

Abstract
Electrocatalysis is often thought of solely in the inorganic realm, most often applied to energy conversion in fuel cells. However, the ever-growing field of bioelectrocatalysis has made great strides in advancing technology for both biofuel cells as well as biological detection platforms. Within the context of bioelectrocatalytic detection systems, DNA-based platforms are especially prevalent. One subset of these platforms, the one we have developed, takes advantage of the inherent charge transport properties of DNA. Electrocatalysis coupled with DNA-mediated charge transport has enabled specific and sensitive detection of lesions, mismatches and DNA-binding proteins. Even greater signal amplification from these platforms is now being achieved through the incorporation of a secondary electrode to the platform both for patterning DNA arrays and for detection. Here, we describe the evolution of this new DNA sensor technology.
1. Introduction
Bioelectrocatalysis has become an invaluable tool in applications ranging from biofuel cells and electrochemical processing of small molecules to electrochemical biosensing. Redox-active biomolecules may be wired directly to an electrode surface, or instead may rely upon a redox mediator. Proteins, nucleic acids (ribozymes and deoxyribozymes), organelles (mitochondria and thylakoid membranes), whole cells (bacteria and fungi), and even intact tissues (spinach leaves) have all been used as key components in electrode-driven biocatalysis [1–8].
As biological targets may be present in only trace amounts, electrocatalysis offers a simple and effective method of signal amplification, thereby greatly increasing the sensitivity and on/off signal differential of an electrochemical assay. Our work has focused on nucleic acid-based bioelectrocatalysis for biomarker detection. The chemical structure of DNA is readily amenable to functionalization for biocatalytic applications; DNA is easily prepared and manipulated using standard biochemical methods, and nucleic acid base pairing provides a recognition strategy for capturing target sequences onto electrode surfaces. The DNA polyanion can be used to recruit and concentrate redox-active cations and intercalators into DNA films for use as electrochemical reporter molecules, and the redox chemistry of the bases themselves even permits direct electrochemical readout at an electrode surface.
To date, most electrochemical nucleic acid sensors have focused on the detection of hybridization events. Often the DNA is labeled with a redox-active probe molecule (ferrocene, for example) that subsequently participates in an electrocatalytic cycle [5,9,10]. Alternatively, DNA sensors may exploit non-covalent electrostatic interactions between the negatively charged DNA backbone and redox-active cations such as Ru(NH3)63+ [11,12]; here, the electrochemical signal of the bound cation serves as the reporter in a “phosphate-counting” assay to signal hybridization. Electrochemical impedance spectroscopy using Fe(CN)63− in solution also has been employed to measure surface hybridization to electrode-bound probe sequences by providing a direct measure of differential surface passivation by single- versus double-stranded DNA [13].
In each of these assays, DNA serves primarily as a scaffold to bring (or block, in the case of impedance) redox-active probe molecules into direct contact with an electrode surface, but does not participate directly in the electron-transfer event. In contrast, in our laboratory we have focused on developing electrochemical detection schemes in which the π-stack itself is an integral component of the charge-transfer system. By exploiting the remarkable ability of the double helix to function as medium for long range charge transport, we have been able to extend electrochemical nucleic acid sensors beyond hybridization probes to a wider range of detection applications, including DNA mismatches, lesions, point mutations, and DNA-binding proteins.
Our earliest work depended upon direct electrochemical signals of redox probes intercalated into DNA films self-assembled onto electrode surfaces. By coupling those direct reactions to electrocatalytic cycles, we were able to improve both the selectivity and detection limits of the DNA sensing assays. Most recently, we have developed a two-electrode DNA addressing and detection platform that allows for multiplexed electrochemical detection of different analytes on the same electrode surface, opening up the possibility of identifying multiple biomarkers in cell lysates in parallel with high throughput and specificity.
2. Sensing through DNA-Mediated Charge Transport
Duplex DNA features aromatic bases stacked in a helical column, enabling π-orbital overlap that provides a conduit for charge migration. Accordingly, efficient DNA charge transport (CT) has been demonstrated experimentally using probe molecules both in their ground and excited states [14]. In the electronic ground state, electrochemical reduction of the redox-active dye molecule Nile blue covalently bound to DNA at a distance more than 34 nm (100 base pairs) from the electrode surface established the ability of DNA to function as a molecular wire over exceptionally long distances; the decay factor, β, for DNA CT is exceptionally small, estimated at only < 0.05 Å−1 [15].
Yet despite this wire-like behavior, DNA CT is extraordinarily sensitive to even small perturbations that disrupt the π stack. As first demonstrated electrochemically using daunomycin as the redox probe, covalently bound and intercalated into monolayers of duplex DNA, the presence of an intervening base mismatch is sufficient to turn off DNA CT nearly completely. This same effect has been shown at the single-molecule level using measurements of DNA conductivity in carbon nanotube devices [16]. By inserting a DNA strand functionalized at either end for covalent attachment into a carbon nanotube device, duplex DNA conductivity could be measured relative to the carbon nanotube once a complementary DNA strand was associated with the DNA strand held in the device. Significant current was measured through the well-paired DNA duplex, but when an alternative complementary strand was hybridized to the covalently bound strand to generate a CA or GT mismatch, the current was significantly attenuated. Notably, guanine-containing mismatches are often difficult to detect through conventional hybridization assays due to their thermodynamic stability; indeed assays based on hybridization are limited ultimately by the sequence-dependent thermodynamics of duplex binding. However mismatch detection based upon DNA CT does not depend upon the differential thermodynamics of base pairing. It depends instead upon the dynamics of the base pairs and how perturbations in these base dynamics affect π stacking [14]. Thus DNA-mediated charge transport provides an ideal signaling platform for electrochemical sensors that can detect even small perturbations in the integrity of the π stack.
