

Abstract
Activating transcription factor 3 (ATF3) is a member of the mammalian activation transcription factor/cAMP, physiologically important in the regulation of pro- and anti-inflammatory target genes. We compared the induction of ATF3 protein as measured by Western blot analysis with single-molecule localization microscopy dSTORM to quantify the dynamics of accumulation of intranuclear ATF3 of triglyceride-rich (TGRL) lipolysis product-treated HAEC (Human Aortic Endothelial Cells). The ATF3 expression rate within the first three hours after treatment with TGRL lipolysis products is about 3500/h. After three hours we detected 33,090 ± 3,491 single-molecule localizations of ATF3. This was accompanied by significant structural changes in the F-actin network of the cells at ~3-fold increased localization precision compared to widefield microscopy after treatment. Additionally, we discovered a cluster size of approximately 384 nanometers of ATF3 molecules. We show for the first time the time course of ATF3 accumulation in the nucleus undergoing lipotoxic injury. Furthermore, we demonstrate ATF3 accumulation associated with increased concentrations of TGRL lipolysis products occurs in large aggregates.
Introduction
In the past two decades a number of far-field superresolution methods have been developed and published 1-5. Using fluorescence microscopy techniques, investigators discovered that dye molecules can exhibit a bright fluorescent state and non-fluorescent dark state1, 2, 6. By exploiting the reversible transition between these states, resolutions below the diffraction limit (~250 nm), postulated by Ernst Abbe, can be achieved. Thus, resolving nano-scaled (a few tens of nanometers) intracellular structures can be achieved.
Many variations of localization and calculation-based techniques are available such as (d)STORM ((direct) Optical Reconstruction Microscopy)2, 7, 8, PALM (Photo-Activated Localization Microscopy)3, 9, and SOFI (Superresolution Optical Fluctuation Imaging)4, 10-14. (d)STORM 15 and PALM 1 detect single-molecules based on approximating the fluctuating PSF (pointspread functions) by 2D Gaussians and the subsequent determination of their center of mass on each frame of a movie of thousands of images taken at millisecond time scale15 (~ 10-50 ms). These coordinates are then used to reconstruct an image with subdiffraction resolution. In the present work, we used these techniques to quantify the number of single-molecule signals or localizations. A single-molecule localization is a calculated set of 2D coordinates extracted from a nonlinear fitting process of a gaussian function to a molecule's PSF after a fixed number of iterations. Since these are approximations, the calculations result in a slight overestimation of fluorescent molecules due to multiple localizations.
Elevation of blood triglyceride-rich lipoproteins (TGRL) is a known atherosclerotic cardiovascular disease risk factor and can induce endothelial dysfunction and inflammation. TGRL are hydrolyzed on the endothelial cell surface by lipoprotein lipase (LpL), and increased concentrations of TGRL lipolysis products stimulate the expression of multiple proinflammatory, procoagulant, and proapoptotic genes in cultured endothelial cells16-18. TGRL lipolysis releases neutral and oxidized fatty acids that induce endothelial cell inflammation19. Also, TGRL lipolysis products alter endothelial cells by increasing very low-density lipoprotein remnant deposition in the artery wall, augmented endothelial monolayer permeability, perturbed zonula occludens-1, polymerization of F-actin, and induction of apoptosis20, 21. Further, TGRL lipolysis products significantly increased the production of reactive oxygen species in endothelial cells and altered lipid raft morphology22. Thus, TGRL lipolysis products in high physiological and pathophysiological concentrations have multiple proinflammatory actions on endothelial cells23. Our studies have shown that TGRL lipolysis products stimulate strong up regulation of activating transcription factor 3 (ATF3), which appears to be a key regulating transcription factor inducing endothelial cell proinflammatory response genes. ATF3 is a member of the cAMP 24, 25 responsive element-binding (CREB) protein family of transcription factors. It contains a ZIP protein region facilitating interactions with related proteins that form a suite of AP-1 sequence binding transcription complexes stimulating a variety of cell and context specific gene responses. We have shown that ATF3 accumulates in the nucleus of TGRL lipolysis product treated endothelial cells in vitro and in vivo and that translational inhibition of ATF3 diminishes lipolysis product induction of pro-inflammatory responses 23 In this paper we rely on the ability of dSTORM 15 to quantitate ATF3 at the single-molecule 26 level. By precisely localizing molecular point spread functions (PSF), we are able to determine an upper boundary of the total number of antibodies attached to transcription factor molecules in the nucleus of the endothelial cell and therefore provide a good measure for the actual number27 of ATF3 molecules present. Additionally, dSTORM provides us with optical resolution below the diffraction limit 28 to enable the investigation of structural changes at the nanoscale and to perform a cluster analysis of ATF3 molecules to describe molecular interactions or foci of accumulation. These experiments cannot be performed directly with conventional biological reference methods and therefore dSTORM may provide a significant improvement in the molecular biology toolbox.
