

A dimeric structure of the sodium–proton antiporter NhaA provides insight into the roles of Asp163 and Lys300 in the transport mechanism.
Abstract
Sodium–proton antiporters rapidly exchange protons and sodium ions across the membrane to regulate intracellular pH, cell volume, and sodium concentration. How ion binding and release is coupled to the conformational changes associated with transport is not clear. Here, we report a crystal form of the prototypical sodium–proton antiporter NhaA from Escherichia coli in which the protein is seen as a dimer. In this new structure, we observe a salt bridge between an essential aspartic acid (Asp163) and a conserved lysine (Lys300). An equivalent salt bridge is present in the homologous transporter NapA, but not in the only other known crystal structure of NhaA, which provides the foundation of most existing structural models of electrogenic sodium–proton antiport. Molecular dynamics simulations show that the stability of the salt bridge is weakened by sodium ions binding to Asp164 and the neighboring Asp163. This suggests that the transport mechanism involves Asp163 switching between forming a salt bridge with Lys300 and interacting with the sodium ion. pKa calculations suggest that Asp163 is highly unlikely to be protonated when involved in the salt bridge. As it has been previously suggested that Asp163 is one of the two residues through which proton transport occurs, these results have clear implications to the current mechanistic models of sodium–proton antiport in NhaA.
INTRODUCTION
Electrogenic sodium–proton antiporters use the proton gradient to drive sodium ions out of the cell, usually under conditions of salt stress at alkaline pH (Padan et al., 2004). NhaA from Escherichia coli is the prototypical electrogenic sodium–proton antiporter, with two protons transported for every sodium ion (Taglicht et al., 1993). For a transporter, NhaA is extremely fast, exchanging up to 1,500 ions/s (Taglicht et al., 1991). Its activity is regulated by pH: it is inactive at acidic pH, partially active between pH 6.0 and 7, and fully active at pH 8.0 (Padan et al., 2004). The crystal structure of NhaA was determined at a resolution of 3.45 Å in an inward-facing conformation at low pH where the protein is inactive (Hunte et al., 2005). NhaA is made up of two five-transmembrane (TM) topology-inverted repeats that intertwine to form two distinct structural domains: a core (translocation) domain and a dimer (interface) domain (Hunte et al., 2005; Padan, 2008; Fig. 1). In the crystal structure, a deep cytoplasmic-facing cavity, containing many negatively charged residues, is located between the core and dimer domains. The core domain is characterized by two discontinuous helices that cross over at the center of the protein (Fig. 1). The two well-conserved aspartates Asp163 and Asp164 are located near the crossover region. Sodium ion binding to Asp163 and Asp164 at high pH is believed to elicit a switch of the transporter from the inward- to outward-facing conformation, although the details of the mechanism are unclear (Padan, 2008; Appel et al., 2009; Maes et al., 2012; Kozachkov and Padan, 2013).
Figure 1.

Schematic diagram of the NhaA structure. The core and dimer domains are illustrated with blue and beige shadowing, respectively. TM 4 (red) and TM 11 (yellow) are discontinuous and cross over in the center of the protein. Asp133 and Lys300 have been proposed to neutralize the positively and negatively charged helix dipoles of the discontinuous helices. Asp163 and Asp164 are thought to interact with the sodium ion. Coordinates are from Protein Data Bank accession number 1ZCD (Hunte et al., 2005).
Recently, the structure of another sodium–proton antiporter (NapA) was solved at high pH in the outward-facing conformation (Lee et al., 2013). In this new structure, the equivalent residue to Asp164 (Asp157 in NapA) was found to be accessible from a large cavity to the outside, rather than the inside as seen in NhaA. Furthermore, in the NapA structure, the equivalent residue to Asp163 in NhaA interacts with a well-conserved lysine (Lys305 in NapA, Lys300 in NhaA), which is thought to be important in charge neutralization and pH activation (Hunte et al., 2005; Maes et al., 2012). Here, we describe a new crystal form of NhaA that, like the previous NhaA structure, is in an inward-facing conformation at low pH, but shows the protein packing as a dimer, which is the physiological oligomeric state for sodium–proton antiporters in general (Fafournoux et al., 1994; Hilger et al., 2005; Goswami et al., 2011; Lee et al., 2013). We show that the salt-bridge interaction observed in the outward-facing structure of NapA is also evident between Asp163 and Lys300 residues in the new inward-facing NhaA structure. To probe this salt-bridge interaction, further molecular dynamics (MD) simulations were performed in a model membrane bilayer, systematically changing the protonation states of charged residues in the ion-binding site.
MATERIALS AND METHODS
Expression, purification, and stabilization of NhaA
NhaA-GFP-His8 was obtained from the previously constructed membrane protein GFP fusion library (Daley et al., 2005). The NhaA-GFP-His8 fusion protein was overexpressed and purified as described previously (Sonoda et al., 2011). To confirm the sequence assignment of TM 10, Leu296 was mutated to methionine using the QuickChange II XL Site-Directed Mutagenesis kit (Agilent Technologies) in a variant of NhaA containing two stabilizing point mutations (A109T and Q277G) as shown below. The A109T, Q277G construct is referred to as the “double mutant,” and A109T, Q277G, L296M is referred to as the “triple mutant.” Expression of the mutants was performed using MemStar (Lee et al., 2014). In brief, the E. coli strain pLemo(DE3) was cultured in the auto-induction medium for selenomethionine labeling (PASM-5052) but also included an IPTG induction step at mid-log phase. Cultures were harvested after o/n growth at 25°C and were supplemented with kanamycin at 50 µg/ml throughout (Studier, 2005; Wagner et al., 2008).
Preparation of stable mutant of NhaA.
The L296M mutation was introduced into a variant of NhaA that had been stabilized by the introduction of two mutations. These stabilizing point mutants were identified by screening ∼100 random mutations of NhaA. Mutations were generated by error-prone PCR using the GeneMorph II Random Mutagenesis kit (Agilent Technologies). NhaA mutants were selected based on expression levels (>1 mg/L based on GFP fluorescence) and those that had >50% extraction efficiency in 2% wt/vol octyl-β-d-thioylglucoside (OG) from total membranes, at a concentration of 3 mg/ml, after 1-h incubation at 4°C. This procedure was performed because we observed a correlation between the stability of membrane proteins, as judged by their unfolding rate in LDAO at 40°C (Sonoda et al., 2011), and their solubilization efficiency in OG.
Functional characterization of the thermostabilized NhaA mutant.
NhaA wild-type and the thermostabilized mutant were co-reconstituted with purified ATP synthase from E. coli with an ∼2:1 molar ratio (NhaA/ATP synthase) in MME buffer (10 mM MOPS-NaOH, pH 8.5, 2.5 mM MgCl2, and 100 mM KCl) as described previously (Lee et al., 2013). Typically, 50 µl proteoliposomes were diluted into 1.5 ml MME buffer containing 3 nM 9-aino-6-chloro-2-methoxyacridine (ACMA) and 140 nM valinomycin. Fluorescence was monitored at 480 nm using an excitation wavelength of 410 nm in a fluorescence spectrophotometer (Cary Eclipse; Agilent Technologies). An outward-directed pH gradient (acidic inside) was established by the addition of 2 mM ATP, as followed by a change in ACMA fluorescence. After an ∼2-min equilibration, the activity of NhaA wild-type and thermostabilized mutant was assessed by the dequenching of ACMA fluorescence after the addition of the indicated concentrations of NaCl or LiCl. The addition of 20 mM NH4Cl leads to near complete dequenching. By measuring ACMA dequenching by the addition of 10 mM NaCl or LiCl at pH 6.5, 7.0, 7.5, 8.0, 8.5, and 9.0, respectively, the effect of pH to NhaA wild-type and thermostabilized mutant activity was assessed. Each experiment was performed in triplicate.
Crystallization
Crystals of wild-type NhaA and the triple mutant were grown by mixing equal volumes of a protein solution containing 8 mg/ml of pure protein in 20 mM sodium citrate, pH 3.5, 150 mM NaCl, 0.03% high α-n-dodecyl-β-d-malotoside (α-DDM), and 1% heptyl-thiol-β-d-glucoside with reservoir solution containing 0.1 M sodium citrate, pH 3.5, 0.1 M LiS04, and 26% PEG 400. For the triple mutant, 1% Facade-EM (Avanti Polar Lipids, Inc.) was included as an additive. The crystals were dehydrated by transferring the cover slides sequentially to wells with 2% increments of PEG 400 to a final concentration of 32%. Crystals were soaked with 1 µl of reservoir solution containing 1% high α-DDM and 40% PEG 400 and flash frozen in liquid nitrogen before data collection.
Data collection, processing, and refinement
Wild-type NhaA.
Data were collected at Diamond Light Source beamline I24 and were processed and scaled using HKL2000 (Otwinowski and Minor, 1997). Molecular replacement was performed using Phaser (McCoy et al., 2007) as part of the CCP4 package (Collaborative Computational Project, Number 4, 1994), with the previously published monomeric structure as the search model (Protein Data Bank accession no. 1ZCD; Hunte et al., 2005). Inspection of the solution showed two dimers to be present in the asymmetric unit. Refinement was performed against data extending to 3.7 Å using the PHENIX package (Adams et al., 2010) starting from a model in which the β hairpin (Pro45 to Asn58) and other surface loops were omitted. The maps were improved by averaging the four molecules of the asymmetric unit using the RAVE package (Kleywegt et al., 2001) with B-factor sharpening as described by DeLaBarre and Brunger (2006). Rebuilding was performed in O (Jones and Kjeldgaard, 1997). The structure was refined with one B factor for each residue TLS (Winn et al., 2001), grouped into chains, secondary structure restraints, and noncrystallographic symmetry (NCS) restraints between the four molecules of the asymmetric unit. During refinement, the register of TM 10 was moved by one turn of the helix. This improved the fit of the helix to the density and the geometry of the residues in this region. To confirm the assignment, Leu296 on TM 10 was changed to a methionine and data were collected from selenomethionine derivatized crystals (see next section). As this dataset was of higher resolution than the wild type, the model was refined first against these data (see below) before being refined against the wild-type data in a final round of refinement with dihedral restraints to the higher resolution model.
