

Abstract
Hydrogen sulfide (H2S) exerts a host of biological effects ranging from cytotoxicity to cytoprotection. Cytotoxicity of H2S in neurodegenerative diseases may be mediated by N-methyl-D-aspartate receptor (NMDAR) activation. To exploit cytoprotective effects of H2S while minimizing its toxicity, we synthesized a series of H2S-releasing NMDAR antagonists and examined their effects against 1-methyl-4-phenylpyridinium (MPP+)-induced cell death, a cellular model of Parkinson’s disease. We observed that cytoprotective effect of H2S-releasing NMDAR antagonists correlated with their ability to increase intracellular sulfane sulfur, but not H2S, levels. These studies suggest that H2S-donor compounds that increase intracellular sulfane sulfur are potentially useful neuroprotective agents against neurodegenerative diseases.
1. Introduction
Hydrogen sulfide (H2S) is an endogenously-produced gaseous signaling molecule.1 In mammalian tissues, H2S is generated by cystathionine β-synthase (CBS), cystathionine gamma-lyase (CSE) and 3-mercaptopyruvate sulfurtransferase (3MST).2, 3 On the other hand, H2S is serially oxidized to persulfide, sulfite (SO32−), thiosulfate (S2O32−), and sulfate (SO42−) in reactions catalyzed by several enzymes.4 H2S or its metabolites can also be converted to polysulfides that contain sulfane sulfur, a sulfur atom with six valence electrons but no charge (represented as S0).5, 6 H2S and sulfane sulfur coexist and recent work suggests that sulfane sulfur containing polysulfides, derived from H2S, may be the actual signaling molecules.5-8 Nonetheless, measurement of H2S or sulfur metabolites in biological samples has been technically challenging and the role of sulfane sulfur in the cytoprotective effects of H2S is incompletely understood.6, 9-11
Parkinson’s disease (PD) is one of the most common neurodegenerative diseases.12 It is characterized by a slow and progressive degeneration of dopaminergic neurons in the substantia nigra.13 We found that inhaled H2S protected mice from neurodegeneration induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), a toxin which causes mitochondrial dysfunction in dopaminergic neurons of mammalian midbrain leading to Parkinson’s disease-like symptoms.14 Recently, we have reported that a novel H2S-releasing hybrid N-methyl-D-aspartate receptor (NMDAR) antagonist derivative, S-memantine, is more effective in protecting neurons against ischemic brain injury than H2S donor compound alone (i.e. H2S donor without NMDAR antagonist) or memantine.15 S-mematine is a hybrid derivative of a NMDAR antagonist memantine chemically combined with a slow releasing H2S donor. However, mechanisms responsible for the cytoprotective effects of S-memantine are incompletely defined.
In the present study, we synthesized a series of H2S-releasing NMDAR antagonists (Figure 1) to compare their protective effects and abilities to increase H2S, or sulfane sulfur levels in culture cells, or medium. We hypothesized that the protective effects of H2S-releasing NMDAR antagonists may be related to the ability to increase intracellular levels of sulfane sulfur while inhibiting NMDAR. To address this hypothesis, we examined the cytoprotective effect and the ability to increase intracellular levels of sulfane sulfur of H2S-releasing NMDAR antagonists and their parent H2S donor. Unexpectedly, we observed that the protective effects of H2S-releasing NMDAR antagonists against 1-methyl-4-phenylpyridinium (MPP+, a metabolite of MPTP)-induced cell death were similar to that of the parent H2S donor compounds (H2S-releasing compounds without NMDAR antagonist) alone. However, our study revealed that the cytoprotective effects of H2S donor compounds correlated with their ability to increase intracellular levels of sulfane sulfur, but not H2S.
Figure 1. Structures of NMDAR antagonist, H2S donor, or H2S-releasing NMDAR antagonist.