2.1 DNA-modified Electrodes
DNA-modified electrodes are typically formed by covalently tethering DNA to an electrode surface through a short alkane linker (Figure 1). In our initial work, we modified DNA with an alkylthiol linker, then simply deposited a solution of labeled duplexes onto gold electrodes where the DNA self-assembled into monolayers; depending on the DNA surface coverage the underlying gold was subsequently passivated by backfilling with mercaptohexanol. Films prepared in this fashion have been extensively characterized and, in particular, examined using atomic force microscopy (AFM) under electrochemical control [17]. Interestingly, the film morphology is highly dependent upon applied electric fields: at potentials positive of the pzc (potential of zero charge) the individual helices of loosely packed monolayers lie flat on the surface, while at potentials negative of the pzc the helices stand straight up owing to electrostatic repulsion of the phosphate backbone with the negatively charged electrode surface. Thus, our work typically features reductive activation of probe molecules to ensure that the assay operates in potential regimes where the DNA is oriented normal to the surface. Operating in this potential regime insures that there are no direct interactions between the probe molecules and the electrode. Moreover these potentials are within a window where no direct oxidation or reduction of the DNA itself occurs; during the course of sensing, the DNA is not damaged or chemically modified in any way [8].
Figure 1.

Schematic illustration of a DNA-modified electrode based on DNA CT. Duplex DNA modified with a thiol linker self-assembles onto a gold electrode surface, forming covalent gold-thiol bonds. Regions of the surface that are not covered with DNA are subsequently passivated with mercaptohexanol. The density of the DNA on the surface can be controlled by the addition or omission of magnesium ion to the assembly solution. Mg 2+ shields the negative charges on the DNA backbone, enabling tighter packing of the DNA molecules. Non-covalent methylene blue (MB+) is a common intercalative redox reporter for DNA monolayers and non-specifically intercalates into the DNA base stack.
The DNA concentration at the surface can be controlled somewhat by adjusting the concentration of magnesium ions in the deposition mixture to alter the degree of Debye screening of the negatively charged phosphate groups. Yet with the exception of close-packed films, the DNA monolayers appear to be largely inhomogeneous [18, 19]; during the course of fabrication of the surface, the DNA, despite its anionic character, tends to associate with the surface in clumps, assembling as DNA islands with the helices associated one with another.
More control over the DNA surface coverage, as well as the spacing of individual duplexes within the monolayer, can be accomplished by adding an appropriately modified DNA sequence to a pre-formed, mixed alkylthiol monolayer. One such approach involves copper-free azide/alkyne coupling. With this technique, DNA labeled with a cyclooctyne moiety is added to a pre-formed monolayer of β-mercaptoethanol and 6-azido-1-mercaptohexanol on a gold electrode [20]. Because the cyclooctyne moiety couples only to the alkylthiols that feature an azide head group, the concentration of DNA in the film correlates directly with the fraction of azide within the underlying monolayer. AFM images of the resulting films indicate a significantly more homogenous film than is obtained using thiol-modified DNA. Accordingly, the additional space between individual helices in films prepared this way allows for greater access by proteins and other DNA-binding molecules.
Extending this approach, alkyne-labeled DNA sequences also can be patterned onto mixed monolayers through electrochemically-activated Cu(I) click chemistry at a secondary electrode [21]. First, a mixture of mercaptoundecanephosphoric acid and azidododecane thiol is self-assembled onto the gold electrode. A secondary microelectrode is then positioned ~ 50 μm above the monolayer, and the alkyne-labeled duplex and catalytic precursor ([Cu(phendione)2][SO4]; phendione is 1,10- phenanthroline-5,6-dione) are introduced into the electrolyte solution. Electrochemical reduction of Cu(II) to Cu(I) initiates the azide/alkyne coupling only at sites within the diffusion radius of the electrogenerated Cu(I). Although this process results in non-specific copper adsorption onto the secondary electrode, the composition and morphology of the DNA-labeled surface remain unaffected. This technique allows for the addressing of multiple DNA probe sequences onto an extremely small area of a single-electrode surface.
2.2 Electrochemical reporters for DNA CT-based Detection
To evaluate systematically the ability of DNA films to mediate long-range CT reactions, we studied the electrochemistry of daunomycin (DM, a redox-active chemotherapeutic) that was site specifically cross-linked to guanine residues within the individual helices of the monolayer [22]. Significantly, the rate of DNA CT was invariant over the entire 35-Å span of the 15 base-pair duplexes studied. In fact the rate of CT was limited by the length of the alkane linker, not by the substantially longer DNA length. However incorporation of a single CA base mismatch was sufficient to shut off CT nearly completely. This result demonstrated that interrogating the efficiency of DNA CT could be used as an unusually sensitive electrochemical signal for a wide range of potential DNA lesions and analytes that perturb the integrity of the π-stack.
To avoid the time-consuming chemical cross-linking step, as well as the problem of cross-linking DM to multiple guanine sites within the same sequence, we set out to evaluate potential probes that associate by non-covalent interactions with the DNA films. Both redox-active cations (e.g., Ru(NH3)63+) that associate in the grooves of DNA and intercalators (methylene blue, MB) bind strongly to DNA-modified surfaces through non-covalent interactions. As illustrated in Figure 2, MB and Ru(NH3)63+ each undergo rapid, chemically reversible reductions at DNA-modified gold electrodes and exhibit electrochemical features consistent with surface-bound species (e.g., a linear increase in peak current as a function of scan rate) [23]. Integrating these signals yields respective surface concentrations for MB and Ru(NH3)63+ of 55 pmol/cm2 and 430 pmol/cm2. Comparing these values to the surface density of DNA helices within the film, ~50 pmol/cm2 (as determined separately by radioactive-tagging experiments), yields a probe:DNA binding stoichiometry of ~1:1 for MB and ~ 8:1 for Ru(NH3)63+. The 1:1 binding ratio for MB:DNA is consistent with limited access of MB into the interior base steps of the tightly packed DNA film, while the ~ 8:1 ratio for Ru:DNA suggests that nearly all of the negative charges of the 15-base-pair duplexes are condensed by Ru(NH3)63+ through ion pairing to the phosphate backbone.
Figure 2.

