

Abstract
Non-alcoholic fatty liver disease affects approximately one-third of the population worldwide, and its incidence continues to increase with the increasing prevalence of other metabolic disorders such as type 2 diabetes. As non-alcoholic fatty liver disease can progress to liver cirrhosis, its treatment is attracting greater attention. The pathogenesis of non-alcoholic fatty liver disease is closely associated with insulin resistance and dyslipidemia, especially hypertriglyceridemia. Increased serum levels of free fatty acid and glucose can cause oxidative stress in the liver and peripheral tissue, leading to ectopic fat accumulation, especially in the liver. In this review, we summarize the mechanism underlying the progression of hepatic steatosis to steatohepatitis and cirrhosis. We also discuss established drugs that are already being used to treat non-alcoholic fatty liver disease, in addition to newly discovered agents, with respect to their mechanisms of drug action, focusing mainly on hepatic insulin resistance. As well, we review clinical data that demonstrate the efficacy of these drugs, together with improvements in biochemical or histological parameters.
Keywords: Non-alcoholic fatty liver disease, Insulin resistance, Drugs, Pathogenesis
Core tip: In this review, we summarize the pathogenesis underlying the progression of hepatic steatosis to steatohepatitis and cirrhosis. We also discuss established drugs that are already being used to treat non-alcoholic fatty liver disease, in addition to newly discovered agents, with respect to their mechanisms of drug action, focusing mainly on hepatic insulin resistance. As well, we review clinical data that demonstrate the efficacy of these drugs, together with improvements in biochemical or histological parameters. Furthermore, we introduced future treatment option for non-alcoholic fatty liver disease.
INTRODUCTION
Non-alcoholic fatty liver disease (NAFLD), the accumulation of lipid within hepatocytes, is a common disease[1]. The worldwide prevalence of NAFLD is estimated to be 20%-30%[2], although increasing to 57%-74% among obese patients[3]. NAFLD refers to a wide spectrum of fatty degenerative disorders of the liver in the absence of alcohol intake, ranging from simple steatosis to steatohepatitis and cirrhosis[4]. Nonalcoholic steatohepatitis (NASH) is histologically characterized by inflammatory cell recruitment. NASH is a significant risk factor for hepatic cirrhosis, compared with simple steatosis[5], and 4%-27% of cases of NASH progress to hepatocellular carcinoma after the development of cirrhosis[6]. In one study, NAFLD was present in 75% of obese [body mass index (BMI)≥ 30 kg/m2] patients, 16% of non-obese patients, and 34%-74% of patients with type 2 diabetes[7]. Another study reported diagnoses of fatty liver in 39% of obese (BMI ≥ 30 kg/m2) patients, 41% of patients with known type 2 diabetes, and 32% of patients with dyslipidemia[8]. Patients with NAFLD are not only insulin resistant, but also tend to present with alterations in plasma triglyceride levels[9]. NAFLD is strongly associated with metabolic syndrome, especially insulin resistance, central obesity, and dyslipidemia. Therefore, NAFLD is regarded as a difficult to treat component of metabolic syndrome[10]. In this review, we investigate the mechanisms of hepatic fat accumulation, focusing on the role of insulin resistance therein, and review current therapeutic options and new candidate drugs for the treatment of NAFLD.
PATHOGENESIS
Insulin resistance - free fatty acid flux and hyperinsulinemia
Hepatic steatosis is caused by an imbalance in triglyceride movement through the liver cell. Triglyceride is composed of free fatty acid (FFA) and glycerol. Total FFA is derived from three sources, the diet (15%), de novo synthesis (26%), and circulating FFA (56%)[11]. A high-fat diet is known to lead to the development of hepatic steatosis. However, estimates suggest that approximately 60% of liver fat is derived from circulating nonesterified fatty acids (NEFAs) in individuals who eat a normal fat-containing diet[11]. Obesity is associated with insulin resistance and an elevated leptin level. In particular, increased visceral fat correlates with peripheral and hepatic insulin resistance[12,13]. Insulin resistance in skeletal muscle and adipose tissue results in increased levels of NEFAs through increased lipid oxidation in adipose tissue (Figure 1). Accordingly, NEFA flux plays an important role in hepatic fat accumulation[14]. An increase in hepatocellular diacylglycerol is associated with decreased tyrosine phosphorylation of insulin receptor substrate 2 (IRS-2)[15,16]. In turn, the decreased activity of IRS-2 and PI3K leads to increased hepatic glucose production[17]. Hyperinsulinemia also arises in response to insulin resistance in adipose tissue, leading not only to downregulation of IRS-2 in the liver, but also to a continued increase in the level of sterol regulatory element binding protein-1c (SREBP-1c) via the insulin signaling pathway involving AKT2, liver X receptor (LXR) and mammalian target of rapamycin[18,19]. Elevated levels of SREBP-1c up-regulate lipogenic gene expression, increase fatty acid synthesis, and accelerate hepatic fat accumulation[20]. Additionally, overexpression of SREBP-1c represses IRS-2 expression[21]. Glucose-stimulated lipogenesis is mediated by carbohydrate-responsive element-binding protein (ChREBP) in the liver. Like SREBP-1c, ChREBP increases lipogenesis by inducing lipogenic gene expression during consumption of a diet high in carbohydrates[22,23].
Figure 1.
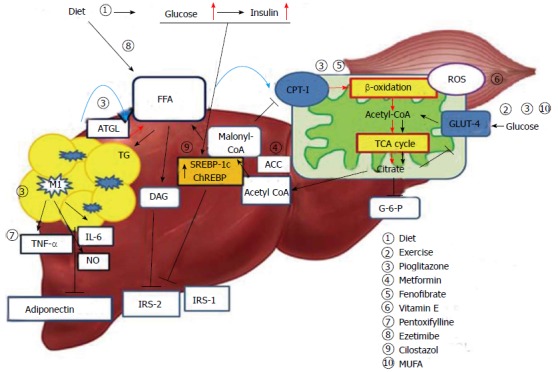
Mechanism of hepatic insulin resistance and the key pathway of drug action. Delivery of FFAs to the liver and skeletal muscle is increased in insulin resistance conditions, and these are metabolized via mitochondrial β-oxidation. Consequently, hyperglycemia and increased hepatic FFA uptake reduce glucose uptake and oxidation in skeletal muscle. Diet and exercise are the main treatment strategies for this pathogenesis; insulin sensitizers and MUFA may contribute to reducing peripheral insulin resistance. Pioglitazone and fenofibrate act on β-oxidation of mitochondria and reduce hepatic steatosis. Accelerated β-oxidation also causes increased production of ROS. Vitamin E can reduce oxidative stress. Adipose tissue inflammation of the liver leads to inflammatory activation of hepatic Kupffer cells via classic response (M1) and produce inflammatory cytokines. This is also associated with decreased adiponectin levels and promotes hepatic steatohepatitis. Pentoxifylline inhibits TNF-α and alleviates steatohepatitis. Hyperglycemia caused by insulin resistance up-regulates lipogenic gene expression, such as SREBP-1c and ChREBP, and induces lipogenesis in hepatocytes. Cilostasol may inhibit SREBP-1c. FFA: Free fatty acid; TG: Triglyceride; CPT-I: Carnitine palmitoyltransferase-I; ACC: Acetyl-CoA carboxylase; ATGL: Adipose triglyceride lipase; ChREBP: Carbohydrate responsive element binding protein; SREBP-1c: Sterol regulatory element binding protein-1c; TCA: Tricarboxylic acid; ROS: Reactive oxygen species; IRS: Insulin receptor substrate; DAG: Diacylglycerol; G-6-P: Glucose 6-phosphate; TNF-α: Tumor necrosis factor- α; MUFA: Monosaturated fatty acids; M1: Kupffer cells activated via classic pathway.
Endoplasmic reticulum stress
The endoplasmic reticulum (ER) is an intracellular organelle that plays an important role in the synthesis, folding, and trafficking of proteins. Cellular nutrient status and energy condition highly influence the function of the ER, and dysfunction in the ER causes accumulation of unfolded proteins therein, triggering an unfolded protein response (UPR)[24]. Under stress, such as hypoxia, inflammation and energy excess, UPR is characterized by adaptive cellular processes of increased degradation of proteins and translational arrest of protein synthesis to restore normal function of the ER. As well, UPR mediates metabolic and immune responses that aggravate insulin resistance[25-27]. Both PKR-like kinase and the α-subunit of translation initiation factor 2 (eIF2α), well-known ER stress markers, are increased in hepatocytes of ob/ob mice, compared with control mice[26]. Obesity causes ER stress that leads to suppression of insulin signaling through serine phosphorylation of insulin receptor substrate-1 (IRS-1) and activation of the c-Jun N-terminal kinase (JNK) pathway[26]. Among subjects with metabolic syndrome, those with NASH showed higher levels phosphorylated JNK protein, compared to subjects with simple hepatic steatosis. Furthermore, subjects with NASH did not generate spliced manipulation of X-box–binding protein-1 (sXBP-1), which is a key regulator in ER stress in relation to insulin action[24,26]. Additionally, weight reduction in obese subjects has been shown to induce improvement in ER stress via suppression of phosphorylated JNK and eIF2α in adipose tissue and the liver[28].
