

Abstract
Muscle wasting is frequently observed in patients with kidney disease, and low muscle strength is associated with poor outcomes in these patients. However, little is known about the effects of skeletal muscle growth per se on kidney diseases. In this study, we utilized a skeletal muscle-specific, inducible Akt1 transgenic (Akt1 TG) mouse model that promotes the growth of functional skeletal muscle independent of exercise to investigate the effects of muscle growth on kidney diseases. Seven days after Akt1 activation in skeletal muscle, renal injury was induced by unilateral ureteral obstruction (UUO) in Akt1 TG and wild-type (WT) control mice. The expression of atrogin-1, an atrophy-inducing gene in skeletal muscle, was upregulated 7 days after UUO in WT mice but not in Akt1 TG mice. UUO-induced renal interstitial fibrosis, tubular injury, apoptosis, and increased expression of inflammatory, fibrosis-related, and adhesion molecule genes were significantly diminished in Akt1 TG mice compared with WT mice. An increase in the activating phosphorylation of eNOS in the kidney accompanied the attenuation of renal damage by myogenic Akt1 activation. Treatment with the NOS inhibitor L-NAME abolished the protective effect of skeletal muscle Akt activation on obstructive kidney disease. In conclusion, Akt1-mediated muscle growth reduces renal damage in a model of obstructive kidney disease. This improvement appears to be mediated by an increase in eNOS signaling in the kidney. Our data support the concept that loss of muscle mass during kidney disease can contribute to renal failure, and maintaining muscle mass may improve clinical outcome.
Loss of skeletal muscle mass is a hallmark of patients with CKD.1 Muscle wasting in CKD is induced by the changes in amino acid and lipid metabolism, reduced food intake, endocrine dysfunction, reduced physical activity, and altered anabolic intracellular signaling pathways.2 Muscle wasting is independently associated with increased mortality in CKD.3 Numerous studies demonstrate that supervised exercise therapy has beneficial effects, including improvement of peak VO2,4 BP control,5 and quality of life6 in patients with CKD. On the basis of these observations, exercise training is now recommended as a complementary therapeutic intervention for patients with CKD.7
Regarding the mode of exercise, studies show that conventional aerobic endurance training is beneficial for patients with CKD.8,9 In addition to endurance training, resistance training is reported to be beneficial for patients with CKD. For example, resistance training is highly effective in enhancing mitochondrial content in patients with moderate-to-severe CKD.10 Resistance training is effective against the catabolism of a low-protein diet and uremia as well as systemic inflammation in patients with predialysis CKD.11,12 Another study also showed that resistance training combined with aerobic training produces greater effectiveness compared with aerobic exercise alone in patients with CKD who receive dialysis.13 These studies clearly demonstrate the beneficial effects of exercise training on kidney diseases; however, the mechanisms are largely unknown. Although exercise may be a reasonable treatment for patients with CKD, it is not clear whether improvements in muscle mass per se can affect kidney function or whether the beneficial effects of exercise are the consequence of general systemic improvements, such as an accompanying increase in cardiovascular function.
Akt is a serine-threonine protein kinase that is activated by various extracellular stimuli through the phosphatidylinositol 3 kinase pathway and mediates a variety of cellular responses such as metabolism, growth, and proliferation.14 In skeletal muscle, Akt is preferentially activated by resistance training and plays a central role in fast/glycolytic fiber hypertrophy.15 Consistent with these observations, muscle atrophy in CKD is associated with reduced Akt signaling in skeletal muscle tissue.16 We previously generated conditional, skeletal muscle–specific Akt1 transgenic (TG) mice that can induce functional skeletal muscle growth in the absence of exercise training by using the Tet-On system of gene activation.17 We previously used these mice as a model to demonstrate that Akt1-mediated skeletal muscle growth improves metabolic parameters in obese mice17 and attenuates cardiac remodeling after myocardial infarction.18 In this study, we utilized these mice to assess the effects of skeletal muscle growth, independent of exercise, on renal damage in a model of kidney disease.
Results
Myogenic Akt1 Activation Inhibits the Muscle Atrophy Program after Renal Injury
To investigate the effects of skeletal muscle growth on renal pathology, renal injury was induced by unilateral ureteral obstruction (UUO) in Akt1 transgenic (Akt1 TG) and wild-type (WT) control mice at 7 days after doxycycline treatment (Figure 1A). As shown in Figure 1B, obstructed kidneys exhibited global renal atrophy and thinning at 1 week after surgery. At this time point, mice were euthanized to assess the degree of renal damage. In this tissue-specific conditional TG system, the Akt1 transgene was detected only in skeletal muscle but not in the kidney and heart18 after doxycycline treatment (Figure 1C). We previously showed that hemodynamic parameters including heart rate, arterial pressure, and cardiac systolic function do not differ between control and Akt1 TG mice after transgene induction.18 As shown in Figure 1D, activation of Akt1 signaling in myofibers led to an increase in skeletal muscle mass, assessed by gastrocnemius muscle weight at 14 days after doxycycline treatment. Skeletal muscle weight was not different between sham- and UUO-treated mice at 7 days after surgery in WT mice (data not shown). However, the expression of atrogin-1, an atrophy-related gene in skeletal muscle, was upregulated at 7 days after UUO in WT mice but not in Akt1 TG mice (Figure 1E). It was previously reported that the increase in atrogin-1 expression in skeletal muscle of a CKD mouse model is associated with decreased Akt1 signaling.16 As shown in Figure 1F, Akt1 phosphorylation in skeletal muscle tissue was significantly decreased in WT mice at 7 days after UUO compared with control mice. Restoring Akt signaling by forced activation of Akt1 significantly decreased atrogin-1 expression in UUO-treated Akt1 TG mice (Figure 1E). These results suggest that the skeletal muscle atrophy program was initiated before the appearance of muscle wasting, and could be blocked by myogenic Akt1 activation in this renal injury model.
Figure 1.

