

Abstract
The Ca2+-sensing receptor (CaSR) regulates Ca2+ homeostasis in the body by monitoring extracellular levels of Ca2+ ([Ca2+]o) and amino acids. Mutations at the hinge region of the N-terminal Venus flytrap domain (VFTD) produce either receptor inactivation (L173P, P221Q) or activation (L173F, P221L) related to hypercalcemic or hypocalcemic disorders. In this paper, we report that both L173P and P221Q markedly impair the functional positive cooperativity of the CaSR as reflected by [Ca2+]o–induced [Ca2+]i oscillations, inositol-1-phosphate (IP1) accumulation and extracellular signal-regulated kinases (ERK1/2) activity. In contrast, L173F and P221L show enhanced responsiveness of these three functional readouts to [Ca2+]o. Further analysis of the dynamics of the VFTD mutants using computational simulation studies supports disruption in the correlated motions in the loss-of-function CaSR mutants, while these motions are enhanced in the gain-of-function mutants. Wild type (WT) CaSR was modulated by L-Phe in a heterotropic positive cooperative way, achieving an EC50 similar to those of the two activating mutations. The response of the inactivating P221Q mutant to [Ca2+]o was partially rescued by L-Phe, illustrating the capacity of the L-Phe binding site to enhance the positive homotropic cooperativity of CaSR. L-Phe had no effect on the other inactivating mutant. Moreover, our results carried out both in silico and in intact cells indicate that residue Leu173, which is close to residues that are part of the L-Phe-binding pocket, exhibited impaired heterotropic cooperativity in the presence of L-Phe. Thus, Pro221 and Leu173 are important for the positive homo- and heterotropic cooperative regulation elicited by agonist binding.
Introduction
The human calcium (Ca2+)-sensing receptor (CaSR) is a seven transmembrane, G protein-coupled receptor (GPCR) that is expressed at the highest levels in the parathyroid glands and kidneys [1]. The principal role of CaSR is to sense alterations of the extracellular calcium concentration ([Ca2+]o) and to maintain Ca2+ homeostasis by regulating parathyroid hormone (PTH) secretion as well as renal Ca2+ reabsorption. Like other members of family C in the GPCR superfamily, the CaSR possesses a large extracellular domain (ECD) consisting of more than 600 amino acids [2]. Ca2+, the principal physiological agonist for CaSR, is thought to bind to the ECD of the dimeric form of the receptor resulting in activation of phospholipase C (PLC), with an attendant accumulation of inositol 1,4,5-triphosphate (IP3) followed by a resultant increase in the cytosolic calcium ([Ca2+]i) concentration [3].
Fluctuations of the plasma levels of amino acids can regulate the rate of hormone synthesis and secretion as well as Ca2+ metabolism, among other processes [4]. CaSR is present throughout the gastrointestinal tract [5], [6]. L-amino acids, especially aromatic amino acids, are known to enhance the sensitivity of CaSR to [Ca2+]o, which could be one potential explanation for how dietary protein modulates [Ca2+]o homeostasis in normal individuals as well as in patients with chronic renal failure [4], [7].
Mutations of the CaSR can perturb intracellular signaling events (e.g., intracellular calcium responses) and disrupt regulation of PTH secretion from the parathyroid chief cell and Ca2+ reabsorption in the renal tubule. Mutations that inactivate CaSR (i.e., result in loss-of-function) cause familial hypocalciuric hypercalcemia (FHH) and neonatal severe primary hyperparathyroidism (NSHPT) [8], [9]. On the other hand, activating mutations of the CaSR lead to autosomal dominant hypocalcemia with hypercalciuria (ADHH) [10]. These mutations, including the four studied here that involve the two “toggle” residues, Leu173 and Pro221, where mutations of the same residue can either activate or inactivate the CaSR, serve as a rich source of structure-function information.
Interestingly, among thirty-four identified ECD missense mutations in patients with FHH, NSHPT and ADHH, 18 were located within 10 Å of one or more of the five Ca2+-binding sites predicted in our previous studies [11], particularly site 1, which is proposed to be the principal site for Ca2+-binding during receptor activation as well as for regulating both the homotropic and heterotropic cooperativity of CaSR [12] [13]. Our group has also reported a putative L-Phe binding site composed of residues S170, Y218, L51, S272 and T145, which are located adjacent to the predicted calcium binding pocket 1 at the hinge region of the CaSR ECD [12]. The location of the L-Phe binding site is pivotal in cooperatively regulating the activity of the CaSR [12].
Our previous computational studies showed that movements of Ca2+-binding site 1 are dynamically correlated with those of the other predicted calcium-binding sites. Mutations in Ca2+-binding site 1 could, therefore, affect both the homotropic and heterotropic cooperativity of the CaSR. These results led us to hypothesize that residues 173 and 221 are essential for the functional cooperativity orchestrated by calcium and L-Phe binding, and that related disease-associated mutations are the result of alteration or perturbation of the receptor's associated molecular connectivity.
In the present studies, we show that the loss-of-function mutations, L173P and P221Q, but not the gain-of-function mutations, L173F and P221L, alter the cooperativity of the CaSR though analyzing changes in functional readouts in response to alterations in [Ca2+]o. Furthermore, L173P and P221Q disrupt the strong correlated motions among the various calcium-binding sites as demonstrated by molecular dynamics (MD) simulations. L-Phe induced positive heterotropic cooperativity of the P221Q mutant, but had only a limited effect in potentiating the functional activity of L173P due to its restricted molecular dynamical features. These in vitro and in silico results provide important insights into how Ca2+ and L-Phe interact at their respective binding sites in the cleft of the VFTD, which are essential for the maintenance of calcium homeostasis that is required for normal physiological function.
Methods
Cell culture and transfection
Human embryonic kidney cells (HEK293) (ATCC) were cultured under standard condition (5% CO2, 37°C) in High Glucose Dulbecco's modified Eagle's medium (DMEM) (Sigma Chemicals) containing 10% fetal bovine serum with 100 µg/ml penicillin-streptomycin. The CaSR mutations were introduced using site-directed mutagenesis (QuikChange, Stratagene, La Jolla, CA, USA) [14]. Cells were transfected with either pEGFP-N1-WT-CaSR or mutant CaSRs using Lipofectamine 2000 in reduced serum Opti-MEM medium following the manufacturer's instructions (Invitrogen). After 4∼6 hours, the medium was changed to High Glucose DMEM, and the cells were incubated for an additional 48 hours to increase receptor expression. Transfection efficiency and expression levels were confirmed by analyzing the fluorescence intensity of the EGFP-tagged CaSR by fluorescence microscopy (Leica, DMI600B).
Measurement of [Ca2+]i responses in single cells transfected with WT or mutant CaSRs
Measurement of [Ca2+]i was carried out as described by Jiang, et al. [15]. Briefly, wild type CaSR or its mutants were transiently transfected into HEK293 cells grown on coverslips and cultured for 48 h. The cells were subsequently loaded with Fura-2 by incubation with 4 µM Fura-2 AM in 2 mL physiological saline buffer (10 mM HEPES, 140 mM NaCl, 5 mM KCl, 1.0 mM MgCl2, 1 mM CaCl2 and pH 7.4). The coverslips were mounted in a bath chamber on the stage of a Leica DM6000 fluorescence microscope. The cells were alternately illuminated with 340 or 380 nm light, and the fluorescence at an emission wavelength of 510 nm was recorded in real time as the concentration of [Ca2+]o was increased in a stepwise manner in the presence or absence of 5 mM L-Phe. The ratio of emission fluorescence intensity from both excitation wavelengths was then monitored as a function of [Ca2+]o and utilized as a parameter reflecting changes in [Ca2+]i All experiments were performed at room temperature. In addition to the use of the cell population assay as a means of calculating EC50 for Ca2+ o or L-Phe, the signals from 30 to 60 single cells were recorded for each measurement in a single cell [Ca2+]i assay. In the latter assay, [Ca2+]i oscillations in individual cells were defined as three or more successive fluctuations in [Ca2+]i from the baseline after the initial peak.
Immunostaining and Western blotting of CaSR
pcDNA3.1-CaSRs were used in the immunostaining and Western blotting experiments. This CaSR construct contains a flag-tag between Asp371 and Thr372. For immunostaining, 48 hours after transfection, cells were fixed with 3.7% formaldehyde for 15 min at room temperature, and subsequently labeled with mouse anti-Flag monoclonal antibody (1∶3000) overnight at 4°C. The next day cells were washed with PBS and stained with goat anti-mouse Alexa488-conjugated secondary antibodies. Images were collected on a Zeiss LSM700 confocal microscope (Carl Zeiss, German). For Western blotting, cells were lysed with RIPA buffer (Millipore, MA). Protein concentrations were determined using a Bradford assay (Bio-Rad laboratories, Hercules, CA). Lysates containing 40 µg of protein were separated by 7.5% SDS-PAGE. After gel electrophoresis, the proteins were transferred to a nitrocellulose membrane, probed with anti-flag monoclonal antibody (1∶3000) in 3% milk/TBST overnight at 4°C. Blots were subsequently probed with alkaline phosphatase-conjugated secondary antibody (1∶3000) for 1 hour at room temperature. After washing, the signals were detected by standard enhanced chemiluminescence. The signals were quantitated using ImageJ.
