium‐mediated transformation. To study the effects of the N‐ and C‐terminal extensions, the localization of recombinant peroxidase was determined using biochemical and molecular techniques.
• Key Results Transgenic tobacco plants can exhibit a ten‐fold increase in peroxidase activity compared with wild‐type tobacco levels, and the majority of this activity is located in the symplast. The N‐terminal extension is essential for the production of high levels of recombinant protein, while the C‐terminal extension has little effect. Differences in levels of enzyme activity and recombinant protein are reflected in transcript levels.
• Conclusions There is no evidence to support either preferential secretion or vacuolar targeting of recombinant peroxidase in this heterologous expression system. This leads us to question the postulated targeting roles of these peptide extensions. The N‐terminal extension is essential for high level expression and appears to influence transcript stability or translational efficiency. Plants have been generated with greatly elevated cytosolic peroxidase activity, and smaller increases in apoplastic activity. These will be valuable for exploring the role of these enzymes in stress amelioration and plant development.
Key words: Armoracia rusticana, Nicotiana tabacum, horseradish peroxidase, protein targeting, N‐terminal and C‐terminal extensions
INTRODUCTION
Peroxidases are involved in many important plant processes, such as the crosslinking of molecules to constituent polymers of the cell wall, auxin oxidation, and responses to biotic and abiotic stresses (Fukuda, 1996; Kristensen et al., 1997; Amaya et al., 1999). The most extensively studied plant peroxidases are those of horseradish (Veitch and Smith, 2001), Armoracia rusticana, which include the first for which the amino acid sequence was determined (Welinder, 1979). The gene encoding this isoenzyme, horseradish peroxidase C, HRP‐C (EC 1.11.1.7), has since been sequenced (Fujiyama et al., 1988) and subsequently a synthetic version was produced and expressed in heterologous systems as diverse as Escherichia coli (Smith et al., 1990) and tobacco (Pellegrineschi et al., 1995). An important feature of HRP‐C is that the pre‐protein has both N‐terminal (30 residues) and C‐terminal (15 residues) amino acid extensions (Fujiyama et al., 1988). N‐terminal extensions, normally 20–50 amino acids in length with an internal stretch of at least six hydrophobic amino acids, are known to act as signal peptides targeting proteins to the endoplasmic reticulum (ER), whence they are delivered to the tonoplast or the cell wall (Chrispeels, 1991). This process involves cleavage of the signal peptide and translocation of the protein through the membrane into the cisternal space of the ER. The protein is retained in the ER lumen if it has a carboxy‐terminal domain with the four amino acids HDEL or KDEL (Wandelt et al., 1992). Correctly folded proteins then enter the Golgi. The Golgi apparatus plays a major role in post‐translation modifications of secretory protein, such as oligomers being transformed to glycans or proteins previously glycoslyated in the ER being modified to more complex glycans using several glycosidases and glycosyl‐transferases (Staehelin and Moore, 1995). From the Golgi the protein can be transported either to the vacuole (Chrispeels and Raikhel, 1992) or to the extracellular space.
If there is no retention signal, or further targeting signals, then the most likely fate of the protein is secretion by the ‘bulk flow’ pathway to the plasma membrane in secretory vesicles (Bednarek et al., 1990). However, other evidence suggests that the absence of retention signals does not necessarily lead to secretion (Neuhaus et al., 1991).
In a tobacco cell‐culture system it has been demonstrated recently that the N‐ and C‐terminal extensions on HRP‐C are associated with vacuolar targeting (Matsui et al., 2003). To investigate the roles of these sequences in intact plants, a number of chimeric constructs were produced and introduced into tobacco. The constructs were based on a synthetic HRP isoenzyme C gene produced by Smith et al. (1990), with or without the N‐ or C‐terminal extensions and under the control of the constitutive cauliflower mosaic virus (CaMV) 35S, or leaf‐specific RUBISCO small subunit (SSU), promoters as described by Pellegrineschi et al. (1995). In addition to providing valuable information on peroxidase targeting, it was hoped that these studies would provide transgenic material differentially expressing recombinant peroxidase in different compartments, which would be valuable for unraveling the role of peroxidases in plant development. For example, previous investigations have shown that over‐expression of horseradish peroxidase in transgenic tobacco can stimulate growth (Kawaoka et al., 1994).
