

Abstract
Purpose of review
Increased emergence of bacterial resistance and the decline in newly developed antibiotics have necessitated the reintroduction of previously abandoned antimicrobial agents active against multidrug-resistant bacteria. Having never been subjected to contemporary drug development procedures, these ‘old’ antibiotics require redevelopment in order to optimize therapy. This review focuses on colistin as an exemplar of a successful redevelopment process and briefly discusses two other old antibiotics, fusidic acid and fosfomycin.
Recent findings
Redevelopment of colistin led to an improved understanding of its chemistry, pharmacokinetics and pharmacodynamics, enabling important steps towards optimizing its clinical use in different patient populations. A scientifically based dosing algorithm was developed for critically ill patients, including those with renal impairment. As nephrotoxicity is a dose-limiting adverse event of colistin, rational combination therapy with other antibiotics needs to be investigated.
Summary
The example of colistin demonstrated that state-of-the-art analytical, microbiological and pharmacokinetic/pharmacodynamic methods can facilitate optimized use of ‘old’ antibiotics in the clinic. Similar methods are now being applied to fosfomycin and fusidic acid in order to optimize therapy. To improve and preserve the usefulness of these antibiotics rational approaches for redevelopment need to be followed.
Keywords: colistin, fosfomycin, fusidic acid, pharmacodynamics, pharmacokinetics
Introduction
The worldwide problem of increasing antibiotic resistance is well documented [1–6]. At the same time, anti-infective development programmes are declining [3,7], forcing the reintroduction of previously abandoned ‘old’ antibiotics never subjected to the battery of contemporary drug development procedures now mandated by international drug regulatory authorities. This has resulted in a lack of important pharmacokinetic, pharmacodynamic and other scientific information with which to inform their rational use. To achieve optimal therapy, ‘older’ agents need to be subjected to scientific investigation akin to that required of their modern counterparts.
The most striking example of the reintroduction of previously abandoned antibiotics involves the polymyxins (colistin and polymyxin B). Having fallen out of favour in the 1970s due to reports of nephrotoxicity and neurotoxicity [8,9], a resurgence in their use began in the late 1980s when colistin (the most commonly used polymyxin) was reintroduced to manage infection or colonization by Pseudomonas aeruginosa in patients with cystic fibrosis (CF) [10]. More recently, as colistin (and polymyxin B) retains significant in-vitro activity against Gram-negative ‘superbugs’ [11], its use in critically ill patients as a last-line therapy has increased dramatically [10]. Over the past decade colistin in particular has been subjected to new investigations generating much information required to optimize clinical use. Other increasingly used ‘old’ antibiotics include fosfomycin, isepamicin, chloramphenicol, rifampicin and fusidic acid [12–14]. This review will focus primarily on colistin as the exemplar for the redevelopment process, highlighting experiences with this drug in order to provide insight that may assist in the redevelopment of other old agents. Fusidic acid and fosfomycin will be examined briefly.
The Redevelopment of Colistin
Colistin is a cationic antimicrobial peptide (Fig. 1a). Two different forms are commercially available: colistin sulphate (hereafter referred to as colistin) and sodium colistin methanesulphonate (CMS; Fig. 1b). CMS is ‘less toxic’ than colistin when administered parenterally [15] and hence it is CMS that is present in all parenteral (and most inhalational) formulations. In aqueous solutions CMS undergoes conversion in vivo [16–18] and in vitro [19] to form a complex mixture of partially sulphomethylated derivatives and colistin. Unfortunately, in Europe, product labels express the contents of CMS vials in international units (IU; there are ∼12 500IU/mg of CMS [20]), whereas in North America, Australia, East Asia and some other regions vials are labelled with ‘colistin base activity’ (CBA); such confusing labelling significantly complicates the clinical use of CMS [21].
Figure 1.

(a) Structures of colistin A and B; (b) structures of colistin A and B methanesulphonate (CMS). Fatty acyl: 6-methyloctanyl for colistin A and 6-methylheptanyl for colistin B; Thr, threonine; Leu, leucine; Dab, α,γ-diaminobutyric acid. α and γ indicate the respective −NH2 involved in the peptide linkage.
Colistin Pharmacokinetics and Pharmacodynamics
A striking example of the lack of information to guide rational dosing of CMS/colistin was the antibacterial activity assigned to each of CMS and colistin; the former was routinely reported as having reduced antibacterial activity compared with colistin [22–24]. It was only recently that CMS was revealed to be an inactive prodrug of colistin, showing separate determination of CMS and formed colistin concentrations are essential to fully understand the pharmacology of CMS/colistin [25]. However, pharmacokinetic studies conducted prior to 2001 were undertaken using microbiological assay methods for the measurement of ‘colistin’ concentrations; such methods are neither able to separately quantify CMS and colistin nor differentiate between colistin present in a sample at collection from that formed by ongoing conversion from CMS during the incubation period [25]. Importantly, dosage regimens in manufacturers' package inserts even today were developed decades ago based upon pharmacokinetic and pharmacodynamic information derived using microbiological assays. The dearth of essential pharmacological information changed with the development of specific analytical methods capable of discriminating between CMS and colistin [26–32], the application of which over the past decade has greatly expanded our ability to define the pharmacokinetics, pharmacodynamics and integrated pharmacokinetics/pharmacodynamics of CMS/colistin.
