

Abstract
Palladium-catalyzed acetoxylation of allylic C–H bonds has been the subject of extensive study. These reactions proceed via allyl-palladium(II) intermediates that react with acetate to afford the allyl acetate product. Benzoquinone and molecular oxygen are two common oxidants for these reactions. Benzoquinone has been shown to promote allyl acetate formation from well-defined π-allyl palladium(II) complexes. Here, we assess the ability of O2 to promote similar reactions with a series of “unligated” π-allyl palladium(II) complexes (i.e., in the absence of ancillary phosphorus, nitrogen or related donor ligands). Stoichiometric and catalytic allyl acetate formation is observed under aerobic conditions with several different alkenes. Mechanistic studies are most consistent with a “pull” mechanism in which O2 traps the Pd0 intermediate following reversible C–O bond-formation from an allyl-palladium(II) species. A “push” mechanism, involving oxidatively induced C–O bond formation, does not appear to participate. These results and conclusions are compared with benzoquinone-promoted allylic acetoxylation, in which a “push” mechanism seems to be operative.
Introduction
Palladium-catalyzed acetoxylation of allylic C–H bonds provides an appealing method for anti-Markovnikov functionalization of alkenes (Scheme 1).1–3 The vast majority of these reactions employ benzoquinone (BQ) as the stoichiometric oxidant; however, reactions with molecular oxygen, hypervalent iodine and other oxidants have also been reported.2,3
Scheme 1.
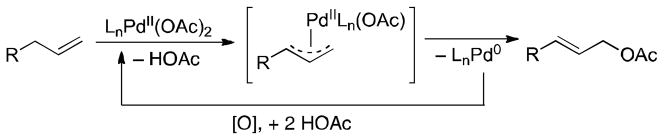
Pd-Catalyzed Oxidative Allylic Acetoxylation Reactions
Fundamental studies have shown that BQ promotes C–O reductive elimination4 from well-defined π-allyl-palladium(II) species to form allyl acetates.1f,g,i,5,6 These observations potentially explain the beneficial effect of BQ in allylic acetoxylation reactions.1–2 BQ could promote the acetoxylation of π-allyl-PdII intermediates via two possible pathways: (1) BQ coordination to the π-allyl-PdII intermediate could withdraw electron density from the PdII center in an oxidatively-induced reductive elimination pathway (“push” mechanism, Scheme 2),7 or (2) BQ could trap the Pd0 intermediate that forms in a reversible C–O reductive elimination step (“pull” mechanism, Scheme 2). In both cases, BQ also serves as the oxidant for conversion of Pd0 to PdII.8 The former mechanism is commonly invoked in the literature, and tentative NMR spectroscopic evidence has been offered in support of this pathway.5c On the other hand, Bercaw and coworkers observed reversible C–O reductive elimination from a bipyrimidine-ligated allyl-PdII species in the absence of BQ and proposed a “pull” mechanism.1i
Scheme 2.

“Push” versus “Pull” Mechanisms to Account for the Promotion Effect of BQ in Pd-Catalyzed Acetoxylation of Allylic C–H Bonds.
Allylic acetoxylation reactions would be more appealing if BQ could be replaced with O2 as the stoichiometric oxidant,9 and several groups have recently reported progress toward this goal. Kaneda and coworkers reported aerobic allylic acetoxylation in N,N-dimethylacetamide as the solvent under elevated pressures (6 atm) of O2 (eq 1).10 Liu and coworkers achieved aerobic allylic imidation under similar conditions by employing 40 mol % of maleic anhydride as an additive in the reaction (eq 2).11 Finally, we reported that use of 4,5-diazafluorenone as an ancillary ligand enables aerobic allylic acetoxylation of alkenes under 1 atm O2 (eq 3).12
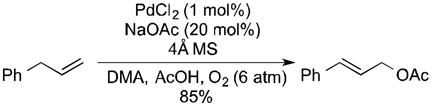 |
(1) |
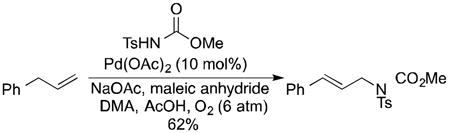 |
(2) |
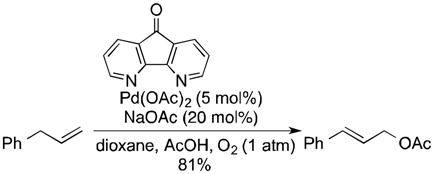 |
(3) |
In connection with our previous study,12 we sought to probe the potential role of O2 in promoting CO reductive elimination from π-allyl-PdII complexes. Such reactivity could be combined with the ability of O2 to oxidize palladium(0) to palladium(II)13 to achieve catalytic aerobic allylic C–H acetoxylation. Here, we describe the reactivity of “unligated” π-allyl-PdII complexes (i.e., lacking a well-defined ancillary neutral donor ligand) in the presence of O2. Selected π-allyl-PdII complexes undergo stoichiometric acetoxylation in the presence of O2, and these observations have been extended to catalytic reactivity. Kinetic and mechanistic studies of the stoichiometric reactions suggest that O2 promotes C–O reductive elimination by a “pull”-type mechanism.12 These observations are compared to reactions involving BQ.
