

Abstract
A catalyst composed of Pd2(dba)3 and (S)-Siphos-PE provides excellent results in Pd-catalyzed alkene carboamination reactions between aniline derivatives bearing pendant alkenes and aryl or alkenyl halides. These transformations generate tetrahydroquinolines and tetrahydroquinoxalines that contain quaternary carbon stereocenters with high levels of asymmetric induction. In addition this catalyst also effects the asymmetric synthesis of tetrahydroisoquinolines via related transformations of 2-allylbenzylamines. In contrast to most other approaches to the asymmetric synthesis of these compounds, which frequently involve functional group interconversion or a single C–C or C–N bond-forming event, the carboamination reactions generate both a C–N bond, a C–C bond, and a stereocenter.
Introduction
Benzo-fused six-membered saturated nitrogen heterocycles such as tetrahydroquinolines and tetrahydroquinoxalines are prominent in a variety of natural products and biologically active compounds.i As such there has been considerable interest in the development of enantioselective reactions for the construction of these structures. The most common approaches towards enantioenriched benzo-fused heterocycles involve the asymmetric hydrogenation of the corresponding aromatic heterocycles.ii,iii Although efficient, these transformations do not lead to the formation of either a C–N bond, a C–C bond, or the heterocyclic ring. Other strategies employed to access these compounds include asymmetric hydroamination reactions,iv asymmetric imine addition reactions,v C–H functionalizations,vi asymmetric alkylations,vii asymmetric N-allylations,viii and asymmetric N-arylations.ix However, these transformations in most cases are limited to the formation of tertiary carbon stereocenters. The use of asymmetric catalysis to generate heterocycles bearing quaternary stereocenters adjacent to the nitrogen atom with high levels of asymmetric induction is very rare.vb,ix
We envisioned an alternative approach to the enantioselective synthesis of a variety of benzo-fused heterocycles via asymmetric palladium catalyzed alkene carboamination reactions between aryl or alkenyl halides and aniline derivatives bearing pendant alkenes.x,xi,xii These transformations would allow for stereocontrolled synthesis of these heterocycles in a manner that effects ring formation, C–C bond formation, and C–N bond formation. Moreover, use of substrates bearing 1,1-disubstituted alkenes would result in the generation of heterocyclic products bearing quaternary stereocenters.xiii In this communication we describe the application of this strategy to the enantioselective synthesis of tetrahydroquinolines, tetrahydroquinoxalines, and tetrahydroisoquinolines. These transformations are the first asymmetric carboamination reactions between aryl/alkenyl halides and alkenes bearing pendant nucleophiles that generate quaternary stereocenters, and are also the first highly enantioselective (>95:5 er) transition metal catalyzed C-N bond forming reactions involving addition to 1,1-disubstituted alkenes.xiv
Results and Discussion
To probe the feasibility of constructing tetrahydroquinolines via asymmetric carboamination reactions we first examined the coupling of substrate 1a with 1-bromo-4-tert-butylbenzene using a catalyst composed of Pd2(dba)3 and (S)-Siphos-PE, which provides good results in other enantioselective alkene carboamination reactions.xi Unfortunately, although product 2a was formed (as judged by 1H NMR analysis of the crude reaction mixture), a complex mixture of inseparable side products was also generated and efforts to improve this result were unsuccessful. The main side products observed in this reaction appeared to derive from competing β-hydride elimination that occurred after the alkene aminopalladation step of the catalytic cycle.x It seemed that simply placing a methyl group at the internal carbon of the alkene should prevent this problem. However, in our previous studies on asymmetric carboamination reactions we found that use of substrates bearing 1,1-disubstituted alkenes led to either poor reactivity or low asymmetric induction.xib Gratifyingly, the presence of a methyl group was well tolerated in this case, as the reaction of 1b under these conditions led to the formation of product 2b in 86% yield with 92:8 er [eqn (1)].xv Further efforts to improve asymmetric induction through use of a variety of other chiral ligands led to lower enantioselectivities and/or yields for this transformation.xvi
 |
(1) |
With a proof-of-concept result in hand, we sought to further improve selectivities and also broaden scope by varying the N-substituent on the substrate. We have previously found that Pdcatalyzed carboamination reactions of N-allylureas derivatives bearing electron-poor N-aryl groups proceed with higher enantioselectivity than analogous transformations of substrates that contain electron-rich N-aryl groups.xib,c In order to determine the correlation between nitrogen nucleophilicity and asymmetric induction in the present system, substrates 1b-1e were synthesized and subjected to the Pd/(S)-Siphos-PE conditions described above. As shown in Table 1, the observed trend in the tetrahydroquinoline-forming reactions was opposite to that previously observed with urea derivatives; electron-rich substrates 1b and 1e were transformed with higher er than 1c and 1d. The reactivity of 1e was lower than that of the other substrates, and heating to 125 °C was necessary to achieve complete conversion. However, this relatively high temperature did not lead to a decrease in er. Efforts to employ related substrates bearing N-boc groups were unsuccessful; no desired product was generated in these transformations.