Materials and Methods
Sample preparation
Human TGRL isolation
The protocol for obtaining human TGRL was approved by the Human Subjects Review Committee at the University of California Davis. Postprandial blood samples were obtained 3.5 h after consumption of a moderately high fat meal, which corresponds to the peak elevation in plasma triglyceride concentrations. Triglyceride-rich lipoproteins (TGRL) were isolated from human plasma at a density of less than 1.0063 g/mL following an 18 h centrifugation at 40,000 rpm in a SW41 Ti swinging bucket rotor (Beckman Coulter, Sunnyvale, CA) held at 14°C within a Beckman L8-70M (Beckman) ultracentrifuge. The top fraction (TGRL) was collected and dialyzed in Spectrapor membrane tubing (mol. wt. cut off 3,500; Spectrum Medical Industries, Los Angeles, CA) at 4°C overnight against a saline solution containing 0.01%EDTA. The TGRL lipolysis mix routinely used in experiments was normalized based on triglyceride concentrations and was diluted to contain 150 mg/dl of triglycerides. Total triglyceride content of samples was determined using the serum triglyceride determination kit (Sigma Aldrich, cat # TR0100). The kit converts triglycerides to free fatty acids and glycerol and glycerol is assayed enzymatically.
Reagents and antibodies
Lipoprotein lipase (LpL) (L2254) and monoclonal anti-β-actin antibody (A 5441) were purchased from Sigma, St. Louis, MO. ATF3 (sc-188) antibody was purchased from Santa Cruz Biotechnology, Santa Cruz, CA. F-actin (Alexa Fluor® 488 Phalloidin, A12379) was purchased from Molecular Probes, Eugene, OR.
Cell culture and lipid treatments
Human aortic endothelial cells (HAEC) (passage 6, Cascade Biologics, Portland, OR) were cultured in EBM-2 basal media (cc-3156) supplemented with EGM-2 SingleQuot Kit (cc-4176) (Lonza, Walkersville MD) under an atmosphere of 5% CO2: 95% air at 37°C. Cells were exposed for 1, 2, and 3 h to the following conditions, media or TGRL lipolysis products (TGRL (150 mg/dL = 1.5 mg/mL) + lipoprotein lipase LpL (2 U/mL). The final concentration of TGRL lipolysis products were diluted in media and pre-incubated for 30 minutes at 37°C prior to application.
Western Blotting
To confirm translation of ATF3 mRNA, we performed Western Blot analysis. HAEC were grown to confluence in 6-well plates and then treated as described above for 1, 2, and 3 h. After the incubation, cells were washed twice with PBS, scraped with cold PBS and centrifuged at 3,000 rpm for 10 min at 4°C. Cell pellets were lysed in radioimmune precipitation assay (RIPA) buffer containing 50 mM Tris (pH 7.4), 150 mM NaCl, 1% NP40, 0.25% sodium deoxycholate, 0.1% SDS, 1x Protease inhibitor cocktail set 1 (Calbiochem, La Jolla, CA), 1mM NaF, and 1mM Na3VO4. Protein concentration was determined with the bichinoicacid assay (Pierce), and equal amounts of proteins (60 μg) were separated by NuPAGE® Novex® 4-12% Bis-Trisprotein gels using NuPAGE® MES SDS Running Buffer (Life Technologies, Grand Island, NY). Proteins then were transferred onto 0.2-µm polyvinylidenedifluoride membranes (Bio-Rad,Hercules, CA), which were subsequently blocked with 5% nonfat milk for 1 hand then probed with ATF3 (1:200 dilution) or blotting control mouse monoclonal anti-β-actin (1:5,000) at 4°C overnight. Membranes were then incubated with HRP-conjugated secondary anti-rabbit or anti-mouse antibody (1:10,000). Blots were developed with the enhanced chemiluminescence detection system according to manufacturer's instructions (Amersham).Protein expression levels were determined using a densitometer and Image Quant.