Results and Discussion
Stoichiometric Acetoxylation of π-Allyl-PdCl Complexes in the Presence and Absence of Oxidants
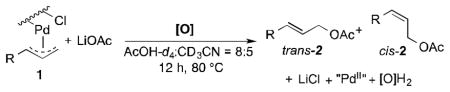
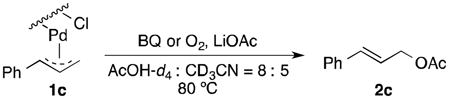
Well-defined π-allyl-PdCl complexes derived from propene (1a), methyl-3-butenoate (1b) and allyl benzene (1c) (Table 1), were prepared according to literature procedures14 and dissolved in a mixture of AcOH-d4 and CD3CN in a ratio of 4:2.5. The use of CD3CN ensured solubility of the Pd complexes and LiOAc, which served as the acetate source. These solutions were then heated for 12 h in the presence of BQ (12 equiv), O2 (2.3 atm) or under an inert (N2) atmosphere. All reactions carried out in the presence of BQ afforded the terminal allylic acetate in good yields (Table 1, entries 1, 4 and 7). For the reactions carried out in the presence of O2, 1a yielded trace acetate product (entry 2), while 1b and 1c reacted to afford allylic acetates 2b and 2c in 94% and 31% yields, respectively. In the reaction with 1c under O2, multiple unidentified by-products started to emerge after 5 hours and formation of the allylic acetate product stopped at 31% yield. Complexes 1a and 1c exhibit negligible reactivity in the absence of BQ or O2 (entries 3 and 9), while the electron-deficient π-allyl-PdCl complex 1b underwent slow acetoxylation under N2, resulting in a 19% yield of 2b after 12 h together with the formation of Pd black (entry 6). No Pd black was observed in the other reactions.
Table 1.
Acetoxylation of π-Allyl-Pd Complexes in the Presence of BQ and O2.a
| ||||
|---|---|---|---|---|
| NMR yield (%) | ||||
|
| ||||
| Entry | π-allyl Pd complex | [O] | trans-2 | cis-2 |
| 1 |
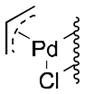 1a |
BQ | 69 | 0 |
| 2 | O2 | 2 | 0 | |
| 3 | None | 1 | 0 | |
| 4 |
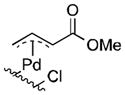 1b |
BQ | 93 | 7 |
| 5 | O2 | 94 | 6 | |
| 6 | None | 19 | 0 | |
| 7 |
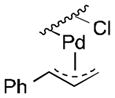 1c |
BQ | 96 | 0 |
| 8 | O2 | 31b | 0 | |
| 9 | None | 1 | 0 | |
Reactions were performed in NMR tubes and monitored by 1H NMR spectroscopy with 1,3,5-tri-tertbutylbenzene as the internal standard. Reaction conditions: [Pd]0 = 2.5 mM, [LiOAc]0 = 12 mM, [O] = 30 mM, AcOH-d4 = 0.4 mL, CD3CN = 0.25 mL, reaction time = 12 h.
Unidentified by-product started to form at 31% conversion, at which point the formation of cinnamyl acetate stopped.