Myogenic Akt1 activation inhibits the muscle atrophy program after renal injury. (A) Schematic illustration of experimental protocol and doxycycline treatment time course. (B) Representative gross appearance of intact and injured sides of the kidney. (C) Transgene expression after the addition of doxycycline. Representative blots of the gastrocnemius muscle and kidney are shown. (D) Gastrocnemius muscle weight/tibial length in WT and Akt1 TG mice at 2 weeks after doxycycline treatment (WT, n=19; TG, n=17). (E) Atrogin-1 mRNA expression in WT and Akt1 TG mice at 1 week after UUO or sham (n=4 mice per experimental group). (F) Representative immunoblots of p-Akt, t-Akt, and β-actin protein expression in 1 week after UUO and control subjects (top). Quantitative analysis of immunoblots (n=4 mice per experimental group) (bottom). Results are presented as the mean±SEM. DOX, doxycycline; p-Akt, phospho-Akt; t-Akt, total Akt.
Akt1-Mediated Skeletal Muscle Growth Attenuates Renal Fibrosis and Tubular Injuries after UUO
Renal tissue sections were stained with periodic acid–Schiff (PAS) to evaluate tubular injuries in WT and skeletal muscle–specific Akt1 TG mice. As shown in Figure 2A, PAS-stained histologic sections revealed that the renal tubular injury score was increased by 1 week of UUO in WT mice, but this score was significantly lower in Akt1 TG mice. The fibrotic area was evaluated by Masson’s trichrome staining of tissue sections. Renal interstitial fibrosis induced by UUO was significantly decreased in Akt1 TG mice compared with WT mice (Figure 2B). Little tubular injuries and renal fibrosis could be detected in intact kidneys of either Akt1 TG or WT mice. Western blot analysis of total kidney lysates revealed that protein expression of collagen I in the injured kidneys was significantly reduced in Akt1 TG mice compared with WT mice (Figure 2C). Consistent with these observations, the upregulation of TGF-β, connective tissue growth factor, collagen I and III, and fibronectin gene expression in the injured kidneys was significantly decreased in Akt1 TG mice compared with WT mice (Figure 2D). Akt1 activation in skeletal muscle did not affect these gene expressions in the contralateral intact kidney.
Figure 2.

Akt1-mediated skeletal muscle growth attenuates renal fibrosis and tubular injuries after UUO. (A) Representative images of PAS-stained kidney sections (top). Quantitative analysis of renal tubular injury in WT and Akt1 TG mice at 1 week after UUO (bottom). (B) Representative images of Masson’s trichrome–stained kidney sections (top). Quantitative analysis of renal fibrosis in WT and Akt1 TG mice at 1 week after UUO (n=4 mice per experimental group) (bottom). (C) Representative immunoblots of collagen I and α-tubulin protein expression in 1 week after UUO (top). Quantitative analysis of immunoblots (n=3 mice per experimental group) (bottom). (D) TGF-β, CTGF, collagen I and III, and fibronectin mRNA expression in WT and Akt1 TG mice at 1 week after UUO (n=11–14 mice per experimental group). Results are presented as the mean±SEM. CTGF, connective tissue growth factor.
Doxycycline, used to activate the transgene in this model, is reported to interfere with the production of fibrosis in pulmonary diseases.19 To exclude the possible involvement of doxycycline independent of the Akt activation on renal fibrosis, we provided normal water or doxycycline-treated water to WT mice 7 days before UUO surgery, and assessed fibrosis-related gene expression 7 days after surgery. There was no significant difference in fibrosis-related gene expression (TGF-β, collagen I and III, α-smooth muscle actin [α-SMA], and smooth muscle protein 22-α) between these groups (data not shown).
Akt1 TG Mice Display Reduced Inflammatory Cell Infiltration into Injured Kidneys
Macrophage infiltration into the kidney was assessed by immunohistologic staining with F4/80. The number of F4/80-positive macrophages was significantly increased in WT kidneys at 7 days after UUO; however, the infiltration of these cells was significantly decreased in Akt1 TG mice (Figure 3A). These findings were confirmed by quantitative real-time PCR of transcripts using specific primers for mouse F4/80 (Figure 3B). UUO surgery led to an increase in the expression of inflammatory-related genes such as IL-6, IL-1β, TNF-α, and monocyte chemoattractant protein-1 in injured kidneys of WT mice, and this upregulation was attenuated in Akt1 TG mice at 7 days after surgery (Figure 3C). The upregulation of the adhesion molecule genes intracellular adhesion molecule-1 and vascular adhesion molecule-1 by UUO was also decreased in Akt1 TG mice compared with WT mice (Figure 3D). In the contralateral uninjured kidney, the low levels of inflammatory cell infiltration and inflammatory-related gene expression were unaffected by Akt transgene induction in skeletal muscle.
Figure 3.

Akt1-mediated skeletal muscle growth attenuates renal fibrosis and tubular injuries after UUO. (A) Representative images of anti-F4/80–stained renal sections (left). Quantitative analysis of F4/80-positive cells in WT and Akt1 TG mice at 1 week after UUO (n=4 mice per experimental group) (right). (B) F4/80 mRNA expression in WT and Akt1 TG mice at 1 week after UUO. (C) IL-6, IL-1β, TNF-α, and MCP-1 mRNA expression in WT and Akt1 TG mice at 1 week after UUO. (D) VCAM-1 and ICAM-1 mRNA expression in WT and Akt1 TG mice at 1 week after UUO. Results are presented as the mean±SEM (n=7–14 mice per experimental group). MCP-1, monocyte chemoattractant protein-1; ICAM-1, intercellular adhesion molecule-1; VCAM-1, vascular adhesion molecule-1.
Myofibroblast Activation and Renal Cell Apoptosis Was Decreased by Akt1 Activation in Skeletal Muscle
Activated myofibroblasts appear to play a crucial role in renal fibrosis.20 Therefore, we evaluated myofibroblast activation by immunohistology and quantitative real-time PCR. As shown in Figure 4A, the α-SMA–positive cell area per field was significantly increased 1 week after UUO in WT mice. However, these changes were significantly attenuated in the Akt1 TG mice. Upregulation of α-SMA and smooth muscle protein 22-α gene expression in response to 1 week of UUO was also attenuated in Akt1 TG mice compared with WT mice (Figure 4B). Akt1 activation in skeletal muscle did not affect the expression of these genes in the intact kidney.