To understand the disposition of CMS and colistin, both clinical and preclinical investigations were required, the latter due to aspects of the overall pharmacokinetics of CMS and formed colistin that are only possible to be revealed by the separate administration of each; such studies are not easily performed in humans. Animal models revealed rapid conversion of administered CMS to colistin [the latter with an apparent elimination half-life (t1/2) approximately twice that of the administered CMS] [17,33], extensive renal tubular reabsorption of colistin [32,34] and net tubular secretion of CMS [17]. Colistin showed potent, concentration-dependent bacterial killing which was subject to a substantial inoculum effect [23,35–39], the latter observation suggesting a potential need for higher colistin exposure or combination regimens to treat deep-seated infections with high inocula. Colistin heteroresistance, the situation in which colistin-resistant subpopulations are present within an isolate considered susceptible based upon minimum inhibitory concentrations (MICs), was also observed [35,38–41]. Regrowth with colistin concentrations well in excess of those safely achievable clinically was a consistent finding of in-vitro studies, with heteroresistance contributing to regrowth via amplification of colistin-resistant subpopulations [35,36,38,39,42,43]. Studies also revealed the area under the unbound concentration–time curve to MIC ratio (fAUC/MIC) was the pharmacokinetic/pharmacodynamic index most predictive of the antibacterial effect [43–45], indicating that time-averaged exposure to colistin is more important than the achievement of high peak concentrations from the administration of larger, less frequent doses.
Initial clinical investigations into the pharmacokinetics of CMS and formed colistin following intravenous administration of CMS were conducted in CF patients and, as with preclinical studies, revealed relatively rapid formation of colistin which, at steady state, had a t1/2 approximately twice that of administered CMS (4.2±1.3 versus 2.1±0.87 h) [16,46]. Importantly, the steady-state plasma Cmax values for colistin were low and, even without considering plasma protein binding, failed to reach the Clinical and Laboratory Standards Institute breakpoint of 2 mg/l for P. aeruginosa and Acinetobacter baumannii [47]. An early case report involving a critically ill adult patient requiring continuous venovenous haemodiafiltration (CVVHDF) highlighted the lack of pharmacokinetic information for CMS and formed colistin in critically ill patients [18]. With no guidance from the product information on whether CMS (or formed colistin) is cleared by dialysis [48–50], it was later shown in this patient that both CMS and colistin were cleared by CVVHDF and total plasma concentrations of formed colistin were below the MIC for the infecting strain for the vast majority of the dosage interval; sadly the patient died. Subsequent studies have confirmed the efficient clearance of both CMS and colistin by intermittent haemodialysis and continuous renal replacement therapy (either CVVHDF or continuous veno-venous haemofiltration) [51■■,52].
Over the past few years the gaps in our knowledge of CMS/colistin have begun to be filled. In critically ill patients formed colistin has a protracted t1/2 (up to ∼18 h) with little fluctuation in plasma colistin concentrations across a dosage interval (Fig. 2) [51■■,53,54,55■■]; such a pharmacokinetic profile is substantially different to that observed in CF patients in whom the t1/2 of formed colistin is substantially shorter [16,46], and emphasizes the need to understand the pharmacokinetics in each target group. Three of the studies in critically ill patients, which included between 10 and 18 patients, found renal function [as measured by creatinine clearance (CrCL)] was not a covariate for the clearance of formed colistin [53,54,55■■] nor was CrCL a covariate for CMS clearance [55■■]. In a much larger population pharmacokinetic study involving 105 patients with a wide range of renal function (including 16 patients receiving renal replacement therapy), Garonzik et al. [51■■] showed an inverse relationship between the average plasma colistin concentration at steady state (CSS,ave) achieved with a given daily dose of CMS and CrCL (Fig. 3). It is most likely that the small sample sizes and relatively narrow range of CrCL values (>∼25 ml/min) for the enrolled patients in the other studies [53,54,55■■] resulted in an inability to detect a relationship between renal function and CMS/colistin clearances.
Figure 2.
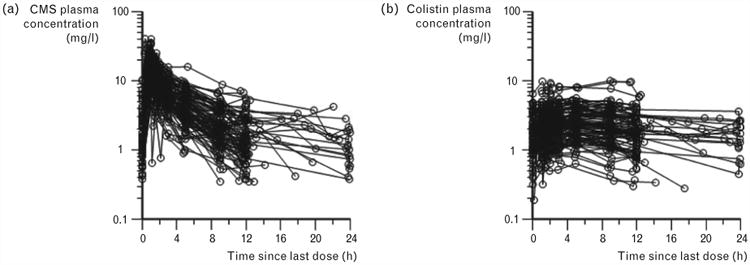
Steady-state plasma concentration–time profiles of (a) CMS or (b) formed colistin in 105 critically ill patients (89 not on renal replacement, 12 on intermittent haemodialysis and 4 on continuous renal replacement therapy) [51■■]. The physician-selected daily dose ranged from 75 to 410 mg colistin base activity; the dosage intervals ranged from 8 to 24 h and hence the inter-dosing blood sampling interval spanned the same range. CMS, colistin methanesulphonate. Reproduced with permission from [51■■].
Figure 3.
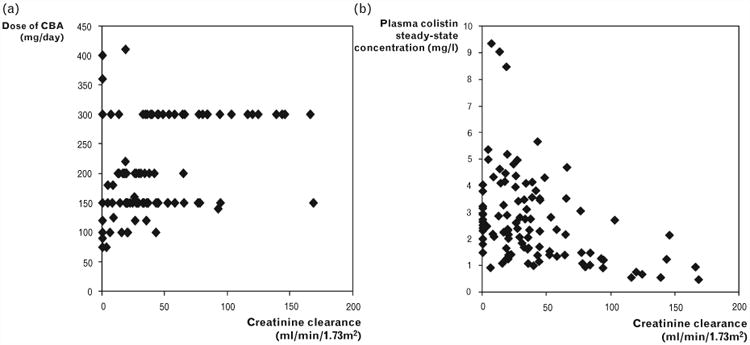
Relationship of (a) physician-selected daily dose of colistin base activity (CBA) and (b) the resultant average steady-state plasma colistin concentration with creatinine clearance in 105 critically ill patients [51■■]. Reproduced with permission from [51■■].