Characterization of the π-Allyl-PdCl Complexes in the AcOH/CH3CN Solvent Mixture
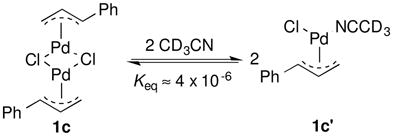
The allylbenzene-derived π-allyl-PdCl complex 1c was selected for further study because it exhibits virtually no reactivity in the absence of an oxidant, but reacts to form cinnamyl acetate 2c in the presence of O2 or benzoquinone. The speciation of 1c in solution was evaluated by UV-visible and 1H NMR spectroscopy. The UV-visible spectrum of the 1c in AcOH exhibits a strong absorption peak at 290 nm (Fig. 2A). Upon addition of CH3CN into the solution, the 290 nm band maximum shifts to 270 nm with a clean isosbestic point (Fig. 1A). A plot of the absorption at 270 nm versus [CH3CN] fits a hyperbolic function (i.e., saturation behavior) (Fig. 1B). A similar titration experiment was evaluated by 1H NMR spectroscopy in deuterated solvent, starting with a somewhat higher concentration of 1c (Fig. 2), and similar behavior was observed. The benzylic proton resonance of the π-allyl ligand shifts downfield upon addition of CD3CN to a solution of 1c in AcOH-d4 (Fig. 3). The data fit a hyperbolic dependence on [CD3CN] up to approximately [CD3CN] = 2 M, and follow a linear trend at high concentrations.
Fig. 2.

The chemical shift of the benzylic proton of 1c ([C6H5CHCHCH2PdCl]2) in AcOH-d4 as a function of [CD3CN] determined by 1H NMR spectroscopy. The curve fit reflects a nonlinear least-squares fit to a modified hyperbolic function of [CD3CN]: δ = [CD3CN]/(c1 + c2[CD3CN]) + c3[CD3CN]) + δ0. Reaction conditions: [1c] = 0.25 mM, solvent = AcOH-d4, total solution volume = 0.65 mL, 24 °C.
Fig. 1.

Titration of CH3CN into a solution of 1c in AcOH determined by UV-Visible spectroscopy, UV-Visible spectra (A) and the absorption of the peak at 270 nm as a function of [CH3CN] (B). Conditions: [1c]0 = 0.025 mM, solvent = AcOH, 22 °C.
Fig. 3.

Kinetic orders of O2-promoted acetoxylation of 1c: dependence of the initial rate on [1c] (A) and [O2] (B). Reaction conditions: AcOH-d4 = 0.4 mL, CD3CN = 0.25 mL, 80 °C, [LiOAc] = 30 mM, (A) [O2] = 13 mM and (B) [1c]0 = 1.5 mM.
π-Allyl-PdCl complexes exist as dimers in AcOH; however, neutral donor ligands, such as pyridine, can coordinate to PdII and convert the dimeric species into monomers.15 The data in Figs. 2 and 3 are consistent with this behavior, and the hyperbolic dependence of the UV-visible absorption and 1H NMR chemical shift is attributed to the conversion of 1c into a monomer 1c′ upon coordination of CH3CN (eq 4). The linear dependence of the 1H NMR resonance observed at high [CD3CN] is attributed to a solvent effect, associated with the bulk change from 100% AcOH to a large fraction of CD3CN as the solvent. Based on the UV-visible spectra, the equilibrium constant of this dimer-monomer equilibrium (Keq) is calculated to be 4.3 × 10−6 M−1 (± 0.2 × 10−6 M−1). An equilibrium constant of 3.7 × 10−6 M−1 (± 0.2 × 10−6 M−1) is calculated on the basis of the 1H NMR spectra (see Supporting Information for details).
 |
(4) |
Mechanistic Studies of Stoichiometric Acetoxylation of the π-Allyl-PdCl Complex 1c
The reaction of 1c proceeds to incomplete conversion to cinnamyl acetate 2c in the presence of O2 (cf. Table 1, entry 8); however, the reaction is well behaved at early times and was investigated by the method of initial rates (eq 5). Formation of 2c was monitored by 1H NMR spectroscopy and quantified by integrating the acetate methylene doublet at 4.76 ppm (C6H5CHCHCH2OAc). The acetoxylation of 1c in the presence of O2 exhibits first order dependence on [1c] and [O2] (Fig. 3). The effect of BQ on the acetoxylation of 1c was determined under similar reaction conditions. The initial rate displays a first order dependence on [1c] and [BQ] (Fig. 4). The slope of the reaction rate as a function of [BQ] is almost 10 times greater relative to the rate as a function of [O2].
Fig. 4.

Kinetic orders of BQ-promoted acetoxylation of 1c: dependence of the initial rate on [1c] (A) and [BQ] (B). Reaction conditions: AcOH-d4 = 0.4 mL, CD3CN = 0.25 mL, 80 °C, (A) [LiOAc] = 12 mM, [BQ] = 30 mM (B) [LiOAc] = 30 mM, [1c]0 = 1.5 mM.
 |
(5) |
A Hammett study was performed to assess electronic effects on the acetoxylation of p-substituted allylbenzene-derived π-allyl-PdII complexes in the presence of BQ and O2 (eq 6). The rho (ρ) values obtained from these reactions, 0.90 and 1.0 for the reactions with BQ and O2, respectively, show that electron-deficient allyl-PdII complexes react more rapidly (Fig. 5). We attribute this effect to ground-state destabilization of the π-allyl-PdII species by electron-withdrawing substituents, resulting in more facile reduction of PdII to Pd0 and nucleophilic attack by acetate on the π-allyl ligand.16
Fig. 5.

