

Abstract
The synthesis of G-N2-(CH2)3-N2-G trimethylene DNA interstrand cross-links (ICLs) in a 5′-CG-3′ and 5′-GC-3′ sequence from oligodeoxynucleotides containing N2-(3-aminopropyl)-2′-deoxyguanosine and 2-fluoro-O6-(trimethylsilylethyl)inosine is presented. Automated solid-phase DNA synthesis was used for unmodified bases and modified nucleotides were incorporated via their corresponding phosphoramidite reagent by a manual coupling protocol. The preparation of the phosphoramidite reagents for incorporation of N2-(3-aminopropyl)-2′-deoxyguanosine is reported. The high-purity trimethylene DNA interstrand cross-link product is obtained through a nucleophilic aromatic substitution reaction between the N2-(3-aminopropyl)-2′-deoxyguanosine and 2-fluoro-O6-(trimethylsilylethyl)inosine containing oligodeoxynucleotides.
Keywords: interstrand cross-links, DNA, N2-(3-aminopropyl)-2′-deoxyguanosine
INTRODUCTION
This unit describes the synthesis of a trimethylene interstrand cross-link (ICL) between the N2-positions of 2′-deoxyguanosine (dG-N2-(CH2)3-N2-dG) in a 5′-GC-3′ and 5′-CG-3′ sequence context. This is a model ICL for the reaction of DNA with acrolein (Kozekov et al., 2003; Kozekov et al., 2010). Bis-electrophiles not only have the capability to form simple mono-adducts with the individual DNA nucleobases, they can also form cross-links between bases within the same strand or opposite strands. Synthetic methods have been developed for the preparation of site-specific oligodeoxynucleotides containing distinct dG-N2-(CH2)3-N2-dG ICLs (Dooley et al., 2001; Dooley et al., 2003; Minko et al., 2008), making it possible to study the effects of such lesions on DNA structure and cellular function (Figure 1). This involved the synthesis of complementary oligodeoxynucleotides containing 2-fluoro-O6-(trimethylsilylethyl)-2′-deoxyinosine and their sequential reaction with 1,3-diaminopropane. One drawback was that the intermediate N2-(3-aminopropyl)-2′-deoxyguanosine containing oligodeoxynucleotide was difficult to purify. An alternative approach is described here, in which the N2-(3-aminopropyl)-2′-deoxyguanosine is incorporated via a phosphoramidite reagent; the resulting oligodeoxynucleotide is significantly easier to purify by HPLC. Following purification, it is reacted with its complementary oligodeoxynucleotide containing a 2-fluoro-O6-(trimethylsilylethyl)-2′-deoxyinosine to afford the dG-N2-(CH2)3-N2-dG ICL.
Figure 1.
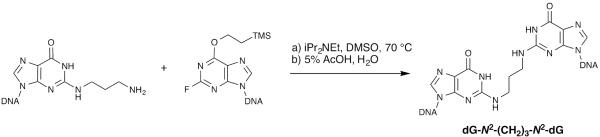
Reaction condition for the synthesis of dG-N2-(CH2)3-N2-dG
Basic Protocol 1 describes the synthesis of the phosphoramidite reagent for incorporation of N2-(3-aminopropyl)-2′-deoxygunaosine into oligodeoxynucleotides. N2-(3-Aminopropyl)-O6-(trimethylsilylethyl)-2′-deoxyinosine was prepared from 2-fluoro-O6-(trimethylsilylethyl)-2′-deoxyinosine as reported by Harris (Harris and Harris, 2000). This section is comprised of three separate procedures: (1) protection of N2-(3-aminopropyl)-O6-(trimethylsilylethyl)-2′-deoxyinosine as a trifluoroacetyl derivative, (2) protection of the 5′-hydroxyl as a 4,4′-dimethoxytrityl (DMT) ether, and (3) phosphitylation of the 3′-hydroxyl to give the phosphoramidite reagent (4) for oligodeoxynucleotide synthesis.
Basic Protocol 2 describes the synthesis of the modified oligodeoxynucleotides by automated, solid-phase DNA synthesis. Unmodified nucleotides were incorporated using standard, commercially available PAC-protect phosphoramidites. An off-line manual coupling protocol was utilized for the modified phosphoramidite prepared in Basic Protocol 1.
Basic Protocol 3 describes the synthesis and purification of ICL oligodeoxynucleotide duplexes. In this section we describe the synthesis of two specific sequences, their purification, and analysis. However, characterization is reported for only one sequence.
BASIC PROTOCOL 1 SYNTHESIS OF N2-[3-[(TRIFLUOROACETYL)AMINO]PROPYL]-5′-O-DIMETHOXYTRITYL-O6-(TRIMETHYLSILYLETHYL)-3′-O-[(2-CYANOETHYL –N,N′-DIISOPROPYL)PHOSPHORAMIDITE]-2′-DEOXYGUANOSINE FROM N2-(3-AMINOPROPYL)-O6-(TRIMETHYLSILYLETHYL)-2′-DEOXYGUANOSINE (4)
This section describes the synthesis of the phosphoramidite reagent for incorporation of N2-(3-aminopropyl)-2′-deoxygunaosine starting from N2-(3-aminopropyl)-O6-(trimethylsilylethyl)-2′-deoxyguanosine (Harris and Harris, 2000), as depicted in Figure 2. It is divided in three basic procedures: (1) protection of 3-aminopropyl moiety as a trifluoroacetyl group (Micheli et al., 2004), (2) protection of the 5′-OH as a 4,4′-dimethoxytrityl ether, and (3) functionalization of the 3′-hydroxyl group with 2-cyanoethyl-N,N,N′N′-tetraisopropylphosphane (DeCorte et al., 1996).
Figure 2.