Figure 4.

Myofibroblast activation was decreased by Akt1 activation in skeletal muscle. (A) Representative images of anti α-SMA–stained renal sections (left). Quantitative analysis of α-SMA–positive cells in WT and Akt1 TG mice at 1 week after UUO (n=4 mice per experimental group) (right). (B) α-SMA and SM22 mRNA expression in WT and Akt1 TG mice at 1 week after UUO (n=11–14 mice per experimental group). Results are presented as the mean±SEM. SM22, smooth muscle protein 22-α.
Apoptosis of renal cells is a hallmark of AKI.21 The number of terminal deoxynucleotidyl transferase–mediated digoxigenin-deoxyuridine nick-end labeling–positive cells was significantly increased in injured kidneys at 7 days after UUO in WT mice; however, the frequency of terminal deoxynucleotidyl transferase–mediated digoxigenin-deoxyuridine nick-end labeling–positive cells was significantly decreased in Akt1 TG mice (Figure 5A). Consistent with these results, the upregulation of cleaved caspase-3 by UUO surgery was significantly reduced in Akt1 TG mice (Figure 5B). By contrast, Akt1 activation in skeletal muscle had no effect on the low levels of renal cell apoptosis and caspase-3 activation in the intact kidneys of these mice.
Figure 5.

Akt1-mediated skeletal muscle growth attenuates renal cell apoptosis after UUO. (A) Representative images of anti-TUNEL–stained renal sections (left). Quantitative analysis of TUNEL-positive cells in WT and Akt1 TG mice at 1 week after UUO (right). (B) Representative immunoblots of caspase-3 and cleaved caspase-3 in kidney 1 week after UUO (left). Quantitative analysis of immunoblots (right). Results are presented as the mean±SEM (n=3 mice per experimental group). TUNEL, terminal deoxynucleotidyl transferase–mediated digoxigenin-deoxyuridine nick-end labeling; HPF, high-power field.
Akt1-Mediated Skeletal Muscle Growth Preserves Renal Function in the Cisplatin Nephrotoxicity Model
We performed a cisplatin nephrotoxicity (CN) model to determine whether Akt1 overexpression in skeletal muscle is effective in a mouse model in which creatinine clearance decreased. Akt1 TG mice and WT mice were injected with 20 mg/kg cisplatin in PBS intraperitoneally 11 days after doxycycline treatment and euthanized 72 hours later (Figure 6A). Skeletal muscle mass of Akt1 TG mice increased at 14 days after doxycycline treatment in the CN model (Figure 6B). As shown in Figure 6C, serum BUN levels were increased in cisplatin-treated WT mice compared with control mice. However, this change was significantly diminished in Akt1 TG mice. Furthermore, a decrease in creatinine clearance at 72 hours after cisplatin administration was reduced in Akt1 TG mice compared with WT mice, although these differences did not reach statistical significance (Figure 6D). These results suggest that skeletal muscle growth protects against renal dysfunction in this model.
Figure 6.

Akt1-mediated skeletal muscle growth preserves renal function in the cisplatin nephrotoxicity model. (A) Schematic illustration of experimental protocol and doxycycline treatment time course of the CN model. (B) Gastrocnemius muscle weight/tibial length in control, WT, and Akt1 TG mice at 2 weeks after doxycycline treatment. (C) Serum BUN levels in control, WT, and Akt1 TG mice at 72 hours after cisplatin administration. (D) Creatinine clearance in control, WT, and Akt1 TG mice at 72 hours after cisplatin administration. Results are presented as the mean±SEM (control, n=4; WT, n=5; TG, n=6). DOX, doxycycline; TL, tibial length; Ccr, creatine clearance.
Myogenic Akt1 Activation Altered the Cytokine Expression Profile
Kidney disease increases the production of cytokines in skeletal muscle and these cytokines can influence metabolic responses of muscle and, potentially, the kidney.22,23 Various cytokines have renoprotective effects and can provide beneficial effects on kidney fibrosis.24,25 To evaluate cytokine production in skeletal muscle in this model, we performed a cytokine array using skeletal muscle tissue of both UUO-treated WT mice and UUO-treated TG mice. This analysis demonstrated that the expression of many cytokines was upregulated in Akt1 TG mice compared with WT mice (Figure 7A). These upregulated cytokines included not only renoprotective cytokines (e.g., IL-2 and IL-10) but also those that potentially have adverse effects (e.g., TNF-α).
Figure 7.
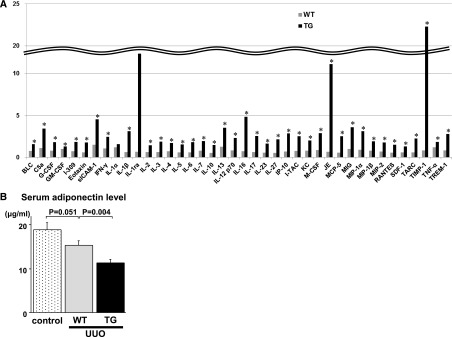
Myogenic Akt1 activation altered cytokine expression profile. (A) Quantitative analysis of protein expression of 40 cytokines by mouse cytokine array panel at 1 week after UUO (n=4 mice per experimental group). BLC, B lymphocyte chemoattractant; C5a, complement component 5a; G-CSF, granulocyte colony-stimulating factor; GM-CSF, granulocyte-macrophage colony-stimulating factor; sICAM-1, soluble ICAM-1; I-TAC, interferon-inducible T-cell alpha chemoattractant; KC, keratinocyte chemoattractant; M-CSF, macrophage colony-stimulating factor; JE/MCP-1, monocyte chemoattractant protein-1; MCP-5, monocyte chemoattractant protein-5; MIG, monokine induced by gamma interferon; MIP, macrophage inflammatory protein; RANTES, regulated upon activation, normal T-cell expressed and secreted; SDF-1, stromal cell-derived factor-1; TARC, thymus and activation-regulated chemokine; TIMP-1, tissue inhibitor of metalloproteinase-1; TNF-α, tumor necrosis factor-α; TREM-1, triggering receptor expressed on myeloid cells-1. (B) Serum adiponectin levels in control, WT, and Akt1 TG mice at 1 week after UUO (control, n=4; WT, n=8; TG, n=13). Results are presented as the mean±SEM.