NhaA triple mutant.
Data were collected on beamline I03 at Diamond Light Source. They were processed with XDS (Kabsch, 2010) through the xia2 pipeline (Winter, 2010). Refinement was performed using the PHENIX package (Adams et al., 2010) as described above. Refinement was performed against data extending to a resolution of 3.5 Å as for the wild type, except that F’ and F” were given for the selenium atoms and the refinement was performed against the anomalous pairs. In the last rounds of refinement, NCS restraints were only applied over the two dimers. This resulted in slightly better R and R-free.
Coordinates and structure factors have been deposited in the Protein Data Bank under accession number 4AU5 for the wild-type structure and 4ATV for the triple mutant.
MD simulations
MD simulations of the NhaA dimer and a monomer in a 1-palmitoyl-2-oleoylphosphatidylcholine (POPC) bilayer were performed with the Gromacs 4.5.3 or 4.6.1 simulation packages (Hess, 2008). All simulations used the OPLS-AA force field (Rizzo and Jorgensen, 1999; Kaminski et al., 2001; Jensen and Jorgensen, 2006) with the TIP4P water model (Jorgensen et al., 1983) and OPLS-UA parameters for POPC lipids (Ulmschneider and Ulmschneider, 2009; provided by M. Ulmschneider and available from the Lipidbook force field repository; see Domański et al., 2010). A POPC bilayer was studied as a generic model of a native-like membrane environment instead of other lipid compositions, as the POPC lipid parameters have been shown to perform well in very long (≥1-µs) simulations (Ulmschneider et al., 2010, 2011). POPC is a reasonable choice as a model membrane because its membrane thickness is comparable to that of the POPE-rich membrane (Rappolt et al., 2003; Murzyn et al., 2005; Kučerka et al., 2006) of E. coli (Raetz, 1986; Cronan, 2003). We used a multi-scale approach to embed the dimer into the membrane (Stansfeld and Sansom, 2011). The protein was first simulated in a coarse-grained representation, and the membrane was allowed to self-assemble around the protein from a random mixture of lipids and water in the simulation box (Scott et al., 2008). In this way, lipids could accumulate in the dimer interface in an unbiased fashion, only driven by the thermodynamics of the partitioning of the random mixture into a water phase and a lipid/membrane phase. After a 200-ns simulation with an integration time step of 20 fs, the bilayer had assembled around either the NhaA dimer or a monomer (based on chain A of the crystal structure). In the second step of the multi-scale approach, the systems were converted to the OPLS-AA atomistic representation with the CG2AT protocol (Stansfeld and Sansom, 2011), and the original crystal structure was inserted in place of the back-translated protein. The dimer simulation system consisted of an orthorhombic simulation box of size of 121 × 121 × 93 Å containing 112,700 atoms in 748 protein residues, 354 lipids, 49 Na+ and 55 Cl− ions, and 27,546 water molecules. The monomer system measured 75 × 75 × 90 Å and contained 52,332 atoms (374 protein residues, 135 POPC lipids, 17 Na+ and 20 Cl− ions, and 9,875 water molecules). The approximate free NaCl concentration was 100 mM in all simulations.
Equilibrium MD simulations were performed with varying protonation states of Asp133, Asp163, Asp164, and Lys300 (see Table 1). The choice of protonation state for other residues was guided by PROPKA (Li et al., 2005) based on their pKa, at pH 7. Asp133 is believed to stabilize the helix dipoles of TMs 4a and 11a, but such a charge–dipole interaction is not encoded in the empirical rules of the PROPKA algorithm (Li et al., 2005). Therefore, we disregarded the predicted value of 7.1 and adopted a charged Asp133, but also performed simulations with Asp133 in its neutral form. Glu82 and Glu252 had predicted pKa values >8, but in the structure and in simulations, they face the water-filled entrance funnel so the charged default states were selected instead of the protonated forms.
Table 1.
MD simulations
| Simulation namea | Assembly | Charge state | Starting structureb | Run length | |||
| D133 | D163 | D164 | K300 | µs | |||
| S1/1 | dimer | — | — | 0 | + | xtal* | 1.1 |
| S1/2 | S1/1@0.1 µs | 0.2 | |||||
| S1/3 | S1/1@0.1 µs | 0.2 | |||||
| S2/1 | dimer | — | — | — | + | xtal* | 1.0 |
| S2/2 | xtal | 1.0 | |||||
| S2/3 | xtal | 1.0 | |||||
| S2/1.1–1.5 | monomer | S2/1 | 5 × 0.1 | ||||
| S2/2.1–-2.2 | 4AU5* | 2 × 0.1 | |||||
| S3/1 | dimer | — | 0 | 0 | + | S2/1* | 0.1 |
| S3/2 | S2/1 | 1.0 | |||||
| S3/3 | xtal* | 0.1 | |||||
| S4/1 | dimer | — | — | — | 0 | S2/1* | 1.0 |
| S4/2 | S4/1@0.1 µs | 1.0 | |||||
| S4/3 | S4/1@0.1 µs | 0.2 | |||||
| S5/1 | dimer | 0 | — | 0 | + | xtal* | 1.1 |
| S5/2 | S5/1@0.1 µs | 0.2 | |||||
| S5/3 | S5/1@0.1 µs | 0.2 | |||||
| S6/1 | dimer | 0 | — | — | + | S2/1* | 0.1 |
| S6/2 | xtal* | 0.1 | |||||
| S7 | dimer | 0 | 0 | 0 | + | xtal* | 0.1 |
| S8 | dimer | 0 | — | — | 0 | S2/1* | 0.1 |
Asterisks denote simulations that were preceded by energy minimization and a 3-ns position restraint MD; simulations without an asterisk were repeats starting from the same initial system conformation as the starred one but with varied initial velocity distribution. The dimer simulation contained 112,700 atoms and the monomer contained 52,332. All simulations were performed with Gromacs 4.6.1 except simulations S2/1, S2/1.1–1.5, and S2/2.1–2.2, which were run in Gromacs 4.5.3. Judging from the RMSD and secondary structure analysis (not depicted), the simulations behaved in the same manner regardless of software version. In total, 10.5 µs of MD simulations was performed. xtal, crystal structure of wild-type NhaA, which was deposited in the Protein Data Bank under accession number 4AU5 after minor refinements; 4AU5, wild-type NhaA as deposited in the Protein Data Bank; S1/1, last frame of simulation S1/1, etc., or frame at 0.1 µs (“@0.1µs”).
Simulations are identified by the protonation states and resulting charge states of Asp133 (D133), Asp163 (D163), Asp164 (D164), and Lys300 (K300), using identifiers S1–S8. Repeat simulations are indicated with a serial number after the identifier.
Starting structure denotes the source for the initial input structure for the simulation.
MD simulations were performed with periodic boundary conditions at constant temperature T = 310 K and pressure P = 1 bar using the velocity-rescaling algorithm for the thermostat (time constant of 0.1 ps) (Bussi et al., 2007) and semi-isotropic weak coupling for the barostat (time constant of 1.0 ps; compressibility of 4.6 × 10−5 bar−1; Berendsen et al., 1984). Long-range corrections for energy and pressure were applied (Hess, 2008). Lennard-Jones interactions were cut off at 10 Å, whereas electrostatic interactions were handled by the smooth particle mesh Ewald (SPME) method (Essmann et al., 1995) that computes Coulomb interactions in real space up to a cutoff of 10 Å and long-range interactions beyond the cutoff in reciprocal space with fast Fourier transforms on a grid with spacing 1.2 Å and fourth-order splines for fitting of the charge density. Bonds to hydrogen atoms were constrained with the P-LINCS algorithm (Hess, 2008) or SETTLE (for water molecules; Miyamoto and Kollman, 1992). The grid-based neighbor list was updated every five steps. The classical equations of motions were integrated with a leapfrog integrator and a time step of 2 fs. Conformations were saved every 1 ps for analysis. The simulation protocol included an initial energy minimization of the atomistic system and a 3-ns equilibrium simulation during which the protein heavy atoms were restrained with a harmonic force constant of 1,000 kJ mol−1 nm−2. Simulations were performed as detailed in Table 1 with at least three repeats of each main simulation. To change protonation states of residues, the Gromacs tool pdb2gmx was used (Hess, 2008), which also rebuilds hydrogens as needed. When a simulation was continued from a previous simulation, another energy minimization and 3-ns equilibrium MD with positional restraints on the protein heavy atoms were performed after changing protonation states. These simulations are marked with an asterisk in Table 1. Repeat simulations started from the last frame of a position restrained simulation were run with different seeds for the random number generator so that the differing initial velocity distributions and the stochastic component of the thermostat (Bussi et al., 2007) would generate independent trajectories. In total, we simulated the dimer system for >10 µs.