The present results indicate that sulfane sulfur, but not H2S, is the intracellular sulfide metabolites that prevent MPP+-induced cytotoxicity and demonstrated the feasibility of drug development against neurodegenerative diseases using H2S- or sulfane sulfur-based compounds.
2. Results and discussion
2.1. Chemistry
4-(3-thioxo-3H-1,2-dithiol-4-yl)benzoic acid (ACS48) and N-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)-4-(3-thioxo-3H-1,2-dithiol-4-yl)-benzamide (S-memantine) were prepared as described previously.15 3-(prop-2-en-1-yldisulfanyl)propanoic acid (ACS81), 4-carbamothioylbenzoic acid (CTBA), 4-(3-thioxo-3H-1,2-dithiol-4-yl)-N-[(1S,3R,5R,7S)-tricyclo[3.3.1.13,7]dec-1-yl]benzamide (ACS48-amantadine), 3-(prop-2-en-1-yldisulfanyl)-N-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)propanamide (ACS81-memantine), and 4-carbamothioyl-N-[(1R,3S,5S,7R)-3,5-dimethyltricyclo[3.3.1.13,7]dec-1-yl]benzamide (CTBA-memantine) were prepared as shown in scheme 1-3.
Scheme 1.

Scheme 3.

2.2. H2S donors prevent cell death induced by 1-methyl-4-phenylpyridinium (MPP+) in SH-SY5Y cells
To examine the protective effects of H2S donors, we measured cell viabilities of SH-SY5Y 24 h after the addition of MPP+ (5 mM) with H2S donor compounds with or without NMDAR antagonist at 20 μM using crystal violet (CV) or 3–(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay as described previously.15 The result showed that CTBA (only in MTT method but not in CV method), ACS48-amantadine, and CTBA-memantine, but not ACS48, ACS81, S-memantine, ACS81-memantine, Na2S, or NMDAR antagonist alone, improved cell viability. However, there was no significant difference in cell viability between SH-SY5Y treated with H2S-releasing NMDAR antagonist and cells treated with its parent H2S donor compounds alone (Figure 2). These observations indicate that, unlike in ischemic neuronal injury,15 combination of NMDAR antagonist with H2S donor may not enhance the cytoprotective effect of H2S donor against MPP+-induced toxicity.
Figure 2. Cell viabilities of SH-SY5Y 24 h after the addition of MPP+ with or without H2S donor.

Cell viabilities were measured by CV or MTT method (panel A or B). N = 4 or 5 each; P < 0.001 Control vs. all the other groups, *, or ** P < 0.05, or 0.01 vs. vehicle.
2.3. H2S donors increase intracellular levels of sulfide and sulfane sulfur in SH-SY5Y cells
To compare the ability of various H2S donors (ACS48, ACS81, CTBA, S-memantine, ACS48-amantadine, ACS81-memantine, CTBA-memantine, and Na2S) to increase H2S or sulfane sulfur levels in the cell or medium in the absence of cells, we measured fluorescent intensity of a novel H2S-specific probe HSip-1 or a sulfane sulfur-specific probe SSP4 loaded in SH-SY5Y cells or medium after the addition of H2S donors at 20 μM to the medium (DMEM/F12, 10 % FBS).10 Since fluorescent intensity of HSip-1 or SSP4 was almost saturated 3 h after the addition of H2S donor (Figure S1 and S2), we considered fluorescent intensity at 3 h after the addition of H2S donor as an index of total amount of H2S or sulfane sulfur increased by H2S donors.
The result showed that S-memantine and ACS48-amantadine increased intracellular H2S levels, while all H2S donors increased H2S levels in the medium (Figure. 3 A and B). ACS48, CTBA, ACS48-amantadine, and CTBA-memantine increased intracellular sulfane sulfur levels, while ACS48, ACS81, S-memantine, ACS48-amantadine, and ACS81-memantine increased sulfane sulfur levels in the medium (Figure 3 C and D). A simple sulfide salt Na2S did not increase sulfane sulfur levels in cells or medium.