MATERIALS AND METHODS
Production of transformation constructs and transgenic tobacco plants
These have been described previously (Pellegrineschi et al., 1995). Briefly, a 120 bp HindIII–HpaI fragment, derived from sequences encoding the N‐terminal half of HRP isoenzyme C, was cloned into pUC18‐HRP to give the plasmid pPUPN which encodes the synthetic HRP gene with the natural N‐terminal extension (Smith et al., 1990). HRP constructs pPUPC, with synthetic C‐terminal coding sequences, and pPUPNC, with both N‐ and C‐terminal coding sequences were also produced (Fig. 1). The HRP construct was excised from the pPUP, pPUPN, pPUPC and pPUPNC and placed under the control of the constitutive cauliflower mosaic virus (CaMV) 35S promoter and a tobacco light‐inducible ribulose 1,5‐bisphosphate carboxylase small subunit (RUBISCO‐SSU) promoter (Mazur and Chui, 1985). The constructs were then transformed into the binary vector pBIN19, which supports the kanamycin resistance gene (nptII) (Pellegrineschi et al., 1995). All cloning methods were performed essentially as described in the standard manual (Sambrook et al., 1989). The constructs were transformed into Agrobacterium tumefaciens (LBA 4404) as described by Bevan (1984), and introduced into Nicotiana tabacum L. ‘Samsun NN’ using the leaf disc transformation procedure of Horsch et al. (1985). The regenerated tobacco shoots were selected on kanamycin (100 mg l–1) and maintained in a growth room at 25 °C with a 16/8 h light/dark photoperiod.

Fig. 1. Horseradish peroxidase (HRP) gene and extension permutations used. The HRP‐C1 transgene was cloned as a BamHI restriction fragment between the CaMV35S promoter (CaMV) and the nopaline synthase (nos) terminator in pBin19. The HRP‐C1 transgene consists of the coding sequence of the synthetic HRP‐C gene described by Smith et al. (1990) and additional coding sequences that specify the N‐terminal and C‐terminal amino acid extensions found on the precursor form but not the processed form of the HRP‐C1 protein. The amino acid sequences of the N‐ and C‐terminal extensions are shown.
Cellular fractionation
A modified form of the method of McDougall (1991) was used. Ten 10‐mm‐diameter leaf discs were cut with a cork borer, weighed and washed in sterile water to remove contaminants. They were then submerged into sterile distilled water (5 ml) and vacuum infiltrated at 15 mmHg for 5 min. They were left for 10 min to allow the water to infiltrate into the evacuated wall and air spaces. The discs were dabbed dry on filter paper and inserted into 5‐ml syringe barrels placed inside 30‐ml universals. After centrifugation at 2400 g for 15 min the infiltrate was collected and used to determine the soluble activity from the cell wall and extracellular space. Another ten discs were prepared, weighed and the above procedure was repeated using 10 mm CaCl2 in place of water. The infiltrate collected was used to determine soluble and ionically bound activity in the cell wall and extracellular space. The leaf material remaining after this extraction was ground in liquid N2 to which 5 ml of 10 mm sodium phosphate, pH 7·2 was added. The homogenate was centrifuged at 25 600 g and the supernatant was used to determine residual (i.e. intracellular) activity. The pellet was retained and digested overnight at 25 °C with an enzyme solution containing 1 g cellulase and 600 mg macerozyme in 100 ml 10 mm sodium phosphate buffer, pH 7·2. After centrifugation at 25 600 g the supernatant was used to determine cell‐wall‐bound activity. Total protein was quantified according to the standard assay (Bradford, 1976) with BSA as the standard protein.
Protoplast isolation
Five large leaves from tobacco plants grown in vitro were cut up into thin strips and placed into a digestive buffer [20 ml K3 (Maliga, 1984) with 0·4 m sucrose, 0·2 g cellulase, 0·06 g macerozyme] overnight. The material was then filtered through nylon mesh (0·45 µm) and the filtrate was used to determine the peroxidase activity released during protoplast isolation. The remaining filtrate was poured into two sterile narrow tubes and spun (300 g, 3 min). Protoplasts floated to the top to form a layer, which was transferred to fresh tubes. An excess of W5 solution (Maliga, 1984) was added, mixed and centrifuged gently for 2 min at 50 g. The supernatant was removed leaving the protoplasts as a pellet.