Role of oxidative stress - mitochondrial dysfunction
The two-hit hypothesis is a key concept of NAFLD pathogenesis. In fatty livers, simple hepatic steatosis (first hit) sensitizes the liver to inflammatory cytokines or oxidative stress (second hit), leading to development of steatohepatitis[29]. Oxidative stress is resulted from a serious imbalance between the limited antioxidant defenses and excessive formation of reactive species such as reactive oxygen species (ROS) or reactive nitrogen species (RNS)[30]. ROS is an integrated term that describes a variety of species of free radicals derived from molecular oxygen, such as superoxide, hydrogen peroxide, and hydroxyl[31]. In cells, mitochondria are a major source of ROS generation. The important factor modulating mitochondrial ROS generation is the redox state of the respiratory chain[32,33] . FFAs are metabolized via the mitochondrial β-oxidation pathway and the tricarboxylic acid (TCA) cycle, which generates citrate that in turn inhibits glycolysis. As a result, glucose oxidation and glucose uptake via glucose transporter type 4 (GLUT4) in skeletal muscle are reduced[34,35]. To compensate for the excessive fat storage in the liver, increased hepatic FFA uptake stimulates hepatic oxidation of fatty acids in obese individuals. Mitochondrial FFA oxidation is maintained until mitochondrial respiration becomes severely impaired[36,37]. However, accelerated β-oxidation not only causes excessive electron flux in the electron transport chain, but also leads to increased production of ROS, and can lead to mitochondrial dysfunction[38]. Excessive ROS production by mitochondria can lead to oxidative damage to the mitochondrial membrane and DNA and can impair mitochondrial metabolic functions[33]. The increase in hepatic lipogenesis in NASH results in increased production of malonyl-CoA. Inhibition of carnitine palmitoyltransferase-I (CPT-1) by malonyl-CoA leads to decreased entry of long chain fatty acid into the mitochondria, and causes reduced β-oxidation and enhanced triglyceride accumulation in the liver[38-40]. The nuclear receptor peroxisome proliferator-activated receptor α (PPAR-α) plays an important role in the transcriptional control of many enzymes involved in mitochondrial fatty acid β-oxidation. Peroxisome proliferator-activated receptor-gamma co-activator (PGC) -1α cooperates with PPAR-α and regulates genes that encode mitochondrial fatty acid oxidation enzymes, such as CPT-1 and medium chain acyl-CoA dehydrogenase[40]. Previously, a PPAR-α-deficient mouse model showed a lack of hepatic peroxisome proliferation and dyslipidemia with obesity and hepatic steatosis[41].
Inflammation and adipokines
Overall obesity is correlated with NAFLD, and accumulation of intra-abdominal fat in particular is believed to play an important role in the development of insulin resistance[12,13]. Meanwhile, hepatic fat accumulation is associated with insulin resistance independent of intra-abdominal fat accumulation and overall obesity. Even in normal weight subjects, hepatic steatosis has been shown to be related to various parameters of insulin resistance, such as basal glucose level or serum FFA level[42]. In addition to being a major organ of triglyceride deposition, adipose tissue acts an endocrine organ that secretes several hormones[43]. Adipocytes secrete adiponectin and leptin, in addition to the other adipokines, such as retinol-binding protein, tumor necrosis factor-α (TNF-α), interleukin 6 (IL-6), and plasminogen activator inhibitor-1[43]. Adiponectin stimulates phosphorylation of AMP-activated protein kinase (AMPK) and acetyl-CoA carboxylase (ACC) in the liver and muscles, thereby, increasing glucose utilization and fatty-acid oxidation[44]. In a previous study, serum adiponectin levels decreased with an increases in obesity, in particular increases in intra-abdominal fat mass[45,46]. In another study, adiponectin knockout mice fed a high-fat diet exhibited increased incidences of obesity, hyperinsulinemia, and steatohepatitis. These experimental data indicate that adiponectin may play a key protective role against the progression of NASH[47]. Reportedly, adipose tissue in obese individuals stimulates a shift in macrophage activation from the alternative response (M2) to the classic response (M1), and these classically activated macrophages secrete a variety of inflammatory cytokines, such as TNF-α, IL-6, and NO[48]. Additionally, studies showed that inflammatory activation of hepatic Kupffer cells in ob/ob mice promotes hepatotoxicity, resulting in hepatic insulin resistance and steatohepatitis[49,50]. Thus, increases in TNF-α and IL-6 in obese subjects may play an important role in insulin resistance and hepatic steatosis[51,52].
Gut-microbial alternation and TLRs stimulation
As mentioned above, obesity is often associated with NASH and systemic inflammation characterized increases in inflammatory cytokine levels. Obesity also can cause increased intestinal mucosa permeability and endotoxin levels in portal circulation that can contribute to hepatocellular damage[53,54]. Kupffer cells in the liver play a key role in clearing endotoxin and are activated through Toll like receptor 2,3,4 and 9 signaling in the presence of endotoxin. In particular, activation of Toll like receptor4 (TLR4) is reportedly associated with stimulation of lipopolysaccharide (LPS)[55-57]. Previously, animal model studies showed that TLRs 2, 4 and 9 may contribute to the pathogenesis of NAFLD[55,58]. Activated Kupffer cells induce expression of pro-inflammatory cytokines, such TNF-α, IL-6, IL-18 and IL-12 as well as anti-inflammatory cytokines[59]. TLRs including TLRs 2,4 and 9 are activated via a MyD88 dependent pathway. This pathway consists of the activation of serine kinase IL-1R-associated kinase and TBF-receptor-associated factor 6 and is involved in the activation of the transcription factor NF-κB, which is related to inflammatory cytokine production[60].
TREATMENT
Life style modification - diet and exercise
Weight loss due to diet and exercise has been demonstrated to alleviate hepatic steatosis[61]. Body weight reduction and exercise are important independent factors for improvement of hepatic steatosis[62]. In obese women, hepatic fat content measured by magnetic resonance imaging was shown to decrease in response to weight loss interventions[63]. Several studies have shown a significant reduction in alanine transaminase (ALT) levels and improvement in biochemical markers following intervention with a calorie-restricted diet combined with exercise[63,64]. A few studies have also shown histologic improvement with increased exercise and weight reduction[65,66] (Table 1). Exercise improves insulin sensitivity in skeletal muscle via GLUT4 expression and increases glucose utilization. Thus, exercise decreases levels of serum glucose and insulin[67]. An improvement in hyperinsulinemia can result in decreased liver fat mass, because hyperinsulinemia stimulates hepatic steatosis via the SREBP-1c pathway[19]. In particular, NAFLD patients with metabolic syndrome show a great improvement in hepatic steatosis after weight loss[68].
Table 1.
Treatment outcomes of variable regimens
| Study | Treatment group | Control group | No. | Study design | Duration (mo) | Histology | Liver enzymes | US |
| Life style modification | ||||||||
| Huang et al[66] | Diet | - | 12 | Open label | 12 | Improved | - | - |
| Ueno et al[65] | Diet/Exercise | Control | 15 | Open label | 3 | Improved | Improved | - |
| Pioglitazone (insulin sensitizer) | ||||||||
| Promrat et al[71] | Pioglitazone | - | 18 | Open label | 12 | Improved | Improved | - |
| Belfort et al[72] | Pioglitazone and Diet | Placebo | 55 | RCT | 6 | Improved | Improved | - |
| Aithal et al[73] | Pioglitazone and diet/Exercise | Placebo | 74 | RCT | 12 | Improved | Improved | - |
| Sanyal et al[74] | Pioglitazone | Placebo | 163 | RCT | 24 | Improved | Improved | - |
| Metformin (insulin sensitizer) | ||||||||
| Garinis et al[100] | Metformin and Diet | Diet | 50 | RCT | 6 | - | - | Improved |
| Haukeland et al[103] | Metformin | Placebo | 48 | RCT | 6 | - | - | - |
| Uygun et al[101] | Metformin and Diet | Control | 50 | Open label | 6 | - | - | Improved |
| Bugianes et al[99] | Metformin | Diet | 53 | RCT | 12 | Improved | Improved | - |
| Bugianes et al[99] | Metformin | Vitamin E | 57 | RCT | 12 | - | Improved | - |
| Vitamin E (antioxidant) | ||||||||
| Bugianesi et al[99] | Vitemin E | Diet | 55 | RCT | 12 | - | - | - |
| Sanyal et al[74] | Vitamin E | Placebo | 167 | RCT | 24 | Improved | Improved | |
| Vajro et al[109] | Vitamin E | Diet | 25 | RCT | 6 | - | - | - |
| Other drugs | ||||||||
| Sanjay et al[112] | Pentoxifylline | - | 18 | Open label | 6 | - | Improved | - |
| Yoneda et al[127] | Ezetimibe | - | 10 | Open label | 6 | Improved | Improved | |
| Vasilios et al[118] | Statin | Control | 437 | Open label | 36 | - | Improved | - |
| Lindor et al[130] | UDCA | Placebo | 166 | RCT | 48 | - | - | - |
| Capani et al[138] | PUFA | Control | 42 | RCT | 12 | - | Improved | Improved |
No.: Number; US: Ultrasonography; RCT: Randomized controlled trial.
Insulin sensitizer-thiazolidinedione, metformin
Thiazolidinedione: Thiazolidinediones (TZDs) are insulin-sensitizing agents that have been shown to improve not only hepatic steatosis, but also whole body insulin resistance[69]. Improvements in insulin resistance and histologic and biochemical parameters were reported with TZD treatment[70-74]. Rosiglitazone is one TZD and is associated with an increased risk of myocardial infarction and cardiovascular death[75]. Meanwhile, pioglitazone is regarded as safe in regards to cardiovascular outcomes and is not associated with increased cardiovascular risk[76,77]. In patients with type 2 diabetes, pioglitazone has been recommended for the treatment of steatohepatitis proven by liver biopsy; however, its role in non-diabetic patients has not been established. The American Association for the Study of Liver Disease (AASLD) introduced pioglitazone as a first-line treatment of NAFLD in patients with type 2 diabetes[78]. TZDs increase glucose utilization of peripheral tissue and improve whole body insulin sensitivity as measured by the hyperinsulinemic euglycemic clamp technique, in patients with type 2 diabetes. Moreover, serum adiponectin levels increase and serum insulin levels decreases after treatment with pioglitazone[79,80]. An increase in serum adiponectin contributes to alleviation of hepatic steatosis and improves hepatic and peripheral insulin resistance[79]. As mentioned above, adiponectin increases lipid oxidation of FFA by ACC phosphorylation in the liver[44], and promotes the activation of anti-inflammatory M2 macrophages rather than M1 macrophages[81]. Obesity is closely related to an increase in NAFLD risk[82]. Increased levels of inflammatory adipose tissue macrophages (ATMs) and their secreted cytokines in a mouse model were shown to be related to systemic insulin resistance, which is associated with NAFLD development[15,83]. According to previous studies, ATMs are increased in obese subjects[84], and pioglitazone treatment results in not only a decrease in ATM content, but also in the inflammatory markers, TNF-α, IL-6, and inducible nitric oxide synthase[85,86]. TZDs also promote the alternative activation of monocytes into macrophages with anti-inflammatory properties, as opposed to the pro-inflammatory phenotype[87]. Although the pathogenesis of NAFLD development is closely related to obesity, the distribution of fat is more important than overall obesity. Excessive visceral fat accumulation plays an important role in the development of insulin resistance and NAFLD by acting as a source of FFA[12]. Pioglitazone is strongly associated with fat redistribution, increases in subcutaneous fat area decreases in visceral fat area (visceral to subcutaneous fat ratio)[88]. Another study showed that the ratio of visceral fat thickness to subcutaneous fat thickness decreases after pioglitazone treatment and is correlated with a change in high sensitivity C-reactive protein levels[89]. TZD treatment results revealed a decrease in serum FFA levels, which in turn reduced FFA supply to the liver and led to a decrease in hepatic triglyceride content[90]. Recent studies have focused on the role of sirtuin-6 (SIRT-6) in the glucose and lipid metabolism associated with TZDs. TZD treatment reduced hepatic fat accumulation and increased expression of SIRT-6 and PGC1-α in rat livers[91]. Also, liver-specific SIRT-6 knock-out mice exhibited fatty liver formation[92], leading to NASH[93].
Metformin: Metformin improves insulin resistance and hyperinsulinemia by increasing peripheral glucose uptake and decreasing hepatic gluconeogenesis[94]. Metformin activates AMP kinase via a LKB-1 dependent mechanism in skeletal muscle. Also it can activate AMPK by stimulating AMP accumulation in hepatocytes. The increase in AMP interferes with glucagon action and decreases cAMP levels, leading to decreased production of hepatic glucose[95,96]. Activation of AMPK results in decreased hepatic triglyceride synthesis and increased fatty acid oxidation[97], as well as attenuated hepatic steatosis due to decreased SREBP-1c activity[98]. A randomized controlled trial showed that subjects treated with metformin exhibit significant improvement in ALT levels, compared with those who were on a restricted diet or were treated with vitamin E, as well as improvements in histology after a 12 mo of treatment[99,100]. Many studies have shown that metformin treatment normalizes transaminase levels and decreases hepatic steatosis as determined by follow-up ultrasound; nevertheless, histologic data remain limited[100-103]. As NASH is closely associated with development of HCC and liver fibrosis, metformin may be limited in the reduction of these severe outcomes, including mortality[104].
Antioxidant - vitamin E (α-tocopherol), pentoxifylline
As mentioned above, oxidative stress contributes to the progression of NASH from simple hepatic steatosis. A recent study reported that subjects who were treated with vitamin E (α-tocopherol) showed improvement in hepatic steatosis and serum aminotransferase levels compared to a placebo group[74]. Vitamin E (α-tocopherol) has been used to treat non-diabetic NASH patients diagnosed by liver biopsy[78]. Meta-analyses of vitamin E have revealed an increase in all-cause mortality with high dose (≥ 400 IU/d) vitamin E supplement use, especially in subjects with chronic disease or at high risk for cardiovascular disease events, such as type 2 diabetes. However, these results are uncertain in healthy subjects[105,106]. Two pilot studies reported improved ALT levels with vitamin E treatment[107,108]. However, two randomized controlled trials failed to show the efficacy of vitamin E treatment in NAFLD[109,110]. Pentoxifylline, a TNF-α inhibitor, has also been considered for the treatment of hepatic steatosis, since it plays an important role in the progression of simple hepatic steatosis to steatohepatitis. In previous studies, administration of pentoxifylline generated improvements in biochemical markers, such as aminotransferase and Homa-IR, in patients with NASH[111,112]. Nevertheless, further study is needed to prove the efficacy of pentoxifylline with respect to histologic improvement of NAFLD.
Lipid-lowering agents - fibrates, ezetimibe and statins
Hypertriglyceridemia is a major component of metabolic syndrome and is strongly associated with NAFLD. Increased FFA delivery to the liver causes accumulation of hepatic fat[9]. Many different lipid-lowering agents have been investigated for the treatment of NAFLD. Patients treated with gemfibrozil, one type of fibrate, showed decreased ALT levels, compared to the control group[113]. However, clofibrate did not show a beneficial effect on NAFLD[114]. PPAR-α modulates not only FFA transport and β-oxidation to decrease triglyceride in hepatocytes, but also glucose and amino acid metabolism in liver and skeletal muscle. PPAR-α activation is involved in lipoprotein metabolism by increasing lipolysis, thus reducing the production of triglyceride-rich particles[115]. Fenofibrate increased levels of PPAR-α and decreased hepatic steatosis in an APOE2KI mouse model that represented diet-induced NASH[116]. A prospective study using atorvastatin reported significant reductions in serum transaminase level[117,118]. Atorvastatin induces hepatic low-density lipoprotein receptor-related protein 1 (LRP-1) that plays an important role in clearance of circulating triglyceride in the liver[119]. In disposal of chylomicron in hepatocytes, interaction of LRP-1 receptors and apolipoprotein E (AposE) play important roles[120]. Thus, ApoE-deficient mice showed development of hepatic steatosis even when they were fed a normal chow-diet. Accordingly, ApoE may play a key role in intracellular metabolism and control of VLDL production by hepatocytes[121]. Statins are very important drugs to treat dyslipidemia in subjects with both insulin resistance and NAFLD. However, there is continued concern about the use of statins in subjects with established liver disease. According to several randomized controlled studies and retrospective studies, statin rarely induces serious liver injury[122-125]. Ezetimibe, a potent inhibitor of cholesterol absorption, has been reported to improve hepatic steatosis in obese Zucker fatty rats[126]. In a randomized controlled study, six months of treatment with ezetimibe led to improvements in serum ALT levels and histologic observations[127,128].
Ursodeoxycholic acid
Ursodeoxycholic acid (UDCA) is widely used in subjects with abnormal liver function. Several studies have investigated the efficacy of UDCA as a treatment drug of NAFLD, reporting that UDCA treatment attenuated hepatic steatosis, including histologic improvement[114,129,130]. However, in a placebo controlled, randomized control trial, UDCA exhibited limited efficacy in histologic improvement in subjects with NASH and improvements in liver enzyme did not differ in the UDCA group, compared to the placebo group[130]. Accordingly, AASLD does not recommend UDCA for the treatment of NAFLD[78].
Other treatment options - future candidates
Cilostazol: SREBP-1c is a key regulator of lipogenic gene expression in hepatocytes. Recent data have shown that cilostazol, a selective type III phosphodiesterase inhibitor, inhibits SREBP-1c expression via the suppression of LXR and Sp1 activity[131]. Cilostazol also decreases serum triglyceride levels by increasing lipoprotein lipase (LPL) activity in STZ-induced diabetic rats[132]. Also, experimental data show that cilostazol stimulates LRP1 promoter activity in hepatocytes, leading to increased hepatic LRP1 expression[133]. In a study that used two experimental NAFLD models, both high-fat/high-calorie (HF/HC) diet mice and the choline-deficient/L-amino acid-defined (CDAA) diet mice, cilostazol generated improvement in hepatic steatosis in both mice models[134]. Cilostazol exhibits the potential for improvement of hepatic steatosis, and further data on its role in NAFLD are needed.
Polyunsaturated fatty acids and monounsaturated fatty acids: Polyunsaturated fatty acids (PUFAs) are found primarily in safflower, corn, soybean, cottonseed, sesame, and sunflower oils. Omega-3 fatty acids are representative of PUFA. A marked increase in long-chain PUFA n-6/n-3 ratio is observed in NAFLD patients and is associated with increased production of pro-inflammatory eicosanoids and dysregulation of liver and adipose tissue function[135]. PPAR-α activity is impaired in conditions in which levels of circulating n-3 PUFA are decreased and the n-6/n-3 fatty acid ratio is increased[136,137]. Treatment with n-3 PUFA was shown to improve biochemical parameters and alleviated hepatic steatosis by ultrasound follow-up[138,139]. Monounsaturated fatty acids (MUFAs) are comprised in olive oil. In a rat model, supplementation with MUFA resulted in improved insulin sensitivity, compared to rats fed a saturated fatty acid (SFA) diet. Additionally, GLUT4 translocation in skeletal muscle was decreased in rats fed a SFA diet, but not in those fed a MUFA diet. Increased GLUT4 translocation is related to an improvement in insulin sensitivity[140]. In obese rats, MUFA diet attenuated hepatic steatosis and altered hepatic fatty acid levels[141]. The beneficial effects of dietary MUFA in NAFLD patients should be investigated.
GLP-1 analogue: Exenatide is the synthetic form of exendin4 and it stimulates endogenous insulin secretion, leading to decreases in blood glucose. In one animal study, treatment of exendin4 resulted in a decrease of hepatic fat content, as well as reduction of fatty acid synthesis, in the liver of ob/ob mice[142]. In patients with type 2 diabetes, an exenatide treatment group showed greater improvements in liver enzymes, attenuating hepatic steatosis, than the metformin treatment group. However, this study had limitations of a lack of histologic confirmation of the liver[143]. To prove the efficacy of glucagon like peptide-1 (GLP-1) analogue in treatment of NAFLD, randomized controlled trials over a longer period are required.
MK615: MK615 is extracted from Japanese apricots, and can suppress the production of inflammatory cytokines such as TNF-α and IL-6 by inactivating NF-κB[144,145]. MK615 is regarded as a hepatoprotective agent, as it has been shown that a MK615 treatment group exhibited greater decreases in liver enzyme levels, compared with control groups. In rat models, MK615 treatment mice showed more improved liver histology than control mice[146]. Thus, further studies are required to clarify the effects of MK615 in subjects with NAFLD.