Hammett plot of the acetoxylation of p-substituted π-allyl-PdCl complexes in the presence of BQ (A) and O2 (B). Reaction conditions: [Pd]0 = 3 mM, [LiOAc] = 30 mM, AcOH-d4 = 0.4 mL, CD3CN = 0.25 mL, 80 °C, (A) BQ = 30 mM; (B) [O2] = 13 mM.
 |
(6) |
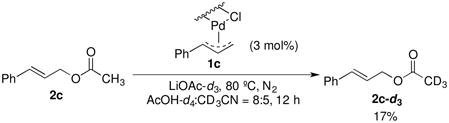
In order to probe whether the reductive C–O bond formation is reversible, we conducted an acetate scrambling experiment with 2c. Cinnamyl acetate, 2c, and a catalytic quantity of 1c (3 mol %) were mixed with LiOAc-d3 under an inert atmosphere (eq 7). After 12 hours, the overall concentration of cinnamyl acetate 2c and 1c remained constant, but 17% of the total cinnamyl acetate 2c was converted to deuterium labeled cinnamyl acetate 2c-d3. Accordingly, the same quantity of free AcOH was formed.
 |
(7) |
Pd(OAc)2-Catalyzed Aerobic Allylic Acetoxylation of Alkenes
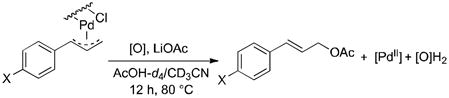
Following these stoichiometric reactions, we investigated catalytic allylic C–H acetoxylation reactions of alkenes with Pd(OAc)2 under the conditions similar to those used in the stoichiometric experiments (cf. Tables 1 and 2). 1-Octene was used as a representative alkyl olefin, and no acetoxylation product was observed with this substrate under 60 psi of O2 after 24 hours (Table 2, entry 1). Instead, isomerization of the C=C bond occurred, resulting in a mixture of octene isomers. In contrast, methyl 3-butenoate and allyl benzene yielded the corresponding trans-allylic acetate products in 75% and 67% yields, respectively (Table 2, entries 2 and 3), together with unreacted alkene. The reaction of β-methylstyrene was also tested and led to a 30% yield of cinnamyl acetate (Table 2, entry 4). The favorable reactivity of allylbenzene relative to β-methylstyrene may be attributed to the increased acidity of the allylic C–H bond and/or the more-favorable binding of the terminal alkene in allylbenzene. Overall, the relative reactivity of these alkenes under catalytic conditions, methyl-3-butenoate > allyl benzene > 1-octene, correlates with the acidity of the allylic C–H bond.17 The catalytic reactivity also tracks with the relative rates of the stoichiometric reactions of π-allyl-PdII complexes (cf. Table 1).
Table 2.
Pd(OAc)2-Catalyzed Aerobic Allylic Acetoxylation Reactions.a
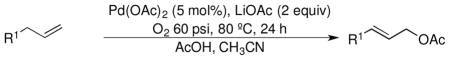
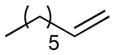
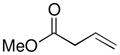
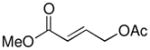
Reaction conditions: [substrate]0 = 0.5 M, [LiOAc] = 1 M, AcOH-d4 = 2 ml, CD3CN = 1.2 ml.
Yields were determined by 1H NMR spectroscopy with 1,3,5-trimethoxybenzene as the internal standard. Only the trans alkene isomer was obtained.
Double bond migration was observed.
Consideration of the Mechanism of O2-Promoted Acetoxylation and Comparison of O2 and BQ as Stoichiometric Oxidants
The stoichiometric and catalytic results presented above demonstrate that O2 can promote stoichiometric and catalytic allylic C–H acetoxylation reactions with “unligated” PdII complexes; however, this effect appears to be limited to activated (i.e., electron deficient) alkenes. The origin of O2 promotion in stoichiometric reactions of π-allyl-PdII complexes could potentially arise from a “push” or a “pull” effect (Scheme 3; see also, Scheme 2).1c Specifically, O2 could coordinate to PdII to generate a more electrophilic or oxidized π-allyl-Pd species that is more susceptible to nucleophilic attack by acetate (“push” mechanism), or O2 could trap the Pd0 species generated in C–O bond-forming step (“pull” mechanism).