Peroxidase assay
Activity was determined using the peroxidase substrate guaiacol. The reaction mixture consisted of 0·28 % guaiacol (Sigma, Dublin, Ireland) and 0·30 % H2O2 (Sigma) in 100 ml 0·05 M Na2HPO4, pH 6·2. A sample (1–20 µl) was added to 1 ml of reaction mixture and the increase in absorbance was measured for 3 min at 30‐s intervals at 420 nm (Zimmerlan and Bolwell, 1993). The rate of this reaction was used to express peroxidase activity.
Electrophoretic protein analysis and immunoblotting
Protein was quantified according to the method of Bradford (1976) with BSA as the standard protein. A modified Bio‐Rad (Hemel Hampstead, UK) protein assay kit was used. Total extracts were separated on 13 % SDS–PAGE gels according to Laemmli (1970) and stained with Commassie Blue G250 or blotted by wet transfer, using the Bio‐Rad ‘mini protean’, onto 0·45 µm PVDF membranes (Amersham, Little Chalfont, UK) for immunodetection. Membranes were probed with a primary antibody directed against HRP (DAKO, Ely, UK) diluted to 1 : 1000 in TBST (50 mm Tris–HCl, pH 7·4, 150 mm NaCl and 0·05 % Tween20). The secondary antibody was alkaline phosphatase conjugated anti‐rabbit IgG (Sigma) diluted 1 : 2000 in TBST. The blots were washed and developed, as described by Harlow and Lane (1988), with nitroblue tetrazoliun and 5‐bromo‐4‐chloro‐3‐indolyl phosphate (BCIP) as substrates.
RNA extraction and Northern analysis
Leaf tissue was collected, frozen in liquid nitrogen and ground to a fine powder. Total RNA was extracted as described by Eggermont et al. (1996). RNA (1·5 µg per sample) was run on a 1 % formaldehyde–agarose gel, blotted onto nitrocellulose and fixed using a transillumintor. A 32P‐labelled HRP‐C probe was prepared according to Feinberg and Vogelstein (1983) and the blot was hybridized, washed and exposed to X‐ray film (Kodak) at –70 °C either for a few hours or overnight.
RESULTS
Subcellular fractionation to assign peroxidase activity to specific cellular compartments.
For each construct analysed in the following experiments two independent transformant lines (except for ROPN‐SSU, where a single line was available) were used and the assays were performed on four separate, clonally propagated, 6‐week‐old plants. Peroxidase activities from the different extracts are illustrated in Fig. 2. In both CaMV 35S‐ and SSU‐promoter plants in which the HRP lacks the N‐terminal extension, there was little or no increase in peroxidase activity levels, compared with control tobacco plants. When the N‐terminal extension is present (i.e. ROPN and ROPNC transgenic lines), a higher level of peroxidase activity was found. Figure 2A shows that soluble peroxidase is present in the apoplast, while the higher activities in Fig. 2B illustrate that there is an additional fraction ionically bound to the cell wall. The values in Fig. 2D represent another cell wall component, a fraction not removed by other extraction methods, and therefore in a tighter (possibly covalent) linkage to the cell wall. The same trend is observed as that found in the other apoplastic fractions. Increased peroxidase levels appear to require the N‐terminal extension, and increases are up to about three‐fold, in comparison with non‐transformed tobacco.