Synthesis of N2-[3-[(trifluoroacetyl)amino]propyl]-5′-O-DMT-O6-(trimethylsilylethyl)-3′-O-[(2-cyanoethyl-N,N′-diisopropyl)phosphoramidite]-2′-deoxyguanosine. a) Ethyl trifluoroacetate and methanol, room temperature, 4 hours; b) 4,4′-dimethoxytrityl chloride, pyridine, room temperature, 8 hours; c) 2-cyanoethyl N,N,N,N-tetraisopropylphosphane, tetrazole, dichloromethane, room temperature, 3 hours.
NOTE: Anhydrous solvents are required for these steps. They can be purchased or dried by distillation (Perrin and Armarego, 1988) and stored under nitrogen.
Materials
N2-(3-aminopropyl)-O6-(trimethylsilylethyl)-2′-deoxyguanosine (Harris and Harris, 2000)
Diisopropylethylamine
Methanol, anhydrous
Ethyl trifluoroacetate
Dimethoxytrityl chloride
Pyridine, anhydrous
Tetrazole (0.45 M in acetonitrile)
Dichloromethane, anhydrous
2-cyanoethyl-N,N,N′,N′-tetraisopropylphosphane
Dry nitrogen
63-to 200-mesh silica gel
Sand
Triethylamine
100 mL round bottom flask with 24/40 joint and and septum
Rotary evaporator
Stir bars
UV light source for visualizing TLC plates
Vacuum system (oil pump) capable of <1 mm Hg pressure, with manifold and cold trap
10 mL glass syringes
100 μL glass syringes
500 μL glass syringes
Heavy walled glass columns for flash chromatography: 3.6 × 40 cm and 1.5 × 20 cm
Protection of aminopropyl NH2
Synthesis
-
1.
Dry N2-(3-aminopropyl)-O6-(trimethylsilylethyl)-2′-deoxyguanosine (1) in an Abderhalden apparatus overnight at 70°C and pressure <1 mm Hg
-
2.
Dissolve 0.2 g of 1 in anhydrous methanol in a 100 mL, oven-dried round bottom flask fitted with a stir bar and rubber septum. (Table 1)
-
3.
Add 56 μL of ethyl trifluoroacetate dropwise to the stirred solution via syringe. (Table 1)
-
4.
Stir at room temperature for 4 hours while monitoring the reaction by TLC.
-
5.
Perform TLC analysis on 0.25-mm silica gel 60F-254 glass plates and 9:1 (v/v) dichloromethane/methanol with containing 1% triethylamine as the mobile phase. The Rf of the product is 0.37 while starting material remains at or near the baseline with Rf <0.1. (see APPENDIX 3D)
Table 1.
Synthesis of N2-[3-[(trifluoroacetyl)amino]propyl]-O6-(trimethylsilylethyl)-2′-deoxyguanosine (2) starting from 1
| Reagent | Amount | MW (g/mol) | Millimoles | Equivalents |
|---|---|---|---|---|
| N2-(3-aminopropyl)-O6-(trimethylsilylethyl)-2′-deoxyguanosine | 0.2 g | 324 | 0.58 | 1 |
| Ethyl trifluoroacetate | 56 μl | 142 | 0.75 | 1.3 |
| Methanol, anhydrous | 6 mL | 32 | 140 | 255 |
Purification of N2-[3-[(trifluoroacetyl)amino]propyl]-O6-(trimethylsilylethyl)-2′-deoxyguanosine (2)
-
6.
When TLC analysis shows that the reaction is complete, remove solvents under reduced pressure using a rotary evaporator.
-
7.
Purify 2 by flash column chromatography using a heavy-walled glass column 3.6 × 40 cm with ~32 g of silica gel eluting with 9:1 (v/v) dichloromethane/methanol with 1% triethylamine. (see APPENDIX 3E)
-
8.
Analyze the fractions by TLC as described in step 4. Collect and rotary evaporate the fractions containing 1.
-
9.
Dry 2 under high vacuum.
5′-O-Protection as a 4,4′-dimethoxytrityl ether
Synthesis
-
10.
Add 2 mL of pyridine to 2 and evaporate using a rotary evaporator.
-
11.
Repeat addition of pyridine and evaporation two additional times.
-
12.
Dry 2 under vacuum overnight with an oil pump capable of <1 mm Hg vacuum.
-
13.
Dissolve 93 mg of 2 in 5 mL of anhydrous pyridine under argon and stir at room temperature. (Table 2) Add 78 mg of solid 4,4′-dimethoxytrityl chloride in one single portion, while keeping the reaction under argon.
-
14.
Stir the reaction at room temperature for 8 hours.
-
15.
Monitor the progression of the reaction by TLC using 9:1 (v/v) dichloromethane/methanol with 1% triethylamine. Rf of the starting material: 0.37, Rf of the product: 0.45 (APPENDIX 3D)
-
16.
When the reaction is complete, removed the solvents under pressure using a rotary evaporator without heating.
-
17.
Dry under vacuum using an oil pump as in step 14.
Table 2.
Synthesis of N2-[3-[(trifluoroacetyl)amino]propyl]-5′-O-DMT-O6-(trimethylsilylethyl)-2′-deoxyguanosine (3) from compound 2
| Reagent | Amount | MW (g/mol) | Millimoles | Equivalents |
|---|---|---|---|---|
| N2-[3-[(trifluoroacetyl)amino]propyl]-O6-(trimethylsilylethyl)-2′-deoxyguanosine | 0.093 g | 522 | 0.18 | 1 |
| 4,4′-Dimethoxytrityl chloride | 0.078 g | 338 | 0.23 | 1.3 |
| Pyridine, anhydrous | 5 mL | 79 | 61 | 344 |
Purification of 3
-
18.