Scheme 3.

Possible Pathways for Oxidant-Promoted Acetoxylation of π-allyl-Pd Complexes, [O] = BQ or O2.
The kinetic data cannot definitively distinguish between these two mechanisms
The rate laws in Scheme 3 show that both mechanisms could lead to a first-order dependence on [1c] and the oxidant, [O2] or [BQ] (i.e., when k4[O] ≪ k-3), as has been observed experimentally (cf. Figs. 3 and 4). The “pull” mechanism could exhibit a saturation or zero-order dependence on the oxidant; however, this kinetic regime may not be readily accessible for the reactions described here. In principle, the mechanisms could be distinguished by the kinetic dependence on [–OAc] because the “push” and “pull” mechanisms are predicted to exhibit a saturation and first-order kinetic dependence on [–OAc], respectively. A saturation kinetic dependence on [-OAc] in the presence of O2 was observed (Figure S5). This result could support a “push” mechanism; however, 1H NMR spectroscopic studies show that acetate coordinates to the ground-state (π-allyl)PdII complex (Figure S4), and this equilibrium binding step could explain a saturation kinetic dependence on [–OAc], even in the case of a “pull” mechanism. This complexity is compounded by an inability to distinguish between inner-sphere versus outer-sphere C–O reductive elimination (i.e., nucleophilic attack by acetate), which also contributes to the ambiguity of the rate-dependence on [OAc].
Despite the ambiguities associated with the kinetic data, several considerations argue in favor a “pull” mechanism for O2 and a “push” mechanism for BQ
The acetate scrambling experiments with 1c show that C–O bond formation is reversible. That this process takes place on a time scale similar to the stoichiometic acetoxylation reaction under O2 (cf. Table 1 and eq 6) is consistent with O2 trapping of the Pd0 product of the reaction (i.e., a “pull” mechanism). In this case, aerobic allylic acetoxylation reactions (e.g., eqs. 1 and 3) will be effective only with catalyst systems or under reaction conditions that enable C–O reductive elimination in the absence of an oxidant. This conclusion would explain the limited scope of catalytic allylic acetoxylation reactions typically observed when O2 is used as the oxidant.
The reactions of the “unligated” π-allyl-PdII complex 1c with BQ are nearly 10-fold faster than those with O2 as the oxidant (Figs. 3 and 4). If BQ and O2 promote the reaction via the same mechanism, this observation would require that (reversible) C–O reductive elimination is much more rapid than the O2-based turnover rate and that BQ is significantly more effective than O2 in trapping the Pd0 species (k4, Scheme 3).18 Alternatively, BQ could directly promote C–O bond formation in a “push” mechanism (k1, Scheme 3). This possibility could explain the broader scope of catalytic allylic acetoxylation reactions typically observed with BQ as the oxidant.
Several observations from allylic acetoxylation reactions mediated by diazafluorenone (DAF)-PdII complexes are relevant to the present discussion.12 π-Allyl-PdII complexes with DAF and 2,2′-bipyridyl (4,4′-tBu2bpy) ancillary ligands were compared in stoichiometric acetoxylation and acetate-scrambling experiments, similar to the experiments shown in Table 1 and eq 6. The results, which are summarized in Fig. 6, show that the DAF-ligated π-allyl-PdII complex forms allyl acetate in the presence of BQ, O2 and even under N2, while the tBu2bpy-ligated complex reacts only in the presence of BQ. The acetate scrambling data show that the DAF-ligated complex undergoes reversible C–O bond formation in the presence of O2 and under an inert atmosphere, while no acetate scrambling occurs with the tBu2bpy-ligated complex under any conditions. Taken together, the observations suggest that O2 can serve as an effective trap for Pd0 (“pull” mechanism) upon C–O reductive elimination from PdII, but it cannot directly promote C–O reductive elimination (“push” mechanism). The ability of BQ to promote allyl acetate formation from the tBu2bpy complex, which does not undergo reversible C–O reductive elimination, is most readily explained by direct promotion of allyl acetate formation by BQ via a “push” mechanism.
Fig. 6.

Comparison between the reactivity of DAF- and tBu2bpy-ligated π-allyl-PdII complexes in the presence of O2 and BQ reported in ref. 12.
The broad scope of aerobic allylic C–H acetoxylation observed with the DAF/Pd(OAc)2 appears to reflect, at least in part, the ability of the DAF ligand to facilitate C–O reductive elimination and thereby promote the “pull” mechanism. This property of DAF seems quite unusual, and ongoing studies seek to gain additional insights into the role of DAF in these reactions. Meanwhile, it seems possible that other ligands could be identified to promote a “push”-type mechanism. For example, Mirica and coworkers have shown that certain preorganized tri- and tetradentate ligands can promote stoichiometric oxidation of organopalladium(II) complexes by O2 and facilitate reductive elimination reactions.19 Incorporation of such concepts into catalytic allylic acetoxylation reactions could lead to further expansion of the scope of reactions compatible with O2.
Conclusion
Overall, this study highlights both opportunities and constraints on the use of O2 as an oxidant in Pd-catalyzed oxidation reactions. Mechanistic studies of allylic C–H acetoxylation with “unligated” PdII complexes and catalysts show that aerobic catalytic turnover is possible when the PdII/substrate-based intermediate undergoes facile reductive elimination to afford a Pd0 intermediate that can react with O2. When reductive elimination from PdII is not facile, oxidants other than O2 (BQ, in the present allylic oxidation reactions) are required to promote product formation via “oxidatively induced” reductive elimination. The identification of ligands, such as 4,5-diazafluorenone, that facilitate reductive elimination from PdII provides one strategy to expand the scope of Pd-catalyzed aerobic oxidation reactions.
Experimental
All commercially available compounds and deuterated solvents were used as received, and were purchased from Sigma-Aldrich. All NMR experiments were acquired on a Varian INOVA-500 MHz spectrometer. The chemical shifts (δ) of 1H NMR spectra are given in parts per million and referenced to the residual acetate proton of AcOH-d4 (2.04 ppm). Unless specified, all experiments used recrystalized 1,3,5-tri-(tert-butyl)benzene as the internal standard. All reactions were conducted in J-Young tubes to determine the dependence the initial rate on [O2], except for the experiments in Fig. 4B, which were performed in thick-wall flame-sealed NMR tubes.
General Procedure for Synthesis of π-Allyl Palladium Complexes
All π-allyl palladium complexes were prepared according to literature procedures.14 A representative procedure for the synthesis of cinnamyl derived π-allyl palladium complex 1c is presented here. To an oven-dried round bottom flask equipped with a magnetic stir bar was added palladium trifluoroacetate (154 mg, 0.5 mmol). The flask was wrapped with aluminum foil, and purged with N2. Dry THF (5 mL) and allyl benzene (66 μL, 0.5 mmol) were added via syringe. The solution was stirred for 1 hour at room temperature, followed by addition of tetrabutylammonium chloride (150 mg, 0.55 mmol). The solution was allowed to stir for another 30 min. After the reaction, the mixture was filtered through Celite to remove palladium black. The resulting yellow solid was purified by silica gel column chromatography, and recrystallized from CH2Cl2 and hexane. The 1H NMR spectrum of this compound is consistent with the literature.14
General Procedure for Pd(OAc)2-Catalyzed Aerobic Allylic Acetoxylation of Alkenes
Allylbenzene (132 μL, 1 mmol), LiOAc (130 mg, 3 mmol) and Pd(OAc)2 (10 mg, 5 mol%) were weighed out in a thick-wall pressure tube. 1.2 ml HOAc and 0.8 ml CH3CN were then added. The tube was equipped with a pressure regulator. The tube was charged with 60 psi of O2 following purging and allowed to stir at 80 °C overnight. The reaction was then cooled to room temperature and trimethoxybenzene (50 mg, 0.3 mmol) was added as an internal standard. Solvent was then removed by rotovap and the sample was analyzed by 1H NMR spectroscopy. Reactions with other alkene substrates applied the same procedure as allylbenzene.
Reversible Ligand Exchange Reaction
The allyl benzene derived π-allyl palladium complex 1c (2.2 mg, 4.0 mmol), cinnamyl acetate 2c (5.5 mg, 5.2 μl, 31 mmol) and LiOAc-d3 (2.7 mg, 40 mmol) were added into a J-Young tube. AcOH-d4 and CD3CN stock solution were added via gas-tight syringe. The NMR tube was immediately placed in an acetone/dry ice bath. Three freeze-pump-thaw cycles were carried out to degas the solvent. N2 (1 atm) was introduced to the tube. The probe of the NMR spectrometer was heated to 80 °C. The NMR tube was placed into the INOVA spectrometer and data were acquired over 12 h.
Supplementary Material
Acknowledgments
We thank Dr. Charlie G. Fry for NMR spectroscopic assistance, and we are grateful for financial support from the NIH (R01 GM067173).