Table 1.
Electronic Effects.a

| |||
|---|---|---|---|
| Entry | Y | Yield (%)b | er |
| 1 | CN (1c) | 51 (2c) | 62:38 |
| 2 | tBu (1d) | 93 (2d) | 87:13 |
| 3 | OMe (1b) | 86 (2b) | 92:8 |
| 4 | NMe2 (1e) | 60 (2e) | 95:5 |
| 5c | NMe2 (1e) | 95 (2e) | 95:5 |
Conditions: 1.0 equiv 1b-1e, 2.0 equiv p-tBu-C6H4-Br, 2.0 equiv NaOtBu, 2 mol % Pd2(dba)3, 6 mol % (S)-Siphos-PE, Toluene (0.1 M), 110 °C, 12 h.
Isolated yield.
The reaction was conducted at 125 °C in xylenes.
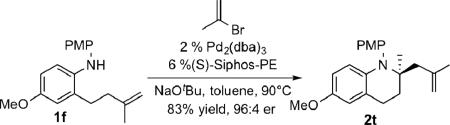
In order to test the versatility of this transformation, substrate 1e was treated with a series of aryl and alkenyl halides using the optimized reaction conditions (Table 2). Transformations of electron rich aryl halides (entries 1-3), electron poor aryl halides (entries 4-6), and a variety of alkenyl bromides (entries 7-9) all gave products in good yield and high enantioselectivities. Similarly, coupling reactions between substrate 1b and various aryl and alkenyl halides also proceeded smoothly (entries 9-16). The nature of the halide (I vs. Br) does not affect the enantioselectivity in the reaction (entries 10-11), which is similar to what has previously been observed in asymmetric pyrrolidine-forming carboamination reactions,xia but contrasts with halide effects in carboamination reactions of N-allylureas.xib,c Although most of these transformations were conducted on a relatively small scale (0.2 mmol), when the coupling of 1b with 4-bromobenzophenone was conducted on a 1.0 mmol scale using half of the standard catalyst loading product 2p was obtained in similar yield and er as with the smaller scale conditions (entry 14). The presence of substituents larger than a methyl group on the alkene had an adverse effect on the reaction outcome. Substrate 1h was transformed to product 2s in good yield, but with only modest enantioselectivity (75:25 er), and substrates 1i and 1j that contain larger alkene substituents were unreactive. However compounds 1f and 1g bearing substituted or fused aromatic rings were converted to 2t and 2u in moderate to good yield with good to excellent stereocontrol [eqn (2-3)].
 |
(2) |
 |
(3) |
Table 2.