Purify 3 by flash column chromatography using a 1.5 cm heavy-walled glass column with ~12 g of silica gel eluting with 99:1 (v/v) dichloromethane/methanol and 1% triethylamine. (see APPENDIX 3E)
-
19.
Combine fractions containing pure 3 and evaporate to dryness with a rotary evaporator, and then with an oil pump.
The product is sensitive to heat and long exposure in solution. It should be dried and kept under nitrogen. It can be stored at 4°C for several months.
The product is a pale yellow solid, obtained 92 mg (63% yield). Unreacted starting material (2) can also be recovered during purification of 3 by flash column chromatography as reported in step 7.
Functionalization of 3′-O with 2-cyanoethyl-N,N,N′,N′-tetraisopropylphosphane (4)
Synthesis
-
20.
Dry compound 3 as reported in steps 12–14.
-
21.
Under argon, dissolve 64 mg of compound 3 in 7 mL of anhydrous dichloromethane. (Table 3)
-
22.
To the stirred solution, simultaneously add tetrazole and 2-cyanoethyl-N,N,N′,N′-tetraisopropylphosphane with two separate syringes.
-
23.
Stir at room temperature for 3 hours.
-
24.
Monitor the progression of the reaction by TLC using 95:5 (v/v) dichloromethane/methanol and 1% triethylamine. Rf product: 0.41; Rf starting material 0.37. (APPENDIX 3D)
-
25.
When the reaction is complete, remove the solvents using a rotary evaporator, then dry under vacuum with an oil pump.
Table 3.
Synthesis of N2--[3-[(trifluoroacetyl)amino]propyl]-5′-O-DMT-O6-(trimethylsilylethyl)-3′-O-[(2-cyanoethyl-N,N′-diisopropyl)phosphoramidite]-2′-deoxyguanosine (4) from compound 3
| Reagent | Amount | MW (g/mol) | Millimoles | Equivalents |
|---|---|---|---|---|
| N2-[3-[(trifluoroacetyl)amino]propyl]-5′-O-DMT-O6-(trimethylsilylethyl)-2′-deoxyguanosine | 0.064 g | 824 | 0.077 | 1 |
| 2-cyanoethyl-N,N,N,N-tetraisopropylphosphane | 32 μl | 301 | 0.1 | 1.5 |
| Tetrazole (0.45M solution in acetonitrile) | 257 μl | 70 | 0.12 | 1.3 |
| Dichloromethane, anhydrous | 7 mL | 84 | 110 | 1428 |
Purification of N2-[3-[(trifluoroacetyl)amino]propyl]-5′-O-DMT-O6-(trimethylsilylethyl)-3′-O-[(2-cyanoethyl-N,N′-diisopropyl)phosphoramidite]-2′-deoxyguanosine (4)
-
26.
Purify 4 by flash column chromatography using a 1.5× 20 cm heavy-walled glass column with 7 g of silica gel eluting with dichloromethane and 1% triethylamine. (see APPENDIX 3E)
-
27.
Combine fractions containing pure 4 and evaporate solvent utilizing a rotary evaporator followed by drying with an oil pump with <1 mm Hg vacuum.
Pure product 4 (48 mg obtained, yield 61%) is a pale yellow solid and is very sensitive to air exposure and heat. Moreover, it is not stable if left in solution. It can be stored under nitrogen or argon at 4°C.
BASIC PROTOCOL 2 SYNTHESIS OF MODIFIED OLIGODEOXYNUCLEOTIDES CONTAINING 2-FLUORO-O6-(TRIMETHYLSILYLETHYL)-2′-DEOXYINOSINE AND N2-(3-AMINOPROPYL)-O6-(TRIMETHYLSILYLETHYL)-2′-DEOXYGUANOSINE
This section describes the synthesis of oligodeoxynucleotides precursors for the synthesis of G-N2-(CH2)3-N2-G interstrand cross-links. The cross-links are synthesized in a 5′-CG-3′ and 5′-GC-′. The sixteen base pair oligodeoxynucleotides in 5′-GC-3′ and 5′-CG-3′ sequences contained the N2-(3-aminopropyl)-O6-(trimethylsilylethyl)-2′-deoxyguanosine (designated as a in Figure 3) as the modified base, whereas the oligodeoxynucleotides designated as b contained the 2-fluoro-O6-(trimethylsilylethyl)-2′-deoxyinosine. The oligodeoxynucleotides were synthesized on a Perseptive Biosystems Model 8909 DNA synthesizer on 1 μmol scale using the UltraMild line of phosphoramidites and solid supports from Glen Research. The manufacturer's standard synthesis protocol was followed except at the incorporation of the modified phosphoramidites, which were accomplished manually off-line (Elmquist et al., 2004). The manual coupling uses less of the modified phosphoramidite since the DNA synthesizer lines do not have to be primed with the reagent solution.
Figure 3.
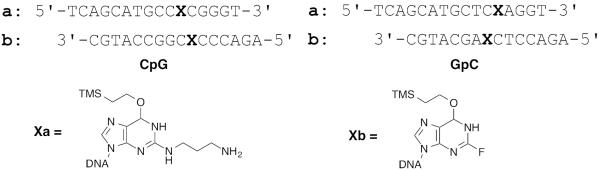
5′-CG-3′ and 5′-GC-3′ oligonucleotides sequences and structures of the modified bases.