Additional important findings have emerged from these recent pharmacokinetic studies in critically ill patients. The attainment of steady-state plasma colistin concentrations was shown to be a prolonged process (up to 3–4 days) [53], with total plasma colistin concentrations below the MIC breakpoint of 2 mg/l for the first few doses of the currently recommended dosing regimen, effectively delaying appropriate therapy. This highlights the importance of administering a loading dose of CMS in order to more rapidly achieve plasma colistin concentrations [51■■,53,55■■], and this is beginning be used clinically [55■■]. Garonzik et al. [51■■] recently published the first scientifically based CMS dosing algorithms for patients with a large range of renal function (including patients on intermittent haemodialysis or continuous renal replacement therapy); the algorithms allowed calculation of the CMS loading and maintenance doses required to achieve a desired target plasma CSS,ave for colistin in critically ill patients. The recent population pharmacokinetic studies in critically ill patients [51■■,53] demonstrated that in patients with moderate to good renal function it is not possible to achieve steady-state plasma colistin concentrations that are likely to be reliably efficacious with a daily dose of CMS at or close to the upper limit of the current product-recommended dose range (300 mg CBA per day) [48,50]; this is particularly the case for strains with MICs in the upper range of the susceptibility breakpoint for colistin (i.e. >1 mg/l) [51■■]. As nephrotoxicity is a dose-limiting adverse effect of CMS in up to approximately 50% of patients [51■■,56,57], increasing the daily dose may not be an acceptable option. Such a situation places patients at risk for clinical failure and the development of colistin resistance [36,38,39,42,43]. These factors suggest CMS/colistin might best be used as part of a highly active combination and highlight the importance of investigating rational colistin combinations.
Where to with Colistin?
Although there is still much to learn about colistin a clearer picture on the appropriateness of currently administered CMS dosage regimens has slowly emerged. The development of a maintenance dosing algorithm for CMS incorporating renal function, translationally derived from murine pharmacokinetic/pharmacodynamic data, has provided clinicians for the first time with scientifically based guidelines to inform CMS administration. As clinical studies progress, the future incorporation of human pharmacokinetic/pharmacodynamic data into such algorithms, including information on colistin plasma protein binding, as well as studies examining colistin monotherapy versus combination therapy, will further optimize colistin therapy.
Fusidic Acid
Fusidic acid has been widely used for uncomplicated skin and skin-structure infections predominantly in Europe and Australia since the 1960s, with standard dosage regimens of 500 mg 12 or 8 hourly [58,59]. Owing to activity against methicillin-resistant Staphylococcus aureus (MRSA) [60■] it is currently undergoing redevelopment in the US [61■■]. As for colistin, there are major knowledge gaps relating to both its pharmacokinetics and pharmacodynamics. Fusidic acid has complex pharmacokinetics which includes a decrease in clearance with increasing dose and with multiple compared to single doses [59,62,63]. Administration with food decreases both the bioavailability (by ∼18%) [63] and the rate of absorption [62]. It also has high between-patient variability in pharmacokinetics, is almost entirely nonrenally eliminated, is highly protein-bound (>97%) and appears unaffected by haemodialysis [59,64]. Population pharmacokinetic modelling of data from 75 healthy volunteers estimated a clearance of approximately 1.28 l/h and apparent t1/2 of 14 h for single doses of up to 400 mg. However, with a substantial decrease in clearance to approximately 0.37 l/h at high concentrations due to auto-inhibition, the time required to reach steady state for a regimen of 500 mg 12 hourly was predicted to be approximately 3 weeks [63]. With such a slow attainment of steady state, efficacious concentrations may not be achieved during the first days of therapy which may lead to resistance. Therefore the use of front-loaded regimens was proposed. Simulations predicted loading doses of 1500 mg at 0 and 12 h followed by 600 mg every 12 h allow attainment of steady state within 24 h [63]. Based upon the population model, various human dosage regimens were simulated in vitro against a MRSA USA300 clinical isolate. The bacterial killing was well correlated with AUC/MIC, with a front-loaded regimen (1500 mg at 0 and 12 h followed by 600 mg 12 hourly) increasing killing and delaying the emergence of resistance (by up to 72 h compared to the 600 mg 12 hourly regimen) [61■■]. The prospectively identified front-loading regimen was subsequently tested in a phase 2 clinical study showing comparable efficacy to linezolid [61■■]. However, S. aureus develops resistance to fusidic acid when used as monotherapy primarily due to mutations in the fusA gene (which encodes elongation factor G, the site of action of fusidic acid) and plasmid-mediated resistance, and therefore, like colistin, may best be used in combination [65]. Rationally designed combination regimens (e.g. with rifampicin) will be critical to preserve the efficacy of fusidic acid into the future [58].
Fosfomycin
Fosfomycin is a broad-spectrum bactericidal antibiotic known for more than 40 years but abandoned in several parts of the world. It displays a unique mechanism of action by inhibiting an early step in bacterial cell wall synthesis [66], making cross-resistance unlikely and allowing fosfomycin to retain significant in-vitro activity against many Gram-positive and Gram-negative pathogens, including multidrug resistant (MDR) strains [67,68]. Fosfomycin is primarily administered orally for treatment of uncomplicated urinary tract infections. Fosfomycin tromethamine, the oral formulation, is rapidly absorbed achieving serum concentrations of approximately 22–32 mg/l following the standard single 3 g oral dose, with very high concentrations (range 1053–4415 mg/l [69]) achieved in urine after 3 h [70]; urinary concentrations remain high for several days. Elimination is almost entirely via glomerular filtration (serum t1/2 2.4–7.3 h) [69]. In some countries fosfomycin disodium is used intra-venously for systemic infections [71]. A Cmax of approximately 250 mg/l is achieved following intra-venous administration of a 4 g dose [72,73]. Given the susceptibility breakpoints for fosfomycin, when specified, range from 32 to 64 mg/l [47,74], oral therapy for systemic infections should be avoided until more information is forthcoming. Fosfomycin is almost entirely unbound in serum [75] with its low molecular weight facilitating good distribution into many tissues (e.g. muscle, lung and bone) [76–78]. Although fosfomycin crosses the blood–brain barrier [79], activity against Gram-negative bacteria is significantly reduced in human cerebrospinal fluid [80].