CONCLUSION
NAFLD is a common disease that can progress to liver cirrhosis. Moreover, NAFLD is strongly associated with type 2 diabetes and insulin resistance. NAFLD is the result of complex interactions among diet, metabolic components, adipose tissue inflammation, and mitochondrial dysfunction. The pathogenesis of hepatic steatosis has not yet been fully determined. In this review, we outlined previously known mechanisms of NAFLD, as well as introduced new mechanisms that have been recently discovered. Above all, we reviewed the mechanisms of drugs matched to the pathogenesis of NAFLD. Furthermore, we introduced future treatment option for NAFLD. TZDs play a key role in restoring insulin sensitivity and decreasing adipose tissue inflammation, generating histologic improvements in hepatic steatohepatitis. Pioglitazone can be used to treat NASH in patients with type 2 diabetes with biopsy-proven NAFLD; meanwhile, non-diabetic patients can be treated with vitamin E. Metformin is a well-known insulin sensitizer; however, further study is needed to prove histologic improvements in patients with NAFLD. Additionally, the cholesterol-lowering agent ezetimibe has also shown histologic improvements. Cilostazol acts on SREBP-1c and can improve dyslipidemia; however, further research is needed to clarify the relationship between NAFLD and cilostazol. Finally, there is an outstanding need for effective preventive and therapeutic regimens to overcome NAFLD.
Footnotes
P- Reviewer: Liang J, Malnick S, Pan Q S- Editor: Tian YL L- Editor: A E- Editor: Wu HL
References
- 1.Teli MR, James OF, Burt AD, Bennett MK, Day CP. The natural history of nonalcoholic fatty liver: a follow-up study. Hepatology. 1995;22:1714–1719. [PubMed] [Google Scholar]
- 2.Milić S, Stimac D. Nonalcoholic fatty liver disease/steatohepatitis: epidemiology, pathogenesis, clinical presentation and treatment. Dig Dis. 2012;30:158–162. doi: 10.1159/000336669. [DOI] [PubMed] [Google Scholar]
- 3.Angulo P. Nonalcoholic fatty liver disease. N Engl J Med. 2002;346:1221–1231. doi: 10.1056/NEJMra011775. [DOI] [PubMed] [Google Scholar]
- 4.Byrne CD, Olufadi R, Bruce KD, Cagampang FR, Ahmed MH. Metabolic disturbances in non-alcoholic fatty liver disease. Clin Sci (Lond) 2009;116:539–564. doi: 10.1042/CS20080253. [DOI] [PubMed] [Google Scholar]
- 5.Argo CK, Northup PG, Al-Osaimi AM, Caldwell SH. Systematic review of risk factors for fibrosis progression in non-alcoholic steatohepatitis. J Hepatol. 2009;51:371–379. doi: 10.1016/j.jhep.2009.03.019. [DOI] [PubMed] [Google Scholar]
- 6.Starley BQ, Calcagno CJ, Harrison SA. Nonalcoholic fatty liver disease and hepatocellular carcinoma: a weighty connection. Hepatology. 2010;51:1820–1832. doi: 10.1002/hep.23594. [DOI] [PubMed] [Google Scholar]
- 7.Browning JD, Szczepaniak LS, Dobbins R, Nuremberg P, Horton JD, Cohen JC, Grundy SM, Hobbs HH. Prevalence of hepatic steatosis in an urban population in the United States: impact of ethnicity. Hepatology. 2004;40:1387–1395. doi: 10.1002/hep.20466. [DOI] [PubMed] [Google Scholar]
- 8.Fan JG, Zhu J, Li XJ, Chen L, Lu YS, Li L, Dai F, Li F, Chen SY. Fatty liver and the metabolic syndrome among Shanghai adults. J Gastroenterol Hepatol. 2005;20:1825–1832. doi: 10.1111/j.1440-1746.2005.04058.x. [DOI] [PubMed] [Google Scholar]
- 9.Cassader M, Gambino R, Musso G, Depetris N, Mecca F, Cavallo-Perin P, Pacini G, Rizzetto M, Pagano G. Postprandial triglyceride-rich lipoprotein metabolism and insulin sensitivity in nonalcoholic steatohepatitis patients. Lipids. 2001;36:1117–1124. doi: 10.1007/s11745-001-0822-5. [DOI] [PubMed] [Google Scholar]
- 10.Zelber-Sagi S, Nitzan-Kaluski D, Halpern Z, Oren R. Prevalence of primary non-alcoholic fatty liver disease in a population-based study and its association with biochemical and anthropometric measures. Liver Int. 2006;26:856–863. doi: 10.1111/j.1478-3231.2006.01311.x. [DOI] [PubMed] [Google Scholar]
- 11.Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest. 2005;115:1343–1351. doi: 10.1172/JCI23621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cnop M, Landchild MJ, Vidal J, Havel PJ, Knowles NG, Carr DR, Wang F, Hull RL, Boyko EJ, Retzlaff BM, et al. The concurrent accumulation of intra-abdominal and subcutaneous fat explains the association between insulin resistance and plasma leptin concentrations : distinct metabolic effects of two fat compartments. Diabetes. 2002;51:1005–1015. doi: 10.2337/diabetes.51.4.1005. [DOI] [PubMed] [Google Scholar]
- 13.Miyazaki Y, Glass L, Triplitt C, Wajcberg E, Mandarino LJ, DeFronzo RA. Abdominal fat distribution and peripheral and hepatic insulin resistance in type 2 diabetes mellitus. Am J Physiol Endocrinol Metab. 2002;283:E1135–E1143. doi: 10.1152/ajpendo.0327.2001. [DOI] [PubMed] [Google Scholar]
- 14.Bugianesi E, Gastaldelli A, Vanni E, Gambino R, Cassader M, Baldi S, Ponti V, Pagano G, Ferrannini E, Rizzetto M. Insulin resistance in non-diabetic patients with non-alcoholic fatty liver disease: sites and mechanisms. Diabetologia. 2005;48:634–642. doi: 10.1007/s00125-005-1682-x. [DOI] [PubMed] [Google Scholar]
- 15.Samuel VT, Liu ZX, Qu X, Elder BD, Bilz S, Befroy D, Romanelli AJ, Shulman GI. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J Biol Chem. 2004;279:32345–32353. doi: 10.1074/jbc.M313478200. [DOI] [PubMed] [Google Scholar]
- 16.Erion DM, Shulman GI. Diacylglycerol-mediated insulin resistance. Nat Med. 2010;16:400–402. doi: 10.1038/nm0410-400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Preiss D, Sattar N. Non-alcoholic fatty liver disease: an overview of prevalence, diagnosis, pathogenesis and treatment considerations. Clin Sci (Lond) 2008;115:141–150. doi: 10.1042/CS20070402. [DOI] [PubMed] [Google Scholar]
- 18.Shimomura I, Matsuda M, Hammer RE, Bashmakov Y, Brown MS, Goldstein JL. Decreased IRS-2 and increased SREBP-1c lead to mixed insulin resistance and sensitivity in livers of lipodystrophic and ob/ob mice. Mol Cell. 2000;6:77–86. [PubMed] [Google Scholar]
- 19.Li S, Brown MS, Goldstein JL. Bifurcation of insulin signaling pathway in rat liver: mTORC1 required for stimulation of lipogenesis, but not inhibition of gluconeogenesis. Proc Natl Acad Sci USA. 2010;107:3441–3446. doi: 10.1073/pnas.0914798107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109:1125–1131. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shimano H. SREBP-1c and TFE3, energy transcription factors that regulate hepatic insulin signaling. J Mol Med (Berl) 2007;85:437–444. doi: 10.1007/s00109-007-0158-5. [DOI] [PubMed] [Google Scholar]
- 22.Uyeda K, Repa JJ. Carbohydrate response element binding protein, ChREBP, a transcription factor coupling hepatic glucose utilization and lipid synthesis. Cell Metab. 2006;4:107–110. doi: 10.1016/j.cmet.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 23.Browning JD, Horton JD. Molecular mediators of hepatic steatosis and liver injury. J Clin Invest. 2004;114:147–152. doi: 10.1172/JCI22422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Puri P, Mirshahi F, Cheung O, Natarajan R, Maher JW, Kellum JM, Sanyal AJ. Activation and dysregulation of the unfolded protein response in nonalcoholic fatty liver disease. Gastroenterology. 2008;134:568–576. doi: 10.1053/j.gastro.2007.10.039. [DOI] [PubMed] [Google Scholar]
- 25.Hummasti S, Hotamisligil GS. Endoplasmic reticulum stress and inflammation in obesity and diabetes. Circ Res. 2010;107:579–591. doi: 10.1161/CIRCRESAHA.110.225698. [DOI] [PubMed] [Google Scholar]
- 26.Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, Görgün C, Glimcher LH, Hotamisligil GS. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306:457–461. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- 27.Schwabe RF, Uchinami H, Qian T, Bennett BL, Lemasters JJ, Brenner DA. Differential requirement for c-Jun NH2-terminal kinase in TNFalpha- and Fas-mediated apoptosis in hepatocytes. FASEB J. 2004;18:720–722. doi: 10.1096/fj.03-0771fje. [DOI] [PubMed] [Google Scholar]