Enantioselective Synthesis of Tetrahydroquinolines.a

| |||||
|---|---|---|---|---|---|
| Entry | Substrate | R1–X | Product | Yield (%)b | er |
| 1 | 1e | p-tBu-C6H4–Br | 2e | 95 | 95:5 |
| 2 | 1e |
|
2f | 87 | 95:5 |
| 3 | 1e | p-MeO-C6H4–Br | 2g | 88 | 95:5 |
| 4 | 1e | p-CF3-C6H4–Br | 2h | 89 | 94:6 |
| 5 | 1e | m-CF3-C6H4–Br | 2i | 94 | 92:8 |
| 6 | 1e | p-PhC(O)C6H4–Br | 2j | 82 | 96:4 |
| 7 | 1e |

|
2k | 86 | 95:5 |
| 8 | 1e |

|
2l | 98 | 95:5 |
| 9 | 1e |
|
2m | 91 | 95:5 |
| 10 | 1b | p-tBu-C6H4–Br | 2b | 86 | 92:8 |
| 11 | 1b | p-tBu-C6H4-I | 2b | 85 | 92:8 |
| 12 | 1b |
|
2n | 81 | 94:6 |
| 13 | 1b | p-Cl-C6H4–Br | 2o | 81 | 89:11 |
| 14 | 1b | p-PhC(O)C6H4–Br | 2p | 82 | 95:5 |
| 15c | 1b | p-PhC(O)C6H4–Br | 2p | 77 | 94:6 |
| 16 | 1b |

|
2q | 96 | 95:5 |
| 17 | 1b |

|
2r | 83 | 96:4 |
| 18 | 1h | Ph–Br | 2s | 86 | 75:25 |
| 19 | 1i | Ph–Br | – | NRd | |
| 20 | 1j | Ph–Br | – | NRd | |
Conditions: 1.0 equiv 1b or 1e, 2.0 equiv R-X, 2.0 equiv NaOtBu, 2 mol % Pd2(dba)3, 6 mol % (S)-Siphos-PE, xylenes or toluene (0.1 M), 90 °C (substrate 1b) or 125 °C (substrate 1e), 12-14 h.
Isolated yield (average of two or more runs).
The reaction was conducted with 1.1 equiv ArX, 1.1 equiv NaOtBu, 1 mol % Pd2(dba)3, 3 mol % (S)-Siphos-PE, toluene (0.2 M), 90 °C, on a 1mmol scale.
No reaction was observed.
The utility of this method for the stereoselective generation of tetrahydroquinoline derivatives prompted us to explore the application of this strategy to the construction of other benzo-fused nitrogen heterocycles. As such, we synthesized substrate 3 and subjected this compound to the optimized asymmetric carboamination reaction conditions (Table 3). This compound was successfully transformed to a number of tetrahydroquinoxaline derivatives (4a-g) in good yield with good to excellent enantioselectivities. The reactions were effective with a number of different aryl bromides (entries 1–4), and use of the alkenyl bromide Z-1-bromobutene afforded 4f in 74% yield with 98:2 er. Substrate 3b, which contains a CH2OBn group on the alkene, was transformed to the correspoinding heterocycle 4g in comparable yield and er as were observed in reactions of 3a. In contrast, phenyl-substituted substrate 3c proved to be unreactive.
Table 3.
Enantioselective Synthesis of Tetrahydroquinoxalinesa

| |||||
|---|---|---|---|---|---|
| Entry | Substrate | R1–X | Product | Yield (%)b | er |
| 1 | 3a | Ph–Br | 4a | 79 | 97:3 |
| 2 | 3a | p-PhC(O)-C6H4-Br | 4b | 78 | 96:4 |
| 3 | 3a |

|
4c | 84 | 93:7 |
| 4 | 3a |
|
4d | 82 | 96:4 |
| 5c | 3a |
|
4e | 70 | 93:7 |
| 6 | 3a |
|
4f | 74 | 98:2 |
| 7 | 3b | Ph–Br | 4g | 79 | 96:4 |
| 8 | 3c | Ph–Br | NR | ||
Conditions: 1.0 equiv 3, 2.0 equiv R–X, 2.0 equiv NaOtBu, 2 mol % Pd2(dba)3, 6 mol % (S)-Siphos-PE, toluene (0.1 M), 110 °C, 12-14 h.
Isolated yield (average of two or more runs).
The reaction was conducted at 120 °C in xylenes.
d No reaction was observed.
