

Abstract
Introduction
Tumor necrosis factor (TNF) and, possibly, lymphotoxin alpha (LTα) signaling contribute to inflammation and rheumatoid arthritis (RA) pathogenesis. Pateclizumab (anti-lymphotoxin- alpha; MLTA3698A) is a humanized monoclonal antibody that blocks and depletes anti-LTα. This phase 2, randomized, head-to-head, active- and placebo-controlled trial examined the safety and efficacy of pateclizumab compared to adalimumab in RA patients with an inadequate response to disease-modifying antirheumatic drugs (DMARD-IR).
Methods
Patients (n = 214) with active RA (≥6 swollen and tender joints, C-reactive protein ≥10 mg/L) on oral DMARDs were randomized (2:2:1) to receive pateclizumab 360 mg, adalimumab 40 mg, or placebo subcutaneously every 2 weeks. The primary endpoint, 4-variable, 28-joint disease activity score erythrocyte sedimentation rate (DAS28(4)-ESR) response, was evaluated at 12 weeks using an analysis of covariance (ANCOVA) model with adjustments for concomitant DMARD use and geographic region. Secondary efficacy endpoints included American College of Rheumatology (ACR) 20, ACR50, and ACR70 responses at Day 85. Pharmacokinetics, pharmacodynamics, and immunogenicity of pateclizumab were assessed.
Results
Pateclizumab reduced the DAS28(4)-ESR response (−1.89) at 12 weeks, however, this did not reach statistical significance compared to placebo (−1.54), while adalimumab (−2.52) differed significantly from both placebo and pateclizumab. Pateclizumab 12-week ACR20, ACR50 and ACR70 response rates (64%, 33%, and 14%) suggested clinical activity but were not statistically significant compared to placebo rates (46%, 24%, and 8%, respectively). CXCL13 serum levels decreased significantly following pateclizumab and adalimumab administration, demonstrating pharmacological target engagement by both drugs. Overall, adverse events (AEs) were comparable among all cohorts. Infections were the most common AE, occurring with comparable frequency in all groups. Serious AEs occurred in 0% of pateclizumab, 5.9% of adalimumab, and 2.3% of placebo patients, with serious infection in 2.3% of adalimumab patients and none in pateclizumab and placebo patients.
Conclusions
Pateclizumab had a good safety profile in patients inadequately responsive to DMARDs, but no statistically significant improvement in RA signs and symptoms after 12 weeks of treatment. Adalimumab demonstrated efficacy and safety comparable to published results in this head-to-head comparison in DMARD-IR RA patients.
Trial registration
ClinicalTrials.gov NCT01225393, Registered 18 October 2010.
Introduction
The risk of disease progression in rheumatoid arthritis (RA) occurs in patients who do not respond adequately to available treatment options [1]. Despite the advances with biological agents for the treatment of RA, there remains a significant unmet need for those who do not respond to these agents.
Synovitis in RA develops as a result of infiltration of innate and adaptive immune cells causing a significant inflammatory response and cytokine release, including, but not limited to tumor necrosis factor alpha (TNF-α). During chronic inflammation these cellular infiltrates organize into de novo lymphoid aggregates. These ectopic lymphoid aggregates are instigated and maintained by lymphotoxin (LT)-mediated pathways. Lymphotoxin alpha (LTα) and beta (LTβ) are two related TNF superfamily (TNFSF) members produced predominately by activated cells of the innate and adaptive immune response. LTα exists as a secreted homotrimeric molecule (LTα3) that signals via TNF receptor 1 (TNFR1) and TNFR2 to promote inflammation; whereas LTα complexed with LTβ (LTα1β2), on the surface of T and B cells, signals through the LTβ receptor (LTβR) [2-4]. Lymphoid aggregates in the synovium of RA patients are associated with LTβ expression, and production of B cell attractant chemokine CXCL13 [5,6]. B cells in ectopic lymphoid tissue samples from the lungs of RA patients with pulmonary complications produce rheumatoid factor (RF) and autoantibodies to citrullinated proteins [7]. Moreover, the CD4 T helper (Th) subsets Th1 and Th17, which have been implicated in RA and other autoimmune diseases, express high levels of surface LTα1β2, and depletion of these cells with a murine anti-LTα-depleting antibody has demonstrated therapeutic efficacy in preclinical murine models of RA [4]. In comparison, mice treated with the LTβR decoy fusion protein, were only afforded efficacy when treated preventively, consistent with the failure of LTβR immunoglobulin (LTβR-Ig) to meet clinical endpoints in clinical trials in RA patients [8,9].
Given the role lymphotoxin, TNF-α, and Th cells may play in inflammation and RA pathogenesis, and the additional role soluble LTα3 may play in disease pathogenesis [10-15], we generated a fully humanized blocking and depleting anti-LTα monoclonal antibody (MLTA3698A, pateclizumab) that blocks LTα3-TNFR interactions, LTα1β2-LTβR interactions, specifically depletes LTα1β2-expressing lymphocytes [4]. In a phase I study in patients with active RA, pateclizumab was well tolerated and provided preliminary evidence of clinical activity [16]. Here, we report on the ALTARA (Anti-LTa Rheumatoid Arthritis) study - a phase 2, randomized, head-to-head, active- and placebo-controlled trial of the safety and efficacy of pateclizumab compared to adalimumab in RA patients who had an inadequate response to disease-modifying antirheumatic drugs (DMARD-IR).
Methods
Study design
The ALTARA study was conducted in 47 centers in 10 countries, including sites in the United States, Europe, and Latin America. The study was conducted in compliance with the Declaration of Helsinki and the International Conference on Harmonization Good Clinical Practice Guidelines. It was approved by the Comités de Ética e Investigación en Salud del Centro de Especialidades Médicas del Sureste S.A. de C.V. (Unidad de Investigacion Biomedica del Cem). All patients provided written informed consent before performance of any study procedures.