Concerns over the potential for rapid resistance development with fosfomycin have resulted in it primarily being used in combination for treatment of systemic infections [81], with in-vitro combination studies displaying excellent activity against both Gram-negative and Gram-positive pathogens [82]; clinical studies demonstrating the effectiveness of combination therapy, however, are lacking. Fosfomycin exhibits in-vitro synergy with carbapenems as well as with aminoglycosides, colistin and tigecycline against serine carbapenemase-producing and extended spectrum β-lactamase (ESBL)-producing Klebsiella pneumoniae [83]. Combinations of fosfomycin with aminoglycosides, colistin or tigecycline were effective against ESBL-producing Escherichia coli and a considerable subset of MDR P. aeruginosa [83]. Against P. aeruginosa biofilms, fosfomycin has shown good in-vitro activity in combination with fluoroquinolones or aminoglycosides, making it a potential option to treat CF patients [84–88]. Activity against biofilms may be due to the ability of fosfomycin to penetrate deeply into the biofilm, along with enhanced activity under anaerobic conditions [87,88]. Given the limited available evidence from clinical and other studies, this old antibiotic may be a viable alternative for treatment of infections due to both Gram-positive and Gram-negative pathogens, especially MDR pathogens. However, as fosfomycin has not been used widely there is a shortage of clinical efficacy and safety data and further research is warranted.
Conclusion
So what lessons can be learned from the colistin experience? The first is that pharmaceutical companies are generally disinterested in funding research for off-patent anti-infectives. Consequently, the redevelopment task will fall to publicly funded granting bodies such as the US National Institutes of Health (NIH). Additionally, the redevelopment process takes time – perhaps a decade or more – so the redevelopment process needs to start early. Fosfomycin and fusidic acid are two promising ‘old’ antibiotics. The example of colistin has shown that much is to be gained from the redevelopment of ‘old’ antibiotics by employing contemporary analytical and pharmacokinetic/pharmacodynamic methods to maximize bacterial killing and minimize the occurrence of adverse events as well as the emergence of resistance.
Key Points.
Due to increased emergence of bacterial resistance and declining development of new antibiotics, previously abandoned ‘old’ compounds active against multidrug-resistant bacteria are being reintroduced into the clinic.
As these ‘old’ antibiotics have never been subjected to the contemporary drug development process, their redevelopment using contemporary analytical and pharmacokinetic/pharmacodynamic methods is critical in order to optimize therapy.
Colistin is an example of successful redevelopment in which the use of new state-of-the-art bioanalytical, microbiological and pharmacokinetic/pharmacodynamic approaches has generated an improved understanding of its clinical pharmacology, and has enabled rational optimization of patient therapy.
Other old antibiotics such as fosfomycin and fusidic acid will benefit from redevelopment based on the lessons learnt with colistin.
Acknowledgments
R.L.N. and J.L. are supported by Award Numbers R01AI098771, R01AI079330 and R01AI070896 from the National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health (NIH). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Allergy and Infectious Diseases or the National Institutes of Health. J.L. is an Australian National Health and Medical Research Council (NHMRC) Senior Research Fellow.
Footnotes
Conflicts of interest: There are no conflicts of interest.
References and Recommended Reading
Papers of particular interest, published within the annual period of review, have been highlighted as:
■ of special interest
■■ of outstanding interest
Additional references related to this topic can also be found in the Current World Literature section in this issue (p. 720).
- 1.Levy SB, Marshall B. Antibacterial resistance worldwide: causes, challenges and responses. Nat Med. 2004;10(12 Suppl):S122–S129. doi: 10.1038/nm1145. [DOI] [PubMed] [Google Scholar]
- 2.Alanis AJ. Resistance to antibiotics: are we in the postantibiotic era? Arch Med Res. 2005;36:697–705. doi: 10.1016/j.arcmed.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 3.Spellberg B, Blaser M, Guidos RJ, et al. Combating antimicrobial resistance: policy recommendations to save lives. Clin Infect Dis. 2011;52(Suppl 5):S397–S428. doi: 10.1093/cid/cir153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boucher HW, Talbot GH, Bradley JS, et al. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin Infect Dis. 2009;48:1–12. doi: 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- 5.Opal SM, Calandra T. Antibiotic usage and resistance: gaining or losing ground on infections in critically ill patients? J Am Med Assoc. 2009;302:2367–2368. doi: 10.1001/jama.2009.1774. [DOI] [PubMed] [Google Scholar]
- 6.Vincent JL, Rello J, Marshall J, et al. International study of the prevalence and outcomes of infection in intensive care units. J Am Med Assoc. 2009;302:2323–2329. doi: 10.1001/jama.2009.1754. [DOI] [PubMed] [Google Scholar]
- 7.Talbot GH, Bradley J, Edwards JE, Jr, et al. Bad bugs need drugs: an update on the development pipeline from the Antimicrobial Availability Task Force of the Infectious Diseases Society of America. Clin Infect Dis. 2006;42:657–668. doi: 10.1086/499819. [DOI] [PubMed] [Google Scholar]
- 8.Koch-Weser J, Sidel VW, Federman EB, et al. Adverse effects of sodium colistimethate. Manifestations and specific reaction rates during 317 courses of therapy. Ann Intern Med. 1970;72:857–868. doi: 10.7326/0003-4819-72-6-857. [DOI] [PubMed] [Google Scholar]
- 9.Ryan KJ, Schainuck LI, Hickman RO, Striker GE. Colistimethate toxicity. Report of a fatal case in a previously healthy child. J Am Med Assoc. 1969;207:2099–2101. doi: 10.1001/jama.207.11.2099. [DOI] [PubMed] [Google Scholar]