Footnotes
Supporting Information Available: Experimental procedures, and characterization data.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.For leading references, see the following: Kikukawa K, Sakai K, Asada K, Matsuda T. J Organomet Chem. 1974;77:131–145.McMurry JE, Kocotovský P. Tetrahedron Lett. 1984;25:4187–4190.Jia C, Müller P, Mimoun H. J Mol Catal A: Chem. 1995;101:127–136.El Firdoussi L, Baqqa A, Allaoud S, Ait Allal B, Karim A, Castanet Y, Mortreux A. J Mol Catal A: Chem. 1998;135:11–22.Grennberg H, Bäckvall JE. Chem Eur J. 1998;4:1083–1089.Chen MS, White MC. J Am Chem Soc. 2004;126:1346–1347. doi: 10.1021/ja039107n.Chen MS, Prabagaran N, Labenz NA, White MC. J Am Chem Soc. 2005;127:6970–6971. doi: 10.1021/ja0500198.Speziali MG, Robles-Dutenhefner PA, Gusevskaya EV. Organometallics. 2007;26:4003–4009.Lin BL, Labinger JA, Bercaw JE. Can J Chem. 2009;87:264–271.Pilarski LT, Selander N, Böse D, Szabó KnJ. Org Lett. 2009;11:5518–5521. doi: 10.1021/ol9023369.Vermeulen NA, Delcamp JH, White MC. J Am Chem Soc. 2010;132:11323–11328. doi: 10.1021/ja104826g.Henderson WH, Check CT, Proust N, Stambuli JP. Org Lett. 2010;12:824–827. doi: 10.1021/ol902905w.Thiery E, Aouf C, Belloy J, Harakat D, Le Bras J, Muzart J. J Org Chem. 2010;75:1771–1774. doi: 10.1021/jo902587u.Check CT, Henderson WH, Wray BC, Vanden Eynden MJ, Stambuli JP. J Am Chem Soc. 2011;133:18503–18505. doi: 10.1021/ja2089102.Strambeanu II, White MC. J Am Chem Soc. 2013;135:12032–12037. doi: 10.1021/ja405394v.
- 2.For examples of reactions that use catalytic BQ in combination with another stoichiometric oxidant, such as MnO2 or CuCl2, see: Heumann A, Reglier M, Waegell B. Angew Chem, Int Ed Engl. 1982;21:366–367.Heumann A, Åkermark B. Angew Chem Int Ed. 1984;23:453–454.Hansson S, Heumann A, Rein T, Åkermark B. J Org Chem. 1990;55:975–984.Principato B, Maffei M, Siv C, Buono G, Peiffer G. Tetrahedron. 1996;52:2087–2096.
- 3.For examples of reactions that use hypervalent iodine and peroxides as the oxidants (in the absence of BQ), see: Alam R, Pilarski L, Pershagen E, Szabo K, Szabo Kln. J Am Chem Soc. 2012;134:8778–8781. doi: 10.1021/ja302457p.Shi E, Shao Y, Chen S, Hu H, Liu Z, Zhang J, Wan X. Org Lett. 2012;14:3384–3387. doi: 10.1021/ol3013606.Zhou Z, Andrus MB. Tetrahedron Lett. 2012;53:4518–4521.Sharma A, Hartwig JF. J Am Chem Soc. 2013;135:17983–17989. doi: 10.1021/ja409995w.
- 4.The term “C–O reductive elimination” here and elsewhere is intentionally ambiguous with respect to inner-sphere vs. outer-sphere attack of an acetate on the allyl ligand. For a leading reference addressing this issues, see: Bäckvall JE, Nordberg RE, Wilhelm D. J Am Chem Soc. 1985;107:6892–6898.
- 5.(a) Bäckvall JE, Bystroem SE, Nordberg RE. J Org Chem. 1984;49:4619–4631. [Google Scholar]; (b) Bäckvall JE, Nystroem JE, Nordberg RE. J Am Chem Soc. 1985;107:3676–3686. [Google Scholar]; (c) Bäckvall JE, Gogoll A. Tetrahedron Lett. 1988;29:2243–2246. [Google Scholar]
- 6.For BQ-induced allylic chlorination, see: Szabó KJ. Organometallics. 1998;17:1677–1686.
- 7.BQ-promoted reductive elimination has been observed with Pd-alkyl and Pd-hydride complexes. See the following: Lanci MP, Remy MS, Kaminsky W, Mayer JM, Sanford MS. J Am Chem Soc. 2009;131:15618–15620. doi: 10.1021/ja905816q.Decharin N, Stahl SS. J Am Chem Soc. 2011;133:5732–5735. doi: 10.1021/ja200957n.