Immunofluorescence analysis
The cellular localization of endogenous, as well as transiently expressed ATF3, was analyzed by fluorescence microscopy. HAEC were grown to confluence on fibronectin-coated 12-mm round coverslips placed in 24-well medical-grade polystyrene plates (BD Falcon) and were treated (n = 3 coverslips per treatment group) as described for 1, 2, and 3 h. After treatment, cells were fixed with 4% paraformaldehyde and washed with PBS. Cells were permeabilized, blocked with Superblock (Pierce, Rockford, IL) containing saponin, incubated with rabbit polyclonal anti-ATF3 (1:100 dilution), and washed with 0.1% SuperBlock to remove unbound material. Subsequently cells were treated with goat anti-rabbit antibody conjugated to Alexa fluor 488; unbound material was removed as above. For F-actin staining, cells were fixed and stained with Alexa Fluor® 488 Phalloidin after treatment with TGRL lipolysis products for 3 h. After cells were mounted, they were evaluated and photographed with the Applied Precision OMX (Ver. 2).
dSTORM measurements
ATF3 quantification
All imaging experiments were performed on the Applied Precision OMX (Optical Microscope Experimental, Applied Precision, Issaquah, WA 98027, Ver. 2) operating in conventional mode (widefield illumination). The imaging settings were set to 532nm excitation at 30ms per frame adjusted to 50 percent laser intensity. All samples were imaged at a resolution of 80nm/px at 512x512 pixels and rendered at a pixel resolution of 30nm with sub-pixel precision using the built-in Fiji plugin quickPALM 29 developed for localization-based superresolution methods. The endothelial cells were mounted on a conventional microscope slide covered with a thin cover slip. Therefore, the number of frames for the movies acquired is limited to 5,000 by the photostability of the fluorescent dye molecules without using any oxygen-scavenging system. Thus, the stochastic single-molecule blinking and the corresponding off-times of the fluorophores are based on triplet-state blinking. As required in all localization-based single-molecule superresolution approaches, the off-times of the fluorescent labels have to be substantially longer than the on-times in order to achieve sufficient separation in time to prevent the PSF from overlapping in space. This can be tuned by the excitation intensity. High laser intensities correlate with a high probability for the fluorophore to populate the triplet state (long-lived dark state) and therefore to switch a fluorescent dye off temporarily. The reconstruction of a single-molecule-based image is done by approximating the detected intensity distributions at given positions with a 2D-Gaussian density function and determining its center of mass for each PSF for the entire image sequence. As a result of this nonlinear fitting operation the coordinates of the molecules then are used to mark the actual position of the molecule on a new image with an increased number of pixels with high accuracy. Detecting single27 ATF3 molecules was accomplished by attaching a polyclonal antibody stain to the targets (see section on sample preparation). To be precise, we counted single-molecule localizations based on the fitted center of mass for each. This number slightly overestimates the actual number of ATF3 molecules depending on the fitting accuracy of the underlying algorithm 8. Each fluorophore attached to the molecule of interest is localized multiple times due to its blinking behavior. This will be partially compensated by the fact that the localization precision of localization-based superresolution techniques are still limited (error on nonlinear fit is larger than the actual distance between two neighboring dyes) and therefore a single localization can be assigned to different nearby fluorophores. Possible background fluorescence signals due to any non-specificity were removed by subtracting the results obtained from the control group (see section on superresolution measurements). F-actin imaging experiments were performed under the exact same conditions as stated above but with 30,000 frames. The reconstruction conditions are maintained.