Measurement of inositol-1-phosphate (IP1) accumulation in CaSR transfected HEK293 cells
HEK293 cells were seeded in 24-well plates at 3×105 cells per well in 500 µl of culture medium. After transfection with WT CaSR or its various mutants, cells were further cultured for 24 hours at 37°C. Cell monolayers were first washed with Ringer's buffer without calcium (121 mM NaCl, 2.4 mM K2HPO4, 0.4 mM KH2PO4, 10 mM HEPES, 5.5 mM glucose, 1.2 mM MgCl2) and then incubated for 1 hour at 37°C in stimulation buffer (140 mM NaCl, 5 mM KCl, 10 mM LiCl2, 0.55 mM MgCl2, 10 mM HEPES) containing varying concentrations of CaCl2. After treatment, cells were lysed for 30 min at 37°C with 50 µl of 2.5% IP1 ELISA Kit Lysis Reagent (CIS Bio International, Gif-sur-Yvette, France). The accumulation of IP1 was measured using an immunoassay based on competition between free IP1 and horseradish peroxidase (HRP) conjugated IP1 for binding to monoclonal anti-IP1 antibody. The results for IP1 were expressed as percentage inhibition of IP1-HRP binding = [1-IP1-HRP binding in stimulated cells/IP1-HRP binding in unstimulated cells] ×100. The EC50s of [Ca2+]o-dependent responses were calculated by fitting the [Ca2+]o concentration-response curves with the Hill equation.
Determination of ERK1/2 phosphorylation
Thirty-six hours post transfection of monolayers of HEK293 cells with CaSR or its mutants, cells were incubated in serum-free high glucose DMEM medium supplemented with 0.2% w/v BSA at 37°C overnight. On the following day, cells were first incubated with HBSS for 30 min, followed by stimulation with varying levels of CaCl2 (0–20 mM) with or without L-Phe (5 mM) for 10 min. At the end of the [Ca2+]o stimulation, cells were lysed with RIPA lysis buffer (Millipore, CA, USA). In total, 150 µg aliquots of lysate protein were loaded into either a 12.5% SDS-gel or a 4%–12.5% gradient gel for PAGE and analyzed by western blotting with an anti-phospho-p44/42 ERK polyclonal antibody (Cell Signaling Technology, Beverly, MA, USA) diluted (1∶2000). A chemiluminescent method (AP Conjugate Substrate Kit) was employed to detect the phospho-(p)44/42 proteins. Quantitative analysis of the results was performed using ImageJ software (National Institutes of Health). The responses were normalized to the maximal effect observed with [Ca2+]o alone. The EC50 of [Ca2+]o-dependent responses was calculated by fitting the [Ca2+]o concentration-response curves with the Hill equation.
Computational analysis and Molecular Dynamics (MD) simulation of CaSR
CLUSTALW was used to align the sequences of the human CaSR ECD (residues 25–530) and the mGluR1 ECD [16]. The structure of the ECD of CaSR was modeled based on the crystal structure of mGluR1 (1EWT, 1EWK and 1ISR) using SWISS-MODEL [17] and MODELLER software [18], and the potential Ca2+-binding sites in the CaSR ECD were predicted using MetalFinder [19], The electrostatic potentials were calculated using Pymol.
MD simulation provides an approach complementary to experiments in live cells for understanding biomolecular structure, dynamics, and function. The initial coordinates for all the simulations were taken from a 2.20 Å resolution x-ray crystal structure with PDBID 1EWK [20]. The AMBER 10 suite of programs [21] was used to carry out all of the simulations in an explicit TIP3P water model [22], using the modified version of the all-atom Cornell et. al. [23] force field and the re-optimized dihedral parameters for the peptide ω-bond [24]. An initial 2 ns simulation was performed using NOE restraint during the equilibration in order to reorient the side chains of residues in the Ca2+-binding site, but no restraints were used during the actual simulation. A total of three MD simulations were carried out for 50 ns each on the apo-form and the ligand-loaded forms. During the simulations, an integration time step of 0.002 ps was used to solve the Newton's equation of motion. The long-range electrostatic interactions were calculated using the Particle Mesh Ewald method, [25], and a cutoff of 9.0 Å was applied for non-bonded interactions. All bonds involving hydrogen atoms were restrained using the SHAKE algorithm [26]. The simulations were carried out at a temperature of 300 K and a pressure of 1 bar. A Langevin thermostat was used to regulate the temperature with a collision frequency of 1.0 ps-1. The trajectories were saved every 500 steps (1ps). The trajectories were analyzed using the ptraj module in AMBER 10.
Statistics
The data are presented as means ± SE of the indicated number of experiments. One-way repeated measures analysis of variance (ANOVA), followed by the Dunnett post hoc test was performed using SPSS (SPSS Inc, Chicago, IL, USA) for multiple comparisons to compare the differences between groups without adding L-Phe. Two-way ANOVA was used for comparison among different groups in the presence of L-Phe. A P value of < 0.05 was considered to indicate a statistically significant difference.
Results
The disease-associated CaSR mutations alter the [Ca2+]o-triggered [Ca2+]i oscillation pattern
The two residues (Leu173, Pro221) involved in the four disease-associated mutations studied here are located in the hinge region near the predicted calcium-binding site 1, which is located between lobe 1 and lobe 2. Meanwhile they are also adjacent (within 10 Å) to the reported L-Phe binding pocket formed by residues K47, L51, W70, T145, G146, S169, S170, I187, Y218, S272, H413 and R415, which can exert effects over a large region of the ECD through positive heterotropic cooperativity as shown in Figure 1a [12]. The immunostaining as well as the Western blotting results suggest that all of the CaSR mutants are expressed at the cell surface at similar levels (Figure 1b, 1d, 1e).
Figure 1. The disease-related mutations are located in the hinge region near Ca2+-binding site 1.

a. Model structure of CaSR ECD based on the mGluR1 crystal structure (PDB entry: 1ISR) was generated using MODELER 9v4 and Swiss-Model. The global view of the ECD is shown in the left panel. Spheres highlighted in red: Ca2+; Purple: residues involved in predicted Ca2+-binding sites. L-phenylalanine (in yellow) is positioned at the hinge region between the two lobes by Autodock vina. Right panel: A zoomed in view of the Ca2+-binding pocket of site 1. Residues involved in Ca2+-binding site 1are highlighted in purple; residues with disease-related mutations are highlighted in pink; residues predicted to interact with L-Phe are presented in green. b. Immunofluorescence analysis of surface expressed WT CaSR and its mutants in HEK293 cells. Immunostaining was done with anti-flag monoclonal antibody, and detection was carried out with Alex Fluor 488-conjugated, goat anti-mouse secondary antibody. Green: CaSR. The images were taken using equal exposure times. c. Schematic figure of calcium and L-Phe induced downstream signaling changes and the principle for the measurement of IP1 accumulation. Red dots represent calcium ions. d. total proteins (40 µg) were applied to SDS-PAGE under non-reducing conditions and blotted with anti-flag antibody. GAPDH was used as an internal control. e. The signals were analyzed using ImageJ and the normalized intensities were compared with WT CaSR.
Analysis of the [Ca2+]o-triggered [Ca2+]i signaling (Figure 1c), especially the pattern of [Ca2+]i oscillations, is a straightforward method to depict the potential alterations in the homotropic cooperativity induced by mutations. Three parameters were employed to analyze the [Ca2+]i oscillation patterns: the starting point, which refers to the [Ca2+]o at which a given cell starts to show at least three continuous, sequential peaks; the frequency, which is defined as the number of peaks of [Ca2+]i per minute, and the ending point, which is the [Ca2+]o at which the [Ca2+]i ceases to oscillate and reaches a plateau. About 53% of the cells transfected with WT CaSR showing oscillatory responses started to oscillate at 3.0 mM [Ca2+]o (Figure 2 & 3). However, the majority of cells transfected with the inactive mutant, P221Q, did not oscillate until [Ca2+]o reached more than 4.0 mM, and the threshold for [Ca2+]i oscillations in cells transfected with the other inactive mutant, L173P, was even higher at 12.5 mM. Conversely, for the gain-of-function mutations (L173F and P221L), the majority of the cells, 79% for L173F and 40% for P221L, started to oscillate at lower levels of [Ca2+]o, 2.0 mM and 2.5 mM, respectively.
Figure 2. Functional studies of disease-related CaSR mutations in individual HEK293 cells.

The various panels show representative oscillation patterns from single cells. HEK-293 cells transfected with CaSR or one of its mutants were loaded with Fura-2 AM for 15 min. [Ca2+]i was assessed by monitoring emission at 510 nm with excitation alternately at 340 or 380 nm as described in Methods. Each experiment was carried out with or without 5 mM L-Phe and began in Ca2+-free Ringer's buffer (10 mM HEPES, 140 mM NaCl, 5 mM KCl, and 1.0 mM MgCl2, pH 7.4), followed by stepwise increases in [Ca2+]o until [Ca2+]i reached a plateau (up to 30 mM [Ca2+]o).
Figure 3. Frequency distribution of oscillation parameters in HEK293 cells transfected with CaSR or its mutants.

The pattern of the [Ca2+]i response in each cell (minimum of 40 cells) was analyzed. a. The [Ca2+]o at which individual cells started to oscillate was recorded. The X-axis comprises 1.0 mM to 8.0 mM [Ca2+]o for WT and for mutants L173F and P221L, 1.5 mM to 10.0 mM [Ca2+]o for P221Q or 7.5 mM to 25.0 mM for mutant L173P. b. The frequency of the individual cell oscillation patterns was investigated. For experiments without L-Phe, the peaks per minute were recorded at the levels of [Ca2+]o at which the majority of the cells (>50%) started oscillating. Specifically, for the gain-of-function mutants, the peaks per minute were recorded at 2.5 mM [Ca2+]o, while for loss-of-function mutants, the frequency was analyzed at 15.0 mM [Ca2+]o for L173P and 5.0 mM [Ca2+]o for P221Q. c. The [Ca2+]o at which the [Ca2+]i oscillations began to reach a plateau was recorded. Empty bar: in the absence of L-Phe; Black bar: in the presence of 5 mM L-Phe.