Dose selection
Pateclizumab was administered as subcutaneous (SC) injections, 360 mg every other week. This study regimen was selected based on the following considerations: 1) the total exposure was, on average, 60% higher than the 3 mg/kg biweekly SC doses evaluated in the pateclizumab phase I study; 2) this regimen was expected to result in a maximal pharmacological effect as suggested by plateaued reductions in serum CXCL13 level in all dose groups at 1-mg/kg or higher doses; and 3) this regimen has 2.2- to 4.5-fold exposure safety coverage by the highest exposure level assessed in the phase I study [16]. An SC biweekly dose of 360 mg pateclizumab was expected to maximize the chance of demonstrating clinical activity while having sufficient safety coverage. Adalimumab was administered as 40-mg SC injections every other week.
Entry criteria
Eligible patients were ≥18 years of age, had a diagnosis of RA for ≥6 months (meeting the American College of Rheumatology (ACR) 1987 revised criteria for RA [17]), and had active disease, defined as ≥6 tender/painful joints (68-joint count) and ≥6 swollen joints (66-joint count) and a C-reactive protein (CRP) level ≥10 mg/L at the screening and baseline visits. Additional key inclusion criteria included failure of at least one DMARD, including methotrexate or leflunomide, with current regimen stable for ≥3 months.
Key exclusion criteria included the following: previous biologic therapy for RA; evidence of hematopoietic disorders at screening or within 3 months; creatinine >1.5 × upper limit of normal (ULN), aspartate aminotransferase (AST) or alanine aminotransferase (ALT) levels 1.5 × ULN; positive QuantiFeron [18] results for latent and/or active Mycobacterium tuberculosis; clinically significant infection(s); and a history of malignancy, with the exception of adequately treated nonmetastatic basal cell or squamous cell cancer of the skin or cervical carcinoma in situ.
Randomization and study treatment
This was a phase 2, randomized, double-blind, active- and placebo-controlled, parallel-group study. Patients were randomized 2:2:1 to receive one of the following treatments: pateclizumab 360 mg, adalimumab 40 mg, or placebo. The study drug was administered SC once every other week for 10 weeks (baseline (Week 0) and Weeks 2, 4, 6, 8, and 10). We were unable to construct a placebo syringe corresponding to the manufactured syringe supplied with adalimumab. As a result, unblinded medical personnel administered the allocated treatment while maintaining a strict blind for the patients and medical personnel associated with safety, joint, and efficacy evaluations.
Concomitant medications
Patients continued on a stable background regimen of RA therapy, which included antimalarial agents, nonsteroidal anti-inflammatory drugs, acetaminophen, and/or oral corticosteroids (<10 mg prednisone or equivalent/day). Patients continued on either methotrexate or leflunomide at protocol-defined stable doses throughout the study. Intra-articular corticosteroids were prohibited in the 6 weeks prior to screening; they were allowed in one joint during the study, but were discouraged between Weeks 10 and 12. For all injected joints, the treated joint was censored from the efficacy analysis from the time of injection up to a maximum of 12 weeks.
Study assessments
Efficacy measures
The primary objective was to compare the efficacy of pateclizumab to that of adalimumab by assessing the change from baseline in the 4-variable, 28-joint disease activity score erythrocyte sedimentation rate (DAS-ESR(4)) on Day 85 (Week 12).
Secondary objectives included the durability of the pateclizumab treatment response, safety of the treatment over 24 weeks, and the efficacy of adalimumab over 12 weeks. The secondary efficacy endpoints analyzed throughout the study included the following: the ACR20, ACR50, and ACR70 response rates, and the proportions of patients achieving disease remission, according to the 3-variable disease activity score in 28 joints (DAS28) [19] using the CRP (DAS28-CRP), or DAS28 score of <2.6, and the least squares mean change in components of the ACR core set of disease activity measures, including the Health Assessment Questionnaire disability index (HAQ-DI) [20], the 36-item Short Form (SF-36) Health Survey [21], and the patient’s assessment of pain and patient’s and physician’s global assessments of disease activity on 0 to 100-mm visual analog scales.
Safety measures
Safety was evaluated by nature, severity, and drug relation of adverse events (AEs), graded according to National Cancer Institute Common Toxicity Criteria for Adverse Events (NCI-CTCAE v3.0) and reported by incidence. Vital signs, physical examination findings, concomitant medications, and laboratory and pregnancy test results were collected every 2 weeks during the study. Incidence of anti-therapeutic antibodies (ATA) to pateclizumab in serum was evaluated using a bridging enzyme-linked immunosorbent assay (ELISA) on Days 57, 85, and 155 or early termination.
Pharmacokinetics and immunogenicity assessment
For pharmacokinetic (PK) assessment, serum samples were obtained at predose Day 1; at 4 hours and at 3 and 6 days after the first dose; predose on Days 15, 29, 57, and 71; 3 and 14 days after final dose, and during drug washout for up to 12 weeks. The PK samples were analyzed using a validated ELISA with a lower limit of quantification of 100 ng/mL pateclizumab.
For ATA assessment, serum samples were obtained at predose and multiple postdose time points (Days 57, 85, 155, or early termination) and assessed using a bridging ELISA [16]. The relative sensitivity of the assay was estimated to be 50 ng/mL using an anti-pateclizumab complementarity-determining region (CDR) antibody. The assay was optimized to minimize interference by RF and by pateclizumab, and could detect 500 ng/mL of the anti-CDR antibody in the presence of 50 μg/mL of pateclizumab.
Pharmacodynamic biomarker assessment
CXCL13 was measured as a pharmacodynamic biomarker, as identified in the phase 1 study [16,22]. Serum samples were obtained predose on Day 1 and postdose on Days 15, 29, 57, 71, 74, 85, 113, 141, and 155. A bridging electrochemiluminescence assay (ECLA) was validated to quantify CXCL13 in RA serum. Briefly, samples were incubated with two anti-CXCL13 antibody reagents, one labeled with biotin. Samples were captured using streptavidin, and measured using standard immunoassay methods (minimum quantifiable concentration, 20 pg/mL). Patients without detectable CXCL13 at baseline (n = 14) or with changes >1,000% of baseline (n = 2) were excluded from analysis.