It is reported that a decrease in adiponectin, an adipose tissue–derived cytokine, attenuates the regulation of monocyte-to-fibroblast transition with suppression of renal fibrosis.26 In this study, serum adiponectin levels of Akt1 TG mice were significantly lower than those of WT mice (Figure 7B), suggesting that decreased serum adiponectin in Akt1 TG mice might be involved in suppression of renal fibrosis. Collectively, these data suggest that the effect of skeletal muscle growth on kidney fibrosis can be mediated by multiple factors including these cytokine candidates.
Protective Effects of Akt1-Mediated Muscle Growth on Renal Damage Is Blocked by Treatment with a Nitric Oxide Synthase Inhibitor
Nitric oxide synthase (NOS) and endothelial nitric oxide synthase (eNOS) signaling were previously shown to play a central role in renal protection.27–29 To investigate the mechanism by which Akt1-mediated skeletal muscle growth attenuates renal damage after UUO surgery, we analyzed eNOS signaling in kidney tissue. As shown in Figure 8A, there was a trend toward increased eNOS phosphorylation at Ser1177 after 1 week after UUO in WT mice, but this was not statistically significant. However, phosphorylation of eNOS was significantly augmented in the injured side of the kidney in Akt1 TG mice. There was no significant difference in eNOS phosphorylation in the intact side of the kidney between WT and Akt1 TG mice.
Figure 8.

Protective effects of Akt1-mediated muscle growth on renal damage is blocked by the treatment with NOS inhibitor. (A) Representative immunoblots of phospho-eNOS , total eNOS, and α-tubulin protein expression in 1 week after UUO (left). Quantitative analysis of immunoblots (right). (B) Schematic illustration of experimental protocol of L-NAME and doxycycline treatment time course. (C) Gastrocnemius muscle weight/tibial length in WT and Akt1 TG mice at 2 weeks after doxycycline treatment. (D) Time course of systolic BP in WT and Akt1 TG mice before and 1 and 2 weeks after L-NAME treatment. (E) Gene expression analysis in WT and Akt1 TG mice at 1 week after UUO (WT, n=15; TG, n=6). Results are presented as the mean±SEM. DOX, doxycycline; TL, tibial length; MCP-1, monocyte chemoattractant protein-1; CTGF, connective tissue growth factor.
To examine whether augmented renal eNOS activation in Akt1 TG mice plays a causal role in renal protection in the situation of acute renal injuries induced by UUO, WT and Akt1 TG mice were treated with Nω-nitro-L-arginine methyl ester (L-NAME), a NOS inhibitor, for 2 weeks (Figure 8B). Akt1-mediated gastrocnemius muscle growth was not affected by L-NAME treatment (Figure 8C). Systolic BP was significantly increased in both WT and Akt1 TG mice at 1 or 2 weeks after L-NAME treatment, but there was no difference in this parameter between WT and Akt1 TG mice (Figure 8D). As shown in Figure 8E, upregulation of inflammatory-, fibrosis-, and myofibroblast differentiation-related gene expression at 1 weeks after UUO was not different between WT and Akt1 TG mice with chronic L-NAME treatment. These results indicate that the protective effects of Akt1-mediated muscle growth on renal injuries were dependent on the activation of eNOS-derived nitric oxide (NO).
Discussion
Muscle wasting is one of the hallmarks of patients with CKD.1 Thus, therapeutic intervention involving exercise is now recommended for these patients. However, it is unclear whether the beneficial effects of exercise are caused by systemic improvements and there has been little investigation regarding the effect of skeletal muscle mass per se on renal pathology, partially because of the lack of an appropriate animal model that can induce skeletal muscle growth in an inducible manner. In this study, we utilized conditional, skeletal muscle–specific Akt1 TG mice and demonstrated that Akt1-mediated skeletal muscle growth, independent of exercise, attenuated renal damage in a mouse model of obstructive renal injury. Akt1 TG mice exhibited attenuated renal fibrosis, inflammation, and apoptosis after UUO surgery compared with WT mice. These changes were accompanied by increased eNOS phosphorylation in the kidney. The renoprotective effects of Akt1-mediated skeletal muscle hypertrophy were abolished by systemic administration of a NOS inhibitor. These data suggest that increasing the skeletal mass, independent of exercise, can determine the degree of renal damage in response to renal injury.
Several animal studies previously showed the beneficial effect of exercise on kidney diseases. For example, low-intensity exercise was shown to attenuate the progression of diabetic nephropathy in KK-A(Y) mice30 and db/db mice.31 Swimming exercise reduced the progression of renal disease in 5/6 nephrectomized rats.32 Previous exercise training also protects against the progression of diabetic nephropathy produced by streptozotocin-induced diabetes in rats.33 Most of these studies were intended to mimic the aerobic endurance type of exercise training. In addition, there were a few studies investigating the effect of skeletal muscle growth in the setting of CKD-related skeletal muscle atrophy. Muscle-overload, resistance exercise attenuates CKD-induced abnormalities in muscle protein metabolism and progenitor cell function in mice.34 Gene expression of myostatin, which is one of the major mediators of muscle atrophy, is upregulated in CKD.35 Pharmacologic inhibition of myostatin reversed muscle wasting in a mouse model of CKD22; however, these studies did not examine the effects of muscle growth on renal pathology. Furthermore, exercise training studies cannot exclude the possibility that favorable effects on the kidney are secondary to the systemic effects of exercise, such as changes in the cardiovascular system. Thus, we utilized inducible, skeletal muscle–specific Akt1 TG mice as a model of resistance training and analyzed renal pathology in the UUO model.