WT CaSR-transfected cells exhibited a normal distribution of oscillation frequencies with an average frequency of 1.0∼1.5 peaks/min at 3.0 mM [Ca2+]o (Figs. 2&3b, Table 1). The two inactivating mutants (L173P and P221Q) responded to changes in [Ca2+]o over a substantially different range. Therefore, the frequencies were measured at the level of [Ca2+]o at which the majority of the cells (>50%) started to oscillate—15 mM for L173P and 5 mM for P221Q. For L173F and P221L, the frequencies were measured at 2.5 mM [Ca2+]o. The oscillation frequency for the two inactivating mutants did not differ from that of the WT (1.3±0.1 peaks/min) and were 1.3±0.1 peaks/min for L173P and 1.5±0.1 peaks/min for P221Q, but there were slight but significant increases for L173F (1.7±0.1 peaks/min) and P221L (1.6±0.1 peaks/min).
Table 1. Summary of cellular responses of HEK293 cells transiently transfected with WT CaSR or disease related mutants.
| Mutants | EC50[Ca2+]O | Hill coefficient | Frequency | |||
| w/o L-Phe | with 5 mM L-Phe | w/o L-Phe | with 5 mM L-Phe | w/o L-Phe | with 5 mM L-Phe | |
| WT | 3.0±0.2 | 1.9±0.3* | 3.7±0.3 | 5.0±0.7* | 1.3±0.1 | 2.1±0.1* |
| L173F | 1.9±0.2# | 1.6±0.1 | 3.3±0.1 | 3.7±0.3 | 1.7±0.1# | 1.9±0.1 |
| P221L | 2.0±0.1# | 1.7±0.1 | 3.2±0.2 | 2.8±0.2 | 1.6±0.1# | 1.8±0.2 |
| L173P (Phase 1) | 3.8±0.3 | 3.1±0.2 | 3.3±0.6 | 2.5±0.2 | 1.3±0.1 | 1.8±0.2* |
| L173P (Phase 2) | 13.0±0.3 | 12.2±0.2* | 3.6±0.4 | 5.8±0.3 | ||
| P221Q | 5.2±0.4# | 3.7±0.4* | 2.4±0.4# | 4.1±0.7* | 1.5±0.1 | 2.2±0.1* |
The average levels of [Ca2+]o at which cells started to exhibit [Ca2+]i oscillations were recorded for WT or each mutant CaSR. For the oscillation frequencies in the absence of L-Phe, peaks per minute were measured at the level of [Ca2+]o at which more than 50% cells started to oscillate; when L-Phe was added, frequencies were recorded at the same [Ca2+]o as their counterparts without L-Phe. Specifically, the frequencies of WT was measured at 3.0 mM [Ca2+]o, at 2.5 mM [Ca2+]o for L173F and P221L; at 15.0 mM [Ca2+]o for L173Pand at 5.0 mM [Ca2+]o for P221Q. Curve-fitting was performed using the Hill equation. The data were obtained from three experiments for each construct. Values are means ± S.E. EC50 and Hill numbers obtained from the cell population assay by fitting plots using the Hill equation. # indicates significance with respect to wild type CaSR in the absence of L-Phe, p<0.05 (ANOVA, Dunnett test); * indicates significance with respect to the corresponding mutants in the absence of L-Phe, p<0.05 (two-way ANOVA).
We also studied the oscillation ending point. About 50% of the oscillatory cells expressing the CaSR reached a [Ca2+]i plateau (i.e., stopped oscillating) at 5.0 mM [Ca2+]o (Figure 3c). The loss-of-function mutations exhibited a dramatic increase in the [Ca2+]o required for a [Ca2+]i plateau as shown in Figure 3c. The majority of the oscillatory cells transfected with L173P were still exhibiting [Ca2+]i fluctuations even at a level of [Ca2+]o of 30.0 mM. Similarly, the ending point for P221Q was substantially elevated at ∼10.0–15.0 mM. On the other hand, quite a few cells transfected with either of the gain-of-function mutants stopped [Ca2+]i oscillations at a [Ca2+]o below 5.0 mM. Therefore, the [Ca2+]o-triggered [Ca2+]i oscillation pattern, including its frequency, the [Ca2+]o required for initiating oscillations, and the oscillation ending point are altered in the four mutations, suggesting changes in [Ca2+]o–elicited positive homotropic cooperativity induced by these mutations.
The disease-associated CaSR mutations affect functional positive homotropic cooperativity as reflected by changes in multiple CaSR-activated signaling pathways
We then investigated how those mutations affect the CaSR-mediated activation of various intracellular signaling pathways. HEK293 cells transfected with WT CaSR exhibited sigmoidal concentration response curves for the [Ca2+]i responses to increases in [Ca2+]o in a cell population assay with a Hill coefficient of 3.7±0.3 and an EC50 of 3.0±0.2 mM, suggesting strong positive homotropic cooperativity among its five predicted Ca2+-binding sites (Figure 4, Table 1). Notably, the two inactivating mutations showed increases in EC50 and disruption of the CaSR's positive homotropic cooperativity, with a change in the monophasic [Ca2+]o-induced [Ca2+]i response curves to a biphasic one (L173P) or a decrease in Hill number for P221Q, accompanied by an increase of EC50 to 5.2±0.4 mM (Figure 4, Table 1).
Figure 4. L-Phe modulates the [Ca2+]o concentration response curves in CaSR-transfected HEK293 cells.
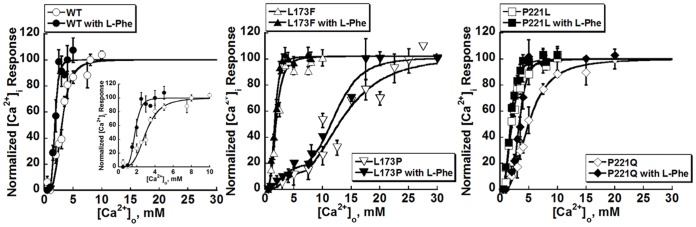
The [Ca2+]i responses of HEK293 cells transiently overexpressing WT CaSR or disease-related mutations were measured using Fura-2AM during stepwise increases in [Ca2+]o from 0.5 to 30 mM with or without L-Phe as above. The ratio of light emitted at 510 nm upon excitation alternately with 340 or 380 nm was normalized to the maximum response. And the average [Ca2+]i responses at various [Ca2+]o were normalized and plotted against [Ca2+]o and further fitted using the Hill equation. Open marker: in the absence of L-Phe; Closed marker: in the presence of 5 mM L-Phe.
Activation of the Gq/11 pathway by GPCRs activates phospholipase C (PLC), which induces an increase in [Ca2+]i caused by the associated accumulation of inositol phosphates (IPs) [3]. Thus the activation of PLC in response to increases in [Ca2+]o in cells transiently transfected with WT CaSR were assessed by measuring the accumulation of IP1 as an index of PLC activation Figure 1c & Figure 5). The EC50 for the stimulation of IP1 accumulation was 2.9±0.3 mM, similar to the EC50 calculated from the [Ca2+]i responses (3.0±0.2). As with their Ca2+ i responses, L173P and P221Q exhibited impaired [Ca2+]o–stimulated IP1 accumulation as reflected by the increases in their EC50s, 10.7±1.0 mM and 6.1±0.3 mM respectively, vs. 2.9±0.3 mM for the WT) (Figure 5, Table 2). In contrast, the L173F and P221L mutants showed substantial decreases in their EC50s to 1.2±0.1 mM and 1.1±0.1 mM, respectively (Table 2).
Figure 5. L-Phenylalanine potentiates [Ca2+]o-induced IP1 accumulation.
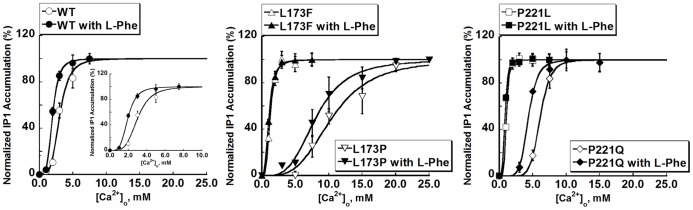
IP1 accumulation was measured using the IP-One ELISA Kit as detailed in Methods. Left: Changes in IP1 accumulation were measured in response to incubation with 0, 1.0, 2.0, 3.0, 5.0 and 7.5 mM [Ca2+]o in HEK293 cells transfected with WT CaSR, or with mutants L173F or P221L. For mutant L173P, IP1 accumulation was measured in response to 0, 3.0, 5.0, 7.5, 10.0, 15.0, 20.0 and 25.0 mM [Ca2+]o; for mutant P221Q, the IP1 responses at 0, 3.0, 5.0, 7.5, 10.0, 15.0 mM [Ca2+]o were recorded. The IP1 response-calcium concentration responses were fitted using Hill equation as described in Methods.