Response of MB and [Ru(NH3)6]3+ on DNA-modified electrodes. MB is capable of intercalation into DNA, resulting in a DNA-mediated signal. In contrast, Ru(NH3)63+ interacts electrostatically with DNA, which merely reports on the number of anionic phosphates, not on the integrity of the DNA. (a) Cyclic voltammogram obtained from free MB interacting with a Watson-Crick paired DNA monolayer. (b) The incorporation of a single-base mismatch into the DNA assembled on the electrode significantly attenuates the signal obtained from the MB reporter. (c) Cyclic voltammogram obtained from the interaction between Ru(NH3)63+ and a DNA-modified electrode. (d) Upon incorporation of a single-base mismatch, no signal loss is observed with the Ru(NH3)63+ reporter. Only electrochemical reporters that interact with the DNA base stack report on DNA-mediated CT.
Notably, when the same probe molecules are bound to DNA monolayers in which the individual helices contain a mismatched base, the MB response drops significantly while the Ru(NH3)63+ signal is virtually unchanged (Figure 2). These results highlight an essential requirement for electrochemical assays based on DNA CT: DNA-mediated assays require efficient electronic coupling of the redox probe into the π stack. Redox reporters that are not coupled directly into the π-stack are unable to differentiate between well-paired DNA versus DNA that contains a lesion, mismatch, or bound protein that disrupts the π stacking.
To expand the range of electrochemical DNA CT reporters, a series of non-covalent redox-active probe molecules that interact with DNA via groove binding or intercalation into the base stack were tested for their mismatch discrimination abilities. The molecules that bind primarily through intercalation, DM, MB, and Ir(bpy)(phen)(phi)3+ (phi = phenanthrenequinone diimine) were found to exhibit substantially smaller electrochemical responses at DNA films containing a mismatch as compared to films containing fully matched DNA [24]. Indeed, among this group the bulkier probes exhibit the best mismatch discrimination, suggesting that the smaller intercalators more readily diffuse within the monolayer and bind beneath the mismatch. In contrast Ru(NH3)63+ and other small cations that bind exclusively through ion-pairing (e.g., Ru(NH3)5Cl2+and Os(NH3)63+) showed no signal attenuation upon mismatch incorporation [25,26]. Thus, for effective DNA CT-based detection using a freely diffusing redox probe, intercalation is a necessity.
If, instead, the redox probe is covalently tethered to the duplex, the probe must maintain conjugation or electronic coupling to the DNA π stack (although many covalent redox probes maintain intercalation as a primary mode of DNA interaction). For example, both covalently tethered methylene blue [27] and DM [22] intercalate into the DNA base stack, while Nile blue is simply electronically conjugated to the DNA through an alkene bond but still reports mismatch discrimination [15]. In fact, covalent attachment without electronic coupling to the π stack does not yield sensitive reporting through DNA CT.
Using non-covalently intercalated DM, we easily detected each of the possible single-base mismatches by cyclic voltammetry. Figure 3 summarizes the cathodic charge passed upon DM reduction through DNA films comprised of 15 base-pair duplexes. Interestingly, the extent of signal attenuation correlates directly with the degree of base-stack perturbation resulting from the mismatch. Thus, the GA mismatch causes only a small drop in the electrochemical response, presumably because this purine-purine base pair is sufficiently well stacked to support efficient CT. Somewhat surprisingly, the thermodynamically stable GT wobble base pair causes a significant drop in the electrochemical signal. This may be a result of increased base dynamics that disrupt π-stacking on the timescale of the electrochemical reaction.
Figure 3.
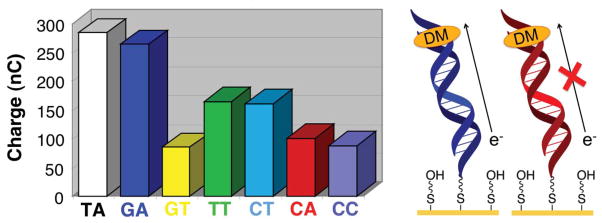
Electrochemistry of daunomycin on DNA-modified electrodes with and without intervening mismatches. The total charge calculated from cyclic voltammograms of electrodes with well matched DNA as well as with DNA containing a variety of mismatches is all shown. Daunomycin shows a preference for intercalation at 5′-GC′-3′ steps in the DNA duplex. As can be seen, the signal differential between well matched DNA and DNA containing various mismatches can be small with this method.
Although it is possible to identify all of the point mismatches using direct electrochemistry, the absolute electrochemical signals are inherently limited by both the electrode size and the amount of DNA that can be assembled onto the electrode surface. Based on the diameter of duplex DNA (20 Å), the number of DNA helices that can be assembled onto a reasonably sized electrode (~2 mm diameter) is limited to the sub-pmol range. This, in turn limits the surface concentration of reporter molecules, resulting in extremely small electrochemical signals, making detection more difficult.
3. Electrocatalysis with DNA CT for DNA Sensing
To address the problem of small electrochemical signals, we initiated a series of experiments to develop an electrocatalytic cycle for signal amplification. The most effective system involved intercalated MB coupled to Fe(CN)63− freely diffusing in solution [24]. Owing to its negative charge, direct electrochemical reduction of Fe(CN)63− is inhibited at the highly (negatively) charged surfaces of DNA-modified electrodes, even at overpotentials as high as 1 V. On the other hand, reduction of Fe(CN)63− by leucomethylene blue (LB, the reduced form of MB), is thermodynamically favored by more than 0.5 eV, ensuring a rapid homogeneous electron transfer reaction. Thus, addition of micromolar MB to electrolyte solutions containing millimolar Fe(CN)63− leads to a dramatic increase in the electrochemical response at a DNA-modified electrode. Importantly, the onset of this response occurs at the reduction potential of MB (indicating MB as the electrochemical mediator), and the reduction is completely irreversible, as the reduced form of MB is oxidized rapidly by Fe(CN)63− and is therefore no longer available for electrochemical oxidation.
3.1 Methylene Blue as Electrocatalyst with Ferricyanide
Based on data collected at rotating disk electrodes [28], we proposed the mechanism illustrated in Figure 4 for this reaction. The cycle begins with MB intercalated into the DNA film. Upon sweeping the potential past the formal MB/LB reduction potential, MB is rapidly reduced through DNA CT to LB, which subsequently dissociates from the film and reduces two equivalents of Fe(CN)63−. Intercalation of regenerated MB back into the film completes the catalytic cycle. The kinetics of this process suggests that the overall catalytic rate is governed by the on/off dynamics of MB/LB into and out of the DNA film. As a consequence, as long as the on/off rates are fast on the electrochemical timescale, the overall current is limited no longer by the surface density of MB in the film, but by the concentration (and diffusion constant) of Fe(CN)63− in solution. Depending on the concentration of Fe(CN)63−, this electrocatalysis results in absolute currents that are roughly an order of magnitude higher than those produced by direct electrochemical reduction of MB.
Figure 4.

Electrocatalytic cycle between free methylene blue and ferricyanide on a DNA-modified electrode. Methylene blue in its oxidized form is intercalated into the DNA base stack. Upon reduction of methylene blue to leucomethylene blue via DNA-mediated charge transport, the affinity of the leucomethylene blue for DNA is lowered and leucomethylene blue is no longer intercalated. The reduced leucomethylene blue is capable of reducing ferricyanide that is freely diffusing in solution. The leucomethylene blue is then reoxidized to methylene blue and can reintercalate into the DNA. The ferricyanide acts as a diffusing electron sink in solution for the redox probe, methylene blue.
3.2 Single Base Mismatch and Lesion Detection with Electrocatalysis
With a successful catalytic cycle in hand, the question now became whether the presence of a mismatch or other DNA lesion would sufficiently attenuate the catalytic response. Several studies were therefore carried out to assess empirically electrocatalytic signal differentials at well matched versus mismatched helices using a series of different electrochemical mediators. These studies reinforced the importance of selecting redox probes with both the right binding mode and binding kinetics for use in DNA CT electrocatalytic assays. Figure 5 illustrates this point by showing the electrochemical response of 1 mM Fe(CN)63− at well matched and mismatched films in the presence of micromolar concentrations of MB, DM, and Ru(NH3)63+ [29]. With relatively fast on/off intercalation dynamics, MB mediates Fe(CN)63− reduction efficiently at well-matched DNA films, but because of attenuated DNA CT, affects a dramatically smaller catalytic reduction at films made up of mismatched helices. This differential signal enables MB to serve as a highly effective reporter for DNA base-stack perturbations using electrocatalysis. In contrast, the tightly intercalated DM probe, with very slow on/off dynamics, is unable to mediate Fe(CN)63− reduction at either type of electrode surface, rendering it unsuitable as an electrocatalyst. Finally, the Ru(NH3)63+ probe, which merely ion pairs to the DNA backbone, exhibits the fastest on/off dynamics, and correspondingly mediates the most efficient Fe(CN)63− reduction. However, because Ru(NH3)63+ reduction is not DNA mediated, the electrocatalytic waves are virtually identical at both matched and mismatched films.
Figure 5.
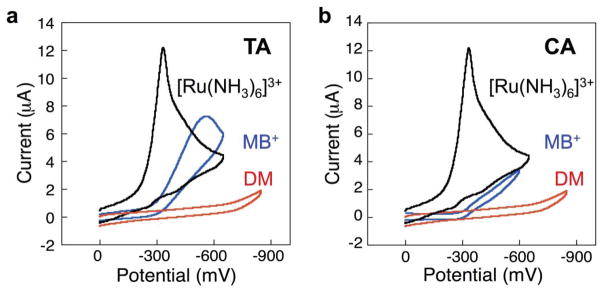
Electrocatalytic signals from DNA-modified electrodes and a variety of redox probes with [Fe(CN)6]3−. In black is the electrocatalytic signal from ruthenium hexammine interacting with [Fe(CN)6]3−; in blue is freely diffusing methylene blue with with [Fe(CN)6]3−, and in red is daunomycin with [Fe(CN)6]3−. In (a) is shown the signals for well-matched DNA, and (b) shows signals for DNA containing a C:A mismatch. As can be seen, no electrocatalytic turnover occurs between daunomycin and [Fe(CN)6]3−, and with ruthenium hexammine, no signal attenuation is observed upon the incorporation of a C:A mismatch. Only methylene blue and [Fe(CN)6]3− produce a DNA mediated signal with electrocatalytic amplification that is attenuated upon incorporation of a mismatch.
Indeed, both the absolute currents for MB-mediated Fe(CN)63− reduction and, more importantly, the differential currents generated at matched versus mismatched DNA films are an order of magnitude greater than the corresponding currents observed for the direct electrochemical reduction of intercalated MB. Moreover, integrating the steady-state catalytic currents as a function of time yields differential charges at matched versus mismatched films that only get larger with time. This effect is illustrated in Figure 6, where the time-dependent electrocatalytic charge is plotted separately for MB-mediated Fe(CN)63− reduction at films featuring either matched or mismatched DNA helices. Significantly, this chronocoulometry assay allows ready detection of all of the possible single-base mismatches, including purine-purine base steps, without any manipulation of hybridization conditions [25]. The improved signal differentiation as a function of time is a direct consequence of the catalytic nature of this assay.
Figure 6.
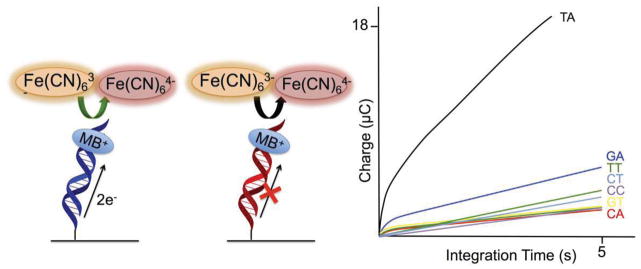
Chronocoulometry of well-matched DNA as well as the same mismatches previously tested with free daunomycin examined with methylene blue and [Fe(CN)6]3−. As can be seen, the difference in charge between well-matched DNA and each of the single base mismatch-containing duplexes is significantly larger for the signals amplified with electrocatalysis than those that do not.
The utility of this electrocatalytic chronocoulometry platform applied to biologically relevant targets was highlighted by the successful detection of different lesion products in DNA, as well as the detection of hot-spot mutations of the human p53 gene [25]. Cellular DNA lesions occur as a result of exposure to reactive-oxygen species and UV light, and this assay proved sensitive enough to differentiate not only between undamaged DNA and DNA containing various lesions but also between the different lesions tested, including an abasic site, 8-oxo-adenine, 5,6-dihydroxy thymine, and deoxy-uracil [25]. Likewise, the assay allowed ready detection of several p53 mutations contained in tumor cell lines using a multiplexed chip featuring microelectrode sensors [25].