In patients with CKD, impaired IGF-1 signaling is observed in skeletal muscle36 and kidneys.37 Intermittent IGF-1 therapy in patients with advanced CKD preserves renal function.37 It is reported that increasing serum IGF-1 levels is positively associated with CKD in a representative sample of United States adults.38 The chronic uremic state impairs basal signaling through the mammalian target of rapamycin anabolic pathway, an abnormality that may contribute to muscle wasting.39 These data suggest the presence of IGF-1 resistance in patients with CKD. In our mouse model, Akt1 can be activated regardless of IGF-1 resistance, because a myristoylated (constitutive active) form of Akt1 is used.40 Because the transgene produced renoprotective effects including reducing interstitial fibrosis and inflammation after UUO, overcoming IGF-1 resistance and activating downstream pathways in skeletal muscle could be a reasonable therapeutic strategy for renal diseases. On the other hand, activation of downstream pathways of IGF-1, including Akt1 and extracellular signal-regulated kinase (ERK) in the kidney, was not different between Akt1 TG mice and WT mice (data not shown), indicating that the renoprotective effect of skeletal muscle growth observed in this study was independent of IGF-1 signaling in kidney tissue.
Reduced production of NO in the kidney is one of the characteristics of CKD41; thus, increasing endogenous NO production is a reasonable strategy. NO production was significantly suppressed in rats with CKD, and both running and swimming training highly upregulated the NO level.42 Treadmill exercise was shown to increase NOS in the kidney and heart in the rat model of myocardial infarction.27 Our data show that Akt1 activation in skeletal muscle increased eNOS phosphorylation in the kidney. Because the Akt1 transgene was restricted in skeletal muscle and was not detected in kidney homogenates, activation of renal eNOS may have been induced by the endocrine regulators that are induced by myogenic Akt1 activation. We previously reported several factors secreted by muscle that are increased by Akt1 overexpression.18,43 In the UUO model, we found that myogenic activation of Akt upregulated serum levels of stromal cell-derived factor-1, IL-10, and IL-17, which are known to activate eNOS.44,45 Further studies will be required to determine whether these factors are involved in renal eNOS activation and renoprotective properties of Akt1-mediated skeletal muscle growth.
During muscle wasting, atrophy-related genes that function to promote proteolytic degradation are upregulated in skeletal muscle.46 Atrogin-1 is robustly upregulated in skeletal muscle in the rodent model of CKD.47 In this study, atrogin-1 gene expression was upregulated before apparent skeletal muscle wasting, suggesting that the muscle atrophy program begins at an early stage of kidney disease. Furthermore, Akt1 activation in skeletal muscle significantly attenuated UUO-induced atrogin-1 gene induction. These results indicate that interventions that promote skeletal muscle Akt activation could reverse the muscle wasting program that is initiated by kidney dysfunction.
In conclusion, Akt1-mediated fast/glycolytic skeletal muscle growth reverses muscle wasting and reduces renal damage in a model of UUO. The improvements in kidney function appear to be mediated by an activation of eNOS signaling in the kidney. Our data suggest that interventions that promote the growth or maintenance of fast/glycolytic skeletal muscle growth could have utility in the treatment of kidney diseases.
Concise Methods
Skeletal Muscle–Specific Conditional Akt1 TG mice
MCK-rtTA TG48 mice were crossed with TRE-myrAkt1 TG mice49 to generate double TG mice (Akt1 TG mice).17 The muscle creatine kinase promoter construct used in the driver line is mutated and transgene expression is expressed in a subset of muscle, but no expression occurs in the kidney.48 For Akt1 transgene expression, Akt1 TG mice were treated with doxycycline in drinking water. In some experiments, mice were given 1 mg/ml L-NAME (Dojindo) in drinking water at 7 days before UUO. The BP of conscious mice was measured by the tail-cuff method (MK-2000ST; Muromachi Kikai Co., Tokyo, Japan) every week.
Experimental Protocol
Control and Akt1 TG mice were subjected to UUO at 10–12 weeks of age. Mice were anesthetized, an incision was made in the abdominal midline, and the left proximal ureter was exposed and ligated with 6-0 silk. The contralateral nonobstructed kidney served as the control.50 Doxycycline treatment was started at 7 days before surgery until the experiments were terminated. Mice were euthanized 7 days after UUO. Mice were anesthetized and bilateral kidneys and skeletal muscles were rapidly excised. Kidneys were fixed in 4% paraformaldehyde for immunohistochemical analysis, and kidneys and skeletal muscle were flash-frozen in liquid nitrogen for further RNA and protein analyses. We used a CN model to evaluate the effect of Akt1-mediated skeletal muscle growth in kidney function. Mice were administered 20 mg/kg of cisplatin (Sigma-Aldrich) by a single intraperitoneal injection. Mice were euthanized 72 hours after the administration of cisplatin, and tissue and blood were collected for further analysis. Doxycycline treatment was started at 11 days before cisplatin administration. All procedures were performed in accordance with the Kumamoto University animal care guidelines (approval reference no. B25-121), which conformed to the US National Institutes of Health Guide for the Care and Use of Laboratory Animals (publication no. 85-23, revised 1996).
Histologic Analyses
Kidney tissues were fixed with 4% paraformaldehyde and embedded in paraffin. Kidney sections were stained with PAS and Masson’s trichrome by standard procedure, and all histologic analyses were made in the cortex. Tubulointerstitial injury scores were graded as follows: 0, 0%–10%; 1+, 11%–25%; 2+, 26%–50%; 3+, 51%–75%; and 4+, 76%–100%, as previously described.51 To evaluate interstitial fibrosis, kidney sections were stained with Masson’s trichrome and the percentage of fibrosis area was quantified digitally. Myofibroblasts and macrophage infiltration were assessed by α-SMA and F4/80 staining, respectively. Kidney sections stained with Masson’s trichrome, α-SMA, and F4/80 were quantified using Lumina Vision analysis software (version 2.2). These analyses were performed by two investigators in a blinded manner.
Quantitative Real-Time PCR
Total RNA was prepared using a Qiagen RNeasy fibrous mini-kit, using the protocol supplied by the manufacturer, and cDNA was produced using PrimeScript RT-PCR Systems (Takara, Otsu, Japan). Quantitative real-time PCR was performed as previously described.18 Transcript expression levels were determined as the number of transcripts relative to those for 18S, and were normalized to the mean value from the control kidney. Table 1 lists the primer sequences used in this study.
Table 1.