Table 2. Summary of EC50s from experiments measuring IP1 accumulation and ERK1/2 phosphorylation.
| Mutants | EC50 (IP1-Elisa) | EC50 (ERK1/2 activity) | ||
| Without L-Phe With L-Phe | With L-Phe | Without L-Phe | With L-Phe | |
| WT | 2.9±0.3 | 2.0±0.1* | 3.0±0.1 | 1.9±0.1* |
| L173F | 1.2±0.1# | 1.0±0.1 | 2.7±0.1# | 1.6±0.1* |
| P221L | 1.1±0.1# | 0.9±0.1 | 2.2±0.3# | 1.7±0.2 |
| L173P | 10.7±1.0# | 9.0±0.9 | 14.9±1.0# | 13.5±0.8 |
| P221Q | 6.1±0.3# | 4.5±0.4* | 11.9±0.3# | 9.5±0.4* |
HEK293 cells were transiently transfected with the WT CaSR or disease-associated CaSR mutants. Cells were then treated with various levels of [Ca2+]o. The EC50s of [Ca2+]o-IP1 responses and the -[Ca2+]o-ERK1/2 phosphorylation activity responses were obtained from curve fitting using the Hill equation as mentioned in Method. # indicates significance with respect to wild type CaSR, p<0.05 (ANOVA, Dunnett test); * indicates significance with respect to the corresponding mutants in the absence of L-Phe, p<0.05 (two-way ANOVA).
We further examined the effects of [Ca2+]o on mitogen-activated protein kinases (MAPK) (ERK1/2) activation. Exposure of WT CaSR-transfected HEK293 cells to increasing concentrations of [Ca2+]o in the range of 0.0-∼2.0 mM for 10 minutes had little effect on the phosphorylation of p44/42 ERK. Greater increases of [Ca2+]o resulted in the accumulation of p44/42 ERK, which exhibited a maximum response at 8.0 mM [Ca2+]o. Immunoblots of [Ca2+]o–induced increases in ERK activity were further quantified using ImageJ and plotted against [Ca2+]o. The data were then fitted using the Hill equation, giving an EC50 of around 3.0±0.1 mM, which is close to the result measured using intracellular calcium responses. The mutants L173P and P221Q also impaired the [Ca2+]o-triggered ERK activity of the CaSR. More than 8.0 mM [Ca2+]o was needed in order to induce the phosphorylation of ERK1/2 for both mutants (Figure 6). For the activating mutation, L173F, a phosphorylated ERK1/2 signal could be detected at 2.0 mM [Ca2+]o, which was in line with its enhanced intracellular calcium response compared to WT CaSR. The mutant P221L also potentiated [Ca2+]o-evoked ERK1/2 activity in the transfected cells, approaching a maximum at 3.0 mM [Ca2+]o.
Figure 6. L-Phe potentiates [Ca2+]o-activated ERK signaling in CaSR-transfected HEK293 cells.

HEK-293 cells transfected with WT CaSR or its mutants were incubated in serum-free high glucose MEM medium containing 0.2% BSA overnight. Cells were washed with Hank's balance salt solution (HBSS) and then incubated in the presence of various Ca2+ concentrations (0.0-∼25.0mM) in the absence or presence of 5 mM L-phenylalanine for 10 min at 37°C. The incubations were stopped by exposure to the lysis buffer and processed for SDS/PAGE and Western blotting as described in the Methods. The Western blot results were further quantified using Image J. All [Ca2+]o-concentration response curves were normalized to the maximum response in each individual experiment. The Hill equation was employed to fit the data. Markers: [Ca2+]o only; closed markers: with L-Phe.
Disease-associated mutations affect the heterotropic positive cooperativity induced by L-Phe on the CaSR
We assessed the impact of L-Phe on [Ca2+]o–induced changes in [Ca2+]i in HEK293 cells transfected with WT CaSR in our recent work [12], however, it was not clear how the disease-associated mutations would influence the heterotropic positive cooperativity generated by L-Phe. For the majority of WT CaSR-transfected HEK293 cells, [Ca2+]i oscillations first began at 1.5 mM [Ca2+]o and ended at 3.0 mM [Ca2+]o in the presence of 5 mM L-Phe, and the oscillation frequency at 3.0 mM [Ca2+]o increased to 2.1±0.1 peaks/min, which was significantly higher than the 1.3±0.1 peaks/min observed in the absence of L-Phe (Figure 3). For the loss-of-function mutants, cells transfected with mutant L173P started oscillating at 10.0 mM [Ca2+]o with 5.0 mM L-Phe (Figure 3). L-Phe increased the frequency of the L173P mutant significantly from 1.3±0.1 to 1.8±0.2 peaks/min at 15.0 mM [Ca2+]o in the presence of L-Phe (Figure 3b & Table 1). In most cells, the [Ca2+]i oscillations stopped at 25.0 mM [Ca2+]o (Figure 3c). In contrast, without L-Phe, 80% of the cells were still oscillating at 30 mM [Ca2+]o. For the P221Q mutant, addition of 5 mM L-Phe at 5.0 mM [Ca2+]o decreased the threshold for initiation of [Ca2+]i oscillations from 5.0 mM to 3.5 mM [Ca2+]o, increased the oscillation frequency from 1.5±0.1 peaks/min to 2.2±0.1 peaks/min, and reduced the [Ca2+]o required to reach the ending point. Regarding the gain-of-function mutations (L173F, P221L), L-Phe lowered the threshold for the termination of [Ca2+]i oscillations, but barely shifted the frequency distribution of their starting points nor significantly increased their frequencies at 2.5 mM [Ca2+]o (Figs. 2 & 3).
Meanwhile, L-Phe facilitated the response of the WT CaSR to [Ca2+]o by significantly decreasing the EC50 for [Ca2+]i responses from 3.0±0.2 mM to 1.9±0.3 mM (p<0.05, two-way ANOVA) and increasing the Hill coefficient to 5.0±0.7 mM (Figure 4). Similar analyses were carried out for the four CaSR mutants. L-Phe shifted the sigmoidal curve to the left producing a lower EC50 (3.7±0.3 mM) with an increase in Hill number (4.1±0.7 mM) for mutant P221Q as measured from the [Ca2+]o-evoked increase in [Ca2+]i. L-Phe did not left-shift the activation of the L173P mutant by [Ca2+]o to the same extent that it did with the other loss-of-function mutant, P221Q, potentially because of the proximity of this residue to the binding site for L-Phe, which might interfere with L-Phe binding. The disruption of the positive homotropic cooperativity induced by [Ca2+]o as a result of the Leu to Pro change, which was reflected as a biphasic concentration response curve, was also not corrected by the addition of L-Phe (Figure 4). On the other hand, the addition of L-Phe barely induced any further left-shift in the concentration-response curves of the two gain-of-function CaSR mutants (p>0.05).
The influence of the L-Phe on the four mutants was also assessed by changes in IP1 production upon ligand stimulation. The addition of 5 mM L-Phe significantly elevated the level of IP1 accumulation in cells transfected with WT CaSR at 2.0 and 3.0 mM [Ca2+]o, resulting in a left-shifted [Ca2+]o-IP1 concentration response curve (EC50 = 2.0±0.1 mM, p<0.05). In the case of the mutant P221Q, the EC50 was reduced from 6.1±0.3 mM to 4.5±0.4 mM in the presence of L-Phe (p<0.05). On the contrary, L-Phe failed to reduce the EC50 of L173P significantly (9.0±0.9 mM). The EC50s of the IP1 responses for the two receptors with activating mutations, L173F and P221L, were not altered by L-Phe either (Figure 5, Table 2). Thus results from the IP1 experiments were consistent with the [Ca2+]i readouts in terms of EC50.
L-Phe also had an impact on the [Ca2+]o-triggered ERK1/2 activities of WT and mutant CaSRs in CaSR transfected cells. As shown in Figure 6 and Table 2, the amount of phosphorylated ERK1/2 was increased significantly by L-Phe at 2.0 mM [Ca2+]o in the WT CaSR. The ERK1/2 activity of the WT CaSR in the presence of L-Phe reached a maximum at 4.0 mM [Ca2+]o. Quantitative analysis of the immunoblots suggests that L-Phe shifted the sigmoidal curves to the left producing a lower EC50s for [Ca2+]o-evoked ERK activity. In accordance with the [Ca2+]i responses and the IP1 accumulation readouts, the addition of 5 mM L-Phe shifted the [Ca2+]o-stimulated p-ERK1/2 concentration response curve of mutant P221Q to the left, resulting in a decrease in the EC50 from 11.9±0.3 mM to 9.5±0.4 mM (p<0.05), but had little influence on the activity of mutant L173P (Figure 6, Table 2). On the other hand, the maximum ERK1/2 activity in cells transfected with the L173F mutant was achieved at 3.0 mM [Ca2+]o with 5.0 mM L-Phe, significantly shifting the [Ca2+]o-triggered [Ca2+]i concentration response curve to the left. For the mutant P221L, the addition of L-Phe failed to activate the ERK1/2 signaling at lower levels of [Ca2+]o, but the maximal p-ERK1/2 activity was enhanced ∼15% in the presence of L-Phe. The differences of the ERK activity results compared with the IP1 readouts suggested that L-Phe may differentially change the heterotropic cooperativity via multiple signaling pathways.