3.3 Tethering Methylene Blue
One drawback of the Fe(CN)63−-based electrocatalytic system is the stringent requirement for thoroughly passivated electrode surfaces: any direct Fe(CN)63− reduction at the bare electrode—even at pinholes—bypasses the DNA CT pathway and renders the assay incapable of sensing π-stack perturbations. This is especially problematic when detecting larger biomarkers, e.g., protein transcription factors, which require lower DNA surface densities in order to gain access to specific sequences within the individual helices [30]. Low-density films additionally require that the redox mediator be prohibited from diffusing down into the DNA sequence and intercalating below the site of π-stack disruption.
A method was therefore developed to covalently tether methylene blue directly to the terminus of the DNA. The covalent MB reporter is coupled to a modified thymine base through a flexible molecular tether which maintains the capacity of MB to intercalate into the DNA base stack and still dissociate upon reduction to LB [27]. In covalent linkage of methylene blue to the DNA, the linker length was of the most concern. The linker must be flexible enough and sufficiently long for the probe to intercalate into the base stack, while not so long that the probe can interact directly with the surface. The length of the tether was optimized to a 6-carbon chain because this length enabled the probe to destack from the bases upon reduction to leucomethylene blue and interact with the diffusing electron sink and to intercalate into the DNA base stack in the oxidized methylene blue form while being sufficiently short to minimize direct surface interactions between the probe and the gold electrode. Additionally, unlike free methylene blue, with the covalent methylene blue probe, there is only a single redox reporter per DNA helix.
We first explored various tethers for MB [27,31]. We found that too short a tether did indeed limit access to solution, but a longer linker gave direct interaction with the gold electrode. We also considered whether intercalation was always intraduplex for the tethered probes. Here, clearly the extent of intraduplex intercalation depended upon how closely packed the DNA helices were. Most importantly, an essential element for all these characterizations was the assay for how effective a mismatch served to attenuate current flow. The key for efficient DNA detection was CT mediated by the full helix, as tested through the inclusion of intervening mismatches.
3.4 Covalent Methylene Blue with Hemoglobin as an Electrocatalysis Pair
To address the problem of direct-electrode Fe(CN)63− reduction, a metalloprotein-based electron sink was additionally employed to provide some inherent shielding from the electrode surface. While a variety of redox-active proteins have been applied generally to electrocatalytic platforms, including glucose oxidase and horseradish peroxidase, many of these generate undesirable reactive oxygen species [32–35]. Hemoglobin, in contrast, is a fairly small protein that does not generate byproducts that can damage DNA.
Because of the iron center shielding afforded by hemoglobin’s native protein conformation, passivation of the DNA-modified electrode is less of a stringent necessity when this protein is used as an electron sink (Figure 7) [36]. The combination of a covalent MB mediator and hemoglobin electron sink enabled the ready detection of restriction-enzyme activity at a low-density DNA film. Notably, using covalent MB and Fe(CN)63−, detection of DNA was limited to greater than 500 fmol on the surface. In contrast, upon incorporation of hemoglobin, DNA was detectable at 5 fmol on the surface, a greater than 100-fold decrease in detection.
Figure 7.
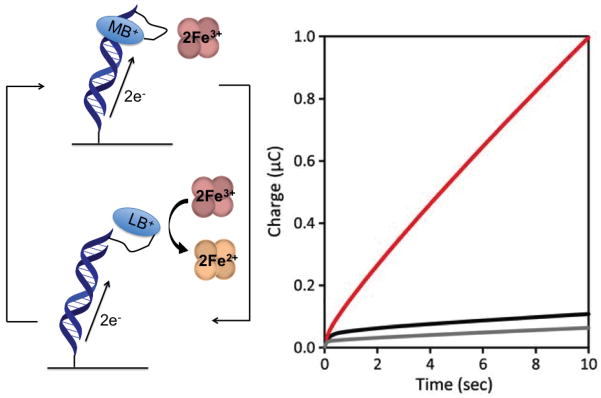
Electrocatalytic cycle between DNA tethered MB and freely-diffusing hemoglobin. As MB is reduced to LB, its affinity for DNA is significantly decreased, resulting in LB dissociation from the duplex. The LB is then reoxidized by hemoglobin in solution while maintaining surface passivation. The amino acids of the hemoglobin shell provide an inherent passivator between the iron center and electrode surface. The chronocoulometry of the system is shown. In red is the signal resulting from electrocatalysis, while in black is the MB-DNA without hemoglobin, and in grey is unmodified DNA duplex.
4. Two-Electrode Platforms for Electrocatalytic Detection
We have recently begun to extend these electrocatalytic detection schemes to a two-electrode format in which a secondary working electrode is positioned above the DNA film to serve as a signaling probe for DNA CT (Figure 8). Such an arrangement offers several significant advantages over a single-electrode detection platform: (i) the catalytic substrate (e.g., Fe(CN)63−) is now regenerated directly at the probe tip, eliminating substrate migration into the depleted diffusion layer as the ultimate limiting factor in the overall electrocatalytic rates; (ii) because detection is accomplished from a secondary electrode that measures turnover of the electron sink, there is no background signal to contend with; and (iii) coupled to in situ Cu(I)-catalyzed DNA film fabrication, the two-electrode platform provides not only spatial resolution of the DNA-modified surface, but also a convenient method to address multiple DNA sequences closely spaced on a single substrate surface.
Figure 8.

Electrocatalytic signal amplification for detection from a secondary electrode. Intercalated methylene blue is reduced through DNA CT to leucomethylene blue, which has a lower affinity for DNA and does not remain intercalated. The leucomethylene blue can be reoxidized to methylene blue by ferricyanide, which is, in turn, reduced to ferrocyanide. Unlike when ferricyanide acts as a simple electron sink, ferrocyanide is now reoxidized to ferricyanide at a secondary electrode (top brown electrode). The current generated at the secondary electrode from the reoxidation of ferricyanide can be measured separately.