Primer sequences used for quantitative real-time PCR
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| α-SMA | GGGAGTAATGGTTGGAATGG | GATGATGCCGTGTTCTATCG |
| Atrogin-1 | TCCAGACCCTCTACACATCCTT | CCTCTGCATGATGTTCAGTTGT |
| Collagen I | GTCCCAACCCCCAAAGAC | CATCTTCTGAGTTTGGTGATACGT |
| Collagen III | TGGTTTCTTCTCACCCTTCTTC | TGCATCCCAATTCATCTACGT |
| CTGF | CTGCCAGTGGAGTTCAAATGC | TCATTGTCCCCAGGACAGTTG |
| Fibronectin | CGAGGTGACAGAGACCACAA | CTGGAGTCAAGCCAGACACA |
| F4/80 | CTTTGGCTATGGGCTTCCAGTC | GCAAGGAGGACAGAGTTTATCGTG |
| ICAM-1 | CTGGTGATGCTCAGGTATCC | AGCTCATCTTTCAGCCACTG |
| IL-1β | GAAGAAGAGCCCATCCTCTG | TCATCTCGGAGCCTGTAGTG |
| IL-6 | AGTCCGGAGAGGAGACTTCA | ATTTCCACGATTTCCCAGAG |
| MCP-1 | CAAGAAGGAATGGGTCCAG | CGGGTCAACTTCACATTCA |
| SM-22 | TTCTTGAAGGCAGCTGAAGA | GCACTGCTGCCATATCCTTA |
| TGF-β | GACGTCACTGGAGTTGTACGG | GCTGAATCGAAAGCCCTGT |
| TNF-α | CCAAAGGGATGAGAAGTTCC | CTCCACTTGGTGGTTTGCTA |
| VCAM-1 | GGAGCCTGTCAGTTTTGAGA | GGATCCTTGGGGAAAGAGTA |
| 18S | CGGCTACCACATCCAAGGAA | GCTGGAATTACCGCGGCT |
CTGF, connective tissue growth factor; ICAM-1, intercellular adhesion molecule-1; MCP-1, monocyte chemoattractant protein-1; SM-22, smooth muscle protein 22-α; VCAM-1, vascular adhesion molecule-1.
Western Blot Analyses
Western blotting was performed with a SDS-PAGE system as previously described.18 Primary antibodies used were as follows: phospho-Akt (Ser473), total Akt, phospho-eNOS (Ser1177), total eNOS, phospho-ERK (Thr202/Tyr204), total ERK, caspase-3, cleaved form of caspase-3, and β-actin (all from Cell Signaling Technology), anti-hemagglutinin (Roche Applied Science), anti-type I collagen (Southern Biotech), and α-tubulin (Calbiochem).
Determination of Circulating Adiponectin Concentrations
Serum adiponectin levels were determined using a mouse adiponectin ELISA kit (Otsuka Pharmaceutical Co., Ltd., Tokushima, Japan).
Cytokine Array Analyses
The expression profile of 40 cytokines was analyzed with a mouse cytokine expression array (R&D Systems, Minneapolis, MN). Blocking, hybridization of the array filters, washing conditions, and chemiluminescent detection steps were performed according to the manufacturer’s instructions.
Statistical Analyses
All data are presented as the mean±SEM. Comparisons between two groups were made using the t test. Differences among more than two groups were analyzed using one-way ANOVA followed by a Bonferroni post hoc test. The significance level of a statistical hypothesis test was 0.05.
Disclosures
None.
Acknowledgments
The authors thank Saeko Tokunaga, Megumi Nagahiro, and Ayuko Tateishi for excellent technical assistance.
This work was supported in part by a Grant-in-Aid for Young Scientists (B-24790768 to Y.I.) and a Grant-in-Aid for Scientific Research (B-25293186 to H.O.) from the Japanese Ministry of Education, Culture, Sports, Science and Technology.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
See related editorial, “Can Muscle-Kidney Crosstalk Slow Progression of CKD?,” on pages 2681–2683.
References
- 1.Workeneh BT, Mitch WE: Review of muscle wasting associated with chronic kidney disease. Am J Clin Nutr 91: 1128S–1132S, 2010 [DOI] [PubMed] [Google Scholar]
- 2.Garibotto G, Bonanni A, Verzola D: Effect of kidney failure and hemodialysis on protein and amino acid metabolism. Curr Opin Clin Nutr Metab Care 15: 78–84, 2012 [DOI] [PubMed] [Google Scholar]
- 3.Kosmadakis GC, Bevington A, Smith AC, Clapp EL, Viana JL, Bishop NC, Feehally J: Physical exercise in patients with severe kidney disease. Nephron Clin Pract 115: c7–c16, 2010 [DOI] [PubMed] [Google Scholar]
- 4.Kouidi E, Albani M, Natsis K, Megalopoulos A, Gigis P, Guiba-Tziampiri O, Tourkantonis A, Deligiannis A: The effects of exercise training on muscle atrophy in haemodialysis patients. Nephrol Dial Transplant 13: 685–699, 1998 [DOI] [PubMed] [Google Scholar]
- 5.Anderson JE, Boivin MR, Jr, Hatchett L: Effect of exercise training on interdialytic ambulatory and treatment-related blood pressure in hemodialysis patients. Ren Fail 26: 539–544, 2004 [DOI] [PubMed] [Google Scholar]
- 6.Kouidi E, Iacovides A, Iordanidis P, Vassiliou S, Deligiannis A, Ierodiakonou C, Tourkantonis A: Exercise renal rehabilitation program: Psychosocial effects. Nephron 77: 152–158, 1997 [DOI] [PubMed] [Google Scholar]
- 7.Howden EJ, Fassett RG, Isbel NM, Coombes JS: Exercise training in chronic kidney disease patients. Sports Med 42: 473–488, 2012 [DOI] [PubMed] [Google Scholar]
- 8.Kosmadakis GC, John SG, Clapp EL, Viana JL, Smith AC, Bishop NC, Bevington A, Owen PJ, McIntyre CW, Feehally J: Benefits of regular walking exercise in advanced pre-dialysis chronic kidney disease. Nephrol Dial Transplant 27: 997–1004, 2012 [DOI] [PubMed] [Google Scholar]
- 9.Mustata S, Groeneveld S, Davidson W, Ford G, Kiland K, Manns B: Effects of exercise training on physical impairment, arterial stiffness and health-related quality of life in patients with chronic kidney disease: A pilot study. Int Urol Nephrol 43: 1133–1141, 2011 [DOI] [PubMed] [Google Scholar]