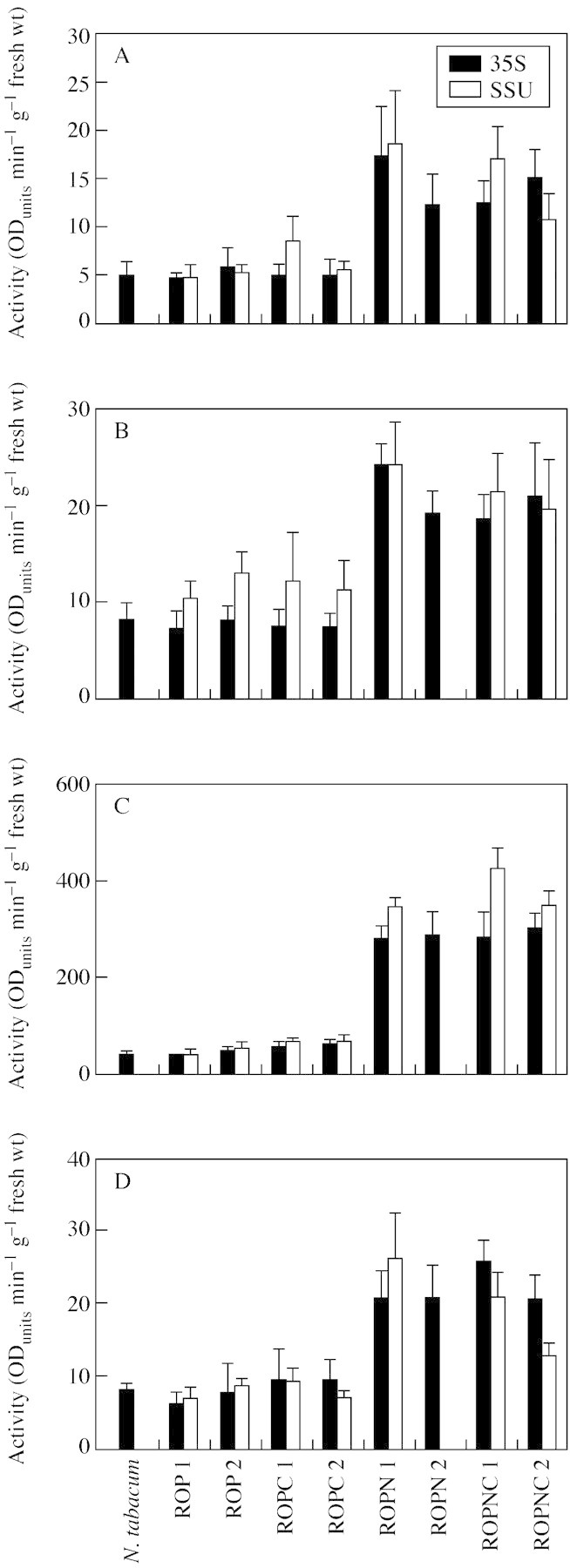
Fig. 2. Subcellular fractionation to determine the peroxidase activity of cellular components. (A) Soluble extracellular peroxidase activity was extracted by vacuum infiltration of leaf material with distilled water. Leaf material was used from two lines for each construct, i.e. ROP, ROPC, ROPN, ROPNC. Wild‐type N. tabacum served as a negative control. Ten 10‐mm leaf discs were submerged in water, vacuum infiltrated at 15 mgHg for 5 min. Discs were dried on filter paper and centrifuged to collect the extracellular fluid. The activity of the fluid was calculated using the peroxidase substrate guaiacol and expressed as ODunits min–1 g–1 f. wt. (B) Soluble plus ionically bound extracellular activity extracted by vacuum infiltrated of leaf material with CaCl2. Another ten 10‐mm leaf discs were cut from the same plant and submerged in CaCl2 and vacuum infiltrated as before. The dried discs were centrifuged at 2400 g for 15 min to collect soluble and ionically bound extracellular peroxidase. The peroxidase activity of the extract was calculated and expressed as ODunits min–1 g–1 f. wt. (C) soluble peroxidase activity remaining in leaf material after the removal of extracellular soluble and ionically bound activity. The discs used in the CaCl2 infiltration step were ground in liquid nitrogen. The powder was homogenized in 5 ml of 10 mm sodium phosphate, pH 7·2. The homogenate was centrifuged at 1800 g and the supernatant was used to calculate the residual peroxidase activity. (D) Peroxidase activity released from the cell wall material. The pellet remaining after grinding leaf material in liquid nitrogen was resuspended in 10 mm sodium phosphate buffer with 1 g cellulase and 600 mg of macerozyme and left overnight. After centrifuging at 25 600 g for 5 min the supernatant was used to calculate the peroxidase activity covalently bound to the cell wall. All results are based on the mean of four replicates and activity is expressed as ODunits min–1 g–1 f. wt.
Figure 2C displays the values for residual activity associated with cytoplasm, cytoplasmically located organelles and vacuoles, i.e. all compartments bounded by the plasmalemma. The same trend was observed as for the apoplastic fractions, except that much higher levels of activity were found. When the N‐terminal was present, this level represented almost a ten‐fold increase on wild‐type N. tabacum, constituting the majority of the total activity.