Analysis and estimation of pKa values
MD trajectories were analyzed with MDAnalysis (Michaud-Agrawal et al., 2011) and Gromacs tools (Hess, 2008). The distance d of the Asp163–Lys300 salt bridge was calculated as the minimum distance between the two carboxyl Oδ1 and Oδ2 atoms with the amine Nζ at each time step; similarly, ion–carboxyl group distances were also calculated as the minimum distance to the two oxygen atoms. After the simulations, the pKa values of titratable residues were estimated with PROPKA 3.1 (Søndergaard et al., 2011) using snapshots of the MD simulations that were sampled every 1 ns. Na+ ions within 6 Å of the protein were taken into account because preliminary calculations showed that the presence of ions close to titratable residues can shift the pKa by up to 2 units. Data were typically split according to (a) the selected protonation states in the simulation and (b) the state of the Asp163–Lys300 salt bridge. The salt bridge was considered formed if d < 4 Å and broken if d ≥ 4 Å. This value is consistent with work by Kumar and Nussinov (1999, 2002) who considered a salt bridge to exist with d < 4 Å between any N–O pair.
The pKa time series data from the individual protomers A and B in each simulation of the dimer and from repeats of individual simulations were aggregated. Distributions of pKa values were modeled with a Gaussian kernel density estimator whereby the kernel width was chosen according to Scott’s criterion (Scott, 1992) as implemented in the scipy.stats.gaussian_kde() function from the SciPy package. Distributions were plotted as violin plots (Hintze and Nelson, 1998) as implemented in the Seaborn package (Waskom, 2014). To assess the effect of the salt bridge on the pKa values of neighboring residues, the distribution f(ΔpKa) of the difference between pKa from frames with a broken salt bridge minus pKa from frames with the intact salt bridge was computed: , where the distributions and of the pKa for the intact and broken salt bridge were computed from the data as Gaussian kernel density estimates as detailed above. The ΔpKa shifts aggregated over all simulations were used to compare the influence on the calculations of the inclusion of Na+ ions, the distance defining whether the salt bridge was formed or broken and the effect of changing the surface dielectric constant εsurface.
Figures were prepared with PyMOL (Schrödinger, Inc.), CCP4mg (Potterton et al., 2004), and VMD (Humphrey et al., 1996), using the Bendix plugin for smoothly bent helices (Dahl et al., 2012). Superpositions were performed in Lsqman (Kleywegt and Jones, 1994), such that all matching Cα pairs were <3.8 Å apart after superposition.
Online supplemental material
The supplemental Discussion gives a more in-depth analysis of the pKa estimations. Table S1 shows the solubilization efficiency of the various NhaA constructs that were screened in searching for a more stable construct. Table S2 shows the root-mean-square deviation (RMSD) in positions of the Cα atoms among the structures. Fig. S1 shows an additional diagram illustrating the difference in the position of Lys300 in this structure relative to the previously published structure (Protein Data Bank accession no. 1ZCD) and provides a stereo diagram of the electron density in this region, complementing Fig. 2. Fig. S2 presents further analysis of the behavior of the protein and lipids during simulation S2/1. Figs. S3–S7 show the analyses of the repeated simulations in different protonation states in a similar fashion to the summary presented in Fig. 7. Fig. S8 shows the variation in the distance between Asp163 and Lys300 plotted over all simulations. The pKa values of Asp163, Asp164, Lys300, and Asp133 are also plotted as a function of time as each of the simulations progressed. Fig. S9 shows a distribution of pKa values for these four residues when the salt bridge is considered intact and when broken as shown in Fig. 8, but each set of simulations is plotted separately. Fig. S10 shows the shift in pKa of these residues when the salt bridge breaks. Videos 1 and 2 show the first 250 ns of the MD simulation S2/1 for protomers A and B, respectively. Online supplemental material is available at http://www.jgp.org/cgi/content/full/jgp.201411219/DC1.
Figure 2.

Electron density. (A) Electron density of TM 10. The 2mFo-DFc map shown in blue (contoured at 1.5 σ) was calculated with phases derived from the model before reassigning the sequence of TM 10 (shown) and averaged over the four molecules of the asymmetric unit. The anomalous difference map shown in red has been calculated from the selenomethionine-derivatized triple mutant and contoured at 3.6 σ. (B) The same maps as in A but with the structure of the wild-type protein (refined before the data of the triple mutant were collected). (C) Stereo view of the superposition of the final refined structure of the triple mutant (green) on that of the published monomeric structure (gray) (Hunte et al., 2005). The anomalous difference map is shown as in A. Met296 is in cyan, and Leu296 from the published structure is in wine red. Overall, there is good correspondence in the position of the helices and methionines between the two structures, in agreement with the similar activity of the mutant to wild type (Fig. 4). An enlarged view of TM 10 is shown in Fig. S1.
Figure 7.

Sodium ion binding and salt-bridge stability in MD simulations with different protonation states of Asp163, Asp164, and Lys300. (Left column; A and B) Asp163 deprotonated and Asp164 and Lys300 protonated (simulation S1/1, protomer B). (Middle column; C and D) Asp163 and Asp164 deprotonated and Lys300 protonated (simulation S2/1, protomer B). (Right column; E and F) Asp163, Asp164, and Lys300 deprotonated (simulation S4/1, protomer B). (Top row; A, C, and E) Distances between the closest sodium ion and Asp163 or Asp164 are plotted as a function of time. Spontaneous Na+ binding to Asp164 was observed when both aspartates were deprotonated. (C) Continuation of the simulation with Lys300 deprotonated (a 3-ns equilibration simulation with position restraints on all heavy protein atoms is symbolized by dashed lines between panels) leads to a rapid change in the Na+-binding mode toward closer interaction with Asp163. (Bottom row; B, D, and F) Distance of the closest Asp163 carboxyl group from the N-amino group of Lys300. Distances <4 Å are indicative of a stable salt-bridge interaction (yellow shaded area), whereas those ≥4 Å are considered a weak or broken salt bridge. Binding of Na+ to Asp164 destabilizes the salt bridge. (D) Lines show data averaged over blocks of 10 ns, with fluctuations in the data indicated as shaded regions encompassing the lower 5 and upper 95% percentile. The snapshots show the Na+ binding event with subsequent rupture of the salt bridge (cytoplasmic view along the axis of helix TM 5). Videos 1 and 2 show this simulation. Repeat simulations (see Table 1) are shown in Figs. S3–S7.
Figure 8.
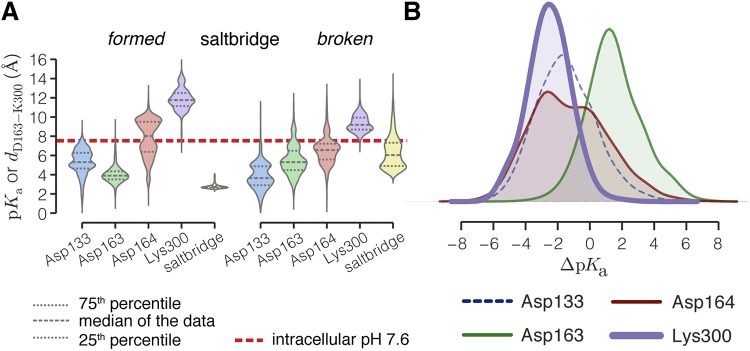
Effect of breaking the Asp163–Lys300 salt bridge on the pKa of conserved residues. (A) Distributions of pKa values estimated with PROPKA 3.1 (Søndergaard et al., 2011) from all MD simulations shown as violin plots. pKa values were calculated from snapshots extracted at 1-ns intervals, with sodium ions included within 6 Å of the protein. For reference, a dotted line has been drawn at pH 7.6. The percentiles of the data are shown as broken lines inside the distributions, which were computed as Gaussian kernel density estimates (see Materials and methods). Data were split depending on the state of the salt bridge (formed if the distance is <4 Å, and broken if it is ≥4 Å; the salt-bridge distance distribution is also shown in yellow). The multimodal distribution of Asp164 when the salt bridge is formed is a result of data from simulations S1 (Fig. S9 A) during which its solvent accessibility is much smaller than during other simulations (see also Fig. S3, M–O). (B) Distributions of pKa shifts (ΔpKa) when the salt bridge is broken (data aggregated over all MD simulations). The pKa of Lys300 is downshifted by −2.5 ± 1.4, and thus the charged form is destabilized. Shifts of the other residues are not significantly different from 0 (also see Table 3).
RESULTS
Structure determination of the NhaA dimer
Crystals of wild-type NhaA were obtained at low pH using protein prepared as a GFP fusion construct (Drew et al., 2001, 2006; Sonoda et al., 2011). These crystals contain two dimers in the asymmetric unit. The structure was solved by molecular replacement and refined at 3.7-Å resolution with noncrystallographic restraints between the four molecules of the asymmetric unit (Table 2). In the previous crystal structure of NhaA, TM helix 10 (TM 10), which contains the conserved Lys300, was reported to be difficult to build into the electron density (Screpanti et al., 2006). During refinement of the NhaA dimer, we observed that the fit could be improved by adjusting the sequence assignment for TM 10 so that it starts at residue 287 and extends to 313, rather than from residue 290 to 316, as was modeled previously (Hunte et al., 2005; Figs. 2 and S1).
Table 2.
Data collection and refinement statistics
| Crystal | Wild type | Triple mutant |
| Wavelength (Å) | 0.9778 | 0.9793 |
| Space group | P21 | P21 |
| Resolution (Å) | 29.6-3.69 (3.80-3.70)a | 56.5-3.5 (3.54-3.50)a |
| Cell dimensions | a = 115.8 Å; b = 100.6 Å; c = 141.6 Å; β = 97.0° | a = 115.8 Å; b = 99.4 Å; c = 140.2 Å; β = 97.4° |
| Number of measured reflections | 117,235 | 313,049 |
| Number of unique reflections | 34,273 | 37,951 |
| Completeness (%) | 98.1 (90.0) | 94.6 (69.5) |
| Redundancy | 3.4 (3.0) | 8.2 (7.2) |
| I/σ(I) | 11.7 (1.2) | 22.0 (1.4) |
| Rmerge (%) | 9.8 (82.0) | 4.4 (111.4) |
| R factor (%) | 31.8 | 28.7 |
| R-freeb (%) | 34.2 | 31.3 |
| RMSD from ideal values | ||
| Bond lengths (Å) | 0.010 | 0.004 |
| Bond angles (°) | 1.28 | 1.48 |
| Ramachandran plot outliersc (%) | 0.7 | 0.7 |
Values in parentheses refer to data in the highest resolution shell.