- 28.Gregor MF, Yang L, Fabbrini E, Mohammed BS, Eagon JC, Hotamisligil GS, Klein S. Endoplasmic reticulum stress is reduced in tissues of obese subjects after weight loss. Diabetes. 2009;58:693–700. doi: 10.2337/db08-1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Day CP, James OF. Steatohepatitis: a tale of two “hits”? Gastroenterology. 1998;114:842–845. doi: 10.1016/s0016-5085(98)70599-2. [DOI] [PubMed] [Google Scholar]
- 30.Robertson G, Leclercq I, Farrell GC. Nonalcoholic steatosis and steatohepatitis. II. Cytochrome P-450 enzymes and oxidative stress. Am J Physiol Gastrointest Liver Physiol. 2001;281:G1135–G1139. doi: 10.1152/ajpgi.2001.281.5.G1135. [DOI] [PubMed] [Google Scholar]
- 31.Halliwell B. Biochemistry of oxidative stress. Biochem Soc Trans. 2007;35:1147–1150. doi: 10.1042/BST0351147. [DOI] [PubMed] [Google Scholar]
- 32.Skulachev VP. Role of uncoupled and non-coupled oxidations in maintenance of safely low levels of oxygen and its one-electron reductants. Q Rev Biophys. 1996;29:169–202. doi: 10.1017/s0033583500005795. [DOI] [PubMed] [Google Scholar]
- 33.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roden M, Price TB, Perseghin G, Petersen KF, Rothman DL, Cline GW, Shulman GI. Mechanism of free fatty acid-induced insulin resistance in humans. J Clin Invest. 1996;97:2859–2865. doi: 10.1172/JCI118742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.García-Ruiz C, Baulies A, Mari M, García-Rovés PM, Fernandez-Checa JC. Mitochondrial dysfunction in non-alcoholic fatty liver disease and insulin resistance: cause or consequence? Free Radic Res. 2013;47:854–868. doi: 10.3109/10715762.2013.830717. [DOI] [PubMed] [Google Scholar]
- 36.Sanyal AJ, Campbell-Sargent C, Mirshahi F, Rizzo WB, Contos MJ, Sterling RK, Luketic VA, Shiffman ML, Clore JN. Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology. 2001;120:1183–1192. doi: 10.1053/gast.2001.23256. [DOI] [PubMed] [Google Scholar]
- 37.Miele L, Grieco A, Armuzzi A, Candelli M, Forgione A, Gasbarrini A, Gasbarrini G. Hepatic mitochondrial beta-oxidation in patients with nonalcoholic steatohepatitis assessed by 13C-octanoate breath test. Am J Gastroenterol. 2003;98:2335–2336. doi: 10.1111/j.1572-0241.2003.07725.x. [DOI] [PubMed] [Google Scholar]
- 38.Rolo AP, Teodoro JS, Palmeira CM. Role of oxidative stress in the pathogenesis of nonalcoholic steatohepatitis. Free Radic Biol Med. 2012;52:59–69. doi: 10.1016/j.freeradbiomed.2011.10.003. [DOI] [PubMed] [Google Scholar]
- 39.McGarry JD, Foster DW. Importance of experimental conditions in evaluating the malonyl-CoA sensitivity of liver carnitine acyltransferase. Studies with fed and starved rats. Biochem J. 1981;200:217–223. doi: 10.1042/bj2000217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vega RB, Huss JM, Kelly DP. The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor alpha in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Mol Cell Biol. 2000;20:1868–1876. doi: 10.1128/mcb.20.5.1868-1876.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Costet P, Legendre C, Moré J, Edgar A, Galtier P, Pineau T. Peroxisome proliferator-activated receptor alpha-isoform deficiency leads to progressive dyslipidemia with sexually dimorphic obesity and steatosis. J Biol Chem. 1998;273:29577–29585. doi: 10.1074/jbc.273.45.29577. [DOI] [PubMed] [Google Scholar]
- 42.Seppälä-Lindroos A, Vehkavaara S, Häkkinen AM, Goto T, Westerbacka J, Sovijärvi A, Halavaara J, Yki-Järvinen H. Fat accumulation in the liver is associated with defects in insulin suppression of glucose production and serum free fatty acids independent of obesity in normal men. J Clin Endocrinol Metab. 2002;87:3023–3028. doi: 10.1210/jcem.87.7.8638. [DOI] [PubMed] [Google Scholar]
- 43.Trayhurn P, Beattie JH. Physiological role of adipose tissue: white adipose tissue as an endocrine and secretory organ. Proc Nutr Soc. 2001;60:329–339. doi: 10.1079/pns200194. [DOI] [PubMed] [Google Scholar]
- 44.Yamauchi T, Kamon J, Minokoshi Y, Ito Y, Waki H, Uchida S, Yamashita S, Noda M, Kita S, Ueki K, et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med. 2002;8:1288–1295. doi: 10.1038/nm788. [DOI] [PubMed] [Google Scholar]
- 45.Arita Y, Kihara S, Ouchi N, Takahashi M, Maeda K, Miyagawa J, Hotta K, Shimomura I, Nakamura T, Miyaoka K, et al. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem Biophys Res Commun. 1999;257:79–83. doi: 10.1006/bbrc.1999.0255. [DOI] [PubMed] [Google Scholar]
- 46.Cnop M, Havel PJ, Utzschneider KM, Carr DB, Sinha MK, Boyko EJ, Retzlaff BM, Knopp RH, Brunzell JD, Kahn SE. Relationship of adiponectin to body fat distribution, insulin sensitivity and plasma lipoproteins: evidence for independent roles of age and sex. Diabetologia. 2003;46:459–469. doi: 10.1007/s00125-003-1074-z. [DOI] [PubMed] [Google Scholar]
- 47.Asano T, Watanabe K, Kubota N, Gunji T, Omata M, Kadowaki T, Ohnishi S. Adiponectin knockout mice on high fat diet develop fibrosing steatohepatitis. J Gastroenterol Hepatol. 2009;24:1669–1676. doi: 10.1111/j.1440-1746.2009.06039.x. [DOI] [PubMed] [Google Scholar]
- 48.Odegaard JI, Chawla A. Alternative macrophage activation and metabolism. Annu Rev Pathol. 2011;6:275–297. doi: 10.1146/annurev-pathol-011110-130138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Odegaard JI, Ricardo-Gonzalez RR, Red Eagle A, Vats D, Morel CR, Goforth MH, Subramanian V, Mukundan L, Ferrante AW, Chawla A. Alternative M2 activation of Kupffer cells by PPARdelta ameliorates obesity-induced insulin resistance. Cell Metab. 2008;7:496–507. doi: 10.1016/j.cmet.2008.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li Z, Diehl AM. Innate immunity in the liver. Curr Opin Gastroenterol. 2003;19:565–571. doi: 10.1097/00001574-200311000-00009. [DOI] [PubMed] [Google Scholar]
- 51.Kern PA, Ranganathan S, Li C, Wood L, Ranganathan G. Adipose tissue tumor necrosis factor and interleukin-6 expression in human obesity and insulin resistance. Am J Physiol Endocrinol Metab. 2001;280:E745–E751. doi: 10.1152/ajpendo.2001.280.5.E745. [DOI] [PubMed] [Google Scholar]
- 52.Carter-Kent C, Zein NN, Feldstein AE. Cytokines in the pathogenesis of fatty liver and disease progression to steatohepatitis: implications for treatment. Am J Gastroenterol. 2008;103:1036–1042. doi: 10.1111/j.1572-0241.2007.01709.x. [DOI] [PubMed] [Google Scholar]
- 53.Brun P, Castagliuolo I, Di Leo V, Buda A, Pinzani M, Palù G, Martines D. Increased intestinal permeability in obese mice: new evidence in the pathogenesis of nonalcoholic steatohepatitis. Am J Physiol Gastrointest Liver Physiol. 2007;292:G518–G525. doi: 10.1152/ajpgi.00024.2006. [DOI] [PubMed] [Google Scholar]
- 54.Li Z, Yang S, Lin H, Huang J, Watkins PA, Moser AB, Desimone C, Song XY, Diehl AM. Probiotics and antibodies to TNF inhibit inflammatory activity and improve nonalcoholic fatty liver disease. Hepatology. 2003;37:343–350. doi: 10.1053/jhep.2003.50048. [DOI] [PubMed] [Google Scholar]
- 55.Rivera CA, Adegboyega P, van Rooijen N, Tagalicud A, Allman M, Wallace M. Toll-like receptor-4 signaling and Kupffer cells play pivotal roles in the pathogenesis of non-alcoholic steatohepatitis. J Hepatol. 2007;47:571–579. doi: 10.1016/j.jhep.2007.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Thobe BM, Frink M, Hildebrand F, Schwacha MG, Hubbard WJ, Choudhry MA, Chaudry IH. The role of MAPK in Kupffer cell toll-like receptor (TLR) 2-, TLR4-, and TLR9-mediated signaling following trauma-hemorrhage. J Cell Physiol. 2007;210:667–675. doi: 10.1002/jcp.20860. [DOI] [PubMed] [Google Scholar]
- 57.Jiang W, Sun R, Wei H, Tian Z. Toll-like receptor 3 ligand attenuates LPS-induced liver injury by down-regulation of toll-like receptor 4 expression on macrophages. Proc Natl Acad Sci USA. 2005;102:17077–17082. doi: 10.1073/pnas.0504570102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Thuy S, Ladurner R, Volynets V, Wagner S, Strahl S, Königsrainer A, Maier KP, Bischoff SC, Bergheim I. Nonalcoholic fatty liver disease in humans is associated with increased plasma endotoxin and plasminogen activator inhibitor 1 concentrations and with fructose intake. J Nutr. 2008;138:1452–1455. doi: 10.1093/jn/138.8.1452. [DOI] [PubMed] [Google Scholar]