Statistical methods
All efficacy analyses were performed using the modified intent-to-treat (mITT) populations; all safety analyses were performed using the as-treated population. The primary efficacy endpoint of change of DAS28(4)-ESR at Day 85 was assessed using an analysis of covariance (ANCOVA) model with adjustment of concomitant DMARD use, geographic region, and baseline value. The secondary efficacy endpoints include ACR20, ACR50, and ACR70 responses at Day 85. Any missing ACR score components were imputed with the last-observation-carried-forward (LOCF) method. The P values for comparing treatment differences in ACR20/50/70 responses were based on the Cochran-Mantel-Haenszel test with adjustment for stratification factors of geographic region and concomitant DMARD use. The P values were not corrected for multiple comparisons.
Results
Patient disposition and baseline characteristics
Overall, 214 patients were randomized to receive study treatment. Of these, 180 received study treatment and completed the study for efficacy evaluations (mITT). The 34 patients withdrawn prior to the completion of the study included 13 patients treated with pateclizumab, 11 treated with adalimumab, and 10 treated with placebo. Reasons for early withdrawal included AE (n = 7), patient decision (n = 17), sponsor decision (n = 8), and loss to follow-up from the pateclizumab cohort (n = 2). An additional 11 patients did not meet eligibility criteria. Thus, of the 214 patients, 170 who received a study treatment were included in efficacy and safety evaluations.
Randomization achieved treatment groups that were well balanced for baseline demographic and clinical characteristics (Table 1). The majority of patients were white and female; the mean age was 50.1 years. The mean duration of RA disease was 7.2 to 9.3 years, and 84 to 87% of patients were on methotrexate at randomization.
Table 1.
Demographic and baseline characteristics
| Pateclizumab | ADA | Placebo | All patients | |
|---|---|---|---|---|
| (n = 85) | (n = 85) | (n = 44) | (n = 214) | |
| Age (yr) | ||||
| Mean (SD) | 50.2 (13.1) | 50.6 (13.3) | 48.8 (14.0) | 50.1 (13.3) |
| Median | 51.0 | 52.0 | 48.5 | 51.0 |
| Range (min, max) | 18-75 | 20-73 | 23-75 | 18-75 |
| Age group | ||||
| <65 yr (%) | 72 (84.7%) | 69 (81.2%) | 36 (81.8%) | 177 (82.7%) |
| ≥65 yr (%) | 13 (15.3%) | 16 (18.8%) | 8 (18.2%) | 37 (17.3%) |
| Sex | ||||
| Female | 78 (91.8%) | 68 (80.0%) | 37 (84.1%) | 183 (85.5%) |
| Male | 7 (8.2%) | 17 (20.0%) | 7 (15.9%) | 31 (14.5%) |
| Race | ||||
| White | 53 (62.4%) | 48 (56.5%) | 29 (65.9%) | 130 (60.7%) |
| Black | 1 (1.2%) | 0 (0.0%) | 0 (0.0%) | 1 (0.5%) |
| American Indian or Alaska Native | 1 (1.2%) | 3 (3.5%) | 0 (0.0%) | 4 (1.9%) |
| Asian | (0.0%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) |
| Native Hawaiian or other Pacific Islander | (0.0%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) |
| Not available | 30 (35.3%) | 34 (40.0%) | 15 (34.1%) | 79 (36.9%) |
| Region | ||||
| US and Western Europe | 12 (14.1%) | 12 (14.1%) | 6 (13.6%) | 30 (14.0%) |
| Latin America and Eastern Europe | 73 (85.9%) | 73 (85.9%) | 38 (86.4%) | 184 (86.0%) |
| BMI (kg/m2) | ||||
| Mean (SD) | 27.114 (5.075) | 27.309 (4.194) | 26.903 (5.547) | 27.148 (4.830) |
| Weight (kg) | ||||
| Mean (SD) | 68.66 (14.88) | 69.52 (12.75) | 69.15 (22.61) | 69.11 (15.96) |
ADA, adalimumab; BMI, body mass index; min, max, minimum, maximum; SD, standard deviation.
Primary efficacy endpoint: DAS(4)-ESR at Week 12
Pateclizumab had minimal clinical activity by DAS28(4)-ESR but this did not reach statistical significance compared to placebo at Week 12. In contrast, adalimumab demonstrated a robust treatment effect at Week 12. The mean changes from baseline in DAS28(4)-ESR at Day 85 were −1.89, −2.52, and −1.54 for the pateclizumab, adalimumab, and placebo groups, respectively (Table 2). The differences between adalimumab and the other two treatment groups in DAS28(4)-ESR mean change from baseline were statistically significant (P <0.01) whereas the difference between the pateclizumab and placebo groups was not (P >0.05).
Table 2.