The total activity of peroxidase secreted into the cell wall was determined by adding together the values for the CaCl2 extraction and the enzymic digestion of the cell wall. These are represented as a percentage of the total activity (Fig. 3). When the N‐terminal extension is present total activity increases by at least four‐fold. However, despite this absolute increase, the proportion of the total peroxidase in the cell walls (Fig. 3B) of ROPN and ROPNC lines is low compared with the rest of the lines.

Fig. 3. Total peroxidase activity and the percentage of total extracted peroxidase that is located in the cell wall and extracellular space. The total values (A) are the pooled values from the CaCl2 extract, the covalently bound (enzymically digested) extract and soluble peroxidase activity remaining in leaf material after the removal of extracellular soluble and ionically bound activity. Percentage wall activity (B) is the CaCl2 extract plus covalently bound extract, expressed as a percentage of the total activity.
Peroxidase release during protoplast production by enzymic digestion of leaf tissue
Protoplasts were isolated by digesting leaf material overnight with the cell‐wall‐degrading enzymes, cellulase and macerozyme. This technique was used to look at the level of peroxidase activity released while removing the cell wall. The gradual release of peroxidase over the time it takes to digest away the cell wall is illustrated in Fig. 4A. As predicted from the low expression levels with the ROP constructs, these treatments show little increase in peroxidase release compared with controls. However, a higher level of peroxidase is released when the N‐terminal extension is present, i.e. in ROPN or ROPNC plants. A high level of activity is retained by purified protoplasts (Fig. 4B). These results confirm the distribution of peroxidase between apoplast and symplast suggested by the fractionation studies.


Fig. 4. Peroxidase activity released during protoplast isolation and the activity in protoplasts derived from transgenic tobacco plants. (A) The measurement of peroxidase activity released into the medium during enzymatic digest of leaf material to isolate protoplasts. Leaf material was digested with 0·2 g of cellulase and 0·06 g of macerozyme overnight. The material was filtered and the filtrate was used to determine the activity of peroxidase released during protoplast isolation. (B) The measurement of peroxidase activity in cultured protoplasts. Preparation of leaf material was as before. This time the filtrate was centrifuged at 300 g for 3 min leaving the protoplasts floating, forming a layer on the top. The layer of protoplasts was carefully removed into fresh tubes with the addition of W5 medium (Maliga, 1984). They were centrifuged at 50 g for 2 min to precipitate the protoplasts. Protoplasts were cultured in Petri dishes and kept under low light. After a set number of days, protein was isolated from the protoplasts and its peroxidase activity was measured.
Western blot analysis
All the different fractionation extracts were subjected to Western analysis to confirm that elevated levels of peroxidase activity were due to recombinant protein (Fig. 5). In all cases, horseradish peroxidase with a molecular weight of 45 kDa was detected in the ROPN and ROPNC lanes. The double band may be due to differences in glycosylation or incomplete removal of the N‐terminal extension. Qualitative differences in band intensity correlate well with the differences in enzyme activity (Fig. 2).

Fig. 5. Immunoblot analysis to show the presence of recombinant protein. (A) Soluble and extracellular space protein extracted by vacuum infiltration with water. Extract remaining from the first step of the cellular fractionation experiment, i.e. vacuum infiltration of ten 10‐mm leaf discs in distilled water followed by centrifugation to collect soluble and extracellular space protein. Twenty micrograms of protein were loaded on to the 13 % SDS gel, run and blotted onto PVDF membrane. Anti‐horseradish peroxidase was used as primary antibody (1 : 1000) and HRP was detected with the secondary antibody (1 : 2000) anti‐rabbit IgG‐alkaline phosphatase conjugated. The blot was developed with NBT and BCIP. Lane 1, wild‐type N. tabacum; lanes 2 and 3, ROP; lanes 4 and 5, ROPC; lanes 6 and 7 (35S) or lane 6 (SSU), ROPN; lanes 8 and 9 (35S) or lanes 7 and 8 (SSU), ROPNC. (B) Soluble, ionically, cell‐wall‐bound and extracellular space protein extracted by vacuum infiltration with CaCl2. Extract remaining from the CaCl2 step of the cellular fractionation experiment was treated as (A). Samples were run in the same order as (A). (C) Residual soluble protein extracted from leaf material after the extraction of extracellular soluble and ionically bound activity by vacuum infiltration with CaCl2. Leaf material left after the extraction of extracellular and ionically bound protein was ground in liquid nitrogen and extracted with 10 mm sodium phosphate buffer, pH 7·2. The extract was treated as (A). (D) Covalently bound cell wall protein extracted by enzymatic degradation from the cell residue remaining after the extract of residual soluble protein from leaf material. Again the extract was treated as in (A).