This strategy was also amenable to the synthesis of substituted tetrahydroisoquinolines, although for these transformations use of a carbamate protecting group was necessary to obtain satisfactory yields as N-aryl substituted substrates were transformed to complex mixtures of products.xvii
After some exploration we found that Pd-catalyzed carboamination reactions of methyl carbamate protected 2-allylbenzylamine 5 provided the desired products 6 in moderate to good yields and good to excellent enantioselectivities (Table 4).xviii Electron rich aryl halides substituted in the para and meta positions were well tolerated (entries 1-2 and 5). However, use of ortho-bromoanisole as an electrophile led to no reaction, and although the coupling of 5 with ortho-iodoanisole did afford tetrahydroisoquinoline 6b (entry 3), this compound was generated in lower yield and selectivity as compared to products 6a and 6c-f.12 Alkenyl bromides were tolerated in the reaction (entry 9) as were aryl iodides. When reactions were conducted at a slightly lower temperature 70 °C the desired products were obtained in similar yields as for reactions at 90 °C, with slightly increased enantioselectivities (entries 7 and 9). Although these transformations were highly effective with terminal alkene substrate 5, efforts to employ an analogous 1,1-disubstituted alkene derivative were unsuccessful. No desired product was generated in reactions of 1,1-disubstituted alkene substrates, and decomposition of the starting material was observed.
Table 4.
Enantioselective Synthesis of Tetrahydroisoquinolinesa

| ||||
|---|---|---|---|---|
| Entry | R–X | Product | Yield (%)b | er |
| 1 | p-MeO-C6H4–Br | 6a | 51 | 93:7 |
| 2 | p-MeO-C6H4–I | 6a | 52 | 93:7 |
| 3 | o-MeO-C6H4–I | 6b | 42 | 80:20 |
| 4 | p-CF3-C6H4–Br | 6c | 72 | 93:7 |
| 5 |
|
6d | 61 | 93:7 |
| 6 | PhBr | 6e | 69 | 94:6 |
| 7c | PhBr | 6e | 64 | 95:5 |
| 8 |
|
6f | 56 | 93:7 |
| 9c |
|
6f | 55 | 94:6 |
Conditions: 1.0 equiv 5, 1.2 equiv R–X, 1.2 equiv NaOtBu, 2 mol % Pd2(dba)3, 6 mol % (S)-Siphos-PE, toluene (0.125 M), 90 °C, 2 h.
Isolated yield (average of two or more runs).
The reaction was conducted at 70 °C for 12-14 hrs.
Conclusions
In conclusion, we have developed a concise enantioselective synthesis of three separate classes of heterocycles: tetrahydroquinolines, tetrahydroquinoxalines, and tetrahydroisoquinolines, via Pd-catalyzed alkene carboamination reactions. A single catalyst system composed of Pd2(dba)3 and (S)-Siphos-PE is effective for all three product classes, and products are generated with good to excellent levels of asymmetric induction. These transformations constitute a fundamentally new approach to the asymmetric synthesis of saturated benzo-fused nitrogen heterocycles bearing quaternary stereocenters, and are also rare examples of highly enantioselective addition reactions between 1,1-disubstituted alkenes and amine nucleophiles.
Supplementary Material
Acknowledgements
The authors acknowledge the NIH-NIGMS (GM-071650) for financial support of this work.
Footnotes
† Electronic Supplementary Information (ESI) available: Experimental procedures, spectroscopic data, and copies of 1H and 13C NMR spectra.
Notes and References
- i.For a review on the synthesis and biological activity of tetrahydroquinolines, see: Sridharan V, Suryavanshi PA, Menéndez JC. Chem. Rev. 2011;111:7157. doi: 10.1021/cr100307m. For reviews on the synthesis and biological activity of tetrahydroisoquinolines, see: Scott JD, Williams RM. Chem. Rev. 2002;102:1669. doi: 10.1021/cr010212u. Bentley KW. Nat Prod. Rep. 2006;23:444. doi: 10.1039/b509523a. Chrzanowska M, Rozwadowska MD. Chem. Rev. 2004;104:3341. doi: 10.1021/cr030692k. For reviews on the synthesis and biological activity of tetrahydroquinoxalines, see: Mamedov VA, Zhukova NA. Prog. Heterocycl. Chem. 2012;24:1. Mamedov VA, Zhukova NA. Prog. Heterocycl. Chem. 2012;24:55.