- 59.Su GL. Lipopolysaccharides in liver injury: molecular mechanisms of Kupffer cell activation. Am J Physiol Gastrointest Liver Physiol. 2002;283:G256–G265. doi: 10.1152/ajpgi.00550.2001. [DOI] [PubMed] [Google Scholar]
- 60.Aderem A, Ulevitch RJ. Toll-like receptors in the induction of the innate immune response. Nature. 2000;406:782–787. doi: 10.1038/35021228. [DOI] [PubMed] [Google Scholar]
- 61.Suzuki A, Lindor K, St Saver J, Lymp J, Mendes F, Muto A, Okada T, Angulo P. Effect of changes on body weight and lifestyle in nonalcoholic fatty liver disease. J Hepatol. 2005;43:1060–1066. doi: 10.1016/j.jhep.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 62.Xiao J, Guo R, Fung ML, Liong EC, Tipoe GL. Therapeutic approaches to non-alcoholic fatty liver disease: past achievements and future challenges. Hepatobiliary Pancreat Dis Int. 2013;12:125–135. doi: 10.1016/s1499-3872(13)60021-1. [DOI] [PubMed] [Google Scholar]
- 63.Tiikkainen M, Bergholm R, Vehkavaara S, Rissanen A, Häkkinen AM, Tamminen M, Teramo K, Yki-Järvinen H. Effects of identical weight loss on body composition and features of insulin resistance in obese women with high and low liver fat content. Diabetes. 2003;52:701–707. doi: 10.2337/diabetes.52.3.701. [DOI] [PubMed] [Google Scholar]
- 64.Palmer M, Schaffner F. Effect of weight reduction on hepatic abnormalities in overweight patients. Gastroenterology. 1990;99:1408–1413. doi: 10.1016/0016-5085(90)91169-7. [DOI] [PubMed] [Google Scholar]
- 65.Ueno T, Sugawara H, Sujaku K, Hashimoto O, Tsuji R, Tamaki S, Torimura T, Inuzuka S, Sata M, Tanikawa K. Therapeutic effects of restricted diet and exercise in obese patients with fatty liver. J Hepatol. 1997;27:103–107. doi: 10.1016/s0168-8278(97)80287-5. [DOI] [PubMed] [Google Scholar]
- 66.Huang MA, Greenson JK, Chao C, Anderson L, Peterman D, Jacobson J, Emick D, Lok AS, Conjeevaram HS. One-year intense nutritional counseling results in histological improvement in patients with non-alcoholic steatohepatitis: a pilot study. Am J Gastroenterol. 2005;100:1072–1081. doi: 10.1111/j.1572-0241.2005.41334.x. [DOI] [PubMed] [Google Scholar]
- 67.Richter EA, Hargreaves M. Exercise, GLUT4, and skeletal muscle glucose uptake. Physiol Rev. 2013;93:993–1017. doi: 10.1152/physrev.00038.2012. [DOI] [PubMed] [Google Scholar]
- 68.Dixon JB, Bhathal PS, Hughes NR, O’Brien PE. Nonalcoholic fatty liver disease: Improvement in liver histological analysis with weight loss. Hepatology. 2004;39:1647–1654. doi: 10.1002/hep.20251. [DOI] [PubMed] [Google Scholar]
- 69.de Souza CJ, Eckhardt M, Gagen K, Dong M, Chen W, Laurent D, Burkey BF. Effects of pioglitazone on adipose tissue remodeling within the setting of obesity and insulin resistance. Diabetes. 2001;50:1863–1871. doi: 10.2337/diabetes.50.8.1863. [DOI] [PubMed] [Google Scholar]
- 70.Neuschwander-Tetri BA, Brunt EM, Wehmeier KR, Oliver D, Bacon BR. Improved nonalcoholic steatohepatitis after 48 weeks of treatment with the PPAR-gamma ligand rosiglitazone. Hepatology. 2003;38:1008–1017. doi: 10.1053/jhep.2003.50420. [DOI] [PubMed] [Google Scholar]
- 71.Promrat K, Lutchman G, Uwaifo GI, Freedman RJ, Soza A, Heller T, Doo E, Ghany M, Premkumar A, Park Y, et al. A pilot study of pioglitazone treatment for nonalcoholic steatohepatitis. Hepatology. 2004;39:188–196. doi: 10.1002/hep.20012. [DOI] [PubMed] [Google Scholar]
- 72.Belfort R, Harrison SA, Brown K, Darland C, Finch J, Hardies J, Balas B, Gastaldelli A, Tio F, Pulcini J, et al. A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N Engl J Med. 2006;355:2297–2307. doi: 10.1056/NEJMoa060326. [DOI] [PubMed] [Google Scholar]
- 73.Aithal GP, Thomas JA, Kaye PV, Lawson A, Ryder SD, Spendlove I, Austin AS, Freeman JG, Morgan L, Webber J. Randomized, placebo-controlled trial of pioglitazone in nondiabetic subjects with nonalcoholic steatohepatitis. Gastroenterology. 2008;135:1176–1184. doi: 10.1053/j.gastro.2008.06.047. [DOI] [PubMed] [Google Scholar]
- 74.Sanyal AJ, Chalasani N, Kowdley KV, McCullough A, Diehl AM, Bass NM, Neuschwander-Tetri BA, Lavine JE, Tonascia J, Unalp A, et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N Engl J Med. 2010;362:1675–1685. doi: 10.1056/NEJMoa0907929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med. 2007;356:2457–2471. doi: 10.1056/NEJMoa072761. [DOI] [PubMed] [Google Scholar]
- 76.Mannucci E, Monami M, Lamanna C, Gensini GF, Marchionni N. Pioglitazone and cardiovascular risk. A comprehensive meta-analysis of randomized clinical trials. Diabetes Obes Metab. 2008;10:1221–1238. doi: 10.1111/j.1463-1326.2008.00892.x. [DOI] [PubMed] [Google Scholar]
- 77.Shah P, Mudaliar S. Pioglitazone: side effect and safety profile. Expert Opin Drug Saf. 2010;9:347–354. doi: 10.1517/14740331003623218. [DOI] [PubMed] [Google Scholar]
- 78.Chalasani N, Younossi Z, Lavine JE, Diehl AM, Brunt EM, Cusi K, Charlton M, Sanyal AJ. The diagnosis and management of non-alcoholic fatty liver disease: practice Guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Hepatology. 2012;55:2005–2023. doi: 10.1002/hep.25762. [DOI] [PubMed] [Google Scholar]
- 79.Bajaj M, Suraamornkul S, Piper P, Hardies LJ, Glass L, Cersosimo E, Pratipanawatr T, Miyazaki Y, DeFronzo RA. Decreased plasma adiponectin concentrations are closely related to hepatic fat content and hepatic insulin resistance in pioglitazone-treated type 2 diabetic patients. J Clin Endocrinol Metab. 2004;89:200–206. doi: 10.1210/jc.2003-031315. [DOI] [PubMed] [Google Scholar]
- 80.Hirose H, Kawai T, Yamamoto Y, Taniyama M, Tomita M, Matsubara K, Okazaki Y, Ishii T, Oguma Y, Takei I, et al. Effects of pioglitazone on metabolic parameters, body fat distribution, and serum adiponectin levels in Japanese male patients with type 2 diabetes. Metabolism. 2002;51:314–317. doi: 10.1053/meta.2002.30506. [DOI] [PubMed] [Google Scholar]
- 81.Lovren F, Pan Y, Quan A, Szmitko PE, Singh KK, Shukla PC, Gupta M, Chan L, Al-Omran M, Teoh H, et al. Adiponectin primes human monocytes into alternative anti-inflammatory M2 macrophages. Am J Physiol Heart Circ Physiol. 2010;299:H656–H663. doi: 10.1152/ajpheart.00115.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Luyckx FH, Desaive C, Thiry A, Dewé W, Scheen AJ, Gielen JE, Lefèbvre PJ. Liver abnormalities in severely obese subjects: effect of drastic weight loss after gastroplasty. Int J Obes Relat Metab Disord. 1998;22:222–226. doi: 10.1038/sj.ijo.0800571. [DOI] [PubMed] [Google Scholar]
- 83.Moraes-Vieira PM, Yore MM, Dwyer PM, Syed I, Aryal P, Kahn BB. RBP4 activates antigen-presenting cells, leading to adipose tissue inflammation and systemic insulin resistance. Cell Metab. 2014;19:512–526. doi: 10.1016/j.cmet.2014.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112:1796–1808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Koppaka S, Kehlenbrink S, Carey M, Li W, Sanchez E, Lee DE, Lee H, Chen J, Carrasco E, Kishore P, et al. Reduced adipose tissue macrophage content is associated with improved insulin sensitivity in thiazolidinedione-treated diabetic humans. Diabetes. 2013;62:1843–1854. doi: 10.2337/db12-0868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Marx N, Kehrle B, Kohlhammer K, Grüb M, Koenig W, Hombach V, Libby P, Plutzky J. PPAR activators as antiinflammatory mediators in human T lymphocytes: implications for atherosclerosis and transplantation-associated arteriosclerosis. Circ Res. 2002;90:703–710. doi: 10.1161/01.res.0000014225.20727.8f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bouhlel MA, Derudas B, Rigamonti E, Dièvart R, Brozek J, Haulon S, Zawadzki C, Jude B, Torpier G, Marx N, et al. PPARgamma activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab. 2007;6:137–143. doi: 10.1016/j.cmet.2007.06.010. [DOI] [PubMed] [Google Scholar]
- 88.Miyazaki Y, Mahankali A, Matsuda M, Mahankali S, Hardies J, Cusi K, Mandarino LJ, DeFronzo RA. Effect of pioglitazone on abdominal fat distribution and insulin sensitivity in type 2 diabetic patients. J Clin Endocrinol Metab. 2002;87:2784–2791. doi: 10.1210/jcem.87.6.8567. [DOI] [PubMed] [Google Scholar]
- 89.Moon JH, Kim HJ, Kim SK, Kang ES, Lee BW, Ahn CW, Lee HC, Cha BS. Fat redistribution preferentially reflects the anti-inflammatory benefits of pioglitazone treatment. Metabolism. 2011;60:165–172. doi: 10.1016/j.metabol.2009.12.007. [DOI] [PubMed] [Google Scholar]