Based on 5% of the reflections
From MolProbity (Chen et al., 2010).
Verification of sequence assignment in a thermostabilized form of NhaA
After repositioning TM 10, Lys300 is now within hydrogen-bonding distance to Asp163 (Fig. 3). To verify the modified sequence assignment of TM 10, Leu296 was substituted to methionine and selenomethionine derivatized protein produced to obtain anomalous difference maps that would show the exact position of the introduced methionine. However, initial attempts at obtaining well-diffracting selenomethionine derivatized crystals were unsuccessful. To improve crystal quality, a thermostabilized NhaA mutant construct was used instead (Fig. 4). The NhaA mutant was generated while developing methodology that can rapidly improve the stabilization of membrane proteins by mutagenesis, in particular, when no high affinity ligands are available. In brief, an error-prone PCR library was generated and NhaA mutants were screened for those that could be extracted with >50% efficiency in 2% OG (see Materials and methods and Table S1), a benchmark based on our observation that many of the most detergent-stable membrane proteins (Sonoda et al., 2011) had >50% extraction efficiency in 2% OG. By incorporating the Leu296Met substitution into an NhaA thermostabilized mutant (Ala109Thr, Gln277Gly), data were obtained extending to a resolution of 3.5 Å after minimal optimization of the crystals (Table 2). Importantly, the thermostable mutant has the same pH-dependent profile and similar transport activity to the wild-type protein (Fig. 4). Anomalous difference maps for the mutant showed peaks in the same position as the methionines in the monomeric wild-type structure (Hunte et al., 2005), consistent with the two proteins having similar conformations. In addition, there was another peak exactly at the position of the introduced methionine for the Leu296Met mutation in the dimer structure (Fig. 2 C). Thus, we were able to confirm that the reassignment of TM 10 was correct.
Figure 3.

Position of Lys300 on TM 10. (A) Cartoon representation of the structure viewed from the periplasmic side of the membrane. The charged residues, Asp133 (TM 4b), Asp163 (TM 5) and Asp164 (TM 5), and Lys300 (TM 10), in the new structure are shown as colored sticks. The position of Lys300 in the previously published structure (Protein Data Bank accession no. 1ZCD) is shown in gray (red text). The structure has been colored from red at the N terminus through to blue at the C terminus. Loop regions except for the breaks between the discontinuous helices have been omitted for clarity. (B) As A, but from the side and zoomed in. The dashed line represents the salt bridge between Asp163 and Lys300.
Figure 4.

Stabilization, characterization, and crystallization of the NhaA mutant. (A) The NhaA double mutant (A109T and Q277G) and NhaA triple mutant (A109T, Q277G, L296M) are more stable in detergent as shown by the longer unfolding half-life (t1/2) in LDAO at 40°C. (B) The ATP synthase and NhaA wild-type and mutants were co-reconstituted in liposomes. ATP-driven proton pumping establishes a ΔpH (acidic inside) as monitored by the quenching of 9-amino-6-chloro-2-fluorescence (ACMA). Proton efflux is initiated by the addition of increasing concentrations of NaCl/LiCl, and apparent ion-binding affinities for NhaA wild type (closed circle) and mutant (open circle) at pH 8.5 were calculated: KMNa+ wild type (mean ± SD): 1.8 ± 0.2; KMNa+ mutant: 1.6 ± 0.1; KMLi+ wild type: 4.1 ± 0.5; KMLi+ mutant: 4.1 ± 0.3. (C) pH dependence of NhaA Na+(Li+)–H+ antiporter activity for wild type (closed circle) and mutant (open circle) were measured in proteoliposomes by the level of ACMA dequenching as in B at the indicated pH values after the addition of saturating NaCl/LiCl at pH 8.5; all experiments were repeated in triplicate and representative traces are shown.
The NhaA mutant structure was refined to an R factor of 28.7% and a corresponding R-free of 31.2% (Table 2). In both the wild-type and mutant NhaA structures, the associated maps are of reasonable quality (Figs. 2, 5, and S1), especially after averaging of the four molecules of the asymmetric unit, although TM 4a, which is thought to move upon binding sodium ions (Appel et al., 2009), was difficult to model. Additional electron density at the dimer interface could not be assigned to protein residues. We have tentatively modeled this by sulfate ions and the detergent α-DDM. At the resolution of the data, there are no obvious differences to the wild-type structure, which was subsequently refined to an R factor of 31.8% and an R-free of 34.2%, with restraints to the higher resolution mutant structure (Table 2).
Figure 5.

Position of β hairpins in the NhaA dimer. (A) 2mFo-DFc electron density averaged over the four molecules of the asymmetric unit (contoured at 1.5 σ). The map was calculated directly after molecular replacement using a search model where the β hairpins and loops had been omitted. (B) Cartoon representation of the NhaA dimer viewed from the periplasmic-facing side of the membrane, with the two protomers shown in light brown and green, respectively. (C) The 14–amino acid β hairpins from each promoter, shown in light brown and green, form a four-stranded antiparallel β sheet, as viewed from the cytoplasmic side of the membrane. The residues facing the membrane are hydrophobic except for Lys57. (D) Comparison of the positions of the β hairpins in each of the determined NhaA structures: crystal structure of the dimer (this work; light brown and green), monomeric crystal structure (Hunte et al., 2005; pale green), and dimer from cryo-EM (Appel et al., 2009; yellow).
The crystal structure of the NhaA dimer
The two dimers of the asymmetric unit are almost identical and superimpose with an RMSD of 0.3 Å (Table S2). The density is well defined for the β sheet, although some of the side chains on the periplasmic side are less ordered (Fig. 5 A). The overall structure of each subunit in the dimer is very similar to the published monomeric structure with an RMSD of 1 Å for 358 out of 374 Cα atoms (Fig. 5 and Table S2). The largest conformational difference lies in the position of the 14 amino acid β hairpins linking TMs 1 and 2, which protrude out along the membrane plane and form the predominant dimer interaction. Relative to the monomeric crystal structure, the hairpins twist by ∼15° to form a flat four-stranded antiparallel β sheet with residues 45–51 involved in hydrogen-bonding interactions between the strands (Fig. 5 C).
The arrangement of the subunits in the dimer is very similar to the cryo–electron microscopy (EM) model (Protein Data Bank accession no. 3FI1; Williams, 2000; Appel et al., 2009), which, in turn, is consistent with electron spin resonance (Hilger et al., 2005) and cross-linking distance measurements (Herz et al., 2009). The exact conformation of the β strands is, however, different. In the structure derived from the cryo-EM data, the β sheet is much more curved, with the tips of the hairpins ∼11 Å above the membrane surface (Appel et al., 2009), placing them parallel, but 7 Å above the β sheet that we observe in the dimeric crystal structure (Fig. 5 D). In this bent position, there are fewer hydrogen-bonding interactions between strands than in the dimer crystal structure.
The interfacial TM preceding the β hairpins and the alternating position of charged and noncharged residues of the four-stranded antiparallel β sheet creates a dimer interface that is amphipathic, with polar residues pointing toward the periplasm and hydrophobic residues toward the protein or the dimer interface (Fig. 5 C). Lys57, at the C terminus of the β hairpin, is the only residue that breaks this trend by pointing into the protein where it makes a salt bridge with Asp65. The Lys57–Asp65 pairing has been proposed to be important, as replacement of either Lys57 or Asp65 by cysteine results in a protein with an apparent Km for Na+ that is four- or 10-fold higher than wild type, respectively (Herz et al., 2009, 2010). These residues are 9 Å apart in the model derived from cryo-EM maps.
Other than the interaction between the β hairpins, there are few direct contacts between monomers. Trp258 of TM 9 makes a bridge to TM 7, and Arg204 of TM 7 may interact with the amide oxygen of Val254. It is likely that lipids would fill the space between the protomers in vivo, and in fact, in the crystal structure a detergent molecule has been modeled in extra density at this position.
MD simulations of the NhaA dimer
To gain further insight into the position of the NhaA dimer in the membrane, the dimer was simulated in a POPC model membrane for 1 µs. As described in Materials and methods, a multi-scale approach (Scott et al., 2008; Stansfeld and Sansom, 2011) was used that provides sufficient sampling of lipid–protein interactions in the absence of any knowledge of the location of the protein in the membrane. The Cα RMSD of each monomer relative to the crystal structure increased to ∼3.4 Å over the time course of a 1-µs simulation (Fig. S2 A). The RMSD of the TM helices alone stabilized around 2.8–3.0 Å, whereas mobile loops contributed substantially to the observed differences from the crystal structure (Fig. S2, B and C). Secondary structure elements such as the β sheet were retained over the whole simulation, although a few regions are less structured: in particular, TM 4a, which was difficult to build in the crystal structure, and to a lesser degree 4b; TM 5 near Asp163 and Asp164 (usually when sodium binding was also observed); and TM 10 near Lys300. NhaA, simulated either as a dimer or as a monomer, sits entirely within the membrane, including the β hairpins (Fig. 6). It does not extend beyond the lipid head groups, as shown by a density profile along the membrane normal (Fig. S2 D). The periplasmic face of the β sheet was fully exposed to solvent and confined the periplasmic interfacial lipids.
Figure 6.