Northern blot analysis
Northern analysis was performed to determine whether the role of the N‐terminal extension in the production of recombinant protein, and enzyme activity, was reflected in the transcript levels. Prominent bands were obtained for the ROPN and ROPNC lines (Fig. 6). Very little mRNA was detected for ROP and none at all for ROPC.

Fig. 6. Northern blot detection of HRP‐C mRNA. Total RNA isolated from ROP, ROPC, ROPNC, ROPN and wild‐type N. tabacum (1·5 µg per sample) was electrophoresed in a formaldehyde gel, and blotted onto a nitrocellulose membrane. A 32P‐labelled HRP‐C probe was used to detect the transgene mRNA. Lane 1, wild‐type N. tabacum; lane 2, ROP; lane 3, ROPN; lane 4, ROPC; lane 5, ROPNC. The arrow indicates the position of a product size of 1·1 kb, the expected size for the HRP‐C transcript.
DISCUSSION
The present study was initiated to evaluate the roles of the N‐ and C‐terminal extensions of horseradish peroxidase C in protein targeting. In the process, it was hoped distinct transgenic tobacco lines would be generated with differential over‐expression of peroxidase enzyme in distinct cellular compartments, specifically cytoplasm, vacuole and apoplast. These could provide an invaluable tool for exploring the role of peroxidases in plant development and stress responses.
Although plants in which the HRP‐C gene was under the control of both constitutive (CaMV 35S) and leaf‐specific (SSU) promoters were included in the study, the characterization reported here was carried out with photosynthetic tissue, and very similar levels of transgene expression were obtained using both promoters. Differences in the presence or absence of the N‐ and C‐terminal extensions, however, had a profound influence on levels of transgene expression. An early report on expression of the same synthetic gene in Escherichia coli (Smith et al., 1990) indicated that neither the N‐ nor C‐terminal extension were necessary for either activity or folding of recombinant protein. However, in a eukaryotic system the N‐terminal extension is responsible for targeting to the secretory pathway via the endoplasmic reticulum. This could explain the lack of both enzyme activity and immunodectable protein in the absence of the N‐terminal extension in the present study. Enzyme activity depends on disulfide bond formation, and immunodetection may require glycosylation, both of which are dependent on targeting to the endoplasmic reticulum. However, the absence of transcript in the ROP and ROPC lines suggests the N‐terminal extension may also influence mRNA stability.
In the absence of detectable protein in ROP and ROPC lines, deductions on the targeting roles of the extensions are dependent on characterization of ROPN and ROPNC lines. C‐terminal extensions can function as vacuolar targeting signals, as has been shown, for example, in tobacco chitinases (Neuhaus et al., 1991) and barley lectin (Bednarek et al., 1990). Vacuolar targeting is clearly a candidate for the role of the C‐terminal extension of horseradish peroxidase. The peroxidase extension shows no sequence similarity with known vacuolar targeting sequences but, as has been observed (Nakamura and Matsuoka, 1993; Bassham and Raikhel, 1997), these sequences in general show little homology. The latter authors suggest that a hydrophobic/acidic motif structure, rather than any specific amino acid sequence, forms the core of the C‐terminal vacuolar‐sorting signal. Such a motif is present in HRP‐C (Welinder, 1979). In the present investigation, the C‐terminal extension had little effect on either transcript levels or peroxidase activity, not significantly influencing the elevated levels found with the N‐terminal extension. These results do not support a vacuolar‐targeting role for the C‐terminal extension, as similar patterns of expression of recombinant protein are found when the N‐terminal extension is present, irrespective of whether the C‐terminal extension is also present. Immunoblot analysis did not reveal any obvious differences between band intensity of ROPN and ROPNC lines. Furthermore, histochemical examination by DAB staining (data not shown), and Immunogold analysis (T.A.Thorpe, pers. comm.) has also failed to reveal preferential localization of peroxidase to the vacuole in ROPNC compared with ROPN transgenic lines. Our data cannot exclude a vacuolar targeting role for the C‐terminal extension in horseradish, which has been supported by studies on cultured tobacco cells (Matsui et al., 2003), but it does not appear to function in that way in this whole plant heterologous expression system. None of our data indicate an alternative function for the C‐terminal extension, which appeared largely superfluous in all the constructs tested.