Cluster analysis of nuclear ATF3 using Ripley's K-Function
Following an unbiased approach by using superresolution localization microscopy, we examined cellular ATF3 dynamics. To investigate the potential interaction of ATF3 molecules in the cell nucleus we performed a cluster analysis of single-molecule localizations30 (Fig. 5 B and E) based on Ripley's K-Function
where γ is the density of points distributed over the observed area. I represents the indicator function with I = 1 if the distance between the i-th and the j-th spot is smaller than a probed distance r, otherwise I is equal to zero. Correcting K(r) for the variance of a Poisson process K(r) is renamed to
In the case of complete spatial randomness (csr) the normalized K-Function
is zero. Briefly, the Ripley's K-Function provides information on the density of localizations as a function (or within) a given distance r. In Fig.5 we evaluate L(r) – r for untreated cells as well as treated ones. Any positive deviation from zero is interpreted as clustering of the molecule of interest. In addition to that there is no ‘cut-off cluster size. Instead, the (normalized) K-Function displays frequently occurring distances between localizations within a region of interest. Negative values account for dispersion. In both samples, treated and media, we found a maximum of the K-Function around 30 nm accounting for unspecific binding of the antibody used for immunostaining (Fig. 5 C and F). In the case of a treated HAEC our analysis reveals three distinct secondary maxima at r = (270 − 22) nm , r = (360 − 40) nm and r = (525 ± 32) nm where major clustering occurs (Figure 4F (inset)). This finding nicely matches the sizes of the clusters shown and labeled with a red dot in the insets (i and ii) of Fig.4E. Inset (ii) of Fig. 5E depicts the blue cross section along one cluster in the digitally magnified image (i). The example aggregate shown in E (ii) can be assigned to the set of clusters predicted by the normalized K-Function at around 360 nm.
Fig. 4.

Quantitative cluster analysis of dSTORM measurements at 30 nanometers per pixel using Ripley's K-function. The control cell shown in A (widefield image) B (dSTORM reconstruction exhibitis a maximum of the normalized K-Function at a cluster size of r=30nm (C) which accounts for non-specific binding of the antibody staining. The dashed-dotted line indicates the K-Function for randomly distributed localizations (C and F). The lipolysis product-treated sample (D) shows significant deviations from a homogeneous distribution of ATF3 in the nucleus (F) at distances larger than 30nm (maximum). At r = 270nm, r = 360nm and r = 525nm, the spatial distribution displays maxima cluster-sizes that visually show up as dense discrete clusters in E (i). Red spots highlight examples of these aggregates. The cross-section indicated by the blue line in E (i) over one arbitrarily chosen cluseter is shown in E (ii). The width was determined to be ~330 nm.
Results and Discussion
ATF3 protein expression by WesternBlot
We compared protein expression of ATF3 by western blotting and immunofluorescence to dSTORM measurements. Western blot analysis showed TGRL lipolysis product treatment induced ATF3 protein expression (Fig 1) in HAEC treated with TGRL + LpL for 1, 2, and 3 h. ATF3 protein express ion was significantly increased at 3h.
Figure 1. ATF3 protein expression after TGRL lipolysis products treatments by Western blotting.