- 10.Balakrishnan VS, Rao M, Menon V, Gordon PL, Pilichowska M, Castaneda F, Castaneda-Sceppa C: Resistance training increases muscle mitochondrial biogenesis in patients with chronic kidney disease. Clin J Am Soc Nephrol 5: 996–1002, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Castaneda C, Gordon PL, Parker RC, Uhlin KL, Roubenoff R, Levey AS: Resistance training to reduce the malnutrition-inflammation complex syndrome of chronic kidney disease. Am J Kidney Dis 43: 607–616, 2004 [DOI] [PubMed] [Google Scholar]
- 12.Castaneda C, Gordon PL, Uhlin KL, Levey AS, Kehayias JJ, Dwyer JT, Fielding RA, Roubenoff R, Singh MF: Resistance training to counteract the catabolism of a low-protein diet in patients with chronic renal insufficiency. A randomized, controlled trial. Ann Intern Med 135: 965–976, 2001 [DOI] [PubMed] [Google Scholar]
- 13.Smart N, Steele M: Exercise training in haemodialysis patients: A systematic review and meta-analysis. Nephrology (Carlton) 16: 626–632, 2011 [DOI] [PubMed] [Google Scholar]
- 14.Datta SR, Brunet A, Greenberg ME: Cellular survival: A play in three Akts. Genes Dev 13: 2905–2927, 1999 [DOI] [PubMed] [Google Scholar]
- 15.Atherton PJ, Babraj J, Smith K, Singh J, Rennie MJ, Wackerhage H: Selective activation of AMPK-PGC-1alpha or PKB-TSC2-mTOR signaling can explain specific adaptive responses to endurance or resistance training-like electrical muscle stimulation. FASEB J 19: 786–788, 2005 [DOI] [PubMed] [Google Scholar]
- 16.Xu J, Li R, Workeneh B, Dong Y, Wang X, Hu Z: Transcription factor FoxO1, the dominant mediator of muscle wasting in chronic kidney disease, is inhibited by microRNA-486. Kidney Int 82: 401–411, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Izumiya Y, Hopkins T, Morris C, Sato K, Zeng L, Viereck J, Hamilton JA, Ouchi N, LeBrasseur NK, Walsh K: Fast/glycolytic muscle fiber growth reduces fat mass and improves metabolic parameters in obese mice. Cell Metab 7: 159–172, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Araki S, Izumiya Y, Hanatani S, Rokutanda T, Usuku H, Akasaki Y, Takeo T, Nakagata N, Walsh K, Ogawa H: Akt1-mediated skeletal muscle growth attenuates cardiac dysfunction and remodeling after experimental myocardial infarction. Circ Heart Fail 5: 116–125, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fujita M, Ye Q, Ouchi H, Harada E, Inoshima I, Kuwano K, Nakanishi Y: Doxycycline attenuated pulmonary fibrosis induced by bleomycin in mice. Antimicrob Agents Chemother 50: 739–743, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meran S, Steadman R: Fibroblasts and myofibroblasts in renal fibrosis. Int J Exp Pathol 92: 158–167, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bengatta S, Arnould C, Letavernier E, Monge M, de Préneuf HM, Werb Z, Ronco P, Lelongt B: MMP9 and SCF protect from apoptosis in acute kidney injury. J Am Soc Nephrol 20: 787–797, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang L, Rajan V, Lin E, Hu Z, Han HQ, Zhou X, Song Y, Min H, Wang X, Du J, Mitch WE: Pharmacological inhibition of myostatin suppresses systemic inflammation and muscle atrophy in mice with chronic kidney disease. FASEB J 25: 1653–1663, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang L, Du J, Hu Z, Han G, Delafontaine P, Garcia G, Mitch WE: IL-6 and serum amyloid A synergy mediates angiotensin II-induced muscle wasting. J Am Soc Nephrol 20: 604–612, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Niedermeier M, Reich B, Rodriguez Gomez M, Denzel A, Schmidbauer K, Göbel N, Talke Y, Schweda F, Mack M: CD4+ T cells control the differentiation of Gr1+ monocytes into fibrocytes. Proc Natl Acad Sci U S A 106: 17892–17897, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mu W, Ouyang X, Agarwal A, Zhang L, Long DA, Cruz PE, Roncal CA, Glushakova OY, Chiodo VA, Atkinson MA, Hauswirth WW, Flotte TR, Rodriguez-Iturbe B, Johnson RJ: IL-10 suppresses chemokines, inflammation, and fibrosis in a model of chronic renal disease. J Am Soc Nephrol 16: 3651–3660, 2005 [DOI] [PubMed] [Google Scholar]
- 26.Yang J, Lin SC, Chen G, He L, Hu Z, Chan L, Trial J, Entman ML, Wang Y: Adiponectin promotes monocyte-to-fibroblast transition in renal fibrosis. J Am Soc Nephrol 24: 1644–1659, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ito D, Ito O, Mori N, Cao P, Suda C, Muroya Y, Hao K, Shimokawa H, Kohzuki M: Exercise training upregulates nitric oxide synthases in the kidney of rats with chronic heart failure. Clin Exp Pharmacol Physiol 40: 617–625, 2013 [DOI] [PubMed] [Google Scholar]
- 28.Kang DH, Nakagawa T, Feng L, Johnson RJ: Nitric oxide modulates vascular disease in the remnant kidney model. Am J Pathol 161: 239–248, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sun D, Wang Y, Liu C, Zhou X, Li X, Xiao A: Effects of nitric oxide on renal interstitial fibrosis in rats with unilateral ureteral obstruction. Life Sci 90: 900–909, 2012 [DOI] [PubMed] [Google Scholar]