- 8.For a study of the conversion of Pd0-BQ to PdII, see: Grennberg H, Gogoll A, Bäckvall JE. Organometallics. 1993;12:1790–1793.
- 9.For reviews of Pd-catalyzed aerobic oxidation reactions, see: Stahl SS. Angew Chem Int Ed. 2004;43:3400–3420. doi: 10.1002/anie.200300630.Stahl SS. Science. 2005;309:1824–1826. doi: 10.1126/science.1114666.Gligorich KM, Sigman MS. Chem Comm. 2009:3854–3867. doi: 10.1039/b902868d.Shi Z, Zhang C, Tang C, Jiao N. Chem Soc Rev. 2012;41:3381–3430. doi: 10.1039/c2cs15224j.
- 10.Takato Mitsudome TU, Nosaka Naoya, Mori Kohsuke, Mizugaki Tomoo, Ebitani Kohki, Kaneda Kiyotomi. Angew Chem Int Ed. 2006;45:481–485. doi: 10.1002/anie.200502886. [DOI] [PubMed] [Google Scholar]
- 11.(a) Liu G, Yin G, Wu L. Angew Chem Int Ed. 2008;47:4733–4736. doi: 10.1002/anie.200801009. [DOI] [PubMed] [Google Scholar]; (b) Yin G, Wu Y, Liu G. J Am Chem Soc. 2010;132:11978–11987. doi: 10.1021/ja1030936. [DOI] [PubMed] [Google Scholar]
- 12.Campbell AN, White PB, Guzei IA, Stahl SS. J Am Chem Soc. 2010;132:15116–15119. doi: 10.1021/ja105829t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.For fundamental studies of Pd0 oxidation by O2, see: Popp BV, Thorman JL, Morales CM, Landis CR, Stahl SS. J Am Chem Soc. 2004;126:14832–14842. doi: 10.1021/ja0459734.Konnick MM, Guzei IA, Stahl SS. J Am Chem Soc. 2004;126:10212–10213. doi: 10.1021/ja046884u.Landis CR, Morales CM, Stahl SS. J Am Chem Soc. 2004;126:16302–16303. doi: 10.1021/ja044674b.Popp BV, Thorman JL, Stahl SS. J Mol Catal A: Chem. 2006;251:2–7.Muzart J. Chem Asian J. 2006;1:508–515. doi: 10.1002/asia.200600202.Popp BV, Wendlandt JE, Landis CR, Stahl SS. Angew Chem Int Ed. 2007;46:601–604. doi: 10.1002/anie.200603667.
- 14.For synthetic routes to various Pd(π-allyl)Cl complexes, see: Trost BM, Metzner PJ. J Am Chem Soc. 1980;102:3572–3577.Tsuji J, Imamura S. Bulletin of the Chemical Society of Japan. 1967;40:197–202. doi: 10.1246/bcsj.40.2154.Imaizumi S, Matsuhisa T, Senda Y. J Organomet Chem. 1985;280:441–448.
- 15.Wolfe S, Campbell PGC. J Am Chem Soc. 1971;93:1499–1501. [Google Scholar]
- 16.(a) Constantine RN, Kim N, Bunt RC. Org Lett. 2003;5:2279–2282. doi: 10.1021/ol034610q. [DOI] [PubMed] [Google Scholar]; (b) Engelin C, Jensen T, Rodriguez-Rodriguez S, Fristrup P. ACS Catalysis. 2013;3:294–302. [Google Scholar]
- 17.The pKa values of allylbenzene and 1-octene in DMSO are 34 and 44, respectively (http://www.chem.wisc.edu/areas/reich/pkatable/index.htm). The pKa of methyl-3-butenoate has not been reported, and is estimated based on ethyl phenylacetate to be 23. See also: Bordwell FG, Fried HE. J Org Chem. 1981;46:4327–4331.
- 18.Direct comparisons between O2 and BQ in fundamental reactions that permit assessment of this proposal are not available. For related considerations, see ref. 13d and the following: Popp BV, Thorman JL, Morales CM, Landis CR, Stahl SS. J Am Chem Soc. 2004;126:14832–14842. doi: 10.1021/ja0459734.Popp BV, Morales CM, Landis CR, Stahl SS. Inorg Chem. 2010;49:8200–8207. doi: 10.1021/ic100806w.
- 19.(a) Tang F, Zhang Y, Rath NP, Mirica LM. Organometallics. 2102;31:6690–6696. [Google Scholar]; (b) Khusnutdinova JR, Rath NP, Mirica LM. J Am Chem Soc. 2012;134:2414–2422. doi: 10.1021/ja210841f. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.