Figure 3. Intracellular or medium levels of H2S or sulfane sulfur.

Intracellular or medium fluorescent intensity (FI) was measured at 3 h after the adition of H2S donor to the medium loaded by a H2S probe HSip-1 (panels A and B) or a sulfane sulfur probe SSP4 (panels C and D). N = 4 or 5; **, or *** P < 0.01, or 0.001 vs. baseline. ##, or ### P < 0.01, or 0.001 vs. Parent H2S donor, ††† P < 0.001.
2.4. Levels of intracellular sulfane sulfur, but not H2S, correlated with the ability of H2S donors to prevent cell death induced by MPP+ in SH-SY5Y cells
To define the role of H2S or sulfane sulfur in the protective effect of H2S donors against MPP+-induced cytotoxicity, we examined the correlation between intracellular fluorescent intensity of HSip-1 or SSP4 and cell viability of SH-SY5Y calculating Spearman’s correlation coefficient. Cell viability which was measured by MTT assay was used for the calculation of correlation since MPP+ caused mitochondrial dysfunction and the mechanism of MTT assay is based on the ability of mitochondria to metabolize MTT into formazan.16 We observed no significant relationship between intracellular or medium fluorescent intensity of HSip-1 and cell viability (Figure 4 A and B). However, we observed the positive relationship between intracellular, but not medium, fluorescent intensity of SSP4 and cell viability (Figure 4 C and D). These observations indicate that the cytoprotective effects of H2S donors against MPP+-induced toxicity correlate with their ability to produce intracellular sulfane sulfur, but not H2S.
Figure 4. Correlation between cell viability and H2S, or sulfane sulfur levels.

Spearman’s correlation coefficient was calculated using cell viability (MTT method) and fluorescent intensity of HSip-1 (panels A and B) or SSP4 (panels C and D).
3. Conclusions
In the present study, we synthesized and characterized the cytoprotective effects of S-memantine and novel H2S-releasing NMDAR antagonists (ACS48-amantadine, ACS81-memantine, and CTBA-memantine). ACS48-amantadine and CTBA-memantine, but not NMDAR antagonists, H2S donor alone, or other H2S-releasing NMDAR antagonists, markedly improved the cell viability of SH-SY5Y cells 24 h after the addition of MPP+. However, we could not observe that any H2S-releasing NMDAR antagonists were significantly more effective than parent H2S donor compounds alone by direct comparisons. Although we have previously reported that S-memantine inhibits NMDAR activation by glutamate, whether or not inhibition of NMDAR by H2S-releasing NMDAR antagonist promotes the beneficial effect of H2S donor against PD remains to be elucidated.15, 17
We measured levels of H2S or sulfane sulfur which was produced from H2S donor compounds added to the medium and examined the relationship between H2S or sulfane sulfur levels and cell viability. These results indicate that intracellular levels of sulfane sulfur, but not H2S, are associated with the protective effects of H2S against MPP+-induced cytotoxicity. Since hydrophobicity of CTBA (octanol/water partition coefficient; cLogP = 1.53, calculated by ChemBioDraw Ultra 13.0, Perkin Elmer, Inc.) is lower than that of ACS48 (cLogP = 2.89) or ACS81 (cLogP = 2.03), the highest level of intracellular sulfane sulfur by the addition of CTBA cannot be explained by hydrophobicity or cell permeability alone. Caliendo and colleagues have reported hypothetic hydrolysis mechanisms of CTBA and ACS48 to release H2S.18 CTBA may undergo hydrolysis more markedly than do ACS48 since the aromatic ring in CTBA makes the formed intermediates stable in the cell.