Materials
5′-O-(DMT)-N2-[3-[(trifluoroacetyl)amino]propyl]-O6-(trimethylsilylethyl)-3′-O-[(2-cyanoethyl-N,N′-diisopropyl)phosphoramidite]-2′-deoxyguanosine (4)
5′-O-(DMT)-2-fluoro-O6-(trimethylsilylethyl)-3′-O-[(2-cyanoethyl-N,N′-diisopropyl)phosphoramidite]-2′-deoxyinosine (Harris and Harris, 2000)
UltraMild phosphoramidites (Glen Research)
Acetonitrile (Glen Research)
Tetrazole, 0.45 N in acetonitrile (Glen Research)
Phenoxyacetyl chloride, 5% solution in tetrahydrofuran (Glen Research, CapA solution)
1-methylimidazole, 10% solution in tetrahydrofuran/pyridine (Glen Reserarch, CapB solution)
1 mL syringes
Argon
Snap cap vials
Synthesis column
Perseptive Biosystems Model 8909 DNA synthesizer
Phenomenex Luna-C18 column (250 × 4.6 mm)
Phenomenex Luna-C18 column (250×10mm)
NaOH 0.1 M
NH4OH
Formic Acid
3 mL glass vials
Stir bars
Ammonium formate buffer 0.1 M
Centrifugal vacuum evaporator
Solid-phase synthesis of 5′-CG-3′ containing an N2-(3-aminopropyl)-2′-deoxyguanosine modified base
-
28.
Weigh 8 mg of phosphoramidite 4, place in a vial, and dissolve in 100 μL of acetonitrile (ACN).
-
29.
Assemble the sequence according to the standard solid-phase synthesis cycle and start the automated DNA synthesis.
-
30.
Remove the column when the manual coupling is to be performed.
-
31.
Fit each end of the column with a syringe, one containing an anhydrous solution of the phosphoramidite 4, and the other syringe containing a solution of 1H-tetrazole.
-
32.
Beginning with the syringe containing 1H-tetrazole, draw the solution through the cassette 5 times each, every 3 minutes, over the course of 30 minutes.
-
33.
Put the column back on the synthesizer and cap with phenoxyacetyl chloride solution.
-
34.
Re-start the standard solid-phase synthesis cycle for the remaining unmodified bases.
-
35.
Cleave the modified oligodeoxynucleotide from the solid support using 1.2 mL of NH4OH at 60°C for one hour.
-
36.
Evaporate under vacuum with a centrifugal evaporator.
Purification
-
37.
Equilibrate the HPLC column (Phenomenex Luna C18(2), 250 × 10mm) for 15 minutes with 1% ammonium formate buffer.
-
38.
Dissolve the residue from step 36 with 200 μL of deionized water.
-
39.
Purify by HPLC using the following gradient: solvent A = 0.1 M ammonium formate buffer, solvent B = acetonitrile. Gradient: 0–20 min, linear gradient from 95% A/5% B to 50% A/50% B (v/v); 20–23 min, linear gradient to 80% A/20% B (v/v); isocratic for 2 minutes at 80% A/20% B; 25–28 min, linear gradient to 95% A/5% B (v/v)
-
40.
Collect all peaks. The desired oligodeoxynucleotides should elute at ~ 13 minutes.
-
41.
Dry the sample on a lyophilizer.
Deprotection from dimethoxytrityl group
-
42.
Dissolve purified compound from step 40 with 0.5 mL of deionized water.
-
43.
Add 1 mL of 2% trifluoroacetic acid and stir for 30 minutes at room temperature.
-
44.
Monitor the reaction by HPLC as in step 39
-
45.
After reaction is complete, neutralize with NH4OH and remove solvent with a centrifugal evaporator
Purification
-
46.
Dissolve oligodeoxynucleotide from step 44 in 300 μL of water.
-
47.
Purify by HPLC using the following gradient: solvent A = 0.1 M ammonium formate buffer, solvent B = acetonitrile. Gradient: 0–20 min, linear gradient from 99% A/1% B to 90% A/10% B (v/v); 20–23 min, linear gradient to 80% A/20% B (v/v); isocratic for 2 minutes at 80% A/20% B; 25–28 min, linear gradient to 20% A/80% B (v/v); isocratic for 2 minutes at 20% A/80% B; 30–33 min, linear gradient to 99% A/1% B (v/v).
-
48.
Collect fraction containing the desired oligodeoxynucleotide, which should elute at ~ 20 minutes.
-
49.
Lyophilize combined fractions.
ALTERNATE PROTOCOL 1
Synthesis of the oligodeoxynucleotide containing the 2-fluoro-O6-(trimethylsilylethyl)-2′-deoxyinosine modified base
The standard protocol for deprotection of synthetic oligodeoxynucleotides and cleavage from the solid support utilize hot NH4OH. The 2-fluoro-O6-(trimethylsilylethyl)-2′-deoxyinosine will undergo nucleophilic aromatic substitution with NH3 under these conditions to give 2′-deoxyguanosine. This would be undesirable as the 2-fluoro functional group is required for the synthesis of the cross-link. Deprotection of the 2-fluoro-O6-(trimethylsilylethyl)-2′-deoxyinosine is therefore achieved with dilute NaOH (0.1 M) solution at room temperature.
Follow steps 29 through 35 except use the 5′-O-(DMT)-2-fluoro-O6-(trimethylsilylethyl)-3′-O-[(2-cyanoethyl-N,N′-diisopropyl)phosphoramidite]-2′-deoxyinosine reagent instead of 4.
-
50.
Cleave the modified oligodeoxynucleotide from the solid support using 1.2 mL of 0.1 M NaOH overnight at room temperature.
Purification
The same HPLC method utilized in Basic Protocol 2 is used for this purification. The desired oligodeoxynucleotide should elute at ~ 14 minutes.
Deprotection from dimethoxytrityl group and purification
Deprotection is carried out exactly as in Basic Protocol 2 (steps 41 to 44).