CaSR mutants exhibit different sensitivities to L-Phe
To determine whether or not the distinctive effects shown by L-Phe on different mutants are due to changes in their apparent affinities for this amino acid, concentration responses for L-Phe at a physiological level of 1.5 mM [Ca2+]o were first compared among cells transfected with different CaSR mutants. Figure 7 shows that at 1.5 mM [Ca2+]o, the loss-of-function mutant L173P exhibited a lower sensitivity (EC50 = 5.5±0.3 mM) to L-Phe compared to WT and the other mutants (all EC50s<5.0 mM). Since the influence of L-Phe might, to some extent, depend on the extracellular calcium concentration, the apparent affinities of L-Phe for the two loss-of-function mutants were further analyzed at levels of [Ca2+]o equal to their respective EC50s for [Ca2+]o, specifically 15.0 mM [Ca2+]o for L173P and 5.0 mM [Ca2+]o for P221Q. P221Q exhibited a reduced EC50 for the L-Phe response (EC50 = 2.5±0.2 mM, p<0.05) at 5.0 mM [Ca2+]o compared with EC50 for L-Phe measured at 1.5 mM [Ca2+]o (EC50 = 4.5±0.4 mM), while L173P retained a similar sensitivity to L-Phe regardless of the extracellular calcium concentration (EC50 = 4.9±0.5 mM, p>0.05) (Figure 7, Table 3).
Figure 7. Sensitivity of various CaSR mutants to L-Phe in HEK293 cells.
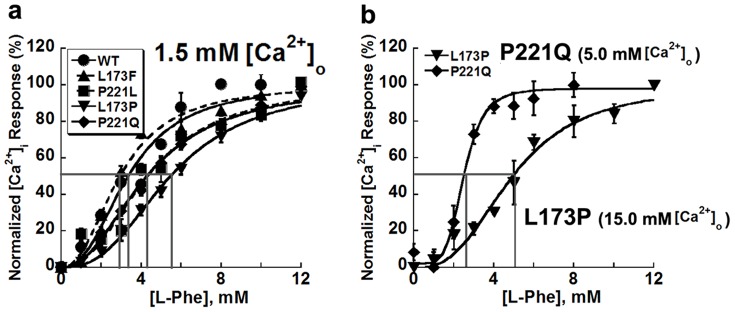
a. HEK-293 cells transfected with CaSR or its mutants were loaded with Fura-2 AM for 15 min. The intracellular Ca2+ level was assessed by monitoring emission at 510 nm with excitation alternately at 340 or 380 nm using fluorescence microscopy as above. Each experiment started with 1.5 mM mM Ca2+ followed by stepwise increases in the level of L-Phe (up to 12.0 mM) while the [Ca2+]o was maintained at 1.5 mM. b. The sensitivity of P221Q and L173P to L-Phe were compared at 5.0 mM [Ca2+]o or 15.0 mM [Ca2+]o, respectively.
Table 3. Summary of EC50s of concentration-response curves for L-Phe at different [Ca2+]o.
| Mutants | EC50 (at 1.5 mM [Ca2+]o) |
| WT | 3.7±0.4 |
| L173F | 3.2±0.2 |
| P221L | 4.3±0.3 |
| L173P | 5.5±0.3# (4.9±0.5) |
| P221Q | 4.5±0.4 (2.5±0.2*) |
The [Ca2+]i responses of HEK-293 cells transfected with CaSR or its mutants upon stepwise increases of L-Phe in the presence of 1.5 mM [Ca2+]o were recorded. For L173P and P221Q, the L-Phe induced intracellular change were also measured in the presence of a level of high [Ca2+]o corresponding to their EC50 values for the [Ca2+]o, specifically 15.0 mM [Ca2+]o for mutant L173P and 5.0 mM for P221Q. The results are shown in the brackets. The EC50s were calculated from the concentration-response curves fitted using the Hill equation. # indicates significance with respect to wild type CaSR, p<0.05 (ANOVA, Dunnett test); * indicates significance with respect to the corresponding mutants in the presence of 1.5 mM [Ca2+]o, p<0.05 (two-way ANOVA).
Computational simulations reveal different dynamic behaviors of the four mutants
The correlated motions in Figure 8 illustrate the molecular dynamic motions of WT CaSR and its mutants obtained from computational simulations. In the modeled WT structure, Site 1 has strong correlated motions with sites 2, 3 and 4, shown in blue (negative correlated motions between a pair of residues signify motions in opposite direction) and red (positive correlations, which are movements in the same direction) (Figure 8, top two panels). We also have reported in our earlier work that the presence of both L-Phe and Ca2+ in site 1 produces greater correlated motions among the Ca2+-binding sites compared with L-Phe alone in site 1 [12]. Interestingly, the loss-of-function mutations, L173P and P221Q, exhibit less correlated motions compared to the WT CaSR as demonstrated by a decrease in the negative (blue) and/or positive correlated motions (Figure 8, bottom panel). On the other hand, the correlated motions of the gain-of-function mutants, L173F and P221L, are dramatically increased in regions similar to those of the WT correlated motions in the presence of L-Phe, indicating an enhancement of correlated motions between the respective residues in these two mutants (Figure 8, middle panel). A closer analysis of the correlated motions of L173F and P221L reveals that the negative correlations between Site 1 and Site 3 and between Site 1 and Site 2 observed in the WT correlation map become positive correlations in the gain-of-function mutants indicating that the mutations might have profound impacts on the dynamic properties of the CaSR-ECD. Especially for P221L, strong negative correlations between lobe 1 and lobe 2 are observed as indicated by the abundant blue area between residues 200–300 and residues 24–170. These results indicate that residues Leu173 and Pro221 play pivotal roles in modulating the molecular connectivity between the binding sites for [Ca2+]o. In order to examine the possibility that the effects of inactivating mutations of the CaSR can be cancelled by those of activating mutations through molecular correlated motions, two double mutations, L173F/P221Q and L173P/P221L have been analyzed. We found that L173F/P221Q behaved like WT CaSR, thereby exhibiting a “cancelling” effect (Figure S1a), while L173P/P221L exhibited oscillation patterns similar to those of the L173P mutant, suggesting that the activating mutant P221L cannot overcome the inactivating effect of L173P (Figure S1b). These results suggest that mutations close to calcium binding Site 1 may play key roles in modulating correlated motions of the CaSR.
Figure 8. Correlation map of the modeled WT and mutant CaSR ECD structures.

The correlation maps are depicted based on molecular dynamic (MD) simulations of the WT and mutant CaSR ECD structures in the apo form or in the presence of Ca2+ and L-Phe. The X axis and Y axis are residue numbers of the CaSR ECD sequence minus twenty four. The strongest negative correlation is given the value -1, while the strongest positive correlation is defined as +1. Residues that have the strongest negative correlated motions are shown in blue, while those involved in positive correlated motions are shown in red. Green indicates no apparent correlated motion between the two residues. The correlated motions between calcium binding site 1 and the other calcium binding sites are mapped onto the CaSR ECD models as shown in the top right panel.
The docking of L-Phe to CaSR mutant P221Q engendered a more correlated molecular dynamics features similar to WT CaSR ECD, suggesting that the receptor is converted into an active conformation. Intriguingly, the other inactive mutant L173P did not exhibited dramatic alterations in its correlated motion in response to L-Phe compared to P221Q. This result suggests that the difference in the influence of L-Phe on potentiating the activity of the CaSR mutants may rely on the intrinsic molecular dynamic connectivity that is disrupted by mutant L173P. It is worth noting that the activating mutant L173F also showed enhanced correlated motions, which could be an explanation for the left shift curve of the ERK activity. The addition of L-Phe was unable to make the profile of mutant P221L into a more dramatically active state, which correlated well with the in vitro studies, suggesting the mutation is already in a “fully active” state upon the binding with Ca2+ similar as WT CaSR with both Ca2+ and L-Phe.
Principal component analysis (PCA) separates out the protein motions into principal modes ranked according to their relative contributions [27]. The analysis was done on the trajectories of the four mutations and the WT CaSR from the molecular dynamics simulations in order to predict the effect of the mutations on the response of L-Phe and Ca2+ at the atomic level (Figure 9).
Figure 9. Principal component analysis (PCA) of the CaSR ECDs.

The trajectories of the molecular dynamics simulations were analyzed using principal component analysis (PCA), which separates out the motions of the CaSR ECD into principal modes ranked according to their relative contributions. The first three principal modes were included in the present study to analyze the four CaSR mutants and the wild type: WT (black), L173F (green), presence P221L (magenta), L173P (red), P221Q (blue).
Projection of the trajectories of the CaSR mutants in both the apo forms (upper panel) and the holo forms (lower panel) onto the first three modes that accounted for the majority of the total fluctuations is shown in Figure 9. In the absence of Ca2+ and L-Phe, the conformations sampled by the CaSR mutants are similar to that of the WT CaSR. However, in the L-Phe and Ca2+-loaded forms, the conformations of the CaSR mutants L173P and L173F are distinguishable from the other mutants and WT CaSR (Figure 9). The results suggest that Ca2+ and L-Phe might shift the population of conformational ensembles of mutant L173P and L173F in a different way than in WT CaSR or in the other two mutants, supporting the essential role for the residue Leu173 in the molecular connectivity underlying the Ca2+ - and Phe-mediated functional cooperativity of CaSR.