- ii.a Wang W-B, Lu S-M, Yang P-Y, Han X-W, Zhou Y-G. J. Am. Chem. Soc. 2003;125:10536. doi: 10.1021/ja0353762. [DOI] [PubMed] [Google Scholar]; b Mršić N, Jerphagnon T, Minnaard AJ, Feringa BL, de Vries JG. Adv. Synth. Catal. 2009;351:2549. [Google Scholar]; c Shi L, Ye Z-S, Cao L-L, Guo R-N, Hu Y, Zhou Y-G. Angew. Chem., Int. Ed. 2012;51:8286. doi: 10.1002/anie.201203647. [DOI] [PubMed] [Google Scholar]; d Iimuro A, Yamaji K, Kandula S, Nagano T, Kita Y, Mashima K. Angew. Chem., Int. Ed. 2013;52:2046. doi: 10.1002/anie.201207748. [DOI] [PubMed] [Google Scholar]; e Rueping M, Antonchick AR, Theissmann T. Angew. Chem., Int. Ed. 2006;45:3683. doi: 10.1002/anie.200600191. [DOI] [PubMed] [Google Scholar]; f Rueping M, Tato F, Schoepke FR. Chem.-Eur. J. 2010;16:2688. doi: 10.1002/chem.200902907. [DOI] [PubMed] [Google Scholar]; h Cartigny D, Berhal F, Nagano T, Phansavath P, Ayad T, Genêt J-P, Ohshima T, Mashima K, Ratovelomanana-Vidal V. J. Org. Chem. 2012;77:4544. doi: 10.1021/jo300455y. [DOI] [PubMed] [Google Scholar]
- iii.For a recent review on asymmetric hydrogenation of heteroarenes see: Wang D-S, Chen Q-A, Lu S-M, Zhou Y-G. Chem. Rev. 2012;112:2557. doi: 10.1021/cr200328h.
- iv.a Han Z-Y, Xiao H, Chen X-H, Gong L-Z. J. Am. Chem. Soc. 2009;131:9182. doi: 10.1021/ja903547q. [DOI] [PubMed] [Google Scholar]; b Patil NT, Wu H, Yamamoto Y. J. Org. Chem. 2007;72:6577. doi: 10.1021/jo0708137. [DOI] [PubMed] [Google Scholar]
- v.a Okamoto S, Teng X, Fujii S, Takayama Y, Sato F. J. Am. Chem. Soc. 2001;123:3462. doi: 10.1021/ja004140k. [DOI] [PubMed] [Google Scholar]; b Hashimoto T, Omote M, Maruoka K. Angew. Chem., Int. Ed. 2011;50:8952. doi: 10.1002/anie.201104017. [DOI] [PubMed] [Google Scholar]; c Shirakawa S, Liu K, Ito H, Le TN, Maruoka K. Adv. Synth. Catal. 2011;353:2614. [Google Scholar]
- vi.Saget T, Cramer N. Angew. Chem., Int. Ed. 2012;51:12842. doi: 10.1002/anie.201207959. [DOI] [PubMed] [Google Scholar]
- vii.a Li X, Coldham I. J. Am. Chem. Soc. 2014;136:5551. doi: 10.1021/ja500485f. [DOI] [PubMed] [Google Scholar]; b Chen Z, Wang Z, Sun J. Chem. Eur. J. 2013;19:8426. doi: 10.1002/chem.201301065. [DOI] [PubMed] [Google Scholar]; c Meyers AI, Gonzalez MA, Struzka V, Akahane A, Guiles J, Warmus JS. Tetrahedron Lett. 1991;32:5501. [Google Scholar]
- viii.Seki T, Tanaka S, Kitamura M. Org. Lett. 2012;14:608. doi: 10.1021/ol203218d. [DOI] [PubMed] [Google Scholar]
- ix.a Yang W, Long Y, Zhang S, Zheng Y, Cai Q. Org. Lett. 2013;15:3598. doi: 10.1021/ol401449b. [DOI] [PubMed] [Google Scholar]; b Zhou F, Guo J, Liu J, Ding K, Yu S, Cai Q. J. Am. Chem. Soc. 2012;134:14326. doi: 10.1021/ja306631z. [DOI] [PubMed] [Google Scholar]
- x.For recent reviews, see: Wolfe JP. Top. Heterocycl. Chem. 2013;32:1–38. Schultz DM, Wolfe JP. Synthesis. 2012;44:351–361. doi: 10.1055/s-0031-1289668.