BASIC PROTOCOL 3 SYNTHESIS OF OLIGODEOXYNUCLEOTIDES WITH AMINOPROPYL CROSS-LINK
This section describes the synthesis and purification of oligodeoxynucleotides containing a G-N2-(CH2)3-N2-G cross-link. We report details for the synthesis of the 5′-GC-3′ sequence, but the same procedure can be applied to the 3′-CG-5′ sequence (Figure 3 and Figure 4). The reaction is carried out at 37°C for 4 days. The desired cross-link product is partially purified by HPLC then further purified by gel electrophoresis. The cross-linked oligodeoxynucleotides are characterized by LC-ESI-MS, MALDI-TOF, and enzyme digestion.
Figure 4.

5′-CG-3′ and 5′-GC-3′ sequences of the trimethylene interstrand cross-link
Materials
5′-TCAGCATGCCXCGGGT-3′ (X= N2-(3-aminopropyl)-2′-deoxyguanosine)
3′-CGTACGGCXCCCAGA-5′ (X= 2-fluoro-O6-(trimethylsilylethyl)inosine)
Borate buffer
Water
Pipettes
Temperature-controlled water bath
Acetic acid 5%
Tris-HCl buffer (50mM pH=7 and 5mM MgCl2)
DNAse I
Phosphodiesterase I
Nuclease P1,
Alkaline phosphatase
Hydroxypicolinic acid
Ammonium formate buffer 0.1M
Acetonitrile
HPLC column: Luna 5u C18(2) 110A 250 × 1.00mm (for LC-MS analysis)
HPLC column: YMC ODS-AQ 250mm × 1.0 mm, 1.5 mL/min flow
Eppendorf vials
Polyacrylamide gel electrophoresis: polyacrylamide, water, 80 mM tris-borate buffer with 1mM
EDTA (see Appendix 3B)
Desalting column: BioSpin 6-Tris column (Bio-Rad)
Synthesis of cross-linked oligodeoxynucleotide duplex
-
51.
Dissolve 71 nmols of 5′-TCAGCATGCCXCGGGT-3′ (X= N2-(3-aminopropyl)-2′-deoxyguanosine modified base) into 300 μL of borate buffer 0.05M pH 9.
-
52.
Add 80 nmols of 3′-CGTACGGCXCCCAGA-5′ (X= 2-fluoro- O6-(trimethylsilylethyl)-2′-inosine modified base).
-
53.
Stir the reaction mixture at 37°C for 4 days.
-
54.
Add 200 μL of 5% acetic acid.
-
55.
Monitor the reaction by HPLC utilizing conditions in step 56.
Purification
-
56.
Purify the desired product by HPLC using the following conditions: flow rate 1.5 mL/min, T = 50°C, solvent A = 0.1 M ammonium formate buffer, solvent B = acetonitrile. Gradient: 0–5 min, isocratic at 95% A/5% B (v/v); 5–20 min, linear gradient to 90.5% A/9.5% B (v/v); 20–23 min, linear gradient to 100% B (v/v); isocratic at 100% B for 3 minutes; 25–28 min, linear gradient to 99% A/1% B (v/v); Collect all peaks for analysis.
-
57.
Dry the samples utilizing a lyophilizer.
-
58.
Dissolve each fraction in 100 μL of water.
-
59.
Desalt samples with an ion-exchange spin column.
-
60.
Analyze fractions by LC-MS.
-
61.
Collect and dry desired cross-linked oligodeoxynucleotide, which should elute at ~ 16.2 minutes.
-
62.
Further purify by polyacrylamide gel electrophoresis as necessary. (Appendix 3B) [Gel size: 38 × 50 cm. Power supply: 80W constant. Run time: 3 hours. After visualizing bands with UV lamp at 254 nm, excise them and elute from crushed gel overnight in 0.05 M TEAA buffer (pH 7.0). Desalt with BioSpin 6-Tris column (Bio-Rad)].
Analysis
LC-ESI-MS
-
63.
Analyze the cross-linked oligodeoxynucleotide by LC-ESI-MS on an Acquity ultraperformance liquid chromatography (UPLC) system (Waters Corp) interfaced to a Finnigan LTQ mass spectrometer (Thermo Scientific Corp, San Jose, CA) operating in the ESI negative ion mode and using an Acquity UPLC system BEH octadecysilane (C18) column (1.7 μm, 2.1 mm × 50 mm). Buffer A contained 10 mM NH4CH3CO2 plus 1% CH3CN (v/v), and buffer B contained 10 mM NH4CH3CO2 plus 90% CH3CN (v/v). The following gradient program was used with a flow rate of 150 μL min−1: 0–4 min, linear gradient from 100% A to 95% A/5% B (v/v); 4–6 min, linear gradient to 80% A/20% B (v/v); 6–7 min, linear gradient to 70% A/30% B (v/v); 7–8 min, linear gradient to 60% A/40% B (v/v); 8–9 min, linear gradient to 50% A/50% B (v/v); 9–13 min, linear gradient to 100% B; 13–15 min, linear gradient to 100% A; 15–16 min, isocratic at 100% A. The spectrum shows a multiply charged species. LC-ESI-MS calcd for [M–5H]−5 1899.03, found 1898.91 (Figure 5a).
Figure 5.

a) LC-ESI-MS of the 5′-GC-3′ cross-link showing a multiply charged species; calculated for[M–5H]−5 m/z 1899.03, found 1898.91. b) MALDI-TOF spectra of the 5′-GC-3′ cross-link showing [M-H]−1 m/z 9499.90. The analysis is performed on a Voyager Elite DE instrument (Perseptive Biosystem) using 3-hydroxypicolinic acid matrix containing ammonium hydrogen citrate. (Table 4)
MALDI-TOF
-
64.
Analyze the cross-linked oligodeoxynucleotide on a Voyager Elite DE instrument (Perseptive Biosystem) using 3-hydroxypicolinic acid matrix containing ammonium hydrogen citrate. (Figure 5b and Table 4)
Table 4.