- 10.Nation RL, Li J. Colistin in the 21st century. Curr Opin Infect Dis. 2009;22:535–543. doi: 10.1097/QCO.0b013e328332e672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gales AC, Jones RN, Sader HS. Contemporary activity of colistin and polymyxin B against a worldwide collection of Gram-negative pathogens: results from the SENTRY Antimicrobial Surveillance Program (2006-09) J Antimicrob Chemother. 2011;66:2070–2074. doi: 10.1093/jac/dkr239. [DOI] [PubMed] [Google Scholar]
- 12.Falagas ME, Kopterides P. Old antibiotics for infections in critically ill patients. Curr Opin Crit Care. 2007;13:592–597. doi: 10.1097/MCC.0b013e32827851d7. [DOI] [PubMed] [Google Scholar]
- 13.Giamarellou H, Poulakou G. Multidrug-resistant Gram-negative infections: what are the treatment options? Drugs. 2009;69:1879–1901. doi: 10.2165/11315690-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 14.Maviglia R, Nestorini R, Pennisi M. Role of old antibiotics in multidrug resistant bacterial infections. Curr Drug Targets. 2009;10:895–905. doi: 10.2174/138945009789108846. [DOI] [PubMed] [Google Scholar]
- 15.Beveridge EG, Martin AJ. Sodium sulphomethyl derivatives of polymyxins. Br J Pharmacol Chemother. 1967;29:125–135. doi: 10.1111/j.1476-5381.1967.tb01946.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li J, Coulthard K, Milne R, et al. Steady-state pharmacokinetics of intravenous colistin methanesulphonate in patients with cystic fibrosis. J Antimicrob Chemother. 2003;52:987–992. doi: 10.1093/jac/dkg468. [DOI] [PubMed] [Google Scholar]
- 17.Li J, Milne RW, Nation RL, et al. Pharmacokinetics of colistin methanesulphonate and colistin in rats following an intravenous dose of colistin methanesulphonate. J Antimicrob Chemother. 2004;53:837–840. doi: 10.1093/jac/dkh167. [DOI] [PubMed] [Google Scholar]
- 18.Li J, Rayner CR, Nation RL, et al. Pharmacokinetics of colistin methanesulfonate and colistin in a critically ill patient receiving continuous venovenous hemodiafiltration. Antimicrob Agents Chemother. 2005;49:4814–4815. doi: 10.1128/AAC.49.11.4814-4815.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li J, Milne RW, Nation RL, et al. Stability of colistin and colistin methanesulfonate in aqueous media and plasma as determined by high-performance liquid chromatography. Antimicrob Agents Chemother. 2003;47:1364–1370. doi: 10.1128/AAC.47.4.1364-1370.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sweetman SC, editor. Martindale. 34th. London, UK: The Pharmaceutical Press; 2005. [Google Scholar]
- 21.Li J, Nation RL, Turnidge JD, et al. Colistin: the re-emerging antibiotic for multidrug-resistant Gram-negative bacterial infections. Lancet Infect Dis. 2006;6:589–601. doi: 10.1016/S1473-3099(06)70580-1. [DOI] [PubMed] [Google Scholar]
- 22.Bergan T, Fuglesang J. Polymyxin antibiotics: chemical and pharmacokinetic properties. Antibiot Chemother. 1982;31:119–144. [PubMed] [Google Scholar]
- 23.Li J, Turnidge J, Milne R, et al. In vitro pharmacodynamic properties of colistin and colistin methanesulfonate against Pseudomonas aeruginosa isolates from patients with cystic fibrosis. Antimicrob Agents Chemother. 2001;45:781–785. doi: 10.1128/AAC.45.3.781-785.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schwartz BS, Warren MR, Barkley FA, Landis L. Microbiological and pharmacological studies of colistin sulfate and sodium colistin methanesulfonate. Antibiot Annu. 1959;7:41–60. [PubMed] [Google Scholar]
- 25.Bergen PJ, Li J, Rayner CR, Nation RL. Colistin methanesulfonate is an inactive prodrug of colistin against Pseudomonas aeruginosa. Antimicrob Agents Chemother. 2006;50:1953–1958. doi: 10.1128/AAC.00035-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dotsikas Y, Markopoulou CK, Koundourellis JE, Loukas YL. Validation of a novel LC-MS/MS method for the quantitation of colistin A and B in human plasma. J Sep Sci. 2011;34:37–45. doi: 10.1002/jssc.201000680. [DOI] [PubMed] [Google Scholar]
- 27.Gobin P, Lemaitre F, Marchand S, et al. Assay of colistin and colistin methanesulfonate in plasma and urine by liquid chromatography-tandem mass spectrometry. Antimicrob Agents Chemother. 2010;54:1941–1948. doi: 10.1128/AAC.01367-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jansson B, Karvanen M, Cars O, et al. Quantitative analysis of colistin A and colistin B in plasma and culture medium using a simple precipitation step followed by LC/MS/MS. J Pharm Biomed Anal. 2009;49:760–767. doi: 10.1016/j.jpba.2008.12.016. [DOI] [PubMed] [Google Scholar]
- 29.Ma Z, Wang J, Gerber JP, Milne RW. Determination of colistin in human plasma, urine and other biological samples using LC-MS/MS. J Chromatogr B Analyt Technol Biomed Life Sci. 2008;862:205–212. doi: 10.1016/j.jchromb.2007.12.009. [DOI] [PubMed] [Google Scholar]
- 30.Li J, Milne RW, Nation RL, et al. A simple method for the assay of colistin in human plasma, using precolumn derivatization with 9-fluorenylmethyl chloroformate in solid-phase extraction cartridges and reversed-phase high-performance liquid chromatography. J Chromatogr B Biomed Sci Appl. 2001;761:167–175. doi: 10.1016/s0378-4347(01)00326-7. [DOI] [PubMed] [Google Scholar]
- 31.Li J, Milne RW, Nation RL, et al. Simple method for assaying colistin methanesulfonate in plasma and urine using high-performance liquid chromatography. Antimicrob Agents Chemother. 2002;46:3304–3307. doi: 10.1128/AAC.46.10.3304-3307.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li J, Milne RW, Nation RL, et al. Use of high-performance liquid chromatography to study the pharmacokinetics of colistin sulfate in rats following intravenous administration. Antimicrob Agents Chemother. 2003;47:1766–1770. doi: 10.1128/AAC.47.5.1766-1770.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marchand S, Lamarche I, Gobin P, Couet W. Dose-ranging pharmacokinetics of colistin methanesulphonate (CMS) and colistin in rats following single intravenous CMS doses. J Antimicrob Chemother. 2010;65:1753–1758. doi: 10.1093/jac/dkq183. [DOI] [PubMed] [Google Scholar]