From the size of the protein detected on immuoblots, and the double bands obtained, the protein appears to be glycosylated, supporting its location in the endoplasmic reticulum. However, any subsequent secretion of the protein is extremely inefficient, as only a small proportion of the recombinant enzyme activity ends up in the cell wall. This observation from the cell fractionation studies gets important support from the measurements on isolated protoplasts, in which the cell wall is completely absent. This inefficient secretion implies a block in the acquisition of transport competence (Chrispeels, 1991). A small proportion of the recombinant protein is, however, secreted, presumably by the ‘bulk flow’ pathway (Bednarek et al., 1990), and leads to a level of peroxidase activity in the cell wall up to 3·5 times higher than that of wild‐type tobacco. These levels can be useful for establishing whether modest increases in wall activity can influence cell wall‐centred peroxidase‐dependent phenomena, such as lignification and stress responses.
ACKNOWLEDGEMENTS
The authors gratefully acknowledge the financial support of Enterprise Ireland (grants SC/91/633/A and SC/97/104) and the European Commission (grant FRMX‐CT98‐0200).
Supplementary Material
Received: 18 July 2003;; Returned for revision: 16 November 2003; Accepted: 3 December 2003, Published electronically: 28 January 2004
References
- AmayaI, Botella MA, de la Calle M, Medina MI, Heredia A, Bressan RA, Hasegawa PM, Quesada MA, Valpuesta V.1999. Improved germination under osmotic stress of tobacco plants overexpressing a cell wall peroxidase. FEBS Letters 457: 80–84. [DOI] [PubMed] [Google Scholar]
- BasshamDC, Raikhel NV.1997. Molecular aspects of vacuole biogenesis. Advances in Botanical Research 23: 43–58. [Google Scholar]
- BednarekSY, Wilkins TA, Dombrowski JE, Raikhel NV.1990. A carboxyl‐terminal propeptide is necessary for proper sorting of barley lectin to vacuoles of tobacco. Plant Cell 2: 1145–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BevanM.1984. Binary Agrobacterium vectors for plant transformation. Nucleic Acids Research 12: 8711–8721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BradfordMM.1976. A rapid and sensitive method for the quantification of microgram quantities of protein utilising the principle of protein‐dye binding. Analytical Biochemistry 72: 248–254. [DOI] [PubMed] [Google Scholar]
- ChrispeelsMJ.1991. Sorting of proteins in the secretory system. Annual Review of Plant Physiology and Plant Molecular Biology 42: 21–53. [Google Scholar]
- ChrispeelsMJ, Raikhel NV.1992. Short peptide domains target proteins to plant vacuoles. Cell 68: 613–616. [DOI] [PubMed] [Google Scholar]
- EggermontK, Goderis IJ, Broekaert WF.1996. High‐throughput RNA extraction from plant samples based on homogenisation by reciprocal shaking in the presence of a mixture of sand and glass beads. Plant Molecular Biology Reporter 14: 273–279. [Google Scholar]
- FeinbergAP, Vogelstein F.1983. A technique for radiolabeling DNA restriction endonuclease fragments to high specific activity. Analytical Biochemistry 132: 6–13. [DOI] [PubMed] [Google Scholar]
- FujiyamaK, Takemura H, Shibayama S, Kobayashi K, Choi J‐K, Shinmyo A, Takano M, Yamada Y, Okada H.1988. Structure of the horseradish peroxidase C genes. European Journal of Biochemistry 173: 681–687. [DOI] [PubMed] [Google Scholar]
- FukudaH.1996. Xylogenesis: initiation, progression, and cell death. Annual Review of Plant Physiology and Plant Molecular Biology 47: 299–325. [DOI] [PubMed] [Google Scholar]
- HarlowE, Lane D.1988.Antibodies: a laboratory manual. Cold Spring Harbor: Cold Spring Harbor Laboratory Press. [Google Scholar]
- HorschRB, Fry JE, Hoffman NL, Eichholtz D, Rogers SG, Fraley RT.1985. A simple and general method for transferring genes into plants. Science 227: 1229–1231. [DOI] [PubMed] [Google Scholar]