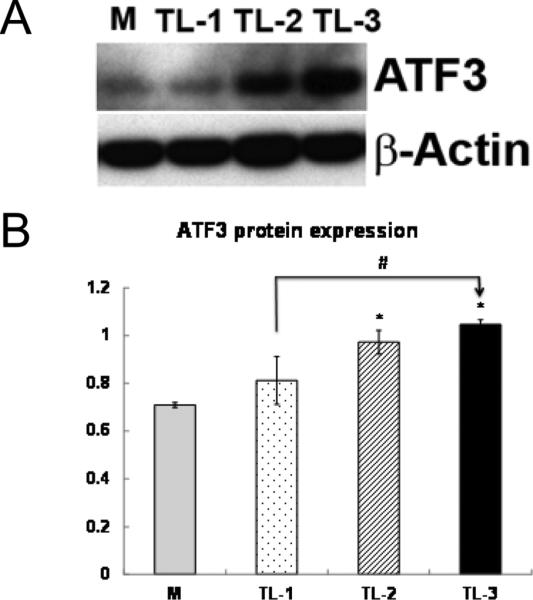
Human aortic endothelial cells were exposed to media (M) or TGRL lipolysis products (TL) for 1 h (TL-1), 2h (TL-2), or 3h (TL-3). Cell lysates were analyzed by Western blotting (A) and (B) densitometry. n=3, * P≤0.05, TL compared with M, #P≤0.05, TL-3 compared with TL-1.
Figure 2 presents the qualitative and quantitative results obtained by single-molecule localization experiments as described in the methods section. A and B show a direct comparison between an untreated cell and one cell treated with lipolysis products for three hours. In A there is no significant fluorescence signal in the nucleus (signal above 30% of maximum intensity) present, indicating only very low concentration of ATF3. A certain amount of false-positive bound antibodies contribute to the signal due to their intrinsic non-specificity. ATF3 molecules are always present in the nucleus and we count 14,027± 1,435 localizations in the nucleus of control cells. A low stress level of growing cell s in culture also can contribute to the expression of the ATF3 mole cules in the nucleus of untreated cells. A reason for this can be found in slight temperature variations and the fixation step itself. Single-molecule signals surrounding the nucleus can be accounted for by non-specific binding of the antibody and baseline levels of ATF3 molecules present in the cytoplasm. The reaction to lipolysis products of a treated cell after three hours is shown in Fig. 2B. ATF3 concentration increased significantly to 33,090±3,491 single-molecules after subtraction of the number of localizations detected in the media control (Fig. 2A). We approximate the up-regulation in the total number of ATF3 molecules to be ~2.5 fold. The e ntire time series suggests a linear increase of the number of localizations of antibody-labeled ATF3 molecules up to three hours. Assuming a linear expression of ATF3 with time for the first three hours, we extracted an expression rate of in the nucleus as Fig. 2C depicts. In Fig. 1C The linear approximation for the first three hours has its y-intercept at about 14,474 ± 350 molecules, which nicely matches the measured number of molecules fo r an untreated cell. Therefore, this parameter works as a good approximation for the media control. We estimated the relative deviation from the measurement to be roughly 3%. This demonstrates that the increase in ATF3 expression follows a linear trend up to three hours. Although the number of labelled epitopes found in the nucleus slightly overestimates the actual number of ATF3 molecules the extraction of an expression rate can be easily justified. As biological reference methods (western blot) also nicely show a linear increase we can be sure of the trend. Additionally, an overestimated number of molecules only affects the position (y-intercept) of the linear function but not its slope.
Figure 2.

Immunostained ATF3 in HAEC cells. A shows the comparison between a conventional widefield image and the dSTORM reconstruction using quickPALM of an untreated cell in media. B. Represents a side-by-side view of the single-molecule reconstruction and the conventional fluorescence image of a cell treated with lipolysis products for three hours. C. Depicts a time series of treatments from one to three hours. The number of labelled epitopes is plotted as a function of incubation time. Error bars represent standard errors at a sample size of five cells. The blue dashed line is a linear approximation serving as a guide for the eye. Red dotted lines represent the standard error interval around the number of localizations detected in the nucleus of the media cell shown in A.
(FWHM=Full Width Half Maximum) obtained from A along the yellow cross sections. Zoom-in squares located below A and B magnify highlighted areas to point-out structural changes in the F-actin network.