Discussion
Loss-of-function mutations impair the functional cooperativity of the CaSR
The CaSR hinge region is considered to be crucial for the sensing of agonists (e.g., polyvalent cations and amino acids) [19], [28]. Mutations near Ca2+-binding site 1 are hypothesized to influence the functional cooperativity of the entire receptor. In vitro studies showed that the L173P and P221Q mutants produce a receptor with reduced positive homotropic cooperativity and impaired capability to sense [Ca2+]o, while the L173F and P221L mutants enhance the sensitivity of the CaSR to [Ca2+]o compared to the WT receptor, but barely change the cooperativity of the receptor. The results with the loss-of-function mutants indicate that Leu173 and Pro221 are important in maintaining the positive homotropic cooperativity among the receptor's calcium-binding sites. Conversely, the gain-of-function mutants show that these residues also are important for restraining the receptor from assuming its most active form (e.g., that seen in the WT CaSR in the presence of both high [Ca2+]o and L-Phe). Thus unlike mutations of residues that cause only gain- or loss-of-function, Leu173 and Pro221 serve dual functions, perhaps reflecting their key locations in the cleft between the receptor's two lobes and their proximities to the binding sites for both [Ca2+]o and L-Phe.
[Ca2+]i oscillations have been postulated to be the result of complex responses from multiple signaling pathways. The [Ca2+]o-triggered [Ca2+]i oscillation pattern is believed to be modulated by the activity of phosphoinositide pathway [29] as well as a negative feedback loop involving the inhibitory effect of protein kinase C (PKC) on IP3 production [30]. Downstream intracellular signaling responses, such as the production of IP1 and the activity of ERK1/2, which will be discussed later, can be reflected in the oscillation patterns associated with the different disease-related mutations. Although most research has focused on intracellular calcium mobilization in HEK293 cells heterologously expressing the CaSR, CaSR-induced [Ca2+]i oscillations have also been found in cells with endogenously expressed CaSR, for instance in parathyroid cells [31], bovine anterior pituitary cells [32], opossum kidney (OK) cells [33] and medullary thyroid carcinoma cells [34]. Oscillations in [Ca2+]i modulate not only the rate of parathyroid hormone (PTH) secretion, but also gene expression and other processes [35]–[38]. The pattern of [Ca2+]i oscillations is thus one of the most important signatures reflecting the state of CaSR activity. In the present study, we showed that the inactive mutants required higher [Ca2+]o to trigger intracellular [Ca2+]i oscillations compared to the WT CaSR, while the active mutants needed lower [Ca2+]o to initiate oscillations. The oscillation frequency of the inactive mutants remained similar to that of the WT CaSR, but higher [Ca2+]i oscillations frequencies were observed in HEK293 cells transfected with gain-of-function mutations. As [Ca2+]o is increased, the [Ca2+]i oscillations cease and reach a plateau in CaSR-transfected cells as the desensitization process occurs and/or there is depletion of [Ca2+]i stores. Although the loss-of-function mutation L173P still oscillated at 30.0 mM [Ca2+]o, considering the elevated and broad range of its sensitivity to [Ca2+]o, it is possible that a plateau would be reached at even higher levels of [Ca2+]o.
Our studies of CaSR-mediated accumulation IP1 (a metabolite of IP3) and activation of the ERK1/2 pathways in HEK293 cells transfected with the various CaSR mutants conveyed two messages: Firstly, the mutations caused changes in the patterns of [Ca2+]i oscillations that at least partially involve activation of the Gαq/11 pathway. The changes in the accumulation of IP1 upon stimulation with various concentrations of [Ca2+]o correlated well with [Ca2+]i changes. Secondly, high [Ca2+]o-evoked increases in ERK1/2 activities of WT-CaSR and its four mutants were in accordance with the accumulation of IP1. Stimulation of the ERK1/2 activity in CaSR-transfected HEK293 cells has been reported to involve PKC-mediated as well as a PTX-sensitive, tyrosine kinase-dependent pathways [39]. Although it is believed that the carboxyl terminus of the protein is involved in the activation of MAPK signaling by interacting with the scaffold protein, filamin A [40], [41], other data showed that that alterations in the extracellular domain can also affect the ERK cascade, which is in agreement with previous studies [42]. Since all the mutations studied here were expressed at similar levels on the cell membrane (Figure 1b), these downstream signaling changes may at least partially be contributed by the disruption of cooperativity among different calcium binding sites introduced by different mutations. On the other hand, the gain-of-function mutations may increase the stability the receptor as suggested by other studies [43], or they possibly induce more correlated motions among the calcium-binding sites as suggested by the dynamic cross-correlation map.
L-Phe-induced heterotropic cooperativity rescues activity of the CaSR
In the current study, we showed that L-Phe could affect one or two of the three parameters depicting the [Ca2+]o triggered [Ca2+]i oscillation patterns in all of the mutants as measured in individual cellular responses (e.g., [Ca2+]i oscillation starting points, ending points and [Ca2+]i oscillation frequencies). The alterations in the oscillation patterns after the introduction of the allosteric activator could be the result of the activation of signaling downstream of Gα12/13 or a combined effect with the positive heterotropic cooperativity between the multiple Ca2+ binding sites induced by L-Phe. The interaction between CaSR and L-Phe has been reported to modulate the Gα12/13-RhoA pathway [44]; however, L-Phe could also potentially enhance [Ca2+]o-induced alterations in intracellular signaling pathways through Gq/11 pathway as reflected by the IP1 accumulation assay.
The present study also confirms that the ERK signaling pathway in CaSR-transfected HEK293 cells can be modulated not only by [Ca2+]o but also by the positive allosteric modulator, L-Phe [45]. It should be noted that the ability of L-Phe to enhance the intracellular responses of the various signaling pathways varied between different mutations. L-Phe enhanced the maximum responses of the ERK signaling pathway to a greater extent than it altered the apparent affinities in the two gain-of-function mutants, although it barely showed any influence on the [Ca2+]o-triggered [Ca2+]i and IP1 responses for these mutants, which may explain, in part, the dramatic changes in correlated motions from the computational simulation results using molecular dynamics. As a signaling messenger further downstream than either IP1 or [Ca2+]i, the phosphorylation of ERK1/2 may not merely reflect activation of the Gq/11 pathway, but could be contributed to by other G-proteins (e.g., Gi). In agreement with the in silico results, L-Phe exhibited less effect on the regulation of the ERK1/2 pathway in mutant L173P. The predicted L-Phe-binding site has been reported to be located near the hinge region between lobe 1 and lobe 2 and to involve residues S169A/S170A/S171A [46]. Leu173 is located within 5 Å of this region. The Leu to Pro change could potentially generate additional steric effects on the closure of lobe 1 and lobe 2 so that addition of exogenous L-Phe has relatively little impact on the CaSR's response to changes in [Ca2+]o. Thus, the hetero-cooperativity between the allosteric modulator and calcium binding sites could be disturbed, resulting in the lack of a left-shifted concentration-response curve in response to [L-Phe].
Disease-associated mutations influence the molecular connectivity between the Ca2+-binding sites of the CaSR as revealed by molecular dynamic simulations
Based on all of the in vitro results and the in silico simulations, we propose a model to illustrate the possible mechanism by which the disease-associated mutations affect the function of CaSR and how their activities can be modulated by Ca2+ and L-Phe through the molecular connectivity that is encoded at the hinge region of the ECD of the protein. As shown in Fig 10, there are four functional states of the CaSR. In the absence of Ca2+, WT CaSR is at the basal state can be converted to the active state upon Ca2+ binding to site 1 at the hinge region. Subsequent binding of L-Phe at the adjacent hinge region further strengthens molecular connectivity and elevates the receptor to a “fully active” status in the presence of high [Ca2+]o by a heterotropic positive cooperative mechanism. Mutations close to the hinge region such as those at Leu173 and Pro221 affect the overall dynamic correlated motions of the respective mutant receptors and the Ca2+-induced homotropic cooperativity, which could further influence the conformation of the receptor. The inactivating mutation L173P impairs the molecular connectivity in the WT CaSR and cannot be rescued by L-Phe. On the other hand, the inactivating mutation P221Q weakens the connectivity but can be partially rescued by binding Phe with a resultant activity similar to the basal activity of the WT CaSR. In contrast, the gain-of-function mutants L173F and P221L may already have been converted to the active state with dynamic properties similar to that of Ca2+- loaded WT CaSR. They can be further activated upon binding extracellular Ca2+ to reach a state close to the “fully active” state (Phe + [Ca2+]o in the WT) that is insensitive to further activation by L-Phe achieving an apparent “ceiling” represented by the maximal achievable activity of the CaSR.
Figure 10. Schematic representation of the mechanisms underlying the effects of the mutations on the CaSR and the modulation of receptor activity by extracellular Ca2+ and L-Phe.

Ca2+ and L-Phe modulate the activity as well as the cooperativity of CaSR (the color changes of the receptor from white to red indicates an increase in functional activity). Elevating [Ca2+]o, e.g., to 3.0 mM, is proposed to change the basal CaSR status into an active form in a positive homotropic cooperative manner and further trigger [Ca2+]i oscillations. L-Phe binds to the hinge region between lobe 1 and lobe 2, modulating the receptor together with Ca2+ in a positive heterotropic cooperative way. This could potentiate conversion of the receptor to a “fully active” form associated with a higher frequency of [Ca2+]i oscillations and a left-shifted EC50. Loss-of-function CaSR mutants (indicated by white color) could cause a disruption of the cooperativity among the various Ca2+-binding sites (dashed arrows). [Ca2+]o at 3.0 mM does not trigger [Ca2+]i oscillations in the mutant CaSRs. The impaired receptor function and the cross-talk between Ca2+-binding sites can be at least partially rescued for some mutants by L-Phe (e.g.P221Q). However, if the mutation interferes the interaction between CaSR and L-Phe, the function of the receptor may not be fully recovered (e.g.L173P). CaSR gain-of-function mutants (left) exhibit enhanced correlated motions (double line arrows) and their activity is not further potentiated by L-Phe, potentially due to a ceiling effect.