MD simulation of the NhaA dimer in a model membrane bilayer. (A) The NhaA dimer is stable in a POPC membrane. Snapshots show the structure after an ∼1-µs MD (simulation S2/1). The periplasmic space is at the top. Protomers are colored wheat and green, and POPC lipids are white. A bound sodium ion is shown as a cyan sphere. Residues Asp163, Asp164, and Lys300 are partially visible in a stick representation. (B) View from the periplasm.
MD simulations of sodium binding with different protonation states of the key residues
The roles of the critical charged residues (Asp164, Asp163, Lys300, and Asp133) have been investigated previously by a combination of biochemical and biophysical experiments and MD simulations (Inoue et al., 1995; Galili et al., 2002; Hunte et al., 2005; Arkin et al., 2007; Kozachkov et al., 2007; Olkhova et al., 2009; Maes et al., 2012). As the outcome of the MD simulations are likely to be affected by the proximity of Lys300 to Asp163 (distance of 2.5 Å in this structure compared with 12 Å in the published structure), we sought to use our new structure in MD simulations to investigate (a) how the protonation states of these four critical residues affect sodium ion binding, and (b) how the protonation state and presence of sodium ions affect the salt-bridge interaction. MD simulations were performed in the presence of sodium ions at a NaCl concentration of ∼100 mM with the residues in different protonation states (Table 1).
First, we examined the most likely situation in the low pH crystal structure, with Asp164 protonated (neutral) and Asp163 deprotonated (negatively charged) because it interacts with the positively charged Lys300 (see Materials and methods). In simulations with these protonation states, no sodium ion binding was observed after 1 µs of sampling (Fig. 7 A). The Asp163–Lys300 salt bridge remained intact with an average distance of ∼2.4 Å (Fig. 7 B). Similar results were obtained in two separate repeat simulations of 100 ns (Fig. S3).
Next, we modeled Asp164 in a deprotonated state, as would be expected at high pH where the protein is active. Three independent simulations of 1 µs of the dimer and seven shorter simulations of 100 ns of just the monomer were performed (Figs. 7, S4, and S5). In all but one protomer of the dimer simulations and one of the monomer repeat simulations, sodium ions were seen to spontaneously enter the vestibule and bind to Asp164. In two cases, Asp163 switched from forming a salt-bridge interaction with Lys300 to interacting with the sodium ion. On the time scales simulated here, the salt bridge between Asp163 and Lys300 typically persisted during the whole length of the simulation, as also observed for the simulations with Asp164 protonated, unless it was destabilized by the presence of a sodium ion (Figs. 7, S4, and S5). With both Asp163 and Asp164 modeled in their protonated (neutral) form, no sodium ion binding was observed during 1 µs of MD and two additional 100-ns simulations (Fig. S6).
We next investigated the effect of changing Asp133 and Lys300 to their neutral states. In the above simulations, Asp133 was deprotonated. When it was protonated (neutral), no spontaneous sodium binding was observed (in one 1.1-µs and two 100-ns simulations; not depicted), indicating that perhaps Asp133 is important to capture ions. Lys300 is unlikely to be deprotonated when involved in a salt-bridge interaction. However, it is possible that disruption of the salt bridge by sodium ion binding enables deprotonation of the lysine (see next section). The simulations also suggest that Lys300 is water accessible, as shown in Figs. S4 and S6. To investigate the consequences of a neutral Lys300 in this situation, the endpoint of the simulation shown in Fig. 7 (C and D) with a bound sodium ion and the salt bridge broken was continued with Lys300 modeled as deprotonated (Fig. 7, E and F). The sodium ion transferred from being bound by Asp164 to binding to Asp163 while the distance between Asp163 and Lys300 increased further, a behavior reproduced in two repeat simulations (Fig. S7).
Estimations of pKa for the charged residues
To obtain estimates of the pKa of the residues putatively involved in sodium ion and proton binding, we used the program PROPKA 3.1 (Søndergaard et al., 2011). Fig. S8 C shows the pKa of the residues in the structures along the time course of the simulation S2/1. While the salt bridge remains intact, the pKa of the four residues remains fairly constant. Upon breaking, however, the pKa of Lys300 drops markedly and that of Asp163 increases. As the heuristic methods used in PROPKA can be quite sensitive to small changes in the environment of the residues, we decided to study the distributions of pKa values of the residues from structures from all of the simulations separated into those where the salt bridge was intact and those where it had broken (Figs. 8 and S9). The largest effect is seen for Lys300, where the pKa of Lys300 is lowered by approximately two to three pH units to pH ∼8.5 (Figs. 8, S9, and S10, and Table 3). A detailed assessment of the effect of breaking the salt bridge together with a sensitivity analysis of the pKa calculations are given in the supplemental Discussion.
Table 3.
Estimated pKa shifts due to breaking of the salt-bridge Lys300–Asp163
| Simulationa | Asp133 | Asp163 | Asp164 | Lys300 |
| S1 | −0.4 ± 0.7 | 0.8 ± 0.7 | −0.1 ± 0.7 | −0.5 ± 0.8 |
| S2 | −1.5 ± 1.8 | 0.6 ± 1.3 | −0.4 ± 1.6 | −1.9 ± 1.6 |
| S4 | −1.0 ± 1.1 | 1.9 ± 1.2 | 0.2 ± 1.4 | −2.1 ± 1.2 |
| S3 | −1.4 ± 0.8 | 3.0 ± 1.5 | 0.6 ± 1.6 | −3.7 ± 1.4 |
| S5 | −0.1 ± 0.7 | 0.6 ± 0.9 | 0.5 ± 0.7 | −1.1 ± 0.8 |
| S6 + S8 | 0.4 ± 2.1 | 2.1 ± 2.1 | −0.0 ± 1.6 | −2.8 ± 1.5 |
| S7 | −0.1 ± 0.6 | 0.3 ± 0.9 | 1.3 ± 1.2 | −1.2 ± 0.8 |
| All data combinedb | −1.5 ± 1.9 | 1.5 ± 1.8 | −1.3 ± 2.4 | −2.5 ± 1.4 |
| All data combined, Na+ excluded from calculationc | −1.3 ± 1.5 | 2.6 ± 1.9 | −1.0 ± 2.1 | −2.4 ± 1.4 |
| All data combined, salt-bridge cutoff 3.5 Åd | −1.4 ± 1.9 | 1.4 ± 1.8 | −1.3 ± 2.4 | −2.5 ± 1.5 |
| All data combined, εsurface = 80e | −1.4 ± 2.0 | 1.5 ± 1.9 | −1.0 ± 2.5 | −2.5 ± 1.5 |
For simulation names, see Table 1. Data were aggregated for all repeats and protomers A and B. Protein conformations were sampled every 1 ns. Na+ ions were included within 6 Å of the protein, and the salt bridge was considered broken if the distance was >4 Å (except where noted).
Distributions of pKa and ΔpKa were recomputed from all trajectory frames of all simulations (i.e., simulated with differing protonation states), assuming that each frame was an independent sample of the protein conformation. This can lead to distributions that are broader than those for individual simulation sets with means that are not just a simple average of individual means.
Na+ ions were not included in the PROPKA 3.1 calculation.
Salt bridge was considered broken if the distance was ≥3.5 Å.
The dielectric constant near the protein surface (corresponding to the solvent-exposed residues) is 160 in the standard PROPKA 3.1 parametrization. A value of 80 was tested, as suggested for calculations on NMR ensembles (Søndergaard et al., 2011).
DISCUSSION
The crystal structure of the NhaA dimer is consistent with the 7-Å cryo-EM structure derived from 2-D crystals grown under more native-like conditions, although a difference in the position of the β sheet is observed. In the crystal structure, the amphipathic β sheet is flat and sits in the plane of the membrane, with one surface exposed to solvent and the other shielding lipids (Figs. 5 C and 6). In this position, unlike in the structure from cryo-EM, Lys57 and Asp65 are close enough to form a salt bridge, an interaction that is reported to be physiologically significant (Herz et al., 2010). Biochemical data indicate that the monomer is the functional unit of the protein, with dimerization probably enhancing its stability in the membrane (Rimon et al., 2007; Mager et al., 2013). In agreement with this, we do not observe significant differences within the TM region of the dimeric structure compared with the previous monomeric structure. The amphipathic β sheet may help to anchor the dimer domain as the core domain undergoes large conformational changes during the transition between outward- and inward-facing states, as suggested by the recent structure of NapA (Lee et al., 2013).
TM 10 was reportedly difficult to build in determining the original crystal structure of NhaA (Screpanti et al., 2006). Based on the density observed in the wild-type dimer structure, we revised the sequence assignment of this helix and subsequently confirmed its reassignment using anomalous scattering of a selenomethionine residue that replaced the naturally occurring Leu296. This process appears to have been greatly facilitated by the use of a thermostabilized construct of NhaA. In the modified structure, Lys300 is displaced 10 Å from its previously modeled position to lie within hydrogen-bonding distance of Asp163, similar to the interaction reported for the active pH NapA structure (Lee et al., 2013; Fig. 9). Although a water-mediated interaction between Lys300 and Asp163 was predicted for NhaA at high pH (Olkhova et al., 2009), a direct interaction observed already at low pH means we must reconsider the most likely steps of the transport cycle.
Figure 9.

Comparison of NhaA with NapA and ASBTNM. (A) Structural comparison of NhaA and NapA. Cartoon representation of TMs 4, 5, 10, and 11 of NhaA superimposed on the same helices of NapA (Lee et al., 2013). Residue numbering is shown for NhaA. K300 in NhaA corresponds to K305 in NapA, and D163 in NhaA corresponds to D156 in NapA. The helices of the TMs in NhaA have been colored as in Fig. 3, and NapA is colored salmon. (B) Structural comparison of NhaA and ASBTNM. Cartoon representation of TMs 4, 10, and 11 of NhaA superimposed on TMs 4, 8, and 9 of ASBTNM (Hu et al., 2011). The helices of the TMs in NhaA have been colored as in A, and ASBTNM is colored gray. The sodium Na2 site in ASBTNM is depicted as a purple sphere, and Lys300 residue in NhaA is shown as a stick model.