A direct comparison of F-actin networks around the cell nucleus of a lipolysis product-treated cell and a corresponding media control is shown in Fig. 3. Our previous study showed that TGRL lipolysis products alter the actin cytoskeleton to form stress fibers and activate the apoptotic cascade21. We applied fluorescence superresolution to resolve the structural response of HAEC cells to lipotoxic stress after a three-hour incubation with li polysis products. Fig. A and B compares an unperturbed network of F-actin filaments of HAEC cells in conventional fluorescence microscopy with superresolved as well as contrast-enhanced imaging. We observe long linear structures in the media-treated c ase (Fig 3A) as expected. Upon treatment with lipolysis produc ts the cells exhibit interrupted filaments as shown in Fig. 3B. Only relatively small filaments of (363.2 ± 31.3) nm length are present. The quantified resolution in terms of the FWHM (Full Width at Half Maximum) can be extracted from C corresponding to the yellow cross-sections depicted in A. We show a ~3-fold increase in localization precision compared to conventional widefield fluoresc ence microscopy. The split-view (Fig. 3A and B) directly show that fine structures can be resolved with great detail with subdiffracti on resolution. Subsequently after treatment with lipolysis product s the cells react with structural changes in their F-actin network, which cannot directly be seen in diffraction-limited microscopy. As the molecular PSF largely overlap (Fig.4), no obvious difference from media-only cells is evident. On increasing the level o f detail with dSTORM, there is a clear change in structure to fragmented filaments forming clumps of F-actin. (Fig 3B).
Figure 3.

A and B show a side-by-side comparison of a conventional fluorescence image and a dSTORM reconstruction of immunostained F-actin filaments in HAEC cells. A. Depicts the control case, whereas B monitors a treated cell. C represents a direct quantification of the determined resolution (localization precision)
cluster analysis of ATF3
In both samples, treated and media, we found a maximum of the K-Function around 30 nm accounting for unspecific binding of the antibody used for immunostaining (Fig. 5 C and F). In the case of a treated HAEC our analysis reveal s three distinct secondary maxima at r = (270 ± 22) nm, r = (360 ± 40) nm and r = (525 ± 32) nm where major clustering occurs (Figure 4F (inset)). This finding nicely matches the sizes of the clusters shown and labeled with a red dot in the i nsets (i and ii) of Fig.4E. Inset (ii) of Fig. 5E depicts the blue cross section along one cluster in the digitally magnified image (i). The example aggregate shown in E (ii) can be assigned to the set of clusters predicted by the normalized K-Function at around 360 nm.
As the clusters found in treated cells show sizes that are larger than the diffraction limit, theoretically they should already be visible in the widefield image. A closer look at Fig. 5D (blue arrows) reveals large white spots corresponding to the clusters determined. Of course in a diffraction-limited image the spots appear blurred and cannot be differentiated very well from the rest of the labelled area. A few bright clusters are highlighted by blue arrows.
Although the untreated cells show a certain amount of localizations due to false-positive binding of the antibodies and a small number of ATF3 molecules expressed, no larger aggregates could be identified (Fig. 5C). for randomly distributed localizations (C and F). The lipolysis product-treated sample (D) shows significant deviations from a homogeneous distribution of ATF3 in the nucleus (F) at distances larger than 30nm (maximum). At r = 270nm, r = 360nm and r = 525nm, the spatial distribution displays maxima cluster-sizes that visually show up as dense discrete clusters in E (i). Red spots highlight examples of these aggre gates. The cross-section indicated by the blue line in E (i) over one arbitrarily chosen cluster is shown in E (ii). The width was determined to be ~330 nm.
Summary and conclusions
In our experiments we were able to demonstrate how HAEC respond to treatment with TGRL lipolysis product-induced lipotoxic injury at the nanoscale. We observed a linear increase of lipolysis product-induced nuclear ATF3 expression for three hours at a rate of increase of ~3,500 single-molecule localizations per hour at the single-molecule level using dSTORM as a localization-based microscopy method. After three hours of incubation with TGRL lipolysis products, we detected over 30,000 immunostained ATF3 localizations (labelled epitopes) in the nucleus. Additionally, we were able to confirm our single-molecule experiments with results of standard western blotting techniques. We also used single-molecule identification to better characterize the previously described remodeling of the actin cytoskele ton in stress fiber formation. We demonstrate remodeling of the pe rinuclear f-actin network towards formation of short fragments and clumps of actin after treatment with TGRL lipolysis products. We determined the F-actin fragments to be (363.2 ± 31.3) nm in length on average with the improved resolution afforded by dSTORM images compared to conventional widefield microscopy. By using subdiffraction single-molecule localization microscopy, we also were able to perform a quantitative cluster analysis. We identified three distinct cluster sizes of ATF3 molecules ranging from 270 nm to 525 nm inside the nucleus of a lipid-treated cell as a reaction to lipotoxicity. Therefore on average we observe small and dense clusters of about 384 nm in diameter. For the first time, we succeeded in visualizing clusters of ATF3 molecules enabling us to further investigate new pathways induced by ATF3 multimers. The biologi c consequences of these clusters remain to be determined. The y may either represent sites of transcription complexes at promoter regions of DNA or alternatively aggregates of transcription factors sequestered by ATF3 splice variants 31.