Primary (DAS28-ESR) and key secondary endpoints at day 85
| Pateclizumab | ADA | Placebo | |
|---|---|---|---|
| (n = 84) | (n = 84) | (n = 43) | |
| DAS28(4)-ESR score at baseline | |||
| Mean (SD) | 6.95 (0.89) | 6.84 (0.90) | 6.80 (0.74) |
| DAS28(4)-ESR score at day 85 | |||
| Mean (SD) | 5.06 (1.52) | 4.31 (1.46) | 5.26 (1.44) |
| DAS28(4)-ESR score change from baseline | |||
| Mean (SD) | –1.89 (1.38) | –2.52 (1.43) | –1.54 (1.34) |
| P value of difference of change scorea | |||
| Versus placebo | 0.5172 | 0.0004 | – |
| Versus ADA | 0.0003 | – | – |
| ACR20 response | |||
| n (%) | 50 (64.1%) | 58 (77.3%) | 17 (45.9%) |
| ACR50 response | |||
| n (%) | 26 (33.3%) | 43 (57.3%) | 9 (24.3%) |
| ACR70 response | |||
| n (%) | 11 (14.1%) | 26 (34.7%) | 3 (8.1%) |
| ACR components, mean (P value vs. placebob) | |||
| Swollen joint count | –8.9 (0.04) | –10.4 (<0.01) | –6.1 |
| Tender joint count | –13.3 (0.10) | –16.3 (<0.01) | –9.8 |
| Patient’s global VAS (mm) | –28.8 (0.27) | –37.0 (<0.01) | –24.0 |
| Physician’s global VAS (mm) | –33.8 (<0.01) | –34.1 (<0.01) | –23.2 |
| Patient’s pain VAS | –26.7 (0.24) | –33.5 (<0.01) | –21.6 |
| CRP | –0.5 (0.08) | –1.2 (<0.01) | 0.3 |
| ESR | –11.9 (0.47) | –22.9 (0.04) | –14.7 |
| HAQ-DI | –0.5 (0.09) | –0.8 (<0.01) | –0.3 |
a P value from analysis of covariance; b P value for least squares mean change from baseline compared with placebo. ACR, American College of Rheumatology; ADA, adalimumab; CRP, C-reactive protein; DAS28(4), 4-variable, 28-joint disease activity score; ESR, erythrocyte sedimentation rate; HAQ-DI, Health Assessment Questionnaire disability index; SD, standard deviation; VAS, visual analog scale (0 to 100).
Secondary efficacy endpoints
The ACR50 response was achieved at Day 85 in 26 (33.3%), 43 (57.3%), and 9 (24.3%) patients from the pateclizumab, adalimumab, and placebo groups, respectively (Table 2). The differences between adalimumab and the two other treatment groups in ACR50 rates were statistically significant (P <0.01), but the difference between pateclizumab and placebo was not (P >0.05). Pateclizumab failed to reach statistical significance for other key secondary efficacy endpoints, including ACR20 and ACR70. Pateclizumab was significantly different from placebo for two components of the composite ACR score: swollen joint count (P <0.04) and physician’s global visual analog scale (VAS) (P <0.01). Adalimumab significantly differed from placebo in each of the ACR components. In addition, SF-36 individual and component summary scores also showed a statistically significant difference between adalimumab and pateclizumab but not between pateclizumab and placebo.
Safety and tolerability
Pateclizumab was generally safe and well tolerated in this study. No deaths occurred. Six patients had serious AEs (SAEs): five patients who received adalimumab and one patient who received placebo. Of the six SAEs, two (pulmonary tuberculosis (TB) and lymphopenia, both in the adalimumab group) were judged by the investigator to be related to the study drug. No patients were withdrawn from treatment due to an SAE.
AEs in the pateclizumab cohort were comparable in frequency to the placebo rates (Table 3). In the vast majority of patients, AEs were Grade 1 or 2 in severity; however, six patients in the pateclizumab group, eight in the adalimumab group, and three in the placebo group experienced AEs of Grade 3 or higher. There was one patient from Peru in the adalimumab-treatment group who had a negative interferon-gamma release assay screening result for latent and/or active TB who presented at randomization on study day 1 with a cough and subsequently developed an active TB infection.
Table 3.
Summary of number and percentage of subjects with treatment-emergent adverse events
| Pateclizumab | ADA | Placebo | |
|---|---|---|---|
| (n = 86) | (n = 85) | (n = 43) | |
| Any AE | 50 (58%) | 65 (76%) | 31 (72%) |
| Any SAE | 0 (0%) | 5 (6%) | 1 (2%) |
| Any AE Grade ≥3 | 6 (7%) | 8 (9%) | 3 (7%) |
| Any drug-related AE | 20 (23%) | 26 (31%) | 10 (23%) |
| Any AE within 24 hr of dosing | 7 (8%) | 27 (32%) | 6 (14%) |
| Any SAE within 24 hr of dosing | 0 (0%) | 0 (0%) | 0 (0%) |
| Any drug-related AE within 24 hr of dosing | 4 (5%) | 12 (14%) | 5 (12%) |
| Any infection AE | 25 (29%) | 32 (38%) | 19 (44%) |
| Any infection SAE | 0 (0%) | 2 (2%) | 0 (0%) |
| Any infection AE Grade 3 or higher | 0 (0%) | 1 (1%) | 0 (0%) |
| Any AE leading to discontinuation of study drug | 2 (2%) | 6 (7%) | 1 (2%) |
| Any death | 0 (0%) | 0 (0%) | 0 (0%) |
ADA, adalimumab; AE, adverse event; SAE, serious adverse event.
The most frequent (≥3%) treatment-emergent AEs are listed in Table 4. From baseline to Week 12, the most frequently reported AEs were pharyngitis, headache, urinary tract infection, hypertension, nasopharyngitis anemia, diarrhea, elevated ALT level, rheumatoid arthritis, and gastroenteritis. No clinically significant changes in laboratory parameters or vital signs were observed in the pateclizumab cohort.
Table 4.
Summary of treatment-emergent adverse events that occurred ≥3% in any treatment group
| Pateclizumab | ADA | Placebo | |
|---|---|---|---|
| (n = 86) | (n = 85) | (n = 43) | |
| Pharyngitis | 7 (8.1%) | 7 (8.2%) | 4 (9.3%) |
| Headache | 3 (3.5%) | 11 (12.9%) | 2 (4.7%) |
| Urinary tract infection | 7 (8.1%) | 7 (8.2%) | 2 (4.7%) |
| Hypertension | 5 (5.8%) | 5 (5.9%) | 1 (2.3%) |
| Nasopharyngitis | 4 (4.7%) | 4 (4.7%) | 3 (7.0%) |
| Anemia | 5 (5.8%) | 3 (3.5%) | 2 (4.7%) |
| Diarrhea | 2 (2.3%) | 5 (5.9%) | 3 (7.0%) |
| Alanine aminotransferase increased | 2 (2.3%) | 3 (3.5%) | 4 (9.3%) |
| Rheumatoid arthritis | 3 (3.5%) | 3 (3.5%) | 3 (7.0%) |
| Gastroenteritis | 3 (3.5%) | 3 (3.5%) | 1 (2.3%) |
ADA, adalimumab.