In MD simulations, sodium ions bind spontaneously to both Asp164 and Asp163, consistent with recent biochemical and biophysical experiments suggesting that both of these residues are likely to coordinate sodium ion binding (Maes et al., 2012). During our simulations, the stability of the salt bridge is clearly affected by the interaction of the sodium ions with the aspartate residues. Asp163 either maintains its interaction with Lys300 or moves toward the positively charged sodium ion, thereby breaking the salt bridge. This suggests a competition between the lysine and sodium ion to bind to Asp163. The TM-embedded Lys300 is important for both activity and pH regulation (Kozachkov et al., 2007). Of the various mutations that have been made, replacement with arginine and histidine, where the positive charge is maintained, causes the least perturbation (Maes et al., 2012). Nevertheless, even these mutations show a marked drop in activity and a substantial decrease in the affinity (KM) for sodium ions (100- and 30-fold for arginine and histidine, respectively), and lysine at this position is clearly preferred.
NhaA transports two protons into the cytoplasm for every sodium ion exported. There has been some discussion in the literature about which residues are likely to form the proton-binding sites. The presence of the Asp163–Lys300 salt bridge necessarily affects this debate. Asp164 is strictly conserved from bacteria to mammals and seems very likely to bind one of the protons (Mager et al., 2011; Maes et al., 2012; Lee et al., 2013). The second proton has been suggested to bind to either Asp133 or Asp163 (Arkin et al., 2007; Padan et al., 2009). Of the two, Asp163 seems the more likely: it is essential for activity (Inoue et al., 1995; Maes et al., 2012), conserved in the electrogenic transporters, and replaced by asparagine in the electroneutral antiporters (Landau et al., 2007; Schushan et al., 2010). Asp133 is not essential for activity (Galili et al., 2002; Maes et al., 2012), is generally less well conserved, and is retained in some electroneutral sodium–proton exchangers (Landau et al., 2007). However, the estimation of pKa by PROPKA 3.1 (Figs. 8 and S9) suggests that Asp163 is unlikely to be protonated when involved in a salt bridge to Lys300. As a salt bridge is observed in both the inward-facing NhaA structure and the outward-facing structure of the homologous NapA, it seems that if Asp163 is involved in the translocation of the proton, this will only be transiently. Another candidate for proton transport, which has not been considered up to now, is the lysine. Lysines have been proposed to be involved in proton translocation in other membrane transporters (Shaffer et al., 2009; Efremov and Sazanov, 2011). Without putting too much emphasis on the absolute values calculated by PROPKA, the analysis of the pKa of Lys300 would suggest that it would be protonated when the salt bridge was present but is more likely to lose its proton when disrupted (Figs. 8 and S10, and Table 3; see also supplemental Discussion). In the electroneutral transporters, the lysine is replaced by an arginine (Landau et al., 2007; Schushan et al., 2010); the pKa of arginines is known to be much less altered by the environment than that of a lysine (Harms et al., 2011; Isom et al., 2011). In our simulations, the protonation state of Lys300 clearly affects the interaction of the sodium ion with the protein, whereby a deprotonated Lys300 facilitates association of the two conserved aspartates with the ion. It is also noteworthy that the amino group of the repositioned lysine is located at the same position as one of the sodium ions in the sodium-dependent bile acid symporter ASBTNM, a structural homologue of NhaA and NapA (Hu et al., 2011; Fig. 9 B). A similar comparison was used to support the role of a lysine in proton transfer in another family of transporters (Shaffer et al., 2009). Given the delicate positioning of charged residues at this site, however, detailed biochemical and structural information needs to be gathered before any firm conclusions can be drawn.
Combining this new structural information on NhaA with the recent structure of NapA allows us to postulate a mechanism whereby the interaction between Asp163 and Lys300, seen in both inward- and outward-facing states, must be taken into consideration. At low pH, as seen in the crystal structure of NhaA, Asp164 is likely to be protonated and Asp163 is involved in a salt-bridging interaction with Lys300 (Fig. 10, 1). As the pH becomes more alkaline, sodium ions will successfully compete with protons for binding to Asp164, in line with a recent electrophysiology study showing that the pH dependence of NhaA can be explained by the competition of sodium ions and protons to bind to the same site (Fig. 10, 2) (Mager et al., 2011). At this point, as demonstrated by the MD simulations, Asp163 can switch from interacting with Lys300 to contribute to binding the sodium ion. The substrate-bound form of the transporter would then switch to the outward-facing conformation where the sodium ion can be released and the salt bridge can reform, as seen in the structure of NapA (Lee et al., 2013; Fig. 10; 3 and 4). In the diagram, we also show the possible deprotonation and protonation of Lys300, as discussed above. Upon proton binding to Asp164, the transporter would then switch back to the inward-facing conformation.
Figure 10.

Proposed schematic model of NhaA transport. The transport mechanism occurs through a reorientation of the protein, alternately exposing a cavity containing Asp164 to the intracellular or the periplasmic space. The core domain is depicted as a blue square, and the panel domain is in beige. (1) The probable situation in the solved NhaA structure at low pH where the protein will be protonated. One of the protons (in orange) is likely to be bound to Asp164, and we hypothesize that the second proton (orange hashed) will interact with Lys300. Sodium ions (blue) will compete with protons for binding to Asp164, causing the Asp163–Lys300 salt bridge to break and possibly Lys300 to lose a proton (2). The sodium ion–bound protein will then switch to the outward-facing state (3). Upon release of the sodium ion, the protein will be reprotonated and the salt bridge between Lys300 and Asp163 reformed (4). The protein will then switch back to the inward-facing conformation.
In summary, the structure of the NhaA dimer here and in particular the repositioning of TM 10 provides critical new information that must be incorporated into the mechanism of electrogenic sodium–proton antiport. Clearly, the crystal structure of the sodium-bound form of NhaA or a homologue, in combination with further biochemical, biophysical, and MD simulation studies, is required to elucidate how binding and unbinding of ions is ultimately coupled to the conformational changes observed during transport.
Supplementary Material
Acknowledgments
We are grateful to Etana Padan, Carola Hunte, and Gunnar von Heijne for useful discussions. The authors are grateful for the use of the Membrane Protein Laboratory funded by the Wellcome Trust (WT089809) at the Diamond Light Source, where data were collected. Computer simulations were carried out on the A2C2 Saguaro supercomputer at Arizona State University.
This work used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation grant OCI-1053575 (allocation MCB130177 to O. Beckstein). This work was supported with grants from The Royal Society through the University Research Fellow scheme (to D. Drew), the Medical Research Council (MRC_G0900990(91997) to A.D. Cameron and D. Drew), the Swedish Research Council (to C. von Ballmoos and D. Drew), and the Biotechnology and Biological Sciences Research Council (BB/G023425/1 to S. Iwata). D. Drew acknowledges the support from EMBO through the EMBO Young Investigator Program. Part of the work was funded by the ERATO Iwata Human Receptor Crystallography Project, Japan Science and Technology Agency and the EU (European Drug Initiative for Channels and Transporters, EDICT grant 201924).
The authors declare no competing financial interests.
Merritt C. Maduke served as editor.