4.1 SECM of DNA-modified Electrodes for Protein Detection
The most widely used two-electrode detection system is the scanning electrochemical microscope [37, 38]. This platform features two working electrodes controlled by a bipotentiostat. Applied to DNA-based detection schemes, a DNA monolayer is assembled onto the substrate electrode, while a scanning probe microelectrode is used to detect electroactive analytes generated at the substrate surface. This technique involves constant-potential amperometry at both the substrate and probe electrodes: a potential sufficiently negative to reduce the redox probe bound to DNA is applied to the substrate electrode, while a potential sufficiently positive to reoxidize the reduced electrocatalytic partner is applied to the probe tip. The current generated at the probe tip from this cycle is then measured as a function of position over the substrate surface. A small tip current signals specific locations within the film that do not support efficient DNA CT (e.g., owing to the presence of a mismatch of DNA lesion), while a large tip current signals locations within the film where the DNA π-stack is intact.
To illustrate this method, scanning electrochemical microscopy (SECM) was used to monitor DNA CT on surfaces in measurements involving DNA covalently labeled with Nile blue and Fe(CN)64− in solution [39]. In these experiments, bulk ferrocyanide in solution is oxidized to ferricyanide at a probe tip to give a steady state, diffusion-controlled current. The probe tip is then physically lowered toward the DNA electrode and the current-distance feedback curve is recorded. If the reduction of tip-generated Fe(CN)63− is blocked by the DNA monolayer, the probe current drops (negative feedback) owing to restricted diffusion of bulk Fe(CN)64− as the tip nears the substrate surface. Alternatively, if the reduction of tip-generated Fe(CN)63− occurs at the substrate electrode due to efficient electrocatalytic DNA CT, the probe current increases (positive feedback), because ferrocyanide at the probe tip is now regenerated more rapidly by electrocatalysis than by diffusion from the bulk solution. Figure 9 shows representative approach curves at two different DNA-modified surfaces in which the covalent Nile blue probe was positioned at the top (positive feedback) and bottom (negative feedback) of the monolayer. Notably, the current detected from DNA modified with Nile blue at the top was significantly larger than with the Nile blue at the bottom, owing to greater access of tip-generated ferricyanide to the top of the films. With Nile blue capable of interacting with the electrocatalytic substrate in solution, the DNA film acts as a conductor.
Figure 9.
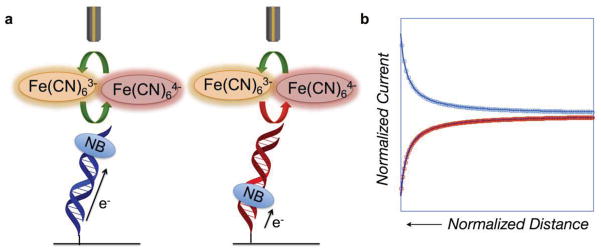
Scanning Electrochemical Microscopy (SECM) detection of DNA-mediated charge transport. (a) DNA monolayers were modified with a covalent Nile blue redox probe at either the top of the monolayer or the bottom. When Nile blue is tethered to the top of the DNA, it is accessible to diffusing [Fe(CN)6]3−, enabling electrocatalysis to occur. When Nile blue is tethered to the base of the DNA monolayer, the [Fe(CN)6]3− is inaccessible, and no interaction between the redox probe and the electron sink occurs. (b) Approach curves for the two monolayers. In blue is the approach curve for the monolayer with Nile blue at the top; it has the shape characteristic of a conductor. In contrast, in red is shown the approach curve for the monolayer modified with Nile blue at the base. The approach curve for this monolayer is characteristic of an insulator, indicating that charge is not flowing from the electrode surface to the microelectrode tip.
To evaluate the ability of this platform to identify biomolecular markers, the transcriptional activator TATA-binding protein (TBP) was added to DNA-modified electrodes with Nile blue probes positioned at the top of the film. TBP binds to a TATA sequence in DNA and kinks the helix 80°, disrupting the integrity of the π-stack. We had seen in earlier experiments the sensitivity of DNA CT to TBP binding [26]. Accordingly, when TBP was added to solution here, a significant decrease in the electrocatalytic tip current was observed.
It is important to note that the SECM images obtained of DNA monolayers show regions of extremely high current, corresponding to imperfections on the surface; these regions do not decrease upon addition of TBP to the surface, and would lead to a falsely high overall signal on a single electrode platform. These studies gave us our first hints of the sensitivity of two-electrode detection to report on DNA held at the surface. Additionally, these studies provided hints as to the heterogeneity of our surfaces and the need for new methods of DNA surface fabrication.
4.2 Two-Electrode Patterned DNA Arrays
With its inherent sensitivity and imaging capabilities, SECM has proven to be an effective platform for DNA biosensing. However its high cost and requirement for highly trained hands-on operation makes the scanning electrochemical microscope illsuited for point-of-care diagnostics. Thus, we became interested in developing a simplified two-electrode platform that maintains the sensitivity and spatial resolution of SECM, but allows for a level of automation and ease of use that is suitable for high-throughput bench-top applications. As a first step toward that goal, we designed a macroscopic SECM-like platform in which multiple DNA probe sequences can be grafted onto a single-electrode surface, and then assayed using DNA CT chemistry to provide parallel detection of multiple analytes from the same solution [21].
The electrodes for this platform were fabricated by vapor deposition of gold onto glass microscope slides. A single square substrate electrode was treated with a solution of 11-mercaptoundecylphosphoric acid doped with 12-azidododecane-1-thiol to form a densely packed mixed monolayer that effectively blocks the direct electrochemical reduction of MB and Fe(CN)63−. Next, a solution of alkyne-labeled DNA and a water-soluble Cu(II) complex was sandwiched between the substrate slide and an “addressing” slide onto which a series of individual interdigitated gold electrodes had been deposited. As shown in Figure 10, electrochemical reduction of Cu(II) at the addressing electrodes allows for efficient grafting of specific DNA sequences at defined locations on the substrate electrode through Cu(I)-catalyzed azide/alkyne coupling.
Figure 10.
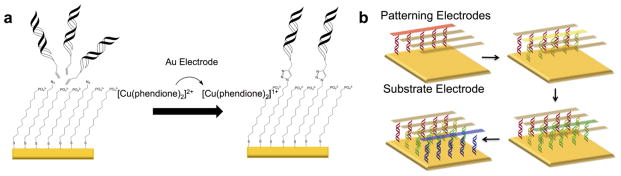
Electrochemically activated click chemistry to pattern DNA onto a single electrode surface. (a) An inert [Cu(phendione)2]2+ catalyst is electrochemically activated to an active [Cu(phendione)2]1+ catalyst that can catalyze the [3+2] cycloaddition between alkyne-modified DNA and an azide-terminated thiol monolayer. (b) Four different sequences of DNA are patterned onto a single substrate pad through sequential catalyst activations from a secondary electrode.
Once formed, the DNA array is electrochemically probed using either a secondary microelectrode mounted to an x,y,z-stage, similar to SECM, or by the stationary interdigitated electrodes on the addressing slide. To assess whether this setup would allow spatial resolution of multiple DNA sequences, we prepared an array of four DNA duplexes, two that were fully matched and two that contained a single-base mismatch. As illustrated in Figure 11, upon addition of MB and Fe(CN)63− to the solution, the probe electrode produced large signals for the reduction of electrocatalytically generated ferrocyanide when positioned over the well-matched DNA sequences, but much smaller signals when positioned over mismatched addresses. Subsequent dehybridization of the substrate electrode, followed by rehybridization of the resulting single strands with complements that swapped the locations of the matched and mismatched sequences now gave small currents at the previously matched addresses, but large currents at the previously mismatched addresses. To highlight the utility of the two-electrode approach, cyclic voltammetry at the substrate electrode instead of the secondary electrode resulted only in an irreversible electrocatalytic response, indicating DNA CT at the surface, but yielding no information about the homogeneity of the film or the DNA sequences present.
Figure 11.

Single electrode versus two electrode readout of a surface patterned with two strips of well matched DNA and two strips that contain a single-base mismatch. (a) Electrochemical readout from a single electrode. The characteristic CV shape of the electrocatalytic process between MB and ferricyanide is evident. (b) Two-electrode electrocatalysis in which a probe microelectrode reduces electrochemically-produced ferrocyanide back to ferricyanide, which no longer limits electrocatalysis by both the amount of electron sink and its speed of diffusion. Additionally, there is differentiation between the two different sequences of DNA: matched (black arrows) and mismatched (red arrows).
As with the previous SECM experiments, this platform also enabled the detection of TBP at concentrations near the dissociation constant of the protein, but because of the ability to graft multiple sequences onto the substrate pad, it additionally allowed spatial resolution of TATA-containing sequences in the presence of alternate DNA probes that did not contain TBP-binding sites. Coupling electrocatalytic DNA CT chemistry to this two-electrode addressing/detection platform thus permits the specificity required for multiple biomarker detection as well as facile multiplexing for quantitative analysis.
5. Conclusions
Electrocatalysis involving biological molecules is advantageous for a variety of applications, from biofuel cells to detection platforms. Our interest lies in platforms that take advantage of the inherent property of DNA, DNA charge transport, that enables DNA to act as a molecular wire, but a fragile wire that reports on the integrity of the DNA itself. While significant strides have been made with such platforms involving redox reporters that interact with DNA alone, the maximum electrochemical signals achievable with such platforms is often insufficient to detect the small changes in signal that are required.
Electrocatalysis between an electron sink and the redox probe that interacts with DNA using DNA CT offers signal amplification without perturbation of the DNA or redox probe. The sole effect of the electron sink is to speed the turnover of the redox probe. Using this platform, the current accumulation from each mismatched base pair can be measured and compared to well-matched DNA. Perturbations to the base pair stack, including those that result from base pair mismatches, common damage products, or binding various proteins, are easily detected.
In order to ensure that charge is transported through the full DNA helix during the course of these experiments, new covalently bound probes needed to be developed. These covalent probes require tethers long enough to allow access to their solution oxidants before catalytic re-reduction, but not so long as to allow direct access to the electrode surface or to an alternate DNA helix. Furthermore, complete passivation of the electrode surface against both the redox reporter and the electron sink can be difficult. A covalently tethered form of methylene blue was therefore developed and paired with a more shielded iron source, hemoglobin, to achieve electrocatalysis and maintain passivation.
Most recently, a two-electrode platform for assembly and detection was developed. Two-electrode detection platforms enable increased signal amplification, allowing for lower detection limits, and provide spatial resolution on an electrode surface. Using this electrocatalytic platform, we can now embark on still more challenging sensing scenarios, testing for DNA-binding proteins and lesions directly in cell lysates, perhaps eventually in samples from single cells.
But it is important to note, what we have described is an evolution of a new sensing technology, one based on detailing mechanistically each step and each obstacle in establishing the electrocatalytic cycle. It is because of this development in mechanistic understanding that our new DNA sensors could be constructed and new devices to detect perturbations in DNA, designed.
Acknowledgments
We are grateful for the financial support of NIH (GM61077) in the evolution of our DNA sensor technology and for the continual great ideas and hard work of our coworkers in creating the many generations of DNA sensors that we explored, some of which was described here. We are grateful also for the great collegiality of John Bercaw over the years, in always asking probing questions to help us focus on mechanism, and thus help clarify the path ahead.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mano N, Mao F, Heller A. J Am Chem Soc. 2003;125:6588–6594. doi: 10.1021/ja0346328. [DOI] [PubMed] [Google Scholar]
- 2.Chen T, Barton SC, Binyamin G, Gao Z, Zhang Y, Kim HH, Heller A. J Am Chem Soc. 2001;123:8630–8631. doi: 10.1021/ja0163164. [DOI] [PubMed] [Google Scholar]
- 3.Kim HH, Mano N, Zhang Y, Heller A. J Electrochemical Soc. 2003;150:A209–A213. [Google Scholar]
- 4.Hickey DP, Giroud F, Schmidtke DW, Glatzhofer DT, Minteer SD. ACS Catalysis. 2013;3:2729–2737. [Google Scholar]
- 5.Patolsky F, Weizmann Y, Willner I. J Am Chem Soc. 2002;124:770–772. doi: 10.1021/ja0119752. [DOI] [PubMed] [Google Scholar]
- 6.Ferapontova EE, Shipovskov S, Gorton L. Biosens Bioelectron. 2007;22:2508–2515. doi: 10.1016/j.bios.2006.09.034. [DOI] [PubMed] [Google Scholar]
- 7.Yamamoto K, Takagi K, Kano K, Ikeda T. Electroanalysis. 2001;13:375–379. [Google Scholar]
- 8.Gorodetsky AA, Buzzeo MC, Barton JK. Bioconj Chem. 2008;19:2285–96. doi: 10.1021/bc8003149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shiddiky MJA, Torriero AAJ, Zeng Z, Spicca L, Bond AM. J Am Chem Soc. 2010;132:10053–10063. doi: 10.1021/ja1021365. [DOI] [PubMed] [Google Scholar]
- 10.Kim E, Kim K, Yang H, Kim YT, Kwak J. Anal Chem. 2003;75:5665–5672. doi: 10.1021/ac034253x. [DOI] [PubMed] [Google Scholar]
- 11.Lapierre-Delvin MA, Asher CL, Taft BJ, Gasparac R, Roberts MA, Kelley SO. Nano Lett. 2005;5:1051–1055. doi: 10.1021/nl050483a. [DOI] [PubMed] [Google Scholar]
- 12.Yang H, Hui A, Pampalakis G, Soleymani L, Liu FF, Sargent EH, Kelley SO. Angew Chem. 2009;48:8461–8464. doi: 10.1002/anie.200902577. [DOI] [PubMed] [Google Scholar]
- 13.Ceres DM, Udit AK, Hill MG, Haley HD, Barton JK. J Phys Chem. 2007;111:663–668. doi: 10.1021/jp066324c. [DOI] [PubMed] [Google Scholar]
- 14.Genereux JG, Barton JK. Chem Rev. 2010;110:1642–1662. doi: 10.1021/cr900228f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Slinker JD, Muren NB, Renfrew SE, Barton JK. Nat Chem. 2011;3:228–233. doi: 10.1038/nchem.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guo X, Gorodetsky AA, Hone J, Barton JK, Nuckolls C. Nature Nanotechnol. 2008;3:163–167. doi: 10.1038/nnano.2008.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kelley SO, Barton JK, Jackson NM, McPherson LD, Potter AB, Spain EM, Allen MJ, Hill MG. Langmuir. 1998;14:6781–84. [Google Scholar]
- 18.Murphy JN, Cheng AK, Yu HZ, Bizzotto DJ. J Am Chem Soc. 2009;131:4042–4040. doi: 10.1021/ja808696p. [DOI] [PubMed] [Google Scholar]
- 19.Mui S, Boon EM, Barton JK, Hill MG, Spain EM. Langmuir. 2001;17:5727–5730.16. [Google Scholar]
- 20.Furst AL, Hill MG, Barton JK. Langmuir. 2013;29:16141–16149. doi: 10.1021/la403262v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Furst AL, Landefeld S, Hill MG, Barton JK. J Am Chem Soc. 2013;135:19099–19102. doi: 10.1021/ja410902j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu T, Barton JK. J Am Chem Soc. 2005;127:10160–10161. doi: 10.1021/ja053025c. [DOI] [PubMed] [Google Scholar]
- 23.Boon EM, Jackson NM, Wightman MD, Kelley SO, Hill MG, Barton JK. J Phys Chem B. 2003;107:11805–11812. [Google Scholar]
- 24.Kelley SO, Boon EM, Barton JK, Jackson NM, Hill MG. Nucl Acids Res. 1999;27:4830–4837. doi: 10.1093/nar/27.24.4830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boon EM, Ceres DM, Drummond TG, Hill MG, Barton JK. Nat Biotechnol. 2000;18:1096–1100. doi: 10.1038/80301. [DOI] [PubMed] [Google Scholar]
- 26.Boon EM, Salas JE, Barton JK. Nature Biotechnol. 2002;20:282–286. doi: 10.1038/nbt0302-282. [DOI] [PubMed] [Google Scholar]
- 27.Pheeney CG, Barton JK. Langmuir. 2012;28:7063–7070. doi: 10.1021/la300566x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boon EM, Barton JK, Bhaghat V, Nerissian M, Wang W, Hill MG. Langmuir. 2003;19:9255–9259. [Google Scholar]
- 29.Boon EM. Electrochemical sensors based on DNA-mediated charge transport chemistry. Cal. Inst. Tech; Pasadena: 2003. [Google Scholar]
- 30.Pheeney CG, Arnold AR, Grodick MA, Barton JK. J Am Chem Soc. 2013;135:11869–11878. doi: 10.1021/ja4041779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pheeney CG, Barton JK. J Am Chem Soc. 2013;135:14944–14947. doi: 10.1021/ja408135g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pelossof G, Tel-Vered R, Elbaz J, Willner I. Anal Chem. 2010;82:4396–4402. doi: 10.1021/ac100095u. [DOI] [PubMed] [Google Scholar]
- 33.Ju H, Shen C. Electroanalysis. 2001;13:789–793. [Google Scholar]
- 34.Xu J, Zhu J, Wu Q, Hu Z, Chen H. Electroanalysis. 2003;15:219–224. [Google Scholar]
- 35.Kuramitz H, Sugawara K, Kawasaki M, Hasebe K, Nakamura H, Tanaka S. Anal Sci. 1999;15:589–592. [Google Scholar]
- 36.Pheeney CG, Guerra LF, Barton JK. Proc Natl Acad Sci. 2012;109:11528–11533. doi: 10.1073/pnas.1201551109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bard AJ, Mirkin MV. Scanning Electrochemical Microscopy. Marcel Dekker, Inc; New York: 2001. [Google Scholar]
- 38.Bard AJ, Denuault G, Friesner RA, Dornblaser BC, Tuckerman LS. Anal Chem. 1991;63:1282–1288. doi: 10.1021/ac00013a019. [DOI] [PubMed] [Google Scholar]
- 39.Gorodetsky AA, Hammond WJ, Hill MG, Slowinski K, Barton JK. Langmuir. 2008;24:14282–14288. doi: 10.1021/1a8029243. [DOI] [PMC free article] [PubMed] [Google Scholar]