ACS48-amantadine increased H2S level in the cell or medium as well as S-memantine, while ACS48-amantadine increased sulfane sulfur levels in the cells and medium more markedly than did S-memantine. The structural difference between ACS48-amantadine and S-memantine is only NMDAR antagonist (amantadine or memantine). cLogP of ACS48-amantadine or S-memantine is 4.52 or 5.56, respectively. Therefore, hydrophobicity or cell permeability cannot explain the reason for the higher level of intracellular sulfane sulfur by the addition of ACS48-amantadine. The reason why just two additional methyl moieties in the NMDAR antagonist dramatically enhanced the ability of ACS48-amantadine to increase the intracellular sulfane sulfur levels remains to be elucidated in the future study.
The present study indicates that combination of NMDAR antagonist with H2S donor may not enhance the cytoprotective effect of H2S donors against MPP+-induced toxicity. These observations appear to contradict with our recent finding that S-memantine exerted more potent neuroprotective effects than its parent compounds alone (ACS48 or memantine) against ischemic neuronal injury.15 The reason for this apparent contradiction may be the differing role of NMDAR in ischemic neuronal injury and MPP+-induced cytotoxicity. NMDAR antagonists have been shown to protect neurons from ischemic insults.15 In fact, in the previous study, we observed that memantine per se exerted cytoprotective effects against OGD-induced cell death.15 In contrast, it has been reported that NMDAR antagonism may not be protective against MPP+-induced cytotoxicity since the mechanism of MPP+ toxicity does not involve NMDAR activation.19, 20 We plan to examine cytoprotective effects of the series of H2S-relasing NMDAR antagonist against ischemic neuronal injury in the future studies.
It has been suggested that sulfane sulfur containing polysulfides, but not H2S itself, are the signaling molecules that mediate biological effects of “hydrogen sulfide”. For example, Greiner and colleagues recently reported that polysulfides, but not H2S, induce oxidation of lipid phosphatase and tensin homolog (PTEN) modifying its activity.21 Similarly, Kimura and colleagues showed that polysulfides activate transient receptor potential (TRP) A1 channels at EC50 of 91 nM while H2S activate TRPA1 at EC50 of 116 μM.22 Our current results support the important role of sulfane sulfur-containing polysulfides by showing the correlation of the intracellular levels of sulfane sulfur and the cytoprotective effects of H2S-donor compounds in cellular model of PD. These observations may form a foundation that enables future drug development against PD exploiting the ability of novel compounds that augments intracellular levels of sulfane sulfur.
4. Experimental section
4.1. Chemistry
All moisture-sensitive reactions were performed under an argon atmosphere using ovendried glassware and anhydrous solvents. Unless otherwise noted, commercially available materials were used without purification. Products are purified on Biotage - Isolera Sysyem. Nuclear magnetic resonance (NMR) splitting patterns are described as singlet (s), doublet (d), triplet (t), quartet (q); the value of chemical shifts (δ) are given in ppm relative to residual solvent, and coupling constants (J) are given in hertz (Hz). The mass spectra were recorded on Agilient/Micromass LC/MS.
4.1.1. Synthesis of SSP4
A fluorescent probe for sulfane sulfur SSP4 (shown in Figure S3) was prepared using the same method as SSP2 reported previously.11 1H NMR (300 MHz, DMSO-d6) δ 5.49 (s, 2H), 6.97 (d, J = 9.0 Hz, 2H), 7.12 (d, J = 9.0 Hz, 2H), 7.32 (t, J = 6.0 Hz, 2H), 7.44–7.55 (m, 5H), 7.66 (d, J = 9.0 Hz, 2H), 7.76–7.88 (m, 2H), 8.09 (d, J = 9.0 Hz, 1H), 8.19 (d, J = 6.0 Hz, 2H). 13C NMR (75 MHz, CD3Cl) δ 81.9, 110.9, 116.9, 118.2, 124.3, 124.7, 125.2, 125.5, 129.3, 131.4, 132.5, 133.7, 140.1, 151.8, 152.2, 153.2, 164.9, 169.4; MS (ESI+) m/z 627.6 (M+Na)+.