Footnotes
Abbreviations used in this paper:
- α-DDM
- α-n-dodecyl-β-d-malotoside
- EM
- electron microscopy
- MD
- molecular dynamics
- OG
- octyl-β-d-thioylglucoside
- POPC
- 1-palmitoyl-2-oleoylphosphatidylcholine
- RMSD
- root-mean-square deviation
- TM
- transmembrane
References
- Adams P.D., Afonine P.V., Bunkóczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W., et al. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66:213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appel M., Hizlan D., Vinothkumar K.R., Ziegler C., and Kühlbrandt W.. 2009. Conformations of NhaA, the Na+/H+ exchanger from Escherichia coli, in the pH-activated and ion-translocating states. J. Mol. Biol. 388:659–672 10.1016/j.jmb.2009.03.010 [DOI] [PubMed] [Google Scholar]
- Arkin I.T., Xu H., Jensen M.O., Arbely E., Bennett E.R., Bowers K.J., Chow E., Dror R.O., Eastwood M.P., Flitman-Tene R., et al. 2007. Mechanism of Na+/H+ antiporting. Science. 317:799–803 10.1126/science.1142824 [DOI] [PubMed] [Google Scholar]
- Berendsen H.J.C., Postma J.P.M., van Gunsteren W.F., DiNola A., and Haak J.R.. 1984. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81:3684–3690 10.1063/1.448118 [DOI] [Google Scholar]
- Bussi G., Donadio D., and Parrinello M.. 2007. Canonical sampling through velocity rescaling. J. Chem. Phys. 126:014101 10.1063/1.2408420 [DOI] [PubMed] [Google Scholar]
- Chen V.B., Arendall W.B. III, Headd J.J., Keedy D.A., Immormino R.M., Kapral G.J., Murray L.W., Richardson J.S., and Richardson D.C.. 2010. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66:12–21 10.1107/S0907444909042073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4. 1994. The CCP4 suite: programs for protein crystallography. Acta Cryst. D50:760–763 10.1107/S0907444994003112 [DOI] [PubMed] [Google Scholar]
- Cronan J.E.2003. Bacterial membrane lipids: Where do we stand? Annu. Rev. Microbiol. 57:203–224 10.1146/annurev.micro.57.030502.090851 [DOI] [PubMed] [Google Scholar]
- Dahl A.C.E., Chavent M., and Sansom M.S.P.. 2012. Bendix: intuitive helix geometry analysis and abstraction. Bioinformatics. 28:2193–2194 10.1093/bioinformatics/bts357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daley D.O., Rapp M., Granseth E., Melén K., Drew D., and von Heijne G.. 2005. Global topology analysis of the Escherichia coli inner membrane proteome. Science. 308:1321–1323 10.1126/science.1109730 [DOI] [PubMed] [Google Scholar]
- DeLaBarre B., and Brunger A.T.. 2006. Considerations for the refinement of low-resolution crystal structures. Acta Crystallogr. D Biol. Crystallogr. 62:923–932 10.1107/S0907444906012650 [DOI] [PubMed] [Google Scholar]
- Domański J., Stansfeld P.J., Sansom M.S., and Beckstein O.. 2010. Lipidbook: A public repository for force-field parameters used in membrane simulations. J. Membr. Biol. 236:255–258 10.1007/s00232-010-9296-8 [DOI] [PubMed] [Google Scholar]
- Drew D.E., von Heijne G., Nordlund P., and de Gier J.W.. 2001. Green fluorescent protein as an indicator to monitor membrane protein overexpression in Escherichia coli. FEBS Lett. 507:220–224 10.1016/S0014-5793(01)02980-5 [DOI] [PubMed] [Google Scholar]
- Drew D., Lerch M., Kunji E., Slotboom D.J., and de Gier J.W.. 2006. Optimization of membrane protein overexpression and purification using GFP fusions. Nat. Methods. 3:303–313 10.1038/nmeth0406-303 [DOI] [PubMed] [Google Scholar]
- Efremov R.G., and Sazanov L.A.. 2011. Structure of the membrane domain of respiratory complex I. Nature. 476:414–420 10.1038/nature10330 [DOI] [PubMed] [Google Scholar]
- Essmann U., Perela L., Berkowitz M.L., Darden T., Lee H., and Pedersen L.G.. 1995. A smooth particle mesh Ewald method. J. Chem. Phys. 103:8577–8592 10.1063/1.470117 [DOI] [Google Scholar]
- Fafournoux P., Noël J., and Pouysségur J.. 1994. Evidence that Na+/H+ exchanger isoforms NHE1 and NHE3 exist as stable dimers in membranes with a high degree of specificity for homodimers. J. Biol. Chem. 269:2589–2596. [PubMed] [Google Scholar]
- Galili L., Rothman A., Kozachkov L., Rimon A., and Padan E.. 2002. Trans membrane domain IV is involved in ion transport activity and pH regulation of the NhaA-Na+/H+ antiporter of Escherichia coli. Biochemistry. 41:609–617 10.1021/bi011655v [DOI] [PubMed] [Google Scholar]
- Goswami P., Paulino C., Hizlan D., Vonck J., Yildiz O., and Kühlbrandt W.. 2011. Structure of the archaeal Na+/H+ antiporter NhaP1 and functional role of transmembrane helix 1. EMBO J. 30:439–449 10.1038/emboj.2010.321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harms M.J., Schlessman J.L., Sue G.R., and García-Moreno B.. 2011. Arginine residues at internal positions in a protein are always charged. Proc. Natl. Acad. Sci. USA. 108:18954–18959 10.1073/pnas.1104808108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herz K., Rimon A., Jeschke G., and Padan E.. 2009. β-Sheet-dependent dimerization is essential for the stability of NhaA Na+/H+ antiporter. J. Biol. Chem. 284:6337–6347 10.1074/jbc.M807720200 [DOI] [PubMed] [Google Scholar]
- Herz K., Rimon A., Olkhova E., Kozachkov L., and Padan E.. 2010. Transmembrane segment II of NhaA Na+/H+ antiporter lines the cation passage, and Asp65 is critical for pH activation of the antiporter. J. Biol. Chem. 285:2211–2220 10.1074/jbc.M109.047134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess B.2008. P-LINCS: A parallel linear constraint solver for molecular simulation. J. Chem. Theory Comput. 4:116–122 10.1021/ct700200b [DOI] [PubMed] [Google Scholar]
- Hilger D., Jung H., Padan E., Wegener C., Vogel K.P., Steinhoff H.J., and Jeschke G.. 2005. Assessing oligomerization of membrane proteins by four-pulse DEER: pH-dependent dimerization of NhaA Na+/H+ antiporter of E. coli. Biophys. J. 89:1328–1338 10.1529/biophysj.105.062232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hintze J., and Nelson R.D.. 1998. Violin plots: A Box plot-density trace synergism. Am. Stat. 52:181–184. [Google Scholar]
- Hu N.J., Iwata S., Cameron A.D., and Drew D.. 2011. Crystal structure of a bacterial homologue of the bile acid sodium symporter ASBT. Nature. 478:408–411 10.1038/nature10450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W., Dalke A., and Schulten K.. 1996. VMD: Visual molecular dynamics. J. Mol. Graph. 14:33–38 10.1016/0263-7855(96)00018-5 [DOI] [PubMed] [Google Scholar]
- Hunte C., Screpanti E., Venturi M., Rimon A., Padan E., and Michel H.. 2005. Structure of a Na+/H+ antiporter and insights into mechanism of action and regulation by pH. Nature. 435:1197–1202 10.1038/nature03692 [DOI] [PubMed] [Google Scholar]
- Inoue H., Noumi T., Tsuchiya T., and Kanazawa H.. 1995. Essential aspartic acid residues, Asp-133, Asp-163 and Asp-164, in the transmembrane helices of a Na+/H+ antiporter (NhaA) from Escherichia coli. FEBS Lett. 363:264–268 10.1016/0014-5793(95)00331-3 [DOI] [PubMed] [Google Scholar]
- Isom D.G., Castañeda C.A., Cannon B.R., and García-Moreno B.. 2011. Large shifts in pKa values of lysine residues buried inside a protein. Proc. Natl. Acad. Sci. USA. 108:5260–5265 10.1073/pnas.1010750108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen K.P., and Jorgensen W.L.. 2006. Halide, ammonium, and alkali metal ion parameters for modeling aqueous solutions. J. Chem. Theory Comput. 2:1499–1509 10.1021/ct600252r [DOI] [PubMed] [Google Scholar]
- Jones T.A., and Kjeldgaard M.. 1997. Electron-density map interpretation. Methods Enzymol. 277:173–208 10.1016/S0076-6879(97)77012-5 [DOI] [PubMed] [Google Scholar]
- Jorgensen W.L., Chandrasekhar J., Madura J.D., Impey R.W., and Klein M.L.. 1983. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79:926–935 10.1063/1.445869 [DOI] [Google Scholar]
- Kabsch W.2010. XDS. Acta Crystallogr. D Biol. Crystallogr. 66:125–132 10.1107/S0907444909047337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaminski G.A., Friesner R.A., Tirado-Rives J., and Jorgensen W.L.. 2001. Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J. Phys. Chem. B. 105:6474–6487 10.1021/jp003919d [DOI] [Google Scholar]
- Kleywegt G.J., and Jones T.A.. 1994. A super position. ESF/CCP4 Newsletter. 31:9–14. [Google Scholar]
- Kleywegt G.J., Zou J.Y., Kjeldgaard M., and Jones T.A.. 2001. Around O. International Tables for Crystallography, Volume F. Crystallography of Biological Macromolecules Rossmann M.G. and Arnold E., Kluwer Academic Publishers, Dordrecht: 497–506. [Google Scholar]
- Kozachkov L., and Padan E.. 2013. Conformational changes in NhaA Na+/H+ antiporter. Mol. Membr. Biol. 30:90–100 10.3109/09687688.2012.693209 [DOI] [PubMed] [Google Scholar]
- Kozachkov L., Herz K., and Padan E.. 2007. Functional and structural interactions of the transmembrane domain X of NhaA, Na+/H+ antiporter of Escherichia coli, at physiological pH. Biochemistry. 46:2419–2430 10.1021/bi602393s [DOI] [PubMed] [Google Scholar]
- Kučerka N., Tristram-Nagle S., and Nagle J.F.. 2006. Structure of fully hydrated fluid phase lipid bilayers with monounsaturated chains. J. Membr. Biol. 208:193–202 10.1007/s00232-005-7006-8 [DOI] [PubMed] [Google Scholar]
- Kumar S., and Nussinov R.. 1999. Salt bridge stability in monomeric proteins. J. Mol. Biol. 293:1241–1255 10.1006/jmbi.1999.3218 [DOI] [PubMed] [Google Scholar]
- Kumar S., and Nussinov R.. 2002. Relationship between ion pair geometries and electrostatic strengths in proteins. Biophys. J. 83:1595–1612 10.1016/S0006-3495(02)73929-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landau M., Herz K., Padan E., and Ben-Tal N.. 2007. Model structure of the Na+/H+ exchanger 1 (NHE1): Functional and clinical implications. J. Biol. Chem. 282:37854–37863 10.1074/jbc.M705460200 [DOI] [PubMed] [Google Scholar]