- 90.Mayerson AB, Hundal RS, Dufour S, Lebon V, Befroy D, Cline GW, Enocksson S, Inzucchi SE, Shulman GI, Petersen KF. The effects of rosiglitazone on insulin sensitivity, lipolysis, and hepatic and skeletal muscle triglyceride content in patients with type 2 diabetes. Diabetes. 2002;51:797–802. doi: 10.2337/diabetes.51.3.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Park CY, Park SW. Role of peroxisome proliferator-activated receptor gamma agonist in improving hepatic steatosis: Possible molecular mechanism. J Diabetes Investig. 2012;3:93–95. doi: 10.1111/j.2040-1124.2012.00204.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kim HS, Xiao C, Wang RH, Lahusen T, Xu X, Vassilopoulos A, Vazquez-Ortiz G, Jeong WI, Park O, Ki SH, et al. Hepatic-specific disruption of SIRT6 in mice results in fatty liver formation due to enhanced glycolysis and triglyceride synthesis. Cell Metab. 2010;12:224–236. doi: 10.1016/j.cmet.2010.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Xiao C, Wang RH, Lahusen TJ, Park O, Bertola A, Maruyama T, Reynolds D, Chen Q, Xu X, Young HA, et al. Progression of chronic liver inflammation and fibrosis driven by activation of c-JUN signaling in Sirt6 mutant mice. J Biol Chem. 2012;287:41903–41913. doi: 10.1074/jbc.M112.415182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Marchesini G, Brizi M, Bianchi G, Tomassetti S, Zoli M, Melchionda N. Metformin in non-alcoholic steatohepatitis. Lancet. 2001;358:893–894. doi: 10.1016/s0140-6736(01)06042-1. [DOI] [PubMed] [Google Scholar]
- 95.Shaw RJ, Lamia KA, Vasquez D, Koo SH, Bardeesy N, Depinho RA, Montminy M, Cantley LC. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science. 2005;310:1642–1646. doi: 10.1126/science.1120781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Pernicova I, Korbonits M. Metformin--mode of action and clinical implications for diabetes and cancer. Nat Rev Endocrinol. 2014;10:143–156. doi: 10.1038/nrendo.2013.256. [DOI] [PubMed] [Google Scholar]
- 97.Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Li Y, Xu S, Mihaylova MM, Zheng B, Hou X, Jiang B, Park O, Luo Z, Lefai E, Shyy JY, et al. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. 2011;13:376–388. doi: 10.1016/j.cmet.2011.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bugianesi E, Gentilcore E, Manini R, Natale S, Vanni E, Villanova N, David E, Rizzetto M, Marchesini G. A randomized controlled trial of metformin versus vitamin E or prescriptive diet in nonalcoholic fatty liver disease. Am J Gastroenterol. 2005;100:1082–1090. doi: 10.1111/j.1572-0241.2005.41583.x. [DOI] [PubMed] [Google Scholar]
- 100.Garinis GA, Fruci B, Mazza A, De Siena M, Abenavoli S, Gulletta E, Ventura V, Greco M, Abenavoli L, Belfiore A. Metformin versus dietary treatment in nonalcoholic hepatic steatosis: a randomized study. Int J Obes (Lond) 2010;34:1255–1264. doi: 10.1038/ijo.2010.40. [DOI] [PubMed] [Google Scholar]
- 101.Uygun A, Kadayifci A, Isik AT, Ozgurtas T, Deveci S, Tuzun A, Yesilova Z, Gulsen M, Dagalp K. Metformin in the treatment of patients with non-alcoholic steatohepatitis. Aliment Pharmacol Ther. 2004;19:537–544. doi: 10.1111/j.1365-2036.2004.01888.x. [DOI] [PubMed] [Google Scholar]
- 102.Nar A, Gedik O. The effect of metformin on leptin in obese patients with type 2 diabetes mellitus and nonalcoholic fatty liver disease. Acta Diabetol. 2009;46:113–118. doi: 10.1007/s00592-008-0067-2. [DOI] [PubMed] [Google Scholar]
- 103.Haukeland JW, Konopski Z, Eggesbø HB, von Volkmann HL, Raschpichler G, Bjøro K, Haaland T, Løberg EM, Birkeland K. Metformin in patients with non-alcoholic fatty liver disease: a randomized, controlled trial. Scand J Gastroenterol. 2009;44:853–860. doi: 10.1080/00365520902845268. [DOI] [PubMed] [Google Scholar]
- 104.Vernon G, Baranova A, Younossi ZM. Systematic review: the epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment Pharmacol Ther. 2011;34:274–285. doi: 10.1111/j.1365-2036.2011.04724.x. [DOI] [PubMed] [Google Scholar]
- 105.Miller ER, Pastor-Barriuso R, Dalal D, Riemersma RA, Appel LJ, Guallar E. Meta-analysis: high-dosage vitamin E supplementation may increase all-cause mortality. Ann Intern Med. 2005;142:37–46. doi: 10.7326/0003-4819-142-1-200501040-00110. [DOI] [PubMed] [Google Scholar]
- 106.Bjelakovic G, Nikolova D, Gluud LL, Simonetti RG, Gluud C. Mortality in randomized trials of antioxidant supplements for primary and secondary prevention: systematic review and meta-analysis. JAMA. 2007;297:842–857. doi: 10.1001/jama.297.8.842. [DOI] [PubMed] [Google Scholar]
- 107.Hasegawa T, Yoneda M, Nakamura K, Makino I, Terano A. Plasma transforming growth factor-beta1 level and efficacy of alpha-tocopherol in patients with non-alcoholic steatohepatitis: a pilot study. Aliment Pharmacol Ther. 2001;15:1667–1672. doi: 10.1046/j.1365-2036.2001.01083.x. [DOI] [PubMed] [Google Scholar]
- 108.Lavine JE. Vitamin E treatment of nonalcoholic steatohepatitis in children: a pilot study. J Pediatr. 2000;136:734–738. [PubMed] [Google Scholar]
- 109.Vajro P, Mandato C, Franzese A, Ciccimarra E, Lucariello S, Savoia M, Capuano G, Migliaro F. Vitamin E treatment in pediatric obesity-related liver disease: a randomized study. J Pediatr Gastroenterol Nutr. 2004;38:48–55. doi: 10.1097/00005176-200401000-00012. [DOI] [PubMed] [Google Scholar]
- 110.Kugelmas M, Hill DB, Vivian B, Marsano L, McClain CJ. Cytokines and NASH: a pilot study of the effects of lifestyle modification and vitamin E. Hepatology. 2003;38:413–419. doi: 10.1053/jhep.2003.50316. [DOI] [PubMed] [Google Scholar]
- 111.Adams LA, Zein CO, Angulo P, Lindor KD. A pilot trial of pentoxifylline in nonalcoholic steatohepatitis. Am J Gastroenterol. 2004;99:2365–2368. doi: 10.1111/j.1572-0241.2004.40064.x. [DOI] [PubMed] [Google Scholar]
- 112.Satapathy SK, Garg S, Chauhan R, Sakhuja P, Malhotra V, Sharma BC, Sarin SK. Beneficial effects of tumor necrosis factor-alpha inhibition by pentoxifylline on clinical, biochemical, and metabolic parameters of patients with nonalcoholic steatohepatitis. Am J Gastroenterol. 2004;99:1946–1952. doi: 10.1111/j.1572-0241.2004.40220.x. [DOI] [PubMed] [Google Scholar]
- 113.Basaranoglu M, Acbay O, Sonsuz A. A controlled trial of gemfibrozil in the treatment of patients with nonalcoholic steatohepatitis. J Hepatol. 1999;31:384. doi: 10.1016/s0168-8278(99)80243-8. [DOI] [PubMed] [Google Scholar]
- 114.Laurin J, Lindor KD, Crippin JS, Gossard A, Gores GJ, Ludwig J, Rakela J, McGill DB. Ursodeoxycholic acid or clofibrate in the treatment of non-alcohol-induced steatohepatitis: a pilot study. Hepatology. 1996;23:1464–1467. doi: 10.1002/hep.510230624. [DOI] [PubMed] [Google Scholar]
- 115.Lefebvre P, Chinetti G, Fruchart JC, Staels B. Sorting out the roles of PPAR alpha in energy metabolism and vascular homeostasis. J Clin Invest. 2006;116:571–580. doi: 10.1172/JCI27989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Shiri-Sverdlov R, Wouters K, van Gorp PJ, Gijbels MJ, Noel B, Buffat L, Staels B, Maeda N, van Bilsen M, Hofker MH. Early diet-induced non-alcoholic steatohepatitis in APOE2 knock-in mice and its prevention by fibrates. J Hepatol. 2006;44:732–741. doi: 10.1016/j.jhep.2005.10.033. [DOI] [PubMed] [Google Scholar]
- 117.Kiyici M, Gulten M, Gurel S, Nak SG, Dolar E, Savci G, Adim SB, Yerci O, Memik F. Ursodeoxycholic acid and atorvastatin in the treatment of nonalcoholic steatohepatitis. Can J Gastroenterol. 2003;17:713–718. doi: 10.1155/2003/857869. [DOI] [PubMed] [Google Scholar]
- 118.Athyros VG, Tziomalos K, Gossios TD, Griva T, Anagnostis P, Kargiotis K, Pagourelias ED, Theocharidou E, Karagiannis A, Mikhailidis DP. Safety and efficacy of long-term statin treatment for cardiovascular events in patients with coronary heart disease and abnormal liver tests in the Greek Atorvastatin and Coronary Heart Disease Evaluation (GREACE) Study: a post-hoc analysis. Lancet. 2010;376:1916–1922. doi: 10.1016/S0140-6736(10)61272-X. [DOI] [PubMed] [Google Scholar]