Here we demonstrate dSTORM microscopy as a technique maturing from a proof-of-principle technique to a usable tool to effectively and sensitively study processes that are not yet fully understood. We conclude that fluorescence superresolution microscopy has the ability to extend previous cell bio logic reveal and quantify at least three molecular mechanisms ass ociated with treatment of TGRL lipolysis products: generates much larger nuclear ATF3 aggregates than has previously been reported and these macroaggregates initiate the lipotoxic response.
Supplementary Material
{kind=link}
Acknowledgements
We would like to thank Prof. Dr. Thomas Huser, Department of Physics, University of Bielefeld, Germany for proof reading the manuscript and technical advice.
Sources of Funding
This work was supported by the NIH-HL55667, NIH-NIA AG039094, Richard A. and Nora Eccles Harrison Chair Award (to J.C.R.). The work done by Dr. Idir Yahiatene was supported by the Mouse Metabolic Phenotyping Centers (MMPC)-U24DK092993.
References
- 1.Betzig E PG, Sougrat R, Lindwasser OW, Olenych S, Bonifacino JS, Davidson MW, Lippincott-Schwartz J, Hess HF. Science. 2006;313:1642–1645. doi: 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- 2.Rust MBMJ, Zhuang X. Nature Methods. 2006;3:793–795. doi: 10.1038/nmeth929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shroff H WH, Betzig E. Curr Protoc Cell Biol. 2013 doi: 10.1002/0471143030.cb0421s58. Chapter 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dertinger T CR, Iyer G, Weiss S, Enderlein J. Proc Natl Acad Sci U S A. 2009;106:22287–22292. doi: 10.1073/pnas.0907866106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Loew LM HS. Biophys J. 2013;104:741–743. doi: 10.1016/j.bpj.2013.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vicidomini G SA, Ta H, Han KY, Moneron G, Eggeling C, Hell SW. PLoS One. 2013;8:e54421. doi: 10.1371/journal.pone.0054421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Olivier N KD, Rajan VS, Gönczy P, Manley S. Biomed Opt Express. 2013;4:885–899. doi: 10.1364/BOE.4.000885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wolter S LA, Holm T, Aufmkolk S, Dabauvalle MC, van de Linde S, Sauer M. Nature Methods. 2012;9:1040–1041. doi: 10.1038/nmeth.2224. [DOI] [PubMed] [Google Scholar]
- 9.Matsuda A SL, Boulanger J, Kervrann C, Carlton PM, Kner P, Agard D, Sedat JW. PLoS One. 2010;5:e12768. doi: 10.1371/journal.pone.0012768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dedecker P MG, Dertinger T, Zhang J. Proc Natl Acad Sci U S A. 2012;109:10909–10914. doi: 10.1073/pnas.1204917109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dertinger T CR, Vogel R, Heilemann M, Sauer M, Enderlein J, Weiss S. Adv Exp Med Biol. 2012;733:17–21. doi: 10.1007/978-94-007-2555-3_2. [DOI] [PubMed] [Google Scholar]
- 12.Dertinger T HM, Vogel R, Sauer M, Weiss S. Angew Chem Int Ed Engl. 2010;49:9441–9443. doi: 10.1002/anie.201004138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dertinger T CR, Vogel R, Enderlein J, Weiss S. Opt Express. 2010;18:18875–18885. doi: 10.1364/OE.18.018875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Antelman J EY, Dertinger T, Michalet X, Weiss S. J Phys Chem C Nanomater Interfaces. 2009;113:11541–11545. doi: 10.1021/jp811078e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van de Linde S LA, Klein T, Heidbreder M, Wolter S, Heilemann M, Sauer M. Nature Protocols. 2011;6:991–1009. doi: 10.1038/nprot.2011.336. [DOI] [PubMed] [Google Scholar]
- 16.Norata GD, Grigore L, Raselli S, Seccomandi PM, Hamsten A, Maggi FM, Eriksson P, Catapano AL. Journal of molecular and cellular cardiology. 2006;40:484–494. doi: 10.1016/j.yjmcc.2006.01.022. [DOI] [PubMed] [Google Scholar]
- 17.Norata GD, Grigore L, Raselli S, Redaelli L, Hamsten A, Maggi F, Eriksson P, Catapano AL. Atherosclerosis. 2007;193:321–327. doi: 10.1016/j.atherosclerosis.2006.09.015. [DOI] [PubMed] [Google Scholar]
- 18.Ting HJ, Stice JP, Schaff UY, Hui DY, Rutledge JC, Knowlton AA, Passerini AG, Simon SI. Circulation research. 2007;100:381–390. doi: 10.1161/01.RES.0000258023.76515.a3. [DOI] [PubMed] [Google Scholar]
- 19.Wang L, Gill R, Pedersen TL, Higgins LJ, Newman JW, Rutledge JC. Journal of lipid research. 2009;50:204–213. doi: 10.1194/jlr.M700505-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rutledge JC, Mullick AE, Gardner G, Goldberg IJ. Circulation research. 2000;86:768–773. doi: 10.1161/01.res.86.7.768. [DOI] [PubMed] [Google Scholar]
- 21.Eiselein L, Wilson DW, Lame MW, Rutledge JC. American journal of physiology. Heart and circulatory physiology. 2007;292:H2745–2753. doi: 10.1152/ajpheart.00686.2006. [DOI] [PubMed] [Google Scholar]
- 22.Wang L, Sapuri-Butti AR, Aung HH, Parikh AN, Rutledge JC. American journal of physiology. Heart and circulatory physiology. 2008;295:H237–244. doi: 10.1152/ajpheart.01366.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aung HH, Lame MW, Gohil K, An CI, Wilson DW, Rutledge JC. Arteriosclerosis, thrombosis, and vascular biology. 2013 doi: 10.1161/ATVBAHA.113.301375. DOI: 10.1161/ATVBAHA.113.301375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smith MA, Harris PL, Sayre LM, Perry G. Proc Natl Acad Sci U S A. 1997;94:9866–9868. doi: 10.1073/pnas.94.18.9866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bessesen DH, Richards CL, Etienne J, Goers JW, Eckel RH. Journal of lipid research. 1993;34:229–238. [PubMed] [Google Scholar]
- 26.Yahiatene I. Analytical chemistry. 2012;84:2729–2736. doi: 10.1021/ac2030283. [DOI] [PubMed] [Google Scholar]
- 27.Sauer M. Journal of Cell Science. 2013;126:3505–3513. doi: 10.1242/jcs.123612. [DOI] [PubMed] [Google Scholar]
- 28.Yahiatene Idir, Hennig Simon, Huser Thomas. Chemical Physics Letters. 2013;587:1–6. [Google Scholar]
- 29.Henriques R, Lelek M, Fornasiero EF, et al. Nature Methods. 2010:7. doi: 10.1038/nmeth0510-339. [DOI] [PubMed] [Google Scholar]
- 30.Sebastian M. Malkusch, Barbara Müller, Hans-Georg Krausslich, Mike Heilemann. Histochem Cell Biol. 2013:139. [Google Scholar]
- 31.Thompson MR, Xu D, Williams BRG. Journal of Molecular Medicine. 2009;87:1053–1060. doi: 10.1007/s00109-009-0520-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.