4.1.2. Synthesis of ACS48-amantadine
P-isopropyl benzoic acid (2.0 g, 12 mmol) was dissolved in N,N-dimethylformamide (DMF, 41 ml) and followed by addition of amantadine (2.21 g, 15 mmol) and N,N-diisopropylethylamine (DIPEA, 8.48 ml, 49 mmol) at room temperature (rt) under nitrogen (N2). The reaction mixture was then cooled over ice bath for 10 min. 2-(7-Aza-1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HATU) was added over ice bath and the reaction mixture was stirred for 10 min at this temperature. The reaction mixture was warmed up to rt and stirred overnight. The reaction mixture was quenched with a solution of NaHCO3 (200 ml) and then extracted with EtOAc (100 ml × 3). The combined organic layers were washed with water 100ml × 1 and brine 100 ml × 1, and then dried over Na2SO4. The filtrate was concentrated then residue was purified by Biotage. The desired fractions was concentrated and then dried over oil pump to give 2.43 g pale yellow oil of amide (68 % yield, MS (ESI+) m/z 298 (M+1)).
The amide (1.31 g, 4.4 mmol) was mixed with sulfur (3.94 g, 123 mmol) under N2. The mixture was then heated to 190°C oil bath overnight. The reaction mixture was then cooled down. 50 ml toluene was added and stirred for 10 min. The mixture was then loaded to silica cartridge directly and purified by Biotage with gradient from pure hexanes to 50 % EtOAc/hexanes. The desired fractions was collected and then concentrated to give 0.389 g red solid (23 %). 1H NMR: δ (CDCl3, 400 MHz), 8.47 (s, J= 8.6 Hz, 1H); 7.79 (d, J= 8.6 Hz, 2H); 7.61(d, J= 8.2 Hz, 2H); 5.82 (s, 1H); 2.14 (s, 9H); 1.74 (bs, 6H). MS (ESI+) m/z 388 (M+1).
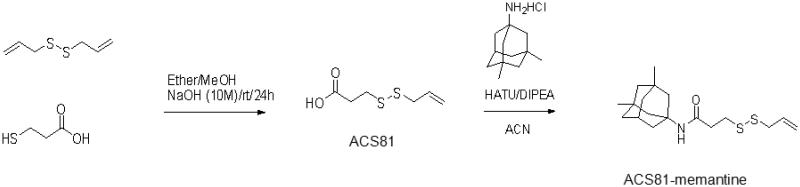
Synthesis of ACS81
To a solution of disulallyl disulfide (5.0 ml, 36.6 mmol) in a mixture of diethyl ether (21 ml) and methanol (44 ml) was added a solution of 3-mercaptpropanoic acid (1.08 g, 10.2 mmol) in diethyl ether (11 ml), followed by a solution of 10M NaOH (1,27 ml, 12.7 mmol). The reaction mixture was stirred at rt overnight. The reaction mixture was then concentrated and the residue was diluted with water (20 ml) and EtOAc (50 ml). A solution of 1N HCl was added to adjust pH=4 and then two layers were separated. Extract aqueous with EtOAc (2 × 50ml) and the combined organic layers were washed with brine (50 ml) followed by drying over Na2SO4. The solid was filtered out and the filtrate was concentrated over rota-evaporator. The residue is purified by Biotage with EtOAc/Hexanes. The desired fractions were combined and concentrated to give 0.52 g pale yellow oil as product (28 % yield, MS (ESI+) m/z 179 (M+1)).