Pharmacokinetics, pharmacodynamics, and immunogenicity
Following six biweekly SC doses of 360 mg pateclizumab, the maximum mean ± standard deviation (SD) observed serum concentration was 41.1 ± 18.4 μg/mL, and occurred on Day 74 (3 days following the sixth dose). The observed steady-state average trough concentration (Ctrough,ss) was 21.3 ± 12.0 μg/mL. The mean trough concentration ratio of Day 85 (14 days after the last dose) to Day 15 (14 days after the first dose) was 1.79, indicating a mild exposure accumulation. The observed PK profile was well aligned with the profile predicted by a population PK model built using the PK data from the pateclizumab phase I study (Figure 1).
Figure 1.
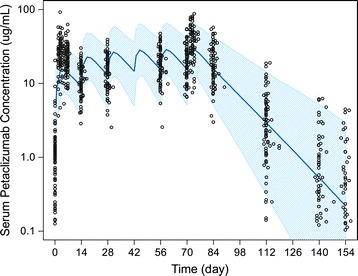
Observed versus population PK model predicted pateclizumab serum concentration-time profiles. Dose was given on study days 0, 14, 24, 42, 56, and 70. The empty symbols represent the observed individual serum concentrations at planned sampling times. The solid line represents the population PK model (built based on phase I PK data) predicted median serum concentration-time profile. The shaded band represents model predicted 90% confidence interval of the concentration-time profile for the study population. PK, pharmacokinetic.
Pre- and postdose levels of serum CXCL13 were evaluated as a biomarker for target modulation. The three treatment groups did not significantly differ in mean (± SD) CXCL13 baseline levels (pg/mL). Serum CXCL13 levels decreased rapidly and significantly in patients treated with pateclizumab (Figure 2), demonstrating evidence of an on-target pharmacological effect (P <0.01, pateclizumab vs. placebo, Day 85). Adalimumab also decreased CXCL13 levels significantly (P <0.01, adalimumab vs. placebo, Day 85), as expected [16]. Serum CXCL13 levels returned to predose levels for both treatment cohorts during the safety follow-up. As a comparison, such a treatment pharmacological effect was not observed in the placebo-treated group.
Figure 2.
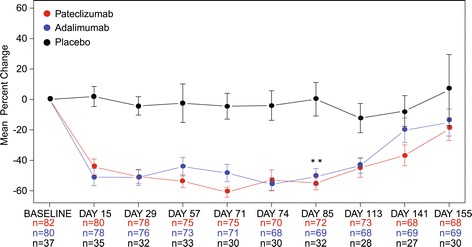
B cell chemoattractant CXCL13 was decreased after treatment with pateclizumab. Mean ± SEM levels of CXCL13, a B cell chemoattractant, are presented over time after treatment with adalimumab, pateclizumab, or placebo. CXCL13 was decreased following treatment with pateclizumab, demonstrating pharmacological engagement of the LTα pathway, as observed in the phase 1 trial of pateclizumab [12]. Adalimumab also modulated CXCL13 levels to a similar degree. Significance was assessed at the efficacy endpoint (Day 85); see Methods. *P <0.05 vs. placebo. LTα, lymphotoxin alpha; SEM, standard error of the mean.
Among the 85 patients who were treated with pateclizumab, four patients were positive for ATA, and all positive ATA signals were detected at a single time point on Day 155, approximately 3 months after the final pateclizumab dose.
Discussion
To our knowledge, ALTARA is the first phase 2 study directly comparing the safety and efficacy of an investigational agent, pateclizumab, with an approved anti-TNF product for the treatment of DMARD-IR patients on background DMARDs. Efficacy was directly compared in a head-to-head study for 3 months of treatment, which is considered a sufficient duration of treatment for such comparisons [23,24]. Since adalimumab combined with the standard-of-care methotrexate is one of the most common therapeutic choices for patients with RA and an inadequate response to DMARDs, it is an appropriate comparator as an anti-TNF class agent. In addition, adalimumab is dosed every 2 weeks subcutaneously like pateclizumab and the choice of this particular anti-TNF agent reduced the complexity of maintaining the blind by reducing the frequency of injections for patients compared to the use of etanercept. The DAS28-ESR and ACR scores reported for adalimumab in the DMARD-IR population in this study are comparable to historical values for this duration of dosing [25-27].
Pateclizumab had a trend for clinical effects on the signs and symptoms of rheumatoid arthritis after 3 months of treatment in patients with active disease and an inadequate response to DMARDs but the differences from placebo responses were not statistically significant. Pateclizumab had an acceptable safety profile, with no SAEs and no increase in infectious AEs compared to placebo likely related to its limited immunosuppressant effects. Injection-site AEs with pateclizumab were uncommon, typically low grade in severity, and comparable to those with placebo.
The pateclizumab dose regimen evaluated in this study provided expected and sufficient exposure during the treatment period as judged by a rapid and sustained decrease in serum CXCL13 levels at all times out to 85 days. The 40 to 50% inhibition of serum CXCL13 levels in the pateclizumab-treated group in this study confirmed that the exposure of 360 mg (approximately 5 mg/kg based on a median body weight of 72 kg) SC biweekly doses is sufficient to produce maximal downstream pharmacological effect in blocking the LTα pathway. However, although such a pharmacodynamic effect was maximized and similar to the CXCL13 reduction observed in the adalimumab-treated cohort, pateclizumab treatment was not associated with significant clinical benefit.