Calculated and Observed masses for 5′-GC-3′ and 5′-CG-3′ ICLs.
| Calculated Mass | Oberserved Mass | Error % | |
|---|---|---|---|
| 5′-GC-3′ cross-link | 9500.30 | 9499.90 | 0.006 |
| 5′-CG-3′ cross-link | 9500.30 | 9503.44 | 0.033 |
Enzymatic digestion
-
65.
Dissolve 10 μg of cross-linked oligodeoxynucleotide in 100 μL of Tris-HCl buffer (50 mM, pH 7, with 5 mM MgCl2).
-
66.
Add 8 units of DNase I, 0.02 units of phosphodiesterase I, 10 units nuclease P1, and 1.7 units of alkaline phosphatase.
-
67.
Incubate for 24 hours at 37° C.
-
68.
Analyze by LC-ESI-MS as in step 63 except using the following UPLC conditions: Solvent A: ammonium formate 0.1 M; solvent B: acetonitrile. Gradient: 0–15 min, linear gradient to 90%A/10%B; 15–20 min, linear gradient to 80%A/20%B; 20–24 min, linear gradient to 100%B; 24–26, linear gradient to 100%A. (Figure 6)
Figure 6.

ESI-LC-MS analysis of enzymatic digestion of the 5′-GC-3′ ICL. MS2 is utilized to monitor m/z corresponding to each base and to the cross-link. On the left, reconstructed ion chromatogram for A, T, G, C and cross-link (from top to bottom). b) Total ion current and mass spectrum for the cross-link standard.
Compound characterization
N2-[3-[(trifluoroacetyl)amino]propyl]-Ox6-(trimethylsilylethyl)-2′-deoxyguanosine (2): 1H NMR
(CD2Cl2) δ 7.67 (s, 1H,H8), 7.30–7.25 (m, 9H, ArH), 6.81–6.78 (m, 4H, ArH), 6.35 (t, 1H, J= 6.7), 4.67 (m, 1H, H3′), 4.55 (m, 2H, OCH2CH2Si), 4.1 (q, 1H, H4′ J=3.6 Hz), 3.79 (s,6H, ArOCH3), 3.54–3.25 (m, 6H, H5′+H5″+-NHCH2CH2CH2NH-), 2.76 (m, 1H, H2″), 2.45 (m, 1H, H2′), 1.84 (m, 2H, NHCH2CH2CH2NH-), 1.3 (m, 2H, CH2Si), 0.11 (s, 9H, Si(CH3)3). 13C NMR 161.2, 158.8, 159.5, 152.7, 138.8, 88.6, 72.4, 64.8, 63.1, 45.9, 39.7, 39.1, 37.4, 28.6, 17.4, 10, 1.9. 19F NMR (CD2Cl2) δ −74.4. ESI-MS calcd for [M-H]+ m/z 521.2150, found 521.2165.
N2-[3-[(trifluoroacetyl)amino]propyl]-5′-O-DMT-O6-(trimethylsilylethyl)-2′-deoxyguanosine (3): 1H NMR
(CD2Cl2) δ 7.67 (s, 1H,H8), 6.35 (t, 1H, J= 6.7), 4.67 (m, 1H, H3′), 4.55 (m, 2H, OCH2CH2Si), 4.1 (q, 1H, H4′ J=3.6 Hz), 3.75 (dd, 1H,H5″, J=3.6 Hz), 3.63 (dd, 1H,H5′, J=3.6 Hz), 3.1 (m, 1H, H2″), 2.32 (m, 1H, H2′), 1.84 (m, 2H, NHCH2CH2CH2NH-), 1.2(m, 2H, CH2Si), 0.11 (s, 9H, Si(CH3)3). 19F NMR (CD2Cl2) δ −73.9 (s, CF3CONH-). 13C NMR (CD2Cl2)161, 160.2, 159.5, 145.1, 138.5, 137.2, 130.9,128.3, 128.1, 127.3, 113.3, 87.2, 84.3, 72.1, 65.1, 64.8, 54.7, 46.6, 40.1, 38.3, 37.4, 29.4, 17.8, 11.6. ESI-MS calcd for [M-H]+ m/z 823.3457, found 823.3433.
N2-[3-[(trifluoroacetyl)amino]propyl]-5′-O-DMT-O6-(trimethylsilylethyl)-3′-O-[(2-cyanoethyl-N,N-diisopropyl)phosphoramidite]-2′-deoxyguanosine (4): 1H NMR
(CD2Cl2): δ 7.71 (s, 1H,H8),7.70 (7.71 (s, 1H,H8) 7.32–7.18 (m, 9H, ArH), 6.82–6.80 (m, 4H, ArH), 6.31 (t, 1H, J= 6.7), 4.79 (m, 1H, H3′), 4.61 (m, 2H, OCH2CH2Si), 4.1 (q, 1H, H4′ J=3.6 Hz), 3.86 (m, 2H, -POCH2CH2CN) 3.79 (m,1H, ArOCH3), 3.63–3.33 (m, 6H, H5′+H5″+-NHCH2CH2CH2NH-), 2.91 (m, 1H, PNCH(CH3)2, 2.76 (m, 1H, H2″), 2.60 (m, 2H, m, 2H, -POCH2CH2CN) 2.45 (m, 1H, H2′), 1.84 (m, 2H, NHCH2CH2CH2NH-), 1.3 (m, 2H, CH2Si), 1.14 (d, 6H, PNCH(CH3)2 J= 6.6 Hz), 0.11 (s, 9H, Si(CH3)3). 19F NMR (CD2Cl2) δ −73.85 (s, CF3CONH-), −73.75 (s, CF3CONH-). 13C NMR (CD2Cl2) 162, 160.2, 159.5, 145.1, 138.5, 137.2, 130.9,128.3, 128.1, 127.3, 126, 119.3, 115, 112, 87.2, 84.3, 84.1, 81, 72.8, 72.1, 65.1, 64.8, 58, 55.7, 46.6, 43, 40.1, 38.3, 37.4, 29.4, 24.6, 19.3, 17.8, 9.6 31P NMR 149.39, 148.51 ESI-MS calcd for [M-H]+ m/z 1023.45, found 1023.23
REAGENTS AND SOLUTIONS
All solvents for the synthetic procedures are anhydrous and HPLC grade solvents are used for HPLC purification and analyses.