The results from PCA may also provide an explanation as to why L-Phe interacts preferably with the P221Q mutant. L-Phe could not effectively shift the inactive population of conformational ensembles of L173P to an active ensemble of conformations as it did with the other inactivating mutant and WT. However, whether the in silico simulations provide a novel method to predict mutational effects on CaSR will ultimately require the kind of structural detail available for the mGluRs, whose structures have been solved. Here, the model structure does not take into consideration the possible interactions between the ECD and the extracellular loops and/or transmembrane domain as well as the intracellular domains. Thus, caution is merited in over-interpreting the in silico results. Nevertheless, the predicted structural motions may help to identify the mutations or ligands that produce increased or diminished coupling among specific structural elements within the CaSR ECD. While this type of analysis is limited at the moment to the environment of the orthosteric site, this strategy could facilitate drug design for modulating the functions of disease-related mutations affecting the CaSR, as well as other members of the family C GPCRs.
The distinctive combinatorial effects of L-Phe and [Ca2+]o on different mutant CaSRs also suggests that ligand selectivity at the orthosteric site and receptor activity at non-orthosteric sites could both contribute to the function of the receptor. This feature of the adjacent orthosteric binding sites and allosteric regulatory sites that cooperatively regulate the molecular connectivity may be shared by other members of the family C GPCRs [47], [48]. In addition to the Ca2+-sensing receptor, [Ca2+]o regulates 14 of the other members of the family C G protein-coupled receptors (GPCRs), including the metabotropic glutamate receptors (mGluR), γ-aminobutyric acid GABAB receptors and receptors for pheromones, amino acids and sweet substances [1], [49]–[55]. The observed molecular modulatory mechanism may be shared by other members of the family C GPCRs [47], [48]. For instance, the analyses of multiple point mutations in the taste receptors (T1R1/T1R3) suggested that the combination of the two distinct determinants selectivity at the ligand binding site and receptor activity at allosteric sites may mediate the ligand specificity of T1R1/T1R3 [56].
In conclusion, through analysis of [Ca2+]i oscillations and the [Ca2+]o- and L-Phe triggered downstream signaling changes, functional positive cooperativity of CaSR was revealed to be disrupted in the identified FHH-associated CaSR mutations, L173P and P221Q, but not in the ADHH related mutations, L173F and P221L. The addition of L-Phe rescued the function of P221Q by inducing heterotropic positive cooperativity. The distinctive correlated motions of the gain-of-function mutations, on the one hand, and the loss-of-function mutations, on the other, and the potential utility revealed by the molecular dynamic simulation in providing insights into the mechanism for the disease-associated functional alterations in the receptor may also apply to other GPCR proteins.
Supporting Information
Functional studies of the CaSR double mutations in individual HEK293 cells. The panels show representative oscillation patterns from single cells. HEK-293 cells transfected with L173F/P221Q or L173P/P221L were loaded with Fura-2 AM for 15 min. [Ca2+]i was assessed by monitoring emission at 510 nm with excitation alternately at 340 or 380 nm as described in Methods. Each experiment began in Ca2+-free Ringer's buffer (10 mM HEPES, 140 mM NaCl, 5 mM KCl, and 1.0 mM MgCl2, pH 7.4), followed by stepwise increases in [Ca2+]o until [Ca2+]i reached a plateau (up to 30 mM [Ca2+]o). a. Cells were transfected with pEGFP-N1-CaSR L173F/P221Q. b. Cells were transfected with pEGFP-N1-CaSR L173P/P221L.
(DOCX)
Acknowledgments
We thank Jie Feng, Li Zhang and Cassandra Lynn Miller for their critical review and assistance.
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the Supporting Information files.
Funding Statement
Funding was provided by National Institutes of Health (http://www.nih.gov/) grants GM081749 and EB007268 to JJY; National Science Foundation (http://www.nsf.gov/) grant MCB-0953061 to DH; and a Center for Diagnostic Technology - Georgia State University (http://cdt.gsu.edu/) fellowship to CZ. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Brown EM, Gamba G, Riccardi D, Lombardi M, Butters R, et al. (1993) Cloning and characterization of an extracellular Ca2+-sensing receptor from bovine parathyroid. Nature 366:575–580. [DOI] [PubMed] [Google Scholar]
- 2. Mun HC, Franks AH, Culverston EL, Krapcho K, Nemeth EF, et al. (2004) The Venus Fly Trap domain of the extracellular Ca2+ -sensing receptor is required for L-amino acid sensing. J Biol Chem 279:51739–51744. [DOI] [PubMed] [Google Scholar]
- 3. Hofer AM, Brown EM (2003) Extracellular calcium sensing and signalling. Nat Rev Mol Cell Biol 4:530–538. [DOI] [PubMed] [Google Scholar]
- 4. Conigrave AD, Franks AH, Brown EM, Quinn SJ (2002) L-amino acid sensing by the calcium-sensing receptor: a general mechanism for coupling protein and calcium metabolism? Eur J Clin Nutr 56:1072–1080. [DOI] [PubMed] [Google Scholar]
- 5. Brown EM, MacLeod RJ (2001) Extracellular calcium sensing and extracellular calcium signaling. Physiol Rev 81:239–297. [DOI] [PubMed] [Google Scholar]
- 6. Conigrave AD, Brown EM (2006) Taste receptors in the gastrointestinal tract. II. L-amino acid sensing by calcium-sensing receptors: implications for GI physiology. Am J Physiol Gastrointest Liver Physiol 291:G753–761. [DOI] [PubMed] [Google Scholar]
- 7. Conigrave AD, Quinn SJ, Brown EM (2000) Cooperative multi-modal sensing and therapeutic implications of the extracellular Ca2+ sensing receptor. Trends Pharmacol Sci 21:401–407. [DOI] [PubMed] [Google Scholar]
- 8. Pidasheva S, Canaff L, Simonds WF, Marx SJ, Hendy GN (2005) Impaired cotranslational processing of the calcium-sensing receptor due to signal peptide missense mutations in familial hypocalciuric hypercalcemia. Hum Mol Genet 14:1679–1690. [DOI] [PubMed] [Google Scholar]
- 9. Thakker RV (2004) Diseases associated with the extracellular calcium-sensing receptor. Cell Calcium 35:275–282. [DOI] [PubMed] [Google Scholar]
- 10. Hannan FM, Nesbit MA, Christie PT, Lissens W, Van der Schueren B, et al. (2010) A homozygous inactivating calcium-sensing receptor mutation, Pro339Thr, is associated with isolated primary hyperparathyroidism: correlation between location of mutations and severity of hypercalcaemia. Clin Endocrinol (Oxf) 73:715–722. [DOI] [PubMed] [Google Scholar]
- 11. Huang Y, Zhou Y, Castiblanco A, Yang W, Brown EM, et al. (2009) Multiple Ca2+-binding sites in the extracellular domain of the Ca2+-sensing receptor corresponding to cooperative Ca2+ response. Biochemistry 48:388–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang C, Huang Y, Jiang Y, Mulpuri N, Wei L, et al. (2014) Identification of an L-phenylalanine binding site enhancing the cooperative responses of the calcium-sensing receptor to calcium. J Biol Chem 289:5296–5309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hannan FM, Nesbit MA, Zhang C, Cranston T, Curley AJ, et al. (2012) Identification of 70 calcium-sensing receptor mutations in hyper- and hypo-calcaemic patients: evidence for clustering of extracellular domain mutations at calcium-binding sites. Hum Mol Genet 21:2768–2778. [DOI] [PubMed] [Google Scholar]
- 14. Pearce SH, Bai M, Quinn SJ, Kifor O, Brown EM, et al. (1996) Functional characterization of calcium-sensing receptor mutations expressed in human embryonic kidney cells. J Clin Invest 98:1860–1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jiang Y, Huang Y, Wong HC, Zhou Y, Wang X, et al. (2010) Elucidation of a novel extracellular calcium-binding site on metabotropic glutamate receptor 1α (mGluR1α) that controls receptor activation. J Biol Chem 285:33463–33474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Higgins DG (1994) CLUSTAL V: multiple alignment of DNA and protein sequences. Methods Mol Biol 25:307–318. [DOI] [PubMed] [Google Scholar]
- 17. Schwede T, Kopp J, Guex N, Peitsch MC (2003) SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res 31:3381–3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Marti-Renom MA, Stuart AC, Fiser A, Sanchez R, Melo F, et al. (2000) Comparative protein structure modeling of genes and genomes. Annu Rev Biophys Biomol Struct 29:291–325. [DOI] [PubMed] [Google Scholar]
- 19. Huang Y, Zhou Y, Yang W, Butters R, Lee HW, et al. (2007) Identification and dissection of Ca2+-binding sites in the extracellular domain of Ca2+-sensing receptor. J Biol Chem 282:19000–19010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kunishima N, Shimada Y, Tsuji Y, Sato T, Yamamoto M, et al. (2000) Structural basis of glutamate recognition by a dimeric metabotropic glutamate receptor. Nature 407:971–977. [DOI] [PubMed] [Google Scholar]
- 21.Case DA, Darden TA, Cheatham TE, Simmerling CL, Wang J, et al. (2008) AMBER 10.