4.1.3. Synthesis of ACS81-memantine
ACS81 (0.2 g, 1.12 mmol) was dissolved in DMF (4ml) and followed by addition of memantane HCl (0.266 g, 1.23 mmol) and DIPEA (0.78ml, 4.49mmol) at rt under N2. The reaction mixture was then cooled over ice bath for 10 min. HATU (0.640 g, 1.68 mmol) was added over ice bath and the reaction mixture was stirred for 10 min at this temperature. The reaction mixture was warmed up to rt and stirred overnight. The reaction mixture was quenched with a solution of NaHCO3 (50 ml) and then extracted with EtOAc (50 ml × 3). The combined organic layers were washed with water 50ml × 1 and brine 50 ml × 1, and then dried over Na2SO4. The filtrate was concentrated then residue was purified by Biotage. The desired fractions was concentrated and then dried over oil pump to give 0.3 g pale yellow solid (79 % yield). 1H NMR: δ (CDCl3, 400 MHz), 5.70-5.85 (m, 1H); 5.30 (bs, 1H), 5.154 (s, 1H), 5.01-5.0151 (m, 1H), 3.15 (m, 2H), 2.73 (t, J= 7.3 Hz, 2H), 2.34 (t J= 7.2 Hz, 2H), 2.14 (m, 1H), 1.84 (m, 2H), 1.65 (m, 4H), 1.38 (m, 2H), 1.29 (m, 2H), 1.10-1.20 (m, 2H), 0.85 (s, 6H). MS (ESI+) m/z 340 (M+1).
4.1.4. Synthesis of CTBA
Under N2, 4-cyano-benzoic acid (0.735 g, 5.0 mml) was dissolved in MeOH (60 ml), a solution of (NH4)2S (3.3 ml, 20 % aqueous solution, 5.5 mmol) was added and the reaction mixture was stirred overnight. The reaction mixture was then concentrated to dryness. The residue was participated in H2O (100 ml) and EtOAc (150 ml). Two layers are separated and the aqueous was extracted with EtOAc. The combined organic layers were washed with water, brine, and then dried over Na2SO4. The filtrate was concentrated then residue was purified by Biotage. The desired fractions was concentrated and then dried over oil pump to give 0.41 g yellow solid (45 % yield). MS (ESI+) m/z 182 (M+1).
4.1.5. Synthesis of CTBA-memantane
4-Cyanobenzoic acid (1.0 g, 6.8 mmol) was dissolved in DMF (23 ml) and followed by addition of memantine HCl (1.76 g, 8.16 mmol) and DIPEA (4.73 ml, 27.19 mmol) at rt under N2. The reaction mixture was then cooled over ice bath for 10 min. HATU (3.88 g, 10.2 mmol) was added over ice bath and the reaction mixture was stirred for 10 min at this temperature. The reaction mixture was warmed up to rt and stirred overnight. The reaction mixture was quenched with a solution of NaHCO3 (100 ml) and then extracted with EtOAc (100 ml × 3). The combined organic layers were washed with water 100 ml × 1 and brine 100 ml × 1, and then dried over Na2SO4. The filtrate was concentrated then residue was purified by Biotage. The desired fractions was concentrated and then dried over oil pump to give 1.7 g pale yellow solid of amide (81 % yield, MS (ESI+) m/z 309 (M+1)). Under a nitrogen atmosphere, the amide (0.616 g, 2.0 mml) was dissolved in MeOH (20 ml), a solution of (NH4)2S (1.31 ml, 20 % aqueous solution, 2.2 mmol) was added and the reaction mixture was stirred overnight. The reaction mixture was then concentrated to dryness. The residue was participated in H2O (50 ml) and EtOAc (50 ml). Two layers are separated and the aqueous was extracted with EtOAc. The combined organic layers were washed with water 100 ml × 1 and brine 100 ml × 1, and then dried over Na2SO4. The filtrate was concentrated then residue was purified by Biotage. The desired fractions was concentrated and then dried over oil pump to give 0.46 g yellow solid (68% yield). 1H NMR: δ(DMSO-d6, 400 MHz), 10.0 (s, 1H); 9.6 (s, 1H); 7.91 (d, J= 8.6 Hz, 2H); 7.80 (d, J= 8.6 Hz, 2H); 2.12-2.15 (m, 1H); 1.92-1.94 (m, 3H), 1.74-1.77 (m, 4H), 1.28-1.39 (m, 4H), 1.16 (s, 2H), 0.86 (s, 6H). MS (ESI+) m/z 343 (M+1).