Ammonium formate buffer
Ammonium formate buffer is prepared by adding 24.53 g of ammonium formate (solid) to 4 L of deionized water. The pH of the buffer is 6.7.
COMMENTARY
Background Information
Interstrand cross-links (ICLs) can form as a consequence of exposure to endogenous, environmental and chemotherapeutic bifunctional electrophiles. ICLs can interfere with replication, transcription, and recombination by covalently linking the two DNA strands thereby preventing strand separation. Examples of agents that form ICLs are α,β-unsaturated aldehydes from lipid peroxidation (acrolein, crotonaldehyde a 4-hydroxynonenal) and some chemotherapeutic agents (nitrogen mustard and mitomycin C) (Noll et al., 2006; Stone et al., 2008). The present trimethylene ICL is a model for the acrolein interstrand cross-link. Other model ICLs have also been reported (Angelov et al., 2009; Wilds et al., 2011).
The G-N2-(CH2)3-N2-G cross-link was previously synthesized from complementary oligodeoxynucleotides containing 2-fluoro-O6-(trimethylsilylethyl)-2′-deoxyinosine (Dooley et al., 2001; Dooley et al., 2003; Minko et al., 2008). One of the oligodeoxynucleotides was reacted with excess 1,3-diaminopropane and purified. The resulting N2-(3-aminopropyl)-2′-deoxyguanosine containing oligodeoxynucleotide was then reacted with a complementary oligodeoxynucleotide containing 2-fluoro-O6-(trimethylsilylethyl)-2′-deoxyinosine to afford the desired ICL. These substrates were used for replication and structural studies (Huang et al., 2009; Minko et al., 2008). Stone et al. reported a detailed structural study of the G-N2-(CH2)3-N2-G cross-link in a 5′-CG-3′ and 5′-GC-3′ sequence context. They observed that the stacking at the cross-linked base pair in the 5′-GC-3′ sequence is highly perturbed, in contrast to 5′-CG-3′, cross-link, which did not show significant perturbation (Huang et al., 2009; Stone et al., 2008). The different structural perturbations of the cross-link may influence their biological processing.
In this protocol we report an alternative synthetic route to the G-N2-(CH2)3-N2-G ICLs. We decided to incorporate an N2-(3-aminopropyl)- 2′-deoxyguanosine into oligodeoxynucleotide via its protected phosphoramidite reagent. A phosphoramidite of N2-(3-aminopropyl)-2′-deoxyguanosine with a different O6-protecting group has been previously reported (Manoharan et al., 1996). The synthesis of the modified nucleoside 1 was previously reported by Harris (Harris and Harris, 2000). The terminal NH2 group was protected with ethyl trifluoroacetate then converted to the corresponding phosphoramidite 4 by established methods (Figure 2). The synthesis of the interstrand cross-link requires several days at 37°C (Figure 1). The removal of the trimethylsilylethyl group is rapid by mild acid treatement. The purification process requires two steps in order to obtain highly pure cross-linked product. HPLC purification allows separation of ICLs from the 2-fluoro-inosine containing oligodeoxynucleotides and recovery of any unreacted N2-(3-aminopropyl)-2′-deoxygunaosine containing oligonucleotide; however, the desired ICL products could only be purified to homogeneity with gel electrophoresis (~25–30% yield).
Critical Parameters and Trouble shooting
It is important that starting materials be thoroughly dried for the synthesis of 4. They can be dried either by co-evaporation with pyridine, or in an Abderhalden apparatus. Fresh reagents were used for automated DNA synthesis, and the modified phosphoramidites must also to be extremely dry. They can be conveniently dried by co-evaporation with pyridine followed by co-evaporation with anhydrous dichloromethane. For the ICL synthesis, it is recommended to use excess of 2-fluoroinosine containing oligodeoxynucleotide, which can be synthesized in higher yield. This oligodeoxynucleotide tends to decompose over the course of the cross-linking reaction, and therefore, cannot be recovered from reaction mixture. Characterization by MALDI-TOF can be challenging due to the fact that DNA cross-linked oligodeoxynucleotides do not ionize very well. Linear mode is recommended instead of reflector mode.
Anticipated Results
Yields of about 25–35% of cross-linked oligodeoxynucleotide after HPLC and PAGE purification can be anticipated. These ICL containing oligodeoxynucleotides can be characterized by enzymatic digestion and LC/MS, and MALDI-TOF MS analysis.
Time Considerations
An approximate time frame for the synthetic part is about a week from compound 1 to compound 4. However, this may vary depending on the time spent on drying, chromatographic separation, and analysis (NMR, ESI-MS). The oligodeoxynucleotides can be synthesized in a few hours using the DNA synthesizer. The purification after cleavage from the solid support takes 2 days, plus one additional day for drying. The subsequent deprotection of the dimethoxytrityl group, purification, and desalting steps require ~3 days. Synthesis and purification of ICLs requires ~7–10 days.