- 34.Ma Z, Wang J, Nation RL, et al. Renal disposition of colistin in the isolated perfused rat kidney. Antimicrob Agents Chemother. 2009;53:2857–2864. doi: 10.1128/AAC.00030-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bergen PJ, Forrest A, Bulitta JB, et al. Clinically relevant plasma concentrations of colistin in combination with imipenem enhance pharmacodynamic activity against multidrug-resistant Pseudomonas aeruginosa at multiple inocula. Antimicrob Agents Chemother. 2011;55:5134–5142. doi: 10.1128/AAC.05028-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bergen PJ, Tsuji BT, Bulitta JB, et al. Synergistic killing of multidrug-resistant Pseudomonas aeruginosa at multiple inocula by colistin combined with doripenem in an in vitro pharmacokinetic/pharmacodynamic model. Antimicrob Agents Chemother. 2011;55:5685–5695. doi: 10.1128/AAC.05298-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bulitta JB, Yang JC, Yohonn L, et al. Attenuation of colistin bactericidal activity by high inoculum of Pseudomonas aeruginosa characterized by a new mechanism-based population pharmacodynamic model. Antimicrob Agents Chemother. 2010;54:2051–2062. doi: 10.1128/AAC.00881-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Poudyal A, Howden BP, Bell JM, et al. In vitro pharmacodynamics of colistin against multidrug-resistant Klebsiella pneumoniae. J Antimicrob Chemother. 2008;62:1311–1318. doi: 10.1093/jac/dkn425. [DOI] [PubMed] [Google Scholar]
- 39.Tan CH, Li J, Nation RL. Activity of colistin against heteroresistant Acinetobacter baumannii and emergence of resistance in an in vitro pharmacokinetic/pharmacodynamic model. Antimicrob Agents Chemother. 2007;51:3413–3415. doi: 10.1128/AAC.01571-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hawley JS, Murray CK, Jorgensen JH. Colistin heteroresistance in acinetobacter and its association with previous colistin therapy. Antimicrob Agents Chemother. 2008;52:351–352. doi: 10.1128/AAC.00766-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Meletis G, Tzampaz E, Sianou E, et al. Colistin heteroresistance in carbapenemase-producing Klebsiella pneumoniae. J Antimicrob Chemother. 2011;66:946–947. doi: 10.1093/jac/dkr007. [DOI] [PubMed] [Google Scholar]
- 42.Bergen PJ, Li J, Nation RL, et al. Comparison of once-, twice- and thrice-daily dosing of colistin on antibacterial effect and emergence of resistance: studies with Pseudomonas aeruginosa in an in vitro pharmacodynamic model. J Antimicrob Chemother. 2008;61:636–642. doi: 10.1093/jac/dkm511. [DOI] [PubMed] [Google Scholar]
- 43.Dudhani RV, Turnidge JD, Nation RL, Li J. fAUC/MIC is the most predictive pharmacokinetic/pharmacodynamic index of colistin against Acinetobacter baumannii in murine thigh and lung infection models. J Antimicrob Chemother. 2010;65:1984–1990. doi: 10.1093/jac/dkq226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dudhani RV, Turnidge JD, Coulthard K, et al. Elucidation of the pharmacokinetic/pharmacodynamic determinant of colistin activity against Pseudomonas aeruginosa in murine thigh and lung infection models. Antimicrob Agents Chemother. 2010;54:1117–1124. doi: 10.1128/AAC.01114-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bergen PJ, Bulitta JB, Forrest A, et al. Pharmacokinetic/pharmacodynamic investigation of colistin against Pseudomonas aeruginosa using an in vitro model. Antimicrob Agents Chemother. 2010;54:3783–3789. doi: 10.1128/AAC.00903-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Reed MD, Stern RC, O'Riordan MA, Blumer JL. The pharmacokinetics of colistin in patients with cystic fibrosis. J Clin Pharmacol. 2001;41:645–654. doi: 10.1177/00912700122010537. [DOI] [PubMed] [Google Scholar]
- 47.Clinical and Laboratory Standards Institute. Performance Standards for Antimicrobial Susceptibility Testing: Twentieth Informational Supplement (M100-S20) CLSI; Wayne, PA, USA: 2010. [Google Scholar]
- 48.Coly-Mycin M Parenteral. Mosman, NSW, Australia: Link Medical Products Pty Ltd; 2006. package insert. [Google Scholar]
- 49.Colomycin Injection. Kent, UK: Forest Laboratories UK Limited; 2004. Summary of product characteristics. [Google Scholar]
- 50.Coly-Mycin M Parenteral. Bristol, TN: Monarch Pharmaceuticals; 2006. package insert. [Google Scholar]
- 51■■.Garonzik SM, Li J, Thamlikitkul V, et al. Population pharmacokinetics of colistin methanesulfonate and formed colistin in critically ill patients from a multicenter study provide dosing suggestions for various categories of patients. Antimicrob Agents Chemother. 2011;55:3284–3294. doi: 10.1128/AAC.01733-10. This is the first study to identify the importance of renal function as a determinant of plasma colistin concentrations. This study also presented the first scientifically based dosing suggestions for various categories of critically ill patients, including those with widely differing creatinine clearance values and those receiving intermittent haemodialysis or continuous renal replacement therapy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Marchand S, Frat JP, Petitpas F, et al. Removal of colistin during intermittent haemodialysis in two critically ill patients. J Antimicrob Chemother. 2010;65:1836–1837. doi: 10.1093/jac/dkq185. [DOI] [PubMed] [Google Scholar]
- 53.Plachouras D, Karvanen M, Friberg LE, et al. Population pharmacokinetic analysis of colistin methanesulphonate and colistin after intravenous administration in critically ill patients with Gram-negative bacterial infections. Antimicrob Agents Chemother. 2009;53:3430–3436. doi: 10.1128/AAC.01361-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Markou N, Markantonis SL, Dimitrakis E, et al. Colistin serum concentrations after intravenous administration in critically ill patients with serious multidrug-resistant, Gram-negative bacilli infections: a prospective, open-label, uncontrolled study. Clin Ther. 2008;30:143–151. doi: 10.1016/j.clinthera.2008.01.015. [DOI] [PubMed] [Google Scholar]