Despite the reports of elevated levels of LTα and LTβ expression in the synovium of RA patients [5,6,28], as well as the presence of lymphoid aggregates, targeting of the LT pathway with pateclizumab did not have insignificant treatment effects compared to adalimumab. Baminercept, an LTβR fusion protein that only blocks LTα1β2-LTβR interaction, was evaluated in two clinical trials and found not to be efficacious for the treatment of the signs and symptoms of RA. In a 14-week, dose-ranging phase 2b trial in DMARD-IR patients (N = 391) [29] and in a 14-week, dose-ranging phase 2b trial in TNF-IR patients (N = 114) [9], baminercept did not have significant treatment effects as assessed by ACR50 response rates. In this second study, baminercept did reduce CXCL13 levels by approximately 50% [9], which is similar to our pharmacodynamic results with pateclizumab in DMARD-IR patients.
Conclusions
Our findings suggest that targeting lymphotoxin signaling, LTα3-TNFR interactions and LTα1β2-LTβR interactions, with pateclizumab on background DMARDs is insufficient to produce a significant reduction in the inflammatory process in RA or to provide superior efficacy over adalimumab, an anti-TNF agent that is commonly used for DMARD-IR RA patients.
Acknowledgements
The authors wish many thanks to all of the patients and the investigators who participated in this study. Acknowledgement is made of the following Institutional Review Boards: US: Quorum Review; Mexico: Comite de Bioetica de la Unidad de Investigacion en Enfermedades Cronico Degenerativas, Comite de Investigacion y Etica del Hospital Centro de Especialidades Medicas del Sureste S.A. de C.V., Comite Bioetico para la Investigacion Clinica S.C. Institutional Review Board, Comite de Etica del Instituto Jalisciense de Investigacion Clinica S.A. de C.V., Comite de Etica Medica e Investigacion del Centro Medico Dalinde, Comite de Etica ICLE SC, Comité de Etica e Investigación del Hospital Aranda de la Parra, Comité Independiente de Investigación y Ética del Centro de Estudios de IBC SC, Comité de Ética en Investigación de Cemsi; Peru: AB PRISMA, Comite de Etica, Comite Institucional de Bioetica (CIB), Universidad Peruana Cayetano Heredia Comite de Etica, Comite Institucional de Bioetica (CIB); Chile: Comite de Evaluacion de Etica Cientifico del Servicio de Salud Metropolitano Oriente; Bulgaria: Komisja Bioetyczna przy Okregowej Radzie Lekarskiej Wielkopolskiej Izby Lekarskiej w Poznaniu; Romania: National Ethics Committee for Clinical Trials on Medicine; Poland: Komisja Bioetyczna przy Okregowej Radzie Lekarskiej Wielkopolskiej Izby Lekarskiej w Poznaniu; Germany: Ethik-Kommission am Fachbereich Humanmedizin Universitätsklinikum Gießen, Ethik-Kommission der Ärztekammer Westfalen-Lippe und der Medizinischen Fakultät der WWU Munster, Ethik-Kommission Kiel der Medizinischen Fakultät der Christian-Albrechts-Universität zu Kiel, Ethik-Kommission der Albert-Ludwigs-Universität Freiburg, Ethik-Kommission der Medizinischen Fakultät der Universität zu Köln, Landesamt für Gesundheit und Soziales Berlin, Ethik-Kommission der Landesärztekammer Thüringen; Spain: CEIC Hospital Universitario de Canarias, CEIC de Galicia (SERGAS), CEIC de Andalucía, CEIC de Asturias, CEIC Hospital Universitario Marques de Valdecill.
Acknowledgement is made of the contributions of Christopher Tehlirian (Pharmaceutical Product Development) to the medical monitoring during the trial and Yehong Wang (Genentech) for assisting in PK data analysis. We thank Linda Phillips who provided copyediting services on behalf of Genentech. This study was financially supported by Genentech, Inc.
Abbreviations
- ADA
adalimumab
- AE
adverse event
- ALT
alanine aminotransferase
- ALTARA
Anti-LTa Rheumatoid Arthritis study
- ANCOVA
analysis of covariance
- ARC
American College of Rheumatology
- AST
aspartate aminotransferase
- ATA
anti-therapeutic antibodies
- CDR
complementarity-determining region
- CRP
C-reactive protein
- DAS-ESR(4)
4-variable, 28-joint disease activity score erythrocyte sedimentation rate
- DAS28
3-variable disease activity score in 28 joints
- DMARDs
disease-modifying antirheumatic drugs
- DMARD-IR
patients who had an inadequate response to DMARDs
- ECLA
electrochemiluminescence assay
- ELISA
enzyme-linked immunosorbent assay
- HAQ-DI
Health Assessment Questionnaire disability index
- Ig
immunoglobulin
- LOCF
last-observation-carried-forward method
- LTα
lymphotoxin alpha
- LTβ
lymphotoxin beta
- LTα1β2
LT-α complexed with LTβ
- LTβR
LTβ receptor
- mITT populations
modified intent-to-treat
- NCI-CTCAE v3.0
National Cancer Institute common toxicity criteria for adverse events
- PK
pharmacokinetic
- RA
rheumatoid arthritis
- RF
rheumatoid factor
- SAE
serious adverse event
- SC
subcutaneous
- SF
superfamily
- SF-36
36-item Short Form Health Survey
- TB
tuberculosis
- Th
T helper
- TNF
tumor necrosis factor
- TNF-α
tumor necrosis factor alpha
- TNFR1 and TNFR2
TNF receptors 1 and 2
- ULN
upper limit of normal
- VAS
visual analog scale
Footnotes
Competing interests
The authors declare that they have no competing interests. FH was an employee and stockholder of Roche at the time of the study. JCD, JLG, AH, PH, WPK CO, MTT, and MT are employees and shareholders of Roche.
Authors’ contributions
WPK contributed to the study design, data interpretation, and coordination of the study. JAS aided in the study design, patient recruitment and data collection, and analysis of the study. CO contributed to the design and coordination of the study. PH contributed to the study design, data collection and study coordination. AH contributed to the design, biomarker collection, and analysis of the study. MT contributed to the design, biomarker data collection and interpretation. MTT contributed to the design, PK data collection and interpretation. JLG contributed to the design of the study. FH contributed to the design, data collection and statistical analysis. JCD contributed to the study design, data collection, and clinical interpretation of data. All authors helped draft the manuscript and participated in manuscript revision. All authors read and approved the final manuscript.
Contributor Information
William P Kennedy, Email: kennedy.william@gene.com.
J Abraham Simon, Email: jabrahamsimon@yahoo.com.mx.
Carolyn Offutt, Email: offutt.carolyn@gene.com.
Priscilla Horn, Email: horn.priscilla@gene.com.
Ann Herman, Email: herman.ann@gene.com.
Michael J Townsend, Email: townsend.michael@gene.com.
Meina T Tang, Email: tang.meina@gene.com.
Jane L Grogan, Email: grogan.jane@gene.com.
Frank Hsieh, Email: hsieh.fang@gene.com.
John C Davis, Jr, Email: davis.john@gene.com.
References
- 1.Smolen JS, Landewé R, Breedveld FC, Buch M, Burmester G, Dougados M, Emery P, Gaujoux-Viala C, Gossec L, Nam J, Ramiro S, Winthrop K, de Wit M, Aletaha D, Betteridge N, Bijlsma JW, Boers M, Buttgereit F, Combe B, Cutolo M, Damjanov N, Hazes JM, Kouloumas M, Kvien TK, Mariette X, Pavelka, van Riel PL, Rubbert-Roth A, Scholte-Voshaar M, Scott DL, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2013 update. Ann Rheum Dis. 2014;73:492–509. doi: 10.1136/annrheumdis-2013-204573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Browning JL. Inhibition of the lymphotoxin pathway as a therapy for autoimmune disease. Immunol Rev. 2008;223:202–220. doi: 10.1111/j.1600-065X.2008.00633.x. [DOI] [PubMed] [Google Scholar]
- 3.Ware CF. Network communications: lymphotoxins, LIGHT, and TNF. Annu Rev Immunol. 2005;23:787–819. doi: 10.1146/annurev.immunol.23.021704.115719. [DOI] [PubMed] [Google Scholar]
- 4.Chiang EY, Kolumam GA, Yu X, Francesco M, Ivelja S, Peng I, Gribling P, Shu J, Lee WP, Refino CJ, Balazs M, Paler-Martinez A, Nguyen A, Young J, Barck KH, Carano RA, Ferrando R, Diehl L, Chatterjea D, Grogan JL. Targeted depletion of lymphotoxin-alpha-expressing TH1 and TH17 cells inhibits autoimmune disease. Nat Med. 2009;15:766–773. doi: 10.1038/nm.1984. [DOI] [PubMed] [Google Scholar]
- 5.Takemura S, Braun A, Crowson C, Kurtin PJ, Cofield RH, O’Fallon WM, Goronzy JJ, Weyand CM. Lymphoid neogenesis in rheumatoid synovitis. J Immunol. 2001;167:1072–1080. doi: 10.4049/jimmunol.167.2.1072. [DOI] [PubMed] [Google Scholar]
- 6.Humby F, Bombardieri M, Manzo A, Kelly S, Blades MC, Kirkham B, Spencer J, Pitzalis C. Ectopic lymphoid structures support ongoing production of class-switched autoantibodies in rheumatoid synovium. PLoS Med. 2009;13:e1. doi: 10.1371/journal.pmed.0060001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rangel-Moreno J, Hartson L, Navarro C, Gaxiola M, Selman M, Randall TD. Inducible bronchus-associated lymphoid tissue (iBALT) in patients with pulmonary complications of rheumatoid arthritis. J Clin Invest. 2006;116:3183–3194. doi: 10.1172/JCI28756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baldassare A, Fiechtner J, Filipowicz-Sosnowska A, Jeka S, O'Gorman J, Weaver M, Beckman E. Preliminary safety and efficacy of baminercept (LTBR-Ig, BG9924) in the treatment of rheumatoid arthritis. Arthritis Rheum. 2007;56:S394. [Google Scholar]
- 9.Genovese MC, Greenwald MW, Alloway JA, Baldassare AR, Chase W, Newman C, Weaver ML. Efficacy and safety of baminercept in the treatment of rheumatoid arthritis (RA) results of the phase 2B study in the TNF-IR population. Arthritis Rheum. 2009;60:417. doi: 10.1002/art.24264. [DOI] [Google Scholar]
- 10.van Vollenhoven R, Harju A, Brannemark S, Klareskog L. Treatment with infliximab (Remicade) when etanercept (Enbrel) has failed or vice versa: data from the STURE registry showing that switching tumour necrosis factor alpha blockers can make sense. Ann Rheum Dis. 2003;62:1195–1198. doi: 10.1136/ard.2003.009589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Laas K, Peltomaa R, Kautiainen H, Leirisalo-Repo M. Clinical impact of switching from infliximab to etanercept in patients with rheumatoid arthritis. Clin Rheumatol. 2008;27:927–932. doi: 10.1007/s10067-008-0880-6. [DOI] [PubMed] [Google Scholar]
- 12.Tracey D, Klareskog L, Sasso EH, Salfeld JG, Tak PP. Tumor necrosis factor antagonist mechanisms of action: a comprehensive review. Pharmacol Ther. 2008;117:244–279. doi: 10.1016/j.pharmthera.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 13.Chatzidionysiou K, van Vollenhoven R. TNF switch after failure of one or more TNF inhibitors: results of an observational study. Arthritis Rheum. 2010;62:S177. [Google Scholar]