4.2. Biology
4.2.1. Cell culture
SH-SY5Y cells were cultured in Eagle’s medium/Ham’s F-12 50/50 Mix (DMEM/F12, Cellgro by Mediatech, Inc.) supplemented with 10 % fetal bovine serum (FBS) and penicillin (100 units/ml)/streptomycin (100 μg/mL). Briefly, cells were seeded into 96 well plates (2×104 cells per well). Cell culture medium was replaced every 2 days, and the cultures were maintained at 37°C in 95% air/ 5% CO2 in a humidified incubator. Cells were used after reaching 80 % confluent.
4.2.2. Cell viability
CV and MTT assays were performed to examine cell viabilities of SH-SY5Y cells 24 h after the addition of MPP+ (5 mM) with vehicle (DMSO, final concentration: 1 %) or H2S donor (20 μM) as reported previously.
4.2.3. Measurement of H2S or sulfane sulfur levels
H2S levels in SH-SY5Y cells or medium (DMEM/F12, 10 % FBS) without cells were measured using H2S-specific fluorescent probe HSip-1 DA for intracellular H2S or HSip-1 for extracellular H2S, respectively, as described previously. Briefly, fluorescent intensity of HSip-1-loaded SH-SY5Y cells or medium without cells was measured by (SpectraMax M5 Microplate reader, Molecular Devices, Inc.) before or 1.5 h, 3 h, or 6 h after the addition of vehicle (DMSO) or H2S donor at 20 μM to the medium. Final concentration of DMSO was adjusted to 1 %. Sulfane sulfur levels in SH-SY5Y cells or medium without cells were measured using sulfane sulfur specific fluorescent probe SSP4 instead of HSip-1 in the protocol of H2S measurement.
4.2.4. Statistical analysis
All data are presented as means ± SE. Data were analyzed by ANOVA using Sigmastat 3.01a (Systat Software Inc., Chicago, IL) and Prism 5 software package (GraphPad Software, La Jolla, CA). Newman-Keuls multiple comparison post hoc test was performed after One-way Anova as required. To determine correlation between cell viability and H2S level, or sulfane sulfur level, Spearman’s correlation coefficient was calculated. Smaller P values than 0.05 were considered significant.
Supplementary Material
Scheme 2.
6. Acknowledgments
This work was supported by National Institutes of Health (NIH) grant HL101930 to F. Ichinose and ACS-Teva USA Scholar Award, and NIH grant HL116571 to M. Xian.
Footnotes
5. Supplementary Material
Refer to Web version on PubMed Central for supplementary material.
Notes and references
- 1.Baumgart K, Radermacher P, Wagner F. Curr Opin Anaesthesiol. 2009;22:168–176. doi: 10.1097/ACO.0b013e328328d22f. [DOI] [PubMed] [Google Scholar]
- 2.Stipanuk MH, Beck PW. Biochem. J. 1982;206:267–277. doi: 10.1042/bj2060267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shibuya N, Tanaka M, Yoshida M, Ogasawara Y, Togawa T, Ishii K, Kimura H. Antioxidants & Redox Signaling. 2008;11:703–714. doi: 10.1089/ars.2008.2253. [DOI] [PubMed] [Google Scholar]
- 4.Hildebrandt TM, Grieshaber MK. FEBS Journal. 2008;275:3352–3361. doi: 10.1111/j.1742-4658.2008.06482.x. [DOI] [PubMed] [Google Scholar]
- 5.Toohey JI. Analytical Biochemistry. 2011;413:1–7. doi: 10.1016/j.ab.2011.01.044. [DOI] [PubMed] [Google Scholar]
- 6.Shen X, Peter EA, Bir S, Wang R, Kevil CG. Free Radical Biology and Medicine. 2012;52:2276–2283. doi: 10.1016/j.freeradbiomed.2012.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Szabo C. Nat Rev Drug Discov. 2007;6:917–935. doi: 10.1038/nrd2425. [DOI] [PubMed] [Google Scholar]
- 8.Ida T, Sawa T, Ihara H, Tsuchiya Y, Watanabe Y, Kumagai Y, Suematsu M, Motohashi H, Fujii S, Matsunaga T, Yamamoto M, Ono K, Devarie-Baez NO, Xian M, Fukuto JM, Akaike T. Proceedings of the National Academy of Sciences. 2014 doi: 10.1073/pnas.1321232111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wintner EA, Deckwerth TL, Langston W, Bengtsson A, Leviten D, Hill P, Insko MA, Dumpit R, VandenEkart E, Toombs CF, Szabo C. British Journal of Pharmacology. 2010;160:941–957. doi: 10.1111/j.1476-5381.2010.00704.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sasakura K, Hanaoka K, Shibuya N, Mikami Y, Kimura Y, Komatsu T, Ueno T, Terai T, Kimura H, Nagano T. Journal of the American Chemical Society. 2011;133:18003–18005. doi: 10.1021/ja207851s. [DOI] [PubMed] [Google Scholar]
- 11.Chen W, Liu C, Peng B, Zhao Y, Pacheco A, Xian M. Chemical Science. 2013;4:2892–2896. doi: 10.1039/C3SC50754H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Twelves D, Perkins KSM, Counsell C. Movement Disorders. 2003;18:19–31. doi: 10.1002/mds.10305. [DOI] [PubMed] [Google Scholar]
- 13.Hirsch E, Graybiel AM, Agid YA. Nature. 1988;334:345–348. doi: 10.1038/334345a0. [DOI] [PubMed] [Google Scholar]
- 14.Kida K, Yamada M, Tokuda K, Marutani E, Kakinohana M, Kaneki M, Ichinose F. Antioxidants & Redox Signaling. 2010;15:343–352. doi: 10.1089/ars.2010.3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marutani E, Kosugi S, Tokuda K, Khatri A, Nguyen R, Atochin DN, Kida K, Van Leyen K, Arai K, Ichinose F. Journal of Biological Chemistry. 2012;287:32124–32135. doi: 10.1074/jbc.M112.374124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Berg K, Hansen MB, Nielsen SE. APMIS. 1990;98:156–162. doi: 10.1111/j.1699-0463.1990.tb01016.x. [DOI] [PubMed] [Google Scholar]
- 17.Hallett PJ, Standaert DG. Pharmacology & Therapeutics. 2004;102:155–174. doi: 10.1016/j.pharmthera.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 18.Caliendo G, Cirino G, Santagada V, Wallace JL. Journal of Medicinal Chemistry. 2010;53:6275–6286. doi: 10.1021/jm901638j. [DOI] [PubMed] [Google Scholar]
- 19.Finiels-Marlier F, Marini AM, Williams P, Paul SM. Journal of Neurochemistry. 1993;60:1968–1971. doi: 10.1111/j.1471-4159.1993.tb13431.x. [DOI] [PubMed] [Google Scholar]
- 20.Michel PP, Agid Y. Brain Research. 1992;597:233–240. doi: 10.1016/0006-8993(92)91479-x. [DOI] [PubMed] [Google Scholar]
- 21.Greiner R, Pálinkás Z, Bäsell K, Becher D, Antelmann H, Nagy P, Dick TP. Antioxidants & Redox Signaling. 2013;19:1749–1765. doi: 10.1089/ars.2012.5041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kimura Y, Mikami Y, Osumi K, Tsugane M, Oka J.-i., Kimura H. The FASEB Journal. 2013;27:2451–2457. doi: 10.1096/fj.12-226415. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.