- xi.For prior studies on asymmetric Pd-catalyzed alkene carboamination reactions between alkenes bearing pendant nitrogen nucleophiles and aryl or alkenyl halides, see: Mai DN, Wolfe JP. J. Am. Chem. Soc. 2010;132:12157. doi: 10.1021/ja106989h. Hopkins BA, Wolfe JP. Angew. Chem., Int. Ed. 2012;51:9886. doi: 10.1002/anie.201205233. Babij NR, Wolfe JP. Angew. Chem., Int. Ed. 2013;52:9247. doi: 10.1002/anie.201302720.
- xii.For asymmetric Pd-catalyzed cascade oxidative cyclizations that proceed via syn-aminopalladation of an alkene, see: He W, Yip K-T, Zhu N-Y, Yang D. Org. Lett. 2009;11:5626. doi: 10.1021/ol902348t. For asymmetric Cu-catalyzed alkene carboamination reactions, see: Zeng W, Chemler SR. J. Am. Chem. Soc. 2007;129:12948. doi: 10.1021/ja0762240. Liwosz TW, Chemler SR. J. Am. Chem. Soc. 2012;134:2020. doi: 10.1021/ja211272v. For a discussion of the influence of alkene aminopalladation mechanism on asymmetric induction, see: Weinstein AB, Stahl SS. Angew. Chem. Int. Ed. 2012;51:11505. doi: 10.1002/anie.201206702.
- xiii.For examples of biologically active benzo-fused heterocycles that contain quaternary stereocenters adjacent to the nitrgen atom, see: Cueva JP, Cai TB, Mascarella SW, Thomas JB, Navarro HA, Carroll FI. J. Med. Chem. 2009;52:7463. doi: 10.1021/jm900756t. Steinhagen H, Corey EJ. Org. Lett. 1999;1:823. doi: 10.1021/ol9908575.
- xiv.To the best of our knowledge, 84% ee is the highest enantioselectivity achieved for transition metal catalyzed addition to a 1,1-disubstituted alkene that leads to C-N bond formation. See: Trost BM, Fandrick DR. J. Am. Chem. Soc. 2003;125:11836. doi: 10.1021/ja037450m. Bovino MT, Sherry R. Chemler. Angew. Chem., Int. Ed. 2012;51:3923. doi: 10.1002/anie.201109044. Du H, Zhao B, Yuan W, Shi Y. Org. Lett. 2008;10:4231. doi: 10.1021/ol801605w.
- xv.The absolute stereochemistry of 2j was established by single crystal x-ray analysis. The absolute stereochemistry of all other products was assigned based on analogy to 2j.
- xvi.For further details on the use of other chiral ligands see the Supporting Information.
- xvii.Analysis of crude reaction mixtures after attempted Pd-catalyzed carboamination reactions of N-aryl derivatives of 5 revealed the formation of mixtures of the desired product (ca. 20%), a regioisomeric side product (ca. 20%), and several other unidentified side products.
- xviii.We have found that nitrogen nucleophilicity/basicity has a large impact on the efficiency of Pd-catalyzed carboamination reactions. See: a reference 10. Bertrand MB, Wolfe JP. Tetrahedron. 2005;61:6447. We estimate the carbamate 5 is similar in pKa to substrates 1 and 3 based on a comparison of pKa values for diphenylamine (25.0 in DMSO) vs. N-Boc-benzylamine (pKa=22.9 in DMSO); see: Bordwell FG, Branca JC, Hughes DL, Olmstead WN. J. Org. Chem. 1980;45:3305. Mita T, Chen J, Sugawara M, Sato Y. Angew. Chem., Int. Ed. 2011;50:1393. doi: 10.1002/anie.201006422.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.