- 55■■.Mohamed AF, Karaiskos I, Plachouras D, et al. Application of a loading dose of colistin methanesulphonate (CMS) in critically ill patients: population pharmacokinetics, protein binding and prediction of bacterial kill. Antimicrob Agents Chemother. 2012;56:4241–4249. doi: 10.1128/AAC.06426-11. This study demonstrated the slow attainment of plasma colistin concentrations if CMS therapy is commenced without a loading dose. The study proposed the administration of a loading dose to more rapidly achieve plasma colistin concentrations that are likely to be effective. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hartzell JD, Neff R, Ake J, et al. Nephrotoxicity associated with intravenous colistin (colistimethate sodium) treatment at a tertiary care medical center. Clin Infect Dis. 2009;48:1724–1728. doi: 10.1086/599225. [DOI] [PubMed] [Google Scholar]
- 57.Kwon JA, Lee JE, Huh W, et al. Predictors of acute kidney injury associated with intravenous colistin treatment. Int J Antimicrob Agents. 2010;35:473–477. doi: 10.1016/j.ijantimicag.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 58.Howden BP, Grayson ML. Dumb and dumber: the potential waste of a useful antistaphylococcal agent: emerging fusidic acid resistance in Staphylococcus aureus. Clin Infect Dis. 2006;42:394–400. doi: 10.1086/499365. [DOI] [PubMed] [Google Scholar]
- 59.Turnidge J. Fusidic acid pharmacology, pharmacokinetics and pharmacodynamics. Int J Antimicrob Agents. 1999;12(Suppl 2):S23–S34. doi: 10.1016/s0924-8579(98)00071-5. [DOI] [PubMed] [Google Scholar]
- 60■.Fernandes P, Pereira D. Efforts to support the development of fusidic acid in the United States. Clin Infect Dis. 2011;52(Suppl 7):S542–S546. doi: 10.1093/cid/cir170. This study describes the scientific, regulatory, legal and political hurdles which need to be overcome to develop an ‘old’ antibiotic and bring it to a new market. [DOI] [PubMed] [Google Scholar]
- 61■■.Tsuji BT, Okusanya OO, Bulitta JB, et al. Application of pharmacokinetic- pharmacodynamic modeling and the justification of a novel fusidic acid dosing regimen: raising Lazarus from the dead. Clin Infect Dis. 2011;52(Suppl 7):S513–S519. doi: 10.1093/cid/cir166. This study describes the development of population pharmacokinetic and mechanism-based mathematical models to identify dosing strategies for fusidic acid that would optimize bacterial eradication and delay the emergence of resistance. A novel front-loaded dosing regimen was identified that was subsequently shown in a phase 2 study to have comparable efficacy to that of linezolid. [DOI] [PubMed] [Google Scholar]
- 62.MacGowan AP, Greig MA, Andrews JM, et al. Pharmacokinetics and tolerance of a new film-coated tablet of sodium fusidate administered as a single oral dose to healthy volunteers. J Antimicrob Chemother. 1989;23:409–415. doi: 10.1093/jac/23.3.409. [DOI] [PubMed] [Google Scholar]
- 63.Bulitta JB, Okusanya OO, Forrest A, et al. Population pharmacokinetics of CEM-102 in healthy subjects (abstract A1-1932); Abstracts of the 49th Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC); San Francisco, California. September 12–15; American Society for Microbiology; 2009. [Google Scholar]
- 64.Brown NM, Reeves DS, McMullin CM. The pharmacokinetics and protein-binding of fusidic acid in patients with severe renal failure requiring either haemodialysis or continuous ambulatory peritoneal dialysis. J Antimicrob Chemother. 1997;39:803–809. doi: 10.1093/jac/39.6.803. [DOI] [PubMed] [Google Scholar]
- 65.Farrell DJ, Castanheira M, Chopra I. Characterization of global patterns and the genetics of fusidic acid resistance. Clin Infect Dis. 2011;52(Suppl 7):S487–S492. doi: 10.1093/cid/cir164. [DOI] [PubMed] [Google Scholar]
- 66.Kahan FM, Kahan JS, Cassidy PJ, Kropp H. The mechanism of action of fosfomycin (phosphonomycin) Ann N Y Acad Sci. 1974;235:364–386. doi: 10.1111/j.1749-6632.1974.tb43277.x. [DOI] [PubMed] [Google Scholar]
- 67.Popovic M, Steinort D, Pillai S, Joukhadar C. Fosfomycin: an old, new friend? Eur J Clin Microbiol Infect Dis. 2010;29:127–142. doi: 10.1007/s10096-009-0833-2. [DOI] [PubMed] [Google Scholar]
- 68.Roussos N, Karageorgopoulos DE, Samonis G, Falagas ME. Clinical significance of the pharmacokinetic and pharmacodynamic characteristics of fosfomycin for the treatment of patients with systemic infections. Int J Antimicrob Agents. 2009;34:506–515. doi: 10.1016/j.ijantimicag.2009.08.013. [DOI] [PubMed] [Google Scholar]
- 69.Patel SS, Balfour JA, Bryson HM. Fosfomycin tromethamine. A review of its antibacterial activity, pharmacokinetic properties and therapeutic efficacy as a single-dose oral treatment for acute uncomplicated lower urinary tract infections. Drugs. 1997;53:637–656. doi: 10.2165/00003495-199753040-00007. [DOI] [PubMed] [Google Scholar]
- 70.Bergan T, Thorsteinsson SB, Albini E. Pharmacokinetic profile of fosfomycin trometamol. Chemotherapy. 1993;39:297–301. doi: 10.1159/000239140. [DOI] [PubMed] [Google Scholar]
- 71.Falagas ME, Giannopoulou KP, Kokolakis GN, Rafailidis PI. Fosfomycin: use beyond urinary tract and gastrointestinal infections. Clin Infect Dis. 2008;46:1069–1077. doi: 10.1086/527442. Review. [DOI] [PubMed] [Google Scholar]
- 72.Goto M, Sugiyama M, Nakajima S, Yamashina H. Fosfomycin kinetics after intravenous and oral administration to human volunteers. Antimicrob Agents Chemother. 1981;20:393–397. doi: 10.1128/aac.20.3.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Forestier F, Salvanet-Bouccara A, Leveques D, et al. Ocular penetration kinetics of fosfomycin administered as a one-hour infusion. Eur J Ophthalmol. 1996;6:137–142. doi: 10.1177/112067219600600207. [DOI] [PubMed] [Google Scholar]
- 74.European Committee on Antimicrobial Susceptibility Testing. Breakpoint tables for interpretation of MICs and zone diameters (Version 2.0, January 1, 2012) [Accessed 25 June 2012]; http://www.eucast.org/clinical_breakpoints.