- KawaokaA, Kawamoto T, Moriki H, Ohta H, Sekine M, Takano M, Shinmyo A.1994. Growth stimulation of tobacco plant introduced the horseradish peroxidase gene prxC1a. Journal of Fermentation and Bioengineering 78: 49–53. [Google Scholar]
- KristensenBK, Brandt J, Boysen K, Thordal‐Christensen H, Kerby KB, Collinge DB, Mikkelsen JD, Rasmussen SK.1997. Expression of a defence‐related intercellular barley peroxidase in transgenic tobacco. Plant Science 122: 173–182. [Google Scholar]
- LaemmliUK.1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227: 680–685. [DOI] [PubMed] [Google Scholar]
- McDougallGJ.1991. Cell‐wall‐associated peroxidase and lignification during growth of flax fibres. Journal of Plant Physiology 139: 182–186. [Google Scholar]
- MaligaP.1984. Cell culture procedures for mutant selection and characterization in Nicotiana plumbaginifolia In: Vasil I. ed. Cell culture and somatic cell genetics of plants Orlando: Academic Press, 552–562. [Google Scholar]
- MatsuiT, Nakayama H, Yoshida K, Shinmyo A.2003. Vesicular transport route of horseradish C1a peroxidase is regulated by N‐ and C‐terminal propeptides in tobacco cells. Applied Microbiology and Biotechnology 62: 517–522. [DOI] [PubMed] [Google Scholar]
- MazurBJ, Chui CF.1985. Sequence of a genomic DNA clone for the small subunit of ribulose bis‐phosphate carboxylase‐oxygenase from tobacco. Nucleic Acids Research 13: 2373–2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NakamuraK, Matsuoka K.1993. Protein targeting to the vacuole in plant cells. Plant Physiology 101: 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NeuhausM, Sticher L, Meins Jr F, Boller T.1991. A short C‐terminal sequence is necessary and sufficient for the targeting of chitinases to the plant vacuole. Proceedings of the National Academy of Sciences of USA 88: 10362–10366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PellegrineschiA, Kis M, Dix I, Kavanagh TA, Dix PJ.1995. Expression of horseradish peroxidase in transgenic tobacco. Biochemical Society Transactions 23: 247–50. [DOI] [PubMed] [Google Scholar]
- SambrookJ, Fritsch EF, Maniatis T.1989.Molecular cloning: a laboratory manual, 2nd edn. Cold Spring Harbor: Cold Spring Harbor Laboratory Press. [Google Scholar]
- SmithAT, Santama N, Dacey S, Edwards M, Bray RC, Thorneley RNF, Burke JF.1990. Expression of a synthetic gene for horseradish peroxidase C in Escherichia coli and folding and activation of the recombinant enzyme with Ca2+ and Heme. Journal of Biological Chemistry 65: 13335–13343. [PubMed] [Google Scholar]
- StaehelinLA, Moore I.1995. The plant Golgi apparatus: structure, functional organization and trafficking mechanisms. Annual Review of Plant Physiology and Plant Molecular Biology 46: 261–288. [Google Scholar]
- VeitchNC, Smith AT.2001. Horseradish peroxidase. Advances in Inorganic Chemistry 51: 107–162. [Google Scholar]
- WandeltCI, Khan MRI, Craig S, Schroeder HF, Spencer D, Higgins TJV.1992. Vicilin with carboxyl‐terminal KDEL is retained in the endoplasmic reticulum and accumulates to high levels in the leaves of transgenic plants. Plant Journal 2: 181–192. [DOI] [PubMed] [Google Scholar]
- WelinderKG.1979. Amino acid sequence studies of horseradish peroxidase: amino and carboxyl termini, cyanogen bromide and tryptic fragments, the complete sequence, and some structural characteristics of horseradish peroxidase C. European Journal of Biochemistry 96: 483–502. [DOI] [PubMed] [Google Scholar]
- ZimmerlanA, Bolwell GP.1993. Characterisations of a cell wall peroxidase from French bean which synthesises diferulate in vitro In: Welinder KG, Rasmussen SK, Penel C, Greppin H, eds. Plant peroxidases: biochemistry and physiology Geneva: University of Geneva, 291–297. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}