- Lee C., Kang H.J., von Ballmoos C., Newstead S., Uzdavinys P., Dotson D.L., Iwata S., Beckstein O., Cameron A.D., and Drew D.. 2013. A two-domain elevator mechanism for sodium/proton antiport. Nature. 501:573–577 10.1038/nature12484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C., Kang H.J., Hjelm A., Qureshi A.A., Nji E., Choudhury H., Beis K., de Gier J.W., and Drew D.. 2014. MemStar: A one-shot Escherichia coli-based approach for high-level bacterial membrane protein production. FEBS Lett. 588:3761–3769 10.1016/j.febslet.2014.08.025 [DOI] [PubMed] [Google Scholar]
- Li H., Robertson A.D., and Jensen J.H.. 2005. Very fast empirical prediction and rationalization of protein pKa values. Proteins. 61:704–721 10.1002/prot.20660 [DOI] [PubMed] [Google Scholar]
- Maes M., Rimon A., Kozachkov-Magrisso L., Friedler A., and Padan E.. 2012. Revealing the ligand binding site of NhaA Na+/H+ antiporter and its pH dependence. J. Biol. Chem. 287:38150–38157 10.1074/jbc.M112.391128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mager T., Rimon A., Padan E., and Fendler K.. 2011. Transport mechanism and pH regulation of the Na+/H+ antiporter NhaA from Escherichia coli: An electrophysiological study. J. Biol. Chem. 286:23570–23581 10.1074/jbc.M111.230235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mager T., Braner M., Kubsch B., Hatahet L., Alkoby D., Rimon A., Padan E., and Fendler K.. 2013. Differential effects of mutations on the transport properties of the Na+/H+ antiporter NhaA from Escherichia coli. J. Biol. Chem. 288:24666–24675 10.1074/jbc.M113.484071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A.J., Grosse-Kunstleve R.W., Adams P.D., Winn M.D., Storoni L.C., and Read R.J.. 2007. Phaser crystallographic software. J. Appl. Cryst. 40:658–674 10.1107/S0021889807021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaud-Agrawal N., Denning E.J., Woolf T.B., and Beckstein O.. 2011. MDAnalysis: A toolkit for the analysis of molecular dynamics simulations. J. Comput. Chem. 32:2319–2327 10.1002/jcc.21787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto S., and Kollman P.A.. 1992. SETTLE: An analytical version of the SHAKE and RATTLE algorithms for rigid water models. J. Comput. Chem. 13:952–962 10.1002/jcc.540130805 [DOI] [Google Scholar]
- Murzyn K., Róg T., and Pasenkiewicz-Gierula M.. 2005. Phosphatidylethanolamine-phosphatidylglycerol bilayer as a model of the inner bacterial membrane. Biophys. J. 88:1091–1103 10.1529/biophysj.104.048835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olkhova E., Kozachkov L., Padan E., and Michel H.. 2009. Combined computational and biochemical study reveals the importance of electrostatic interactions between the “pH sensor” and the cation binding site of the sodium/proton antiporter NhaA of Escherichia coli. Proteins. 76:548–559 10.1002/prot.22368 [DOI] [PubMed] [Google Scholar]
- Otwinowski Z., and Minor W.. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276:307–326 10.1016/S0076-6879(97)76066-X [DOI] [PubMed] [Google Scholar]
- Padan E.2008. The enlightening encounter between structure and function in the NhaA Na+-H+ antiporter. Trends Biochem. Sci. 33:435–443 10.1016/j.tibs.2008.06.007 [DOI] [PubMed] [Google Scholar]
- Padan E., Tzubery T., Herz K., Kozachkov L., Rimon A., and Galili L.. 2004. NhaA of Escherichia coli, as a model of a pH-regulated Na+/H+ antiporter. Biochim. Biophys. Acta. 1658:2–13 10.1016/j.bbabio.2004.04.018 [DOI] [PubMed] [Google Scholar]
- Padan E., Kozachkov L., Herz K., and Rimon A.. 2009. NhaA crystal structure: functional-structural insights. J. Exp. Biol. 212:1593–1603 10.1242/jeb.026708 [DOI] [PubMed] [Google Scholar]
- Potterton L., McNicholas S., Krissinel E., Gruber J., Cowtan K., Emsley P., Murshudov G.N., Cohen S., Perrakis A., and Noble M.. 2004. Developments in the CCP4 molecular-graphics project. Acta Crystallogr. D Biol. Crystallogr. 60:2288–2294 10.1107/S0907444904023716 [DOI] [PubMed] [Google Scholar]
- Raetz C.R.1986. Molecular genetics of membrane phospholipid synthesis. Annu. Rev. Genet. 20:253–291 10.1146/annurev.ge.20.120186.001345 [DOI] [PubMed] [Google Scholar]
- Rappolt M., Hickel A., Bringezu F., and Lohner K.. 2003. Mechanism of the lamellar/inverse hexagonal phase transition examined by high resolution x-ray diffraction. Biophys. J. 84:3111–3122 10.1016/S0006-3495(03)70036-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rimon A., Tzubery T., and Padan E.. 2007. Monomers of the NhaA Na+/H+ antiporter of Escherichia coli are fully functional yet dimers are beneficial under extreme stress conditions at alkaline pH in the presence of Na+ or Li+. J. Biol. Chem. 282:26810–26821 10.1074/jbc.M704469200 [DOI] [PubMed] [Google Scholar]
- Rizzo R.C., and Jorgensen W.L.. 1999. OPLS all-atom model for amines: Resolution of the amine hydration problem. J. Am. Chem. Soc. 121:4827–4836 10.1021/ja984106u [DOI] [Google Scholar]
- Schushan M., Xiang M., Bogomiakov P., Padan E., Rao R., and Ben-Tal N.. 2010. Model-guided mutagenesis drives functional studies of human NHA2, implicated in hypertension. J. Mol. Biol. 396:1181–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott D.W.1992. Multivariate Density Estimation: Theory, Practice, and Visualization. John Wiley & Sons, New York: 317 pp. [Google Scholar]
- Scott K.A., Bond P.J., Ivetac A., Chetwynd A.P., Khalid S., and Sansom M.S.P.. 2008. Coarse-grained MD simulations of membrane protein-bilayer self-assembly. Structure. 16:621–630 10.1016/j.str.2008.01.014 [DOI] [PubMed] [Google Scholar]
- Screpanti E., Padan E., Rimon A., Michel H., and Hunte C.. 2006. Crucial steps in the structure determination of the Na+/H+ antiporter NhaA in its native conformation. J. Mol. Biol. 362:192–202 10.1016/j.jmb.2006.07.019 [DOI] [PubMed] [Google Scholar]
- Shaffer P.L., Goehring A., Shankaranarayanan A., and Gouaux E.. 2009. Structure and mechanism of a Na+-independent amino acid transporter. Science. 325:1010–1014 10.1126/science.1176088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Søndergaard C.R., Olsson M.H.M., Rostkowski M., and Jensen J.H.. 2011. Improved treatment of ligands and coupling effects in empirical calculation and rationalization of pKa values. J. Chem. Theory Comput. 7:2284–2295 10.1021/ct200133y [DOI] [PubMed] [Google Scholar]
- Sonoda Y., Newstead S., Hu N.J., Alguel Y., Nji E., Beis K., Yashiro S., Lee C., Leung J., Cameron A.D., et al. 2011. Benchmarking membrane protein detergent stability for improving throughput of high-resolution X-ray structures. Structure. 19:17–25 10.1016/j.str.2010.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stansfeld P.J., and Sansom M.S.P.. 2011. From coarse grained to atomistic: A serial multiscale approach to membrane protein simulations. J. Chem. Theory Comput. 7:1157–1166 10.1021/ct100569y [DOI] [PubMed] [Google Scholar]
- Studier F.W.2005. Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 41:207–234 10.1016/j.pep.2005.01.016 [DOI] [PubMed] [Google Scholar]
- Taglicht D., Padan E., and Schuldiner S.. 1991. Overproduction and purification of a functional Na+/H+ antiporter coded by nhaA (ant) from Escherichia coli. J. Biol. Chem. 266:11289–11294. [PubMed] [Google Scholar]
- Taglicht D., Padan E., and Schuldiner S.. 1993. Proton-sodium stoichiometry of NhaA, an electrogenic antiporter from Escherichia coli. J. Biol. Chem. 268:5382–5387. [PubMed] [Google Scholar]
- Ulmschneider J.P., and Ulmschneider M.B.. 2009. United atom lipid parameters for combination with the optimized potentials for liquid simulations all-atom force field. J. Chem. Theory Comput. 5:1803–1813 10.1021/ct900086b [DOI] [PubMed] [Google Scholar]
- Ulmschneider M.B., Doux J.P.F., Killian J.A., Smith J.C., and Ulmschneider J.P.. 2010. Mechanism and kinetics of peptide partitioning into membranes from all-atom simulations of thermostable peptides. J. Am. Chem. Soc. 132:3452–3460 10.1021/ja909347x [DOI] [PubMed] [Google Scholar]
- Ulmschneider J.P., Smith J.C., White S.H., and Ulmschneider M.B.. 2011. In silico partitioning and transmembrane insertion of hydrophobic peptides under equilibrium conditions. J. Am. Chem. Soc. 133:15487–15495 10.1021/ja204042f [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waskom M.2014. Seaborn: statistical data visualization. http://web.stanford.edu/∼mwaskom/software/seaborn/ (accessed October 31, 2014).
- Wagner S., Klepsch M.M., Schlegel S., Appel A., Draheim R., Tarry M., Högbom M., van Wijk K.J., Slotboom D.J., Persson J.O., and de Gier J.W.. 2008. Tuning Escherichia coli for membrane protein overexpression. Proc. Natl. Acad. Sci. USA. 105:14371–14376 10.1073/pnas.0804090105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams K.A.2000. Three-dimensional structure of the ion-coupled transport protein NhaA. Nature. 403:112–115 10.1038/47534 [DOI] [PubMed] [Google Scholar]
- Winn M.D., Isupov M.N., and Murshudov G.N.. 2001. Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr. D Biol. Crystallogr. 57:122–133 10.1107/S0907444900014736 [DOI] [PubMed] [Google Scholar]
- Winter G.2010. xia2: an expert system for macromolecular crystallography data reduction. J. Appl. Cryst. 43:186–190 10.1107/S0021889809045701 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.