- 75.Zeitlinger MA, Sauermann R, Traunmuller F, et al. Impact of plasma protein binding on antimicrobial activity using time-killing curves. J Antimicrob Chemother. 2004;54:876–880. doi: 10.1093/jac/dkh443. [DOI] [PubMed] [Google Scholar]
- 76.Joukhadar C, Klein N, Dittrich P, et al. Target site penetration of fosfomycin in critically ill patients. J Antimicrob Chemother. 2003;51:1247–1252. doi: 10.1093/jac/dkg187. [DOI] [PubMed] [Google Scholar]
- 77.Legat FJ, Maier A, Dittrich P, et al. Penetration of fosfomycin into inflammatory lesions in patients with cellulitis or diabetic foot syndrome. Antimicrob Agents Chemother. 2003;47:371–374. doi: 10.1128/AAC.47.1.371-374.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Matzi V, Lindenmann J, Porubsky C, et al. Extracellular concentrations of fosfomycin in lung tissue of septic patients. J Antimicrob Chemother. 2010;65:995–998. doi: 10.1093/jac/dkq070. [DOI] [PubMed] [Google Scholar]
- 79.Kuhnen E, Pfeifer G, Frenkel C. Penetration of fosfomycin into cerebrospinal fluid across noninflamed and inflamed meninges. Infection. 1987;15:422–424. doi: 10.1007/BF01647220. [DOI] [PubMed] [Google Scholar]
- 80.Sauermann R, Schwameis R, Fille M, et al. Cerebrospinal fluid impairs antimicrobial activity of fosfomycin in vitro. J Antimicrob Chemother. 2009;64:821–823. doi: 10.1093/jac/dkp261. [DOI] [PubMed] [Google Scholar]
- 81.Nilsson AI, Berg OG, Aspevall O, et al. Biological costs and mechanisms of fosfomycin resistance in Escherichia coli. Antimicrob Agents Chemother. 2003;47:2850–2858. doi: 10.1128/AAC.47.9.2850-2858.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kastoris AC, Rafailidis PI, Vouloumanou EK, et al. Synergy of fosfomycin with other antibiotics for Gram-positive and Gram-negative bacteria. Eur J Clin Pharmacol. 2010;66:359–368. doi: 10.1007/s00228-010-0794-5. [DOI] [PubMed] [Google Scholar]
- 83.Samonis G, Maraki S, Karageorgopoulos DE, et al. Synergy of fosfomycin with carbapenems, colistin, netilmicin, and tigecycline against multidrug-resistant Klebsiella pneumoniae, Escherichia coli, and Pseudomonas aeruginosa clinical isolates. Eur J Clin Microbiol Infect Dis. 2012;31:695–701. doi: 10.1007/s10096-011-1360-5. [DOI] [PubMed] [Google Scholar]
- 84.Cai Y, Fan Y, Wang R, et al. Synergistic effects of aminoglycosides and fosfomycin on Pseudomonas aeruginosa in vitro and biofilm infections in a rat model. J Antimicrob Chemother. 2009;64:563–566. doi: 10.1093/jac/dkp224. [DOI] [PubMed] [Google Scholar]
- 85.Mikuniya T, Kato Y, Ida T, et al. Treatment of Pseudomonas aeruginosa biofilms with a combination of fluoroquinolones and fosfomycin in a rat urinary tract infection model. J Infect Chemother. 2007;13:285–290. doi: 10.1007/s10156-007-0534-7. [DOI] [PubMed] [Google Scholar]
- 86.Rodriguez-Martinez JM, Ballesta S, Pascual A. Activity and penetration of fosfomycin, ciprofloxacin, amoxicillin/clavulanic acid and co-trimoxazole in Escherichia coli and Pseudomonas aeruginosa biofilms. Int J Antimicrob Agents. 2007;30:366–368. doi: 10.1016/j.ijantimicag.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 87.Mikuniya T, Kato Y, Kariyama R, et al. Synergistic effect of fosfomycin and fluoroquinolones against Pseudomonas aeruginosa growing in a biofilm. Acta Med Okayama. 2005;59:209–216. doi: 10.18926/AMO/31977. [DOI] [PubMed] [Google Scholar]
- 88.Monden K, Ando E, Iida M, Kumon H. Role of fosfomycin in a synergistic combination with ofloxacin against Pseudomonas aeruginosa growing in a biofilm. J Infect Chemother. 2002;8:218–226. doi: 10.1007/s10156-002-0186-6. [DOI] [PubMed] [Google Scholar]