ACKNOWLEDGMENTS
This work was supported by NIH Grants P01 ES05355 and P01 CA160032, and Center grants P30 ES00267 (Center in Molecular Toxicology); P30 CA068485 (Vanderbilt-Ingram Cancer Center).
LITERATURE CITED
- Angelov T, Guainazzi A, Schärer OD. Generation of DNA interstrand cross-links by post-synthetic reductive amination. Org. Lett. 2009;5:661–664. doi: 10.1021/ol802719a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCorte BL, Tsarouhtsis D, S. K, M.D. C, C.M. H, Harris TM. Improved strategies for postoligomerization synthesis of oligodeoxynucleotides bearing structurally defined adducts at the N2 position of deoxyguanosine. Chem. Res. Toxicol. 1996;9:630–637. doi: 10.1021/tx9501795. [DOI] [PubMed] [Google Scholar]
- Dooley PA, Tsarouhtsis D, Korbel GA, Nechev LV, Shearer J, Zegar IS, Harris CM, Stone MP, Harris TM. Structural studies of an oligodeoxynucleotide containing a trimethylene interstrand cross-link in a 5'-(CpG) motif: model of a malondialdehyde cross-link. J. Am. Chem. Soc. 2001;123:1730–1739. doi: 10.1021/ja003163w. [DOI] [PubMed] [Google Scholar]
- Dooley PA, Zhang MZ, Korbel GA, Nechev LV, Harris CM, Stone MP, Harris TM. NMR determination of the conformation of a trimethylene interstrand cross-link in an oligodeoxynucleotide duplex containing a 5'-d(GpC) motif. J. Am. Chem. Soc. 2003;125:62–72. doi: 10.1021/ja0207798. [DOI] [PubMed] [Google Scholar]
- Elmquist CE, Stover JS, Wang Z, Rizzo CJ. Site-specific synthesis and properties of oligonucleotides containing C8-deoxyguanosine adducts of the dietary mutagen IQ. J. Am. Chem. Soc. 2004;126:11189–11201. doi: 10.1021/ja0487022. [DOI] [PubMed] [Google Scholar]
- Harris TM, Harris CM. Synthesis of N2-substituted deoxyguanosine nucleosides from 2-Fluoro-6-O-(trimethylsilylethyl)-2′-deoxyinosine. Curr. Prot. Nucleic Acid Chem. 2000:1.3.1–1.3.19. doi: 10.1002/0471142700.nc0103s00. [DOI] [PubMed] [Google Scholar]
- Huang H, Dooley PA, Harris CM, Harris TA, Stone MP. Differential Base Stacking Interactions Induced by Trimethylene Interstrand DNA Cross-Links in the 5'-CpG-3' and 5'-GpC-3' Sequence Contexts. Chem. Res. Toxicol. 2009;22:1810–1816. doi: 10.1021/tx900225c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozekov ID, Nechev LV, Moseley MS, Harris CM, Rizzo CJ, Stone MP, Harris TM. DNA interchain cross-links formed by acrolein and crotonaldehyde. J. Am. Chem. Soc. 2003;125:50–61. doi: 10.1021/ja020778f. [DOI] [PubMed] [Google Scholar]
- Kozekov ID, Turesky RJ, Alas GR, Harris CM, Harris TM, Rizzo CJ. Formation of deoxyguanosine cross-links from calf thymus DNA treated with acrolein and 4-hydroxy-2-nonenal. Chem. Res. Toxicol. 2010;23:1701–1713. doi: 10.1021/tx100179g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manoharan M, Rasasamy K, Mohan V, Cook P. Oligonucleotides bearing cationic groups: N2-(3-Aminopropyl)deoxyguanosine. Synthesis, enhanced binding properties and conjugation chemistry. Tetrahedron Lett. 1996;37:7675–7678. [Google Scholar]
- Micheli F, Fabio RD, Benedetti R, Capelli AM, Cavallini P, Cavanni P, Davalli S, Donati D, Feriani A, Gehanne S, Hamdan M, Maffeis M, Sabbatini FM, Tranquillini ME, Valeria M, Viziano A. 3-Methyl pyrrole-2,4-dicarboxylic acid 2-propyl ester 4-(1,2,2-trimethylpropyl) ester: an exploration of the C-2 position. Part I. Farmaco (Lausanne) 2004;59:175–183. doi: 10.1016/j.farmac.2003.12.005. [DOI] [PubMed] [Google Scholar]
- Minko IG, Harbut MB, Kozekov ID, Kozekova A, Jakobs PM, Olson SB, Moses RE, Harris TM, Rizzo CJ, Lloyd RS. Role for DNA polymerase κ in the processing of N2-N2-guanine interstrand cross-links. J. Biol. Chem. 2008;283:17075–17082. doi: 10.1074/jbc.M801238200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noll D, Mason T, Miller P. Formation and repair of interstrand cross-links in DNA. Chem. Rev. 2006;106:277–301. doi: 10.1021/cr040478b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrin DD, Armarego WLF. Purification of Laboratory Chemicals. 3rd Ed Butterworth Heinemann; Oxford: 1988. [Google Scholar]
- Stone M, Cho Y-J, Huang H, Kim H-Y, Kozekov ID, Kozekova A, H. W, G. MI, S. LR, M. HT, Rizzo CJ. Interstrand DNA cross-links induced by α,β-unsaturated aldehydes derived from lipid peroxidation and environmental sources. Acc. Chem. Res. 2008:793–804. doi: 10.1021/ar700246x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilds CJ, Booth JDM, Noronha AM. Synthesis of building blocks and oligonucleotides with {G}O6-alkyl-O6{G} cross-Links. Curr. Protoc. Nucleic Acid Chem. 2011:5.9.1–5.9.19. doi: 10.1002/0471142700.nc0509s44. [DOI] [PubMed] [Google Scholar]