- 22. Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML (1983) Comparison of simple potential functions for simulating liquid water. J Chem Phys 79:926–935. [Google Scholar]
- 23. Cornell WD, Cieplak P, Christopher IB, Gould IR, Merz JKM, et al. (1995) A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. J Am Chem Soc 117:5179–5197. [Google Scholar]
- 24. Urmi D, Hamelberg D (2009) Reoptimization of the AMBER Force Field Parameters for Peptide Bond (Omega) Torsions Using Accelerated Molecular Dynamics. J Phys Chem 113:16590–16595. [DOI] [PubMed] [Google Scholar]
- 25. Darden T, York D, Pedersen L (1993) Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J Chem Phys 98:10089–10092. [Google Scholar]
- 26. Ryckaert JP, Ciccotti G, Berendsen HJC (1977) Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Phys 23:327–341. [Google Scholar]
- 27. Levy RM, Srinivasan AR, Olson WK, McCammon JA (1984) Quasi-harmonic method for studying very low frequency modes in proteins. Biopolymers 23:1099–1112. [DOI] [PubMed] [Google Scholar]
- 28. Wang M, Yao Y, Kuang D, Hampson DR (2006) Activation of family C G-protein-coupled receptors by the tripeptide glutathione. J Biol Chem 281:8864–8870. [DOI] [PubMed] [Google Scholar]
- 29. Berridge MJ, Potter BV (1990) Inositol trisphosphate analogues induce different oscillatory patterns in Xenopus oocytes. Cell Regul 1:675–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bird GS, Rossier MF, Obie JF, Putney JW Jr. (1993) Sinusoidal oscillations in intracellular calcium requiring negative feedback by protein kinase C. J Biol Chem. 268:8425–8428. [PubMed] [Google Scholar]
- 31. Ridefelt P, Bjorklund E, Akerstrom G, Olsson MJ, Rastad J, et al. (1995) Ca2+-induced Ca2+ oscillations in parathyroid cells. Biochem Biophys Res Commun 215:903–909. [DOI] [PubMed] [Google Scholar]
- 32. Shorte SL, Stafford SJ, Collett VJ, Schofield JG (1995) Simultaneous measurement of [Ca2+]i and secretion-coupled membrane turnover, by single cell fluorescence microscopy. Cell Calcium 18:440–454. [DOI] [PubMed] [Google Scholar]
- 33. Ward DT, McLarnon SJ, Riccardi D (2002) Aminoglycosides increase intracellular calcium levels and ERK activity in proximal tubular OK cells expressing the extracellular calcium-sensing receptor. J Am Soc Nephrol 13:1481–1489. [DOI] [PubMed] [Google Scholar]
- 34. Ridefelt P, Liu YJ, Rastad J, Akerstrom G, Gylfe E (1994) Calcium sensing by human medullary thyroid carcinoma cells. FEBS Lett 337:243–247. [DOI] [PubMed] [Google Scholar]
- 35. Dolmetsch RE, Xu K, Lewis RS (1998) Calcium oscillations increase the efficiency and specificity of gene expression. Nature 392:933–936. [DOI] [PubMed] [Google Scholar]
- 36. Miedlich S, Gama L, Breitwieser GE (2002) Calcium sensing receptor activation by a calcimimetic suggests a link between cooperativity and intracellular calcium oscillations. J Biol Chem 277:49691–49699. [DOI] [PubMed] [Google Scholar]
- 37. Huang Y, Niwa J, Sobue G, Breitwieser GE (2006) Calcium-sensing receptor ubiquitination and degradation mediated by the E3 ubiquitin ligase dorfin. J Biol Chem 281:11610–11617. [DOI] [PubMed] [Google Scholar]
- 38. Young SH, Rozengurt E (2002) Amino acids and Ca2+ stimulate different patterns of Ca2+ oscillations through the Ca2+-sensing receptor. Am J Physiol Cell Physiol 282:C1414–1422. [DOI] [PubMed] [Google Scholar]
- 39. Kifor O, MacLeod RJ, Diaz R, Bai M, Yamaguchi T, et al. (2001) Regulation of MAP kinase by calcium-sensing receptor in bovine parathyroid and CaR-transfected HEK293 cells. Am J Physiol Renal Physiol 280:F291–302. [DOI] [PubMed] [Google Scholar]
- 40. Awata H, Huang C, Handlogten ME, Miller RT (2001) Interaction of the calcium-sensing receptor and filamin, a potential scaffolding protein. J Biol Chem 276:34871–34879. [DOI] [PubMed] [Google Scholar]
- 41. Hjalm G, MacLeod RJ, Kifor O, Chattopadhyay N, Brown EM (2001) Filamin-A binds to the carboxyl-terminal tail of the calcium-sensing receptor, an interaction that participates in CaR-mediated activation of mitogen-activated protein kinase. J Biol Chem 276:34880–34887. [DOI] [PubMed] [Google Scholar]
- 42. Dong Q, Cheng Z, Chang W, Blackman BE, Conte FA, et al. (2010) Naturally-occurring mutation in the calcium-sensing receptor reveals the significance of extracellular domain loop III region for class C G-protein-coupled receptor function. J Clin Endocrinol Metab 95:E245–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Huang Y, Breitwieser GE (2007) Rescue of calcium-sensing receptor mutants by allosteric modulators reveals a conformational checkpoint in receptor biogenesis. J Biol Chem 282:9517–9525. [DOI] [PubMed] [Google Scholar]
- 44. Breitwieser GE (2006) Calcium sensing receptors and calcium oscillations: calcium as a first messenger. Curr Top Dev Biol 73:85–114. [DOI] [PubMed] [Google Scholar]
- 45. Lee HJ, Mun HC, Lewis NC, Crouch MF, Culverston EL, et al. (2007) Allosteric activation of the extracellular Ca2+ -sensing receptor by L-amino acids enhances ERK1/2 phosphorylation. Biochem J 404:141–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhang Z, Qiu W, Quinn SJ, Conigrave AD, Brown EM, et al. (2002) Three adjacent serines in the extracellular domains of the CaR are required for L-amino acid-mediated potentiation of receptor function. J Biol Chem 277:33727–33735. [DOI] [PubMed] [Google Scholar]
- 47. Brauner-Osborne H, Jensen AA, Sheppard PO, O'Hara P, Krogsgaard-Larsen P (1999) The agonist-binding domain of the calcium-sensing receptor is located at the amino-terminal domain. J Biol Chem 274:18382–18386. [DOI] [PubMed] [Google Scholar]
- 48. Hammerland LG, Krapcho KJ, Garrett JE, Alasti N, Hung BC, et al. (1999) Domains determining ligand specificity for Ca2+ receptors. Mol Pharmacol 55:642–648. [PubMed] [Google Scholar]
- 49. Hu J, Spiegel AM (2003) Naturally occurring mutations of the extracellular Ca2+-sensing receptor: implications for its structure and function. Trends Endocrinol Metab 14:282–288. [DOI] [PubMed] [Google Scholar]
- 50. Hinson TK, Damodaran TV, Chen J, Zhang X, Qumsiyeh MB, et al. (1997) Identification of putative transmembrane receptor sequences homologous to the calcium-sensing G-protein-coupled receptor. Genomics 45:279–289. [DOI] [PubMed] [Google Scholar]
- 51. Hofer AM, Brown EM (2003) Extracellular calcium sensing and signalling. Nat Rev Mol Cell Biol 4:530–538. [DOI] [PubMed] [Google Scholar]
- 52. Wise A, Green A, Main MJ, Wilson R, Fraser N, et al. (1999) Calcium sensing properties of the GABA(B) receptor. Neuropharmacology 38:1647–1656. [DOI] [PubMed] [Google Scholar]
- 53. Wellendorph P, Brauner-Osborne H (2009) Molecular basis for amino acid sensing by family C G-protein-coupled receptors. Br J Pharmacol 156:869–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Galvez T, Urwyler S, Prezeau L, Mosbacher J, Joly C, et al. (2000) Ca2+ requirement for high-affinity gamma-aminobutyric acid (GABA) binding at GABA(B) receptors: involvement of serine 269 of the GABA(B)R1 subunit. Mol Pharmacol 57:419–426. [DOI] [PubMed] [Google Scholar]
- 55. Silve C, Petrel C, Leroy C, Bruel H, Mallet E, et al. (2005) Delineating a Ca2+ binding pocket within the venus flytrap module of the human calcium-sensing receptor. J Biol Chem 280:37917–37923. [DOI] [PubMed] [Google Scholar]
- 56. Toda Y, Nakagita T, Hayakawa T, Okada S, Narukawa M, et al. (2013) Two Distinct Determinants of Ligand Specificity in T1R1/T1R3 (the Umami Taste Receptor). J Biol Chem 288:36863–36877. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Functional studies of the CaSR double mutations in individual HEK293 cells. The panels show representative oscillation patterns from single cells. HEK-293 cells transfected with L173F/P221Q or L173P/P221L were loaded with Fura-2 AM for 15 min. [Ca2+]i was assessed by monitoring emission at 510 nm with excitation alternately at 340 or 380 nm as described in Methods. Each experiment began in Ca2+-free Ringer's buffer (10 mM HEPES, 140 mM NaCl, 5 mM KCl, and 1.0 mM MgCl2, pH 7.4), followed by stepwise increases in [Ca2+]o until [Ca2+]i reached a plateau (up to 30 mM [Ca2+]o). a. Cells were transfected with pEGFP-N1-CaSR L173F/P221Q. b. Cells were transfected with pEGFP-N1-CaSR L173P/P221L.
(DOCX)
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the Supporting Information files.