- 30.Ishikawa Y, Gohda T, Tanimoto M, Omote K, Furukawa M, Yamaguchi S, Murakoshi M, Hagiwara S, Horikoshi S, Funabiki K, Tomino Y: Effect of exercise on kidney function, oxidative stress, and inflammation in type 2 diabetic KK-A(y) mice. Exp Diabetes Res 2012: 702948, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ghosh S, Khazaei M, Moien-Afshari F, Ang LS, Granville DJ, Verchere CB, Dunn SR, McCue P, Mizisin A, Sharma K, Laher I: Moderate exercise attenuates caspase-3 activity, oxidative stress, and inhibits progression of diabetic renal disease in db/db mice. Am J Physiol Renal Physiol 296: F700–F708, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luiz RS, Silva KA, Rampaso RR, Antônio EL, Montemor J, Bocalini DS, Dos Santos L, Moura L, Tucci PJ, de Abreu NP, Schor N: Exercise attenuates renal dysfunction with preservation of myocardial function in chronic kidney disease. PLoS ONE 8: e55363, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Silva KA, Luiz RS, Rampaso RR, de Abreu NP, Moreira ED, Mostarda CT, De Angelis K, de Paulo Castro Teixeira V, Irigoyen MC, Schor N: Previous exercise training has a beneficial effect on renal and cardiovascular function in a model of diabetes. PLoS ONE 7: e48826, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang XH, Du J, Klein JD, Bailey JL, Mitch WE: Exercise ameliorates chronic kidney disease-induced defects in muscle protein metabolism and progenitor cell function. Kidney Int 76: 751–759, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Verzola D, Procopio V, Sofia A, Villaggio B, Tarroni A, Bonanni A, Mannucci I, De Cian F, Gianetta E, Saffioti S, Garibotto G: Apoptosis and myostatin mRNA are upregulated in the skeletal muscle of patients with chronic kidney disease. Kidney Int 79: 773–782, 2011 [DOI] [PubMed] [Google Scholar]
- 36.Zhang L, Wang XH, Wang H, Du J, Mitch WE: Satellite cell dysfunction and impaired IGF-1 signaling cause CKD-induced muscle atrophy. J Am Soc Nephrol 21: 419–427, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kuan Y, Surman J, Frystyk J, El Nahas AM, Flyvbjerg A, Haylor JL: Lack of effect of IGF-I on the glomerular filtration rate in non-diabetic patients with advanced chronic kidney disease. Growth Horm IGF Res 19: 219–225, 2009 [DOI] [PubMed] [Google Scholar]
- 38.Teppala S, Shankar A, Sabanayagam C: Association between IGF-1 and chronic kidney disease among US adults. Clin Exp Nephrol 14: 440–444, 2010 [DOI] [PubMed] [Google Scholar]
- 39.Chen Y, Sood S, McIntire K, Roth R, Rabkin R: Leucine-stimulated mTOR signaling is partly attenuated in skeletal muscle of chronically uremic rats. Am J Physiol Endocrinol Metab 301: E873–E881, 2011 [DOI] [PubMed] [Google Scholar]
- 40.Akasaki Y, Ouchi N, Izumiya Y, Bernardo BL, Lebrasseur NK, Walsh K: Glycolytic fast-twitch muscle fiber restoration counters adverse age-related changes in body composition and metabolism. Aging Cell 13: 80–91, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baylis C: Nitric oxide deficiency in chronic kidney disease. Am J Physiol Renal Physiol 294: F1–F9, 2008 [DOI] [PubMed] [Google Scholar]
- 42.Chen KC, Hsieh CL, Peng CC, Peng RY: Exercise rescued chronic kidney disease by attenuating cardiac hypertrophy through the cardiotrophin-1 → LIFR/gp 130 → JAK/STAT3 pathway. Eur J Prev Cardiol 21: 507–520, 2012 [DOI] [PubMed] [Google Scholar]
- 43.Ouchi N, Oshima Y, Ohashi K, Higuchi A, Ikegami C, Izumiya Y, Walsh K: Follistatin-like 1, a secreted muscle protein, promotes endothelial cell function and revascularization in ischemic tissue through a nitric-oxide synthase-dependent mechanism. J Biol Chem 283: 32802–32811, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zemse SM, Chiao CW, Hilgers RH, Webb RC: Interleukin-10 inhibits the in vivo and in vitro adverse effects of TNF-alpha on the endothelium of murine aorta. Am J Physiol Heart Circ Physiol 299: H1160–H1167, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu AC, Lee M, McManus BM, Choy JC: Induction of endothelial nitric oxide synthase expression by IL-17 in human vascular endothelial cells: Implications for vascular remodeling in transplant vasculopathy. J Immunol 188: 1544–1550, 2012 [DOI] [PubMed] [Google Scholar]
- 46.Thomas SS, Mitch WE: Mechanisms stimulating muscle wasting in chronic kidney disease: The roles of the ubiquitin-proteasome system and myostatin. Clin Exp Nephrol 17: 174–182, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, Walsh K, Schiaffino S, Lecker SH, Goldberg AL: Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 117: 399–412, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Grill MA, Bales MA, Fought AN, Rosburg KC, Munger SJ, Antin PB: Tetracycline-inducible system for regulation of skeletal muscle-specific gene expression in transgenic mice. Transgenic Res 12: 33–43, 2003 [DOI] [PubMed] [Google Scholar]
- 49.Shiojima I, Sato K, Izumiya Y, Schiekofer S, Ito M, Liao R, Colucci WS, Walsh K: Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J Clin Invest 115: 2108–2118, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pulskens WP, Rampanelli E, Teske GJ, Butter LM, Claessen N, Luirink IK, van der Poll T, Florquin S, Leemans JC: TLR4 promotes fibrosis but attenuates tubular damage in progressive renal injury. J Am Soc Nephrol 21: 1299–1308, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kusunoki H, Taniyama Y, Azuma J, Iekushi K, Sanada F, Otsu R, Iwabayashi M, Okayama K, Rakugi H, Morishita R: Telmisartan exerts renoprotective actions via peroxisome proliferator-activated receptor-γ/hepatocyte growth factor pathway independent of angiotensin II type 1 receptor blockade. Hypertension 59: 308–316, 2012 [DOI] [PubMed] [Google Scholar]