- 119.Moon JH, Kang SB, Park JS, Lee BW, Kang ES, Ahn CW, Lee HC, Cha BS. Up-regulation of hepatic low-density lipoprotein receptor-related protein 1: a possible novel mechanism of antiatherogenic activity of hydroxymethylglutaryl-coenzyme A reductase inhibitor Atorvastatin and hepatic LRP1 expression. Metabolism. 2011;60:930–940. doi: 10.1016/j.metabol.2010.08.013. [DOI] [PubMed] [Google Scholar]
- 120.Jiang ZG, Robson SC, Yao Z. Lipoprotein metabolism in nonalcoholic fatty liver disease. J Biomed Res. 2013;27:1–13. doi: 10.7555/JBR.27.20120077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Mensenkamp AR, Havekes LM, Romijn JA, Kuipers F. Hepatic steatosis and very low density lipoprotein secretion: the involvement of apolipoprotein E. J Hepatol. 2001;35:816–822. doi: 10.1016/s0168-8278(01)00249-5. [DOI] [PubMed] [Google Scholar]
- 122.Chalasani N, Aljadhey H, Kesterson J, Murray MD, Hall SD. Patients with elevated liver enzymes are not at higher risk for statin hepatotoxicity. Gastroenterology. 2004;126:1287–1292. doi: 10.1053/j.gastro.2004.02.015. [DOI] [PubMed] [Google Scholar]
- 123.Vuppalanchi R, Teal E, Chalasani N. Patients with elevated baseline liver enzymes do not have higher frequency of hepatotoxicity from lovastatin than those with normal baseline liver enzymes. Am J Med Sci. 2005;329:62–65. doi: 10.1097/00000441-200502000-00002. [DOI] [PubMed] [Google Scholar]
- 124.Chalasani N. Statins and hepatotoxicity: focus on patients with fatty liver. Hepatology. 2005;41:690–695. doi: 10.1002/hep.20671. [DOI] [PubMed] [Google Scholar]
- 125.Browning JD. Statins and hepatic steatosis: perspectives from the Dallas Heart Study. Hepatology. 2006;44:466–471. doi: 10.1002/hep.21248. [DOI] [PubMed] [Google Scholar]
- 126.Deushi M, Nomura M, Kawakami A, Haraguchi M, Ito M, Okazaki M, Ishii H, Yoshida M. Ezetimibe improves liver steatosis and insulin resistance in obese rat model of metabolic syndrome. FEBS Lett. 2007;581:5664–5670. doi: 10.1016/j.febslet.2007.11.023. [DOI] [PubMed] [Google Scholar]
- 127.Yoneda M, Fujita K, Nozaki Y, Endo H, Takahashi H, Hosono K, Suzuki K, Mawatari H, Kirikoshi H, Inamori M, et al. Efficacy of ezetimibe for the treatment of non-alcoholic steatohepatitis: An open-label, pilot study. Hepatol Res. 2010;40:566–573. doi: 10.1111/j.1872-034X.2010.00644.x. [DOI] [PubMed] [Google Scholar]
- 128.Takeshita Y, Takamura T, Honda M, Kita Y, Zen Y, Kato K, Misu H, Ota T, Nakamura M, Yamada K, et al. The effects of ezetimibe on non-alcoholic fatty liver disease and glucose metabolism: a randomised controlled trial. Diabetologia. 2014;57:878–890. doi: 10.1007/s00125-013-3149-9. [DOI] [PubMed] [Google Scholar]
- 129.Leuschner UF, Lindenthal B, Herrmann G, Arnold JC, Rössle M, Cordes HJ, Zeuzem S, Hein J, Berg T. High-dose ursodeoxycholic acid therapy for nonalcoholic steatohepatitis: a double-blind, randomized, placebo-controlled trial. Hepatology. 2010;52:472–479. doi: 10.1002/hep.23727. [DOI] [PubMed] [Google Scholar]
- 130.Lindor KD, Kowdley KV, Heathcote EJ, Harrison ME, Jorgensen R, Angulo P, Lymp JF, Burgart L, Colin P. Ursodeoxycholic acid for treatment of nonalcoholic steatohepatitis: results of a randomized trial. Hepatology. 2004;39:770–778. doi: 10.1002/hep.20092. [DOI] [PubMed] [Google Scholar]
- 131.Jung YA, Kim HK, Bae KH, Seo HY, Kim HS, Jang BK, Jung GS, Lee IK, Kim MK, Park KG. Cilostazol inhibits insulin-stimulated expression of sterol regulatory binding protein-1c via inhibition of LXR and Sp1. Exp Mol Med. 2014;46:e73. doi: 10.1038/emm.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Tani T, Uehara K, Sudo T, Marukawa K, Yasuda Y, Kimura Y. Cilostazol, a selective type III phosphodiesterase inhibitor, decreases triglyceride and increases HDL cholesterol levels by increasing lipoprotein lipase activity in rats. Atherosclerosis. 2000;152:299–305. doi: 10.1016/s0021-9150(99)00480-3. [DOI] [PubMed] [Google Scholar]
- 133.Kim HJ, Moon JH, Kim HM, Yun MR, Jeon BH, Lee B, Kang ES, Lee HC, Cha BS. The hypolipidemic effect of cilostazol can be mediated by regulation of hepatic low-density lipoprotein receptor-related protein 1 (LRP1) expression. Metabolism. 2014;63:112–119. doi: 10.1016/j.metabol.2013.09.006. [DOI] [PubMed] [Google Scholar]
- 134.Fujita K, Nozaki Y, Wada K, Yoneda M, Endo H, Takahashi H, Iwasaki T, Inamori M, Abe Y, Kobayashi N, et al. Effectiveness of antiplatelet drugs against experimental non-alcoholic fatty liver disease. Gut. 2008;57:1583–1591. doi: 10.1136/gut.2007.144550. [DOI] [PubMed] [Google Scholar]
- 135.Scorletti E, Byrne CD. Omega-3 fatty acids, hepatic lipid metabolism, and nonalcoholic fatty liver disease. Annu Rev Nutr. 2013;33:231–248. doi: 10.1146/annurev-nutr-071812-161230. [DOI] [PubMed] [Google Scholar]
- 136.Araya J, Rodrigo R, Videla LA, Thielemann L, Orellana M, Pettinelli P, Poniachik J. Increase in long-chain polyunsaturated fatty acid n - 6/n - 3 ratio in relation to hepatic steatosis in patients with non-alcoholic fatty liver disease. Clin Sci (Lond) 2004;106:635–643. doi: 10.1042/CS20030326. [DOI] [PubMed] [Google Scholar]
- 137.Di Minno MN, Russolillo A, Lupoli R, Ambrosino P, Di Minno A, Tarantino G. Omega-3 fatty acids for the treatment of non-alcoholic fatty liver disease. World J Gastroenterol. 2012;18:5839–5847. doi: 10.3748/wjg.v18.i41.5839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Capanni M, Calella F, Biagini MR, Genise S, Raimondi L, Bedogni G, Svegliati-Baroni G, Sofi F, Milani S, Abbate R, et al. Prolonged n-3 polyunsaturated fatty acid supplementation ameliorates hepatic steatosis in patients with non-alcoholic fatty liver disease: a pilot study. Aliment Pharmacol Ther. 2006;23:1143–1151. doi: 10.1111/j.1365-2036.2006.02885.x. [DOI] [PubMed] [Google Scholar]
- 139.Bouzianas DG, Bouziana SD, Hatzitolios AI. Potential treatment of human nonalcoholic fatty liver disease with long-chain omega-3 polyunsaturated fatty acids. Nutr Rev. 2013;71:753–771. doi: 10.1111/nure.12073. [DOI] [PubMed] [Google Scholar]
- 140.Moon JH, Lee JY, Kang SB, Park JS, Lee BW, Kang ES, Ahn CW, Lee HC, Cha BS. Dietary monounsaturated fatty acids but not saturated fatty acids preserve the insulin signaling pathway via IRS-1/PI3K in rat skeletal muscle. Lipids. 2010;45:1109–1116. doi: 10.1007/s11745-010-3475-3. [DOI] [PubMed] [Google Scholar]
- 141.Hanke D, Zahradka P, Mohankumar SK, Clark JL, Taylor CG. A diet high in α-linolenic acid and monounsaturated fatty acids attenuates hepatic steatosis and alters hepatic phospholipid fatty acid profile in diet-induced obese rats. Prostaglandins Leukot Essent Fatty Acids. 2013;89:391–401. doi: 10.1016/j.plefa.2013.09.009. [DOI] [PubMed] [Google Scholar]
- 142.Ding X, Saxena NK, Lin S, Gupta NA, Anania FA. Exendin-4, a glucagon-like protein-1 (GLP-1) receptor agonist, reverses hepatic steatosis in ob/ob mice. Hepatology. 2006;43:173–181. doi: 10.1002/hep.21006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Fan H, Pan Q, Xu Y, Yang X. Exenatide improves type 2 diabetes concomitant with non-alcoholic fatty liver disease. Arq Bras Endocrinol Metabol. 2013;57:702–708. doi: 10.1590/s0004-27302013000900005. [DOI] [PubMed] [Google Scholar]
- 144.Kawahara K, Hashiguchi T, Masuda K, Saniabadi AR, Kikuchi K, Tancharoen S, Ito T, Miura N, Morimoto Y, Biswas KK, et al. Mechanism of HMGB1 release inhibition from RAW264.7 cells by oleanolic acid in Prunus mume Sieb. et Zucc. Int J Mol Med. 2009;23:615–620. doi: 10.3892/ijmm_00000172. [DOI] [PubMed] [Google Scholar]
- 145.Morimoto Y, Kikuchi K, Ito T, Tokuda M, Matsuyama T, Noma S, Hashiguchi T, Torii M, Maruyama I, Kawahara K. MK615 attenuates Porphyromonas gingivalis lipopolysaccharide-induced pro-inflammatory cytokine release via MAPK inactivation in murine macrophage-like RAW264.7 cells. Biochem Biophys Res Commun. 2009;389:90–94. doi: 10.1016/j.bbrc.2009.08.103. [DOI] [PubMed] [Google Scholar]
- 146.Hokari A, Ishikawa T, Tajiri H, Matsuda T, Ishii O, Matsumoto N, Okuse C, Takahashi H, Kurihara T, Kawahara K, et al. Efficacy of MK615 for the treatment of patients with liver disorders. World J Gastroenterol. 2012;18:4118–4126. doi: 10.3748/wjg.v18.i31.4118. [DOI] [PMC free article] [PubMed] [Google Scholar]