- 14.Buch MH, Conaghan PG, Quinn MA, Bingham SJ, Veale D, Emery P. True infliximab resistance in rheumatoid arthritis: a role for lymphotoxin alpha? Ann Rheum Dis. 2004;63:1344–1346. doi: 10.1136/ard.2003.014878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Neregård P, Krishnamurthy A, Revu S, Engström M, af Klint E, Catrina A. Etanercept decreases synovial expression of tumour necrosis factor-α and lymphotoxin-α in rheumatoid arthritis. Scand J Rheumatol. 2013;43:85–90. doi: 10.3109/03009742.2013.834964. [DOI] [PubMed] [Google Scholar]
- 16.Emu B, Luca D, Offutt C, Grogan JL, Rojkovich B, Williams MB, Tang MT, Xiao J, Lee JH, Davis JC. Safety, pharmacokinetics, and biologic activity of pateclizumab, a novel monoclonal antibody targeting lymphotoxin α: results of a phase I randomized, placebo-controlled trial. Arthritis Res Ther. 2012;144:R6. doi: 10.1186/ar3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, Healey LA, Kaplan SR, Liang MH, Luthra HS, Medsger TA, Jr, Mitchell DM, Neustadt DH, Pinals RS, Schaller JG, Sharp JT, Wilder RL, Hunder GG. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988;31:315–324. doi: 10.1002/art.1780310302. [DOI] [PubMed] [Google Scholar]
- 18.Mazurek GH, Jereb J, Vernon A, LoBue P, Goldberg S, Castro K, IGRA Expert Committee; Centers for Disease Control and Prevention (CDC) Updated guidelines for using interferon gamma release assays to detect mycobacterium tuberculosis infection - United States, 2010. MMWR Recomm Rep. 2010;59(RR-5):1–25. [PubMed] [Google Scholar]
- 19.Prevoo ML, van’t Hof MA, Kuper HH, van Leeuwen MA, van de Putte LB, van Riel PL. Modified disease activity scores that include twenty-eight-joint counts. Development and validation in a prospective longitudinal study of patients with rheumatoid arthritis. Arthritis Rheum. 1995;38:444–448. doi: 10.1002/art.1780380107. [DOI] [PubMed] [Google Scholar]
- 20.Fries JF, Spitz P, Kraines RG, Holman HR. Measurement of patient outcome in arthritis. Arthritis Rheum. 1980;23:137–145. doi: 10.1002/art.1780230202. [DOI] [PubMed] [Google Scholar]
- 21.Ware JE, Jr, Sherbourne CD. The MOS 36-item short-form health survey (SF-36). I. Conceptual framework and item selection. Med Care. 1992;30:473–483. doi: 10.1097/00005650-199206000-00002. [DOI] [PubMed] [Google Scholar]
- 22.Rioja I, Hughes FJ, Sharp CH, Warnock LC, Montgomery DS, Akil M, Wilson AG, Binks MH, Dickson MC. Potential novel biomarkers of disease activity in rheumatoid arthritis patients: CXCL13, CCL23, transforming growth factor alpha, tumor necrosis factor receptor superfamily member 9, and macrophage colony-stimulating factor. Arthritis Rheum. 2008;58:2257–2267. doi: 10.1002/art.23667. [DOI] [PubMed] [Google Scholar]
- 23.Boers M. A new design for registration trials in rheumatoid arthritis allowing secondary head-to-head comparisons with standard of care treatment including biologicals. Ann Rheum Dis. 2010;69:4–6. doi: 10.1136/ard.2009.110007. [DOI] [PubMed] [Google Scholar]
- 24.Boers M. The time has come to limit the placebo period in rheumatoid arthritis trials to 3 months: a systematic comparison of 3- and 6-month response rates in trials of biological agents. Ann Rheum Dis. 2010;69:186–192. doi: 10.1136/ard.2009.115832. [DOI] [PubMed] [Google Scholar]
- 25.Weinblatt ME, Keystone EC, Furst DE, Moreland LW, Weisman MH, Birbara CA, Teoh LA, Fischkoff SA, Chartash EK. Adalimumab, a fully human anti-tumor necrosis factor alpha monoclonal antibody, for the treatment of rheumatoid arthritis in patients taking concomitant methotrexate: the ARMADA trial. Arthritis Rheum. 2003;48:35–45. doi: 10.1002/art.10697. [DOI] [PubMed] [Google Scholar]
- 26.Furst DE, Schiff MH, Fleischmann RM, Strand V, Birbara CA, Compagnone D, Fischkoff SA, Chartash EK. Adalimumab, a fully human anti tumor necrosis factor-alpha monoclonal antibody, and concomitant standard antirheumatic therapy for the treatment of rheumatoid arthritis: results of STAR (Safety Trial of Adalimumab in Rheumatoid Arthritis) J Rheumatol. 2003;30:2563–2571. [PubMed] [Google Scholar]
- 27.van de Putte LB, Atkins C, Malaise M, Sany J, Russell AS, van Riel PL, Settas L, Bijlsma JW, Todesco S, Dougados M, Nash P, Emery P, Walter N, Kaul M, Fischkoff S, Kupper H. Efficacy and safety of adalimumab as monotherapy in patients with rheumatoid arthritis for whom previous disease modifying antirheumatic drug treatment has failed. Ann Rheum Dis. 2004;63:508–516. doi: 10.1136/ard.2003.013052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.O'Rourke KP, O'Donoghue G, Adams C, Mulcahy H, Molloy C, Silke C, Molloy M, Shanahan F, O'Gara F. High levels of Lymphotoxin-Beta (LT-Beta) gene expression in rheumatoid arthritis synovium: clinical and cytokine correlations. Rheumatol Int. 2008;28:979–986. doi: 10.1007/s00296-008-0574-z. [DOI] [PubMed] [Google Scholar]
- 29.Isaacs JD, Genovese MC, Emery P, Scheinberg MA, Spindler AJ, Newman C, Weaver ML, Browning J, O’Gorman J, Cravets M, Hagerty D, Beckman EM, Nirula A. Efficacy and safety of baminercept in the treatment of rheumatoid arthritis (RA). Preliminary results of the phase 2B study in the DMARD-IR population. [abstract] Arthritis Rheum. 2009;60:416. [Google Scholar]