

Abstract
Patients with CKD have an increased risk of cardiovascular mortality from arrhythmias and sudden cardiac death. We used a rat model of CKD (Cy/+) to study potential mechanisms of increased ventricular arrhythmias. Rats with CKD showed normal ejection fraction but hypertrophic myocardium. Premature ventricular complexes occurred more frequently in CKD rats than normal rats (42% versus 11%, P=0.18). By optical mapping techniques, action potential duration (APD) at 80% of repolarization was longer in CKD rats (78±4ms) than normal rats (63±3 ms, P<0.05) at a 200-ms pacing cycle length. Calcium transient (CaT) duration was comparable. Pacing cycle length thresholds to induce CaT alternans or APD alternans were longer in CKD rats than normal rats (100±7 versus 80±3 ms and 93±6 versus 76±4 ms for CaT and APD alternans, respectively, P<0.05), suggesting increased vulnerability to ventricular arrhythmia. Ventricular fibrillation was induced in 9 of 12 CKD rats and 2 of 9 normal rats (P<0.05); early afterdepolarization occurred in two CKD rats but not normal rats. The mRNA levels of TGF-β, microRNA-21, and sodium calcium-exchanger type 1 were upregulated, whereas the levels of microRNA-29, L-type calcium channel, sarco/endoplasmic reticulum calcium–ATPase type 2a, Kv1.4, and Kv4.3 were downregulated in CKD rats. Cardiac fibrosis was mild and not different between groups. We conclude that cardiac ion channel and calcium handling are abnormal in CKD rats, leading to increased vulnerability to early afterdepolarization, triggered activity, and ventricular arrhythmias.
Keywords: cardiovascular, calcium, fibrosis
CKD affects more than 26 million individuals; 15% of American adults have some degree of CKD.1 Cardiovascular events account for 40% of deaths in CKD patients. In patients on dialysis, 25% of cardiovascular deaths are caused by sudden cardiac death, a 100-fold rate compared with the general population.2,3 Although the increased frequency in dialysis patients has often been attributed to electrolyte imbalances induced by the dialysis procedure in patients with underlying cardiac disease, recent data have highlighted the increased risk with CKD in patients not yet on dialysis, indicating that other factors in these patients with CKD may lead to sudden cardiac death.4 The Multicenter Automatic Defibrillator Implantation Trial-II showed that the risk for sudden cardiac death was 17% higher for every 10 ml/min per 1.73 m2 decrease in eGFR.5 The result was comparable even when patients considered low risk for sudden cardiac death were analyzed.6 The pathogenesis by which CKD predisposes to sudden cardiac death remains unknown. Our rat model of CKD, the Cy/+ rat, accurately represents many of the changes associated with human CKD, including hypertension, left ventricular hypertrophy, and CKD–mineral bone disorder. Furthermore, we have occasionally observed spontaneous death in otherwise stable animals, presumably caused by cardiac arrhythmias.7,8 We, therefore, used this model to examine sudden cardiac death-related cardiac remodeling, arrhythmogenesis, and gene expression. The results show abnormalities that may play a pathogenic role in CKD-related sudden cardiac death.
Results
Assessment of CKD
We have previously confirmed that these Cy/+ rats develop slowly progressive and spontaneous CKD.7–10 Biochemical measurements taken at the time of euthanasia in this study are consistent with our previous publications in this model.7,8,11,12 Table 1 shows that the CKD rats had elevated BUN, phosphorus, and fibroblast growth factor 23 (FGF23) compared with normal (NL) rats. However, there was no statistical difference in calcium and potassium concentrations. We have previously shown elevated parathyroid hormone (PTH) levels in this animal model7 but had inadequate plasma to measure it directly in the animals that underwent electrophysiology studies.
Table 1.
Plasma biochemistry results from animals undergoing electrophysiology studies
| Groups | BUN (mg/L) | Phosphorous (mg/L) | FGF23 (pg/ml) | Calcium (mg/L) | Potassium (mg/L) |
|---|---|---|---|---|---|
| NL (n=10) | 24±1 | 4.1±0.5 | 301±56 | 9.5±0.6 | 5.1±0.2 |
| CKD (n=6) | 57±3a | 6.8±0.7a | 1066±80a | 8.2±0.6 | 5.6±0.8 |
Note that, because of sample availability, the potassium results are from different NL and CKD animals at equivalent age.
P<0.05.
Cardiac Morphology and Function
Figure 1A shows representative pictures of hearts from NL and CKD rats. The hearts from CKD rats were enlarged by gross and microscopic appearance. The results evaluated by M-mode echocardiography (time-motion mode) are summarized in Table 2. There was no difference between NL and CKD rats in ejection fraction, suggesting that there was no systolic dysfunction. Hypertrophy of myocardium was identified by significantly increased interventricular septum thickness, left ventricle (LV) posterior wall thickness, and increased LV mass (P<0.001, P<0.01, and P<0.001, respectively). The values of interventricular septum thickness and LV posterior wall thickness were comparable, indicating that the cardiac hypertrophy was concentric.
Figure 1.
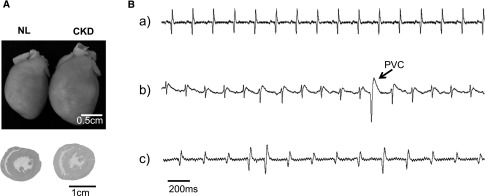
Hearts of CKD rats are hypertrophic and arrhythmogenic. (A) Representative image of hearts from an NL rat and a CKD rat. Note the larger size of the heart from the CKD rat. Cross-sections of heart slices stained by hematoxylin and eosin show thicker myocardium in the CKD rat. (B) Representative surface ECG recording. (a) Normal sinus rhythm recorded from an NL rat. (b) Representative tracing showing occurrence of premature ventricular complex (PVC) during ECG recording on a CKD rat. PVCs were more frequently seen in CKD rats. (c) Spontaneous atrial fibrillation was observed in a CKD rat.
Table 2.
Summary of hypertrophic parameters in echocardiography
| Groups | IVS (cm) | LVPS (cm) | LV mass (g) | EF (%) |
|---|---|---|---|---|
| NL (n=9) | 0.12±0.01 | 0.12±0.01 | 0.91±0.04 | 71±4 |
| CKD (n=12) | 0.20±0.01a | 0.18±0.02b | 1.27±0.05a | 70±2 |
IVS, interventricular septum; LVPW, LV posterior wall; EF, ejection fraction.
P<0.001.
P<0.01.
Spontaneous Arrhythmias in CKD Rats
We observed cardiac rhythm disorders in CKD rats both in vivo and ex vivo. Representative traces from surface electrocardiography (ECG) are shown in Figure 1B. NL rats have normal sinus rhythm without arrhythmia (Figure 1B, a). Premature ventricular complexes (Figure 1B, b) were documented in 5 of 12 CKD rats (42%) and 1 of 9 NL rats (11%), but the difference was not statistically significant (P=0.18). Spontaneous atrial fibrillation was observed in one CKD rat (Figure 1B, c).
Cardiac Electrophysiology
We assessed three major mechanisms for causing arrhythmia: (1) automaticity (the ability of cardiomyocytes to spontaneously depolarize and generate an action potential); (2) afterdepolarization and triggered activity (the depolarizing oscillations in the membrane voltage induced by preceding action potentials); and (3) re-entry (a phenomenon when the wave front circulates around an anatomic or electrophysiological obstacle). Finally, we assessed the differences in the ability to induce ventricular fibrillation (VF) in response to electrical stimuli.
Electrophysiology Results
(1) We assessed the sinoatrial node automaticity by analyzing the heart rate and heart rate variability using the beat to beat intervals on ECG. The beat to beat interval in CKD rats was 193.7±5.8 ms, whereas that of NL rats was 186.4±5.8 ms (P=0.41). The SD of the intervals between normal heart beats in CKD rats was 2.8±0.4 ms, whereas that in NL rats was 3.1±0.8 ms (P=0.70). These data suggest that the sinoatrial node automaticity of the CKD and NL rats is comparable.
(2) Triggered activity describes the generation of action potentials in a cardiomyocyte caused by afterdepolarizations that occur when either the cardiac cells are partially repolarized (early afterdepolarization [EAD]) or completely repolarized (delayed afterdepolarization). When these afterdepolarizations are large enough to induce propagated action potentials, they are called triggered activities. Major causes of EADs and delayed afterdepolarizations are prolonged action potential duration (APD) and large calcium transient duration (CaTD). The intracellular calcium transient (CaT) is determined by the changes in emitted fluorescence during the cardiac cycle over time and provides an assessment of intracellular calcium changes caused by calcium entry and exit through sarcolemma and fluxes of calcium in and out of the sarcoplasmic reticulum (Figure 2A, a and b). Both APD and CaT are affected by the heart rate, and thus, to compare between groups, the hearts are paced at the same fixed pacing cycle lengths (PCLs) when these parameters are assessed. Figure 2A, c shows typical examples of optical action potentials and documents longer APD in CKD rats than NL rats. At 200-ms PCL, APD at 80% of repolarization for CKD and NL rats was 78±4 and 62±3 ms, respectively (P<0.05). At 100-ms PCL, it was 55±3 and 51±2 ms for CKD and NL rats, respectively. In contrast to the APD, the CaTD and the SD of the CaTD between the two groups were comparable (Figure 2C). EAD was identified in two CKD rats (Figure 3B) by optical mapping but not in NL rats. One of them developed nonsustained ventricular tachycardia. Thus, APD prolongation leading to EAD seems to be an important arrhythmogenic mechanism in these CKD rats.
Figure 2.
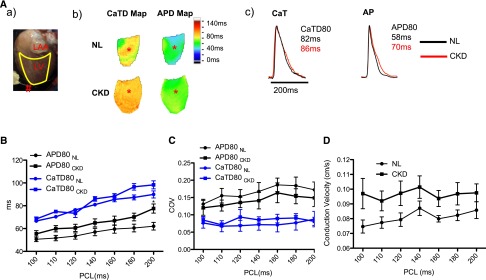
CKD rats have longer APD than NL rats. (A) Representative image showing (a) the position of the imaged heart. #Indicates the site of pacing. LAA indicates the left atrium appendage. LV indicates the left ventricle. (b) Representative APD at 80% of repolarization (APD80) map and CaTD at 80% of repolarization (CaTD80) map at 200-ms PCL. (c) Corresponding action potential (AP) traces and CaT traces sampled at the sites marked by the asterisks in A, b. (B) APD80 and CaTD80 were plotted against PCLs (n=8 and n=11 for NL and CKD, respectively). ANOVA analysis showed that APD80 curves between NL and CKD were statistical different, whereas CaTD80 curves remained comparable. (C) Coefficients of variation (COVs) of APD80 and CaTD80 were similar between NL and CKD rats, suggesting that the heterogeneity was comparable. (D) Conduction velocity was slightly faster for CKD rats, but it did not reach statistical difference.
Figure 3.

CKD rats are more vulnerable to electrical instability and have more phase singularities in ventricular fibrillation than NL rats. (A) PCL thresholds to induce APD alternans and CaT alternans were longer in CKD rats (n=7 and n=11 for NL and CKD, respectively), indicating increased susceptibility. The tracings are CaT and AP recorded at 90-ms PCL. Note that, at this pacing cycle length, the CKD rat developed alternans (nonuniform waveforms) for both CaT and AP, whereas the NL rat showed regular traces from beat to beat. *P<0.05. (B) EAD-induced tachycardia was observed. Yellow arrows indicate the direction of cardiac excitation. When the heart was paced, the cardiac excitation started from the apex and propagated to the base. After EAD occurred, the path of propagation was altered and eventually caused tachycardia. (C, a) Representative tracings of AP and CaT during VF. Note that the pattern of VF between NL and CKD was different. (C, b) Dominant frequency during VF was larger in CKD rats. VF was induced by burst pacing (n=8 and n=10 for NL and CKD, respectively). The frequency was transformed by Fourier transform from traces on pseudo-ECG (insets). The larger dominant frequency indicates slower activation rate for CKD rats. **P<0.01. (D, a) Representative phase singularity map during VF in NL and CKD animals. The scale bar shows the phase from minus π to plus π, where π is the ratio between the circumference and the diameter. In contrast to the NL animals, there is obviously more phase singularity during VF. (D, b) When the phase singularities were quantified every 2 ms for a total of 500 ms during induced VF, the number of phase singularities was larger in CKD (n=8 and n=10 for NL and CKD, respectively). *P<0.05.
(3) The occurrence of re-entry is, in part, determined by the wavelength of excitation (product of APD and conduction velocity).13 In addition, the APD restitution properties (the dependence of APD of the preceding diastolic interval) are also important in determining the generation and maintenance of re-entry.14,15 A steep APD restitution curve (small changes in diastolic interval yielding large changes in APD) is associated with the development of APD and CaT alternans (beat-to-beat variability). It will be more pronounced at rapid heart rates, where there will be a smaller diastolic interval between beats, resulting in greater alternans. At the cellular level, instabilities in membrane voltage (i.e., steep APD restitution slope) and intracellular calcium cycling dynamics cause APD and the CaT to alternate. The alternating APD and CaT are, in part, responsible for the clinical association of cardiac alternans (e.g., T-wave alternans) with arrhythmia risk.16 Alternans of APD often precede the onset of VF. Therefore, if alternans of APD and CaT amplitude occur at longer PCL (slower heart rates), then it implies increased VF susceptibility.17
The coefficient of variation for either APD or CaTD was similar between CKD and NL rats, suggesting a similar degree of heterogeneity (Figure 2C). We did not observe either a slower conduction velocity (Figure 2D) or shorter APD in CKD rats compared with NL rats. However, we found that there is a significantly longer pacing cycle threshold to induce both CaT amplitude alternans (an alternation of the amplitude of CaT from beat to beat) and APD alternans (an alternation of the APD from beat to beat) as shown in Figure 3A. PCL threshold to induce CaT amplitude alternans was 100±7 ms for CKD rats and 80±3 ms for NL rats (P<0.05). For the APD alternans, PCL threshold was 93±6 and 76±4 ms for CKD and NL rats, respectively (P<0.05). The pattern of alternans was similar in CKD and NL rats, starting from a location and spreading throughout the heart when PCL was further reduced. No spatial discordance was identified within the imaged area. The beat with longer APD always accompanied larger CaT amplitude, indicating positive coupling. Thus, the CKD animals are at higher risk of re-entry than NL animals because of CaT amplitude and APD alternans.
We also measured VF susceptibility by further shortening the PCL to determine if VF is easily inducible. After VF is induced, we used optical mapping to measure VF properties. The dominant frequency is used to estimate the rate of activation during VF, whereas the phase singularity is used to describe the sources of re-entrant VF activation.18,19 These signatures of VF are used to determine if there are differential patterns of activation during VF between CKD and NL rats.
The pattern of VF was different between CKD and NL rats (Figure 3C, a). After VF was induced, the dominant frequency was significantly lower in CKD rats (18.60±1.38 Hz) than NL rats (24.69±1.61 Hz, P<0.01) (Figure 3C, b), suggesting that CKD rats have a slower activation rate during VF than NL rats. Phase singularity on the phase map (Figure 3D, a) correlates with the locations of wave breaks and rotors of spiral waves.20 We measured the number of phase singularities for 500 ms during VF and found more phase singularities in CKD rats (150±19) than NL control rats (97±11, P<0.05) (Figure 3D, b).
We attempted VF induction by giving a single premature stimulus (S2) after a train of regularly pacing (S1). The interval between S1 and S2 started at 100 ms and decreased stepwise by 10 ms until VF or effective refractory period was observed. As shown in Figure 4B, a single premature stimulation induced VF in 9 of 12 CKD rats compared with 2 of 9 NL rats (P<0.05). There was a trend to a longer effective refractory period (increased time during which a new action potential cannot occur) in CKD rats (53±8 ms) compared with NL rats (38±3 ms), but the difference was not statistically significant (P=0.12).
Figure 4.
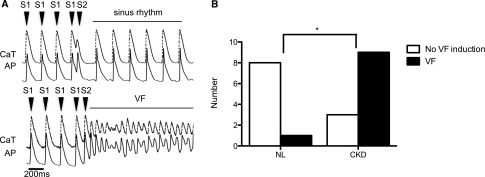
CKD rats are more vulnerable to ventricular fibrillation than NL rats. VF inducibility was evaluated by applying a premature stimulus (S2) after nine consecutive regular stimuli (S1). The CaT is represented by the dashed upper line, and the AP is represented by the solid lower line. (A) Upper panel is a representative tracing showing failure of VF induction, and lower panel is an example of successful induction of VF. (B) The VF inducibility of CKD rats was higher than in NL animals when compared by Fisher exact test (n=9 and n=12 for NL and CKD, respectively). *P<0.05.
Fibrosis
In a previous report from a 5/6 nephrectomy model of CKD, profound fibrosis was identified from hypertrophied hearts.21 The histologic finding has previously been cited as a reason for arrhythmias in CKD.22 We quantified fibrosis using picrosirius red staining grading four randomly selected areas semiquantitatively as we have previously reported for kidney.23 The fibrosis scores for the ventricular septum were 1.45±0.64 for CKD and 1.18±0.04 for NL, and the fibrosis scores for the left ventricular free wall were 1.41±0.38 for CKD and 1.23±0.08 for NL (P=0.20 and P=0.34, respectively). No animal had a score greater than 2.2 (minimal interspersed myocardial fibrosis). We also examined some of the animals after electrophysiology study. Almost one half of the CKD animals (three of seven) and no NL animals (zero of six) had fibrosis (all mild). In three CKD animals that had mild fibrosis, there was no difference in electrophysiological parameters that we measured compared with the CKD rats without fibrosis.
Gene and Protein Expression
Given our findings of APD prolongation, early depolarization, and induction of APD and CaT alternans at longer PCL, we examined molecular changes by real-time PCR. Genes selected included those genes related to fibrosis (TGF-β, microRNA-21 [miR21], and miR29),24,25 intracellular calcium homeostasis (sodium calcium-exchanger type 1 [NCX1], ryanodine receptor type 2, sarco/endoplasmic reticulum calcium-ATPase type 2a [SERCA2a], and L-type calcium channel [Cav1.2]), conduction (connexin 43 and cardiac sodium channel), and repolarization power (potassium voltage-gated channel subfamily A member 4 [Kv1.4], potassium voltage-gated channel subfamily A member 5, potassium voltage-gated channel subfamily D member 3 [Kv4.3], potassium voltage-gated channel subfamily J member 11, potassium voltage-gated channel subfamily Q member 1, and voltage-gated K+ channel subfamily H member 2 [Kv11.1]). Figure 5 shows the results. Altered expression profiles were identified in several genes. The mRNA level of TGF-β, miR21, and NCX1 was upregulated, whereas that of miR29, Cav1.2, SERCA2a, Kv1.4, and Kv4.3 was downregulated. We examined protein expression by Western blotting of key regulators. We examined the major calcium handling protein, SERCA2a, which is known to be causally related to the development of ventricular arrhythmias.26 We also examined protein expression of potassium channel Kv1.4, because the mRNA levels were significantly downregulated in CKD. Furthermore, Kv1.4 encodes the transient outward current (Ito), an important repolarization current for rodents. Reduction of Ito is known to increase the QT interval and the risk of ventricular arrhythmias in mice.27 Kv11.1 was selected, because its mRNA showed comparable expression between CKD and NL rats. As shown in Figure 6, SERCA2a and Kv1.4 (P<0.05) were downregulated, whereas Kv11.1 remained constant, comparable with the real-time PCR results (Figure 5).
Figure 5.

Gene expression profiles differ significantly between NL and CKD rats. Real-time PCR comparison between NL and CKD rats (n=7–15 per group). Cx43, connexin 43; Kir6.2, potassium voltage-gated channel subfamily J member 11 (IKATP); Kv1.5, potassium voltage-gated channel subfamily A member 5 (IKur); Kv7.1, potassium voltage-gated channel subfamily Q member 1 (IKs); Nav1.5, cardiac sodium channel; RYR2, ryanodine receptor type 2. *P<0.05; **P<0.01; ***P<0.001.
Figure 6.

Western blot confirming altered gene expression in CKD rats (n=7 in each group). (A) Representative images. (B) Quantification of the images. Each experiment was repeated three times. *P<0.05; **P<0.01. GAPDH, glyceraldehyde-3-phosphate dehydrogenase
Discussion
The electrophysiology studies in the CKD rats compared with NL control rats showed prolonged APD, increased susceptibility to alternans (electrical instability), occurrence of early depolarization, increased VF inducibility, and increased phase singularities during VF, suggesting increased re-entrant wave fronts and wave breaks during VF. Moreover, spontaneous heart rhythm disorders occurred without any external stimulus in CKD rats. CKD rats exhibited higher frequency of premature ventricular complexes, which has been shown to be associated with increased risk for sudden cardiac death.28 One CKD rat showed spontaneous atrial fibrillation. To our knowledge, this study is the first systematic evaluation of electrophysiology in a CKD model. It should be emphasized that, although the electrophysiology studies are done ex vivo, abnormalities likely reflect the state of uremic toxin exposure at the time that the heart was removed for study.
Ventricular arrhythmias are common in CKD patients, with a progressive increase in the incidence of sudden cardiac death with loss of kidney function.3 CKD patients also suffer from comorbidities that have been proposed to increase the risk of arrhythmias, including coronary artery disease, sympathetic overactivity, hypertension, inflammation, anemia, electrolyte imbalances, and LV hypertrophy (LVH).22 LVH is present in more than 75% of patients with advanced CKD, and it presents with diastolic dysfunction rather than systolic function.29,30 In humans with CKD, biopsy and autopsy studies show interstitial fibrosis and cardiomyocyte hypertrophy. In small studies, the magnitude of the pathologic abnormalities was predictive of subsequent death.30–33 In humans, LVH of any cause may increase the risk of sudden death.34
In our rat model of CKD, we clearly observed the development of LVH but no significant increase in fibrosis by histology. We did, however, observe upregulation of profibrotic TGF-β and miR21 and downregulation of antifibrotic miR29, common in fibrotic pathways in the heart.24,25 This finding implies that our CKD rats were beginning to develop fibrosis. However, the magnitude of fibrosis observed histologically was markedly less than in the 5/6 nephrectomy model.21 Furthermore, because electrophysiology studies in rats with and without the mild fibrosis were comparable, fibrosis is unlikely to be the primary etiology for arrhythmias.22 However, the cardiac remodeling during LVH also results in altered expression and activity of cardiac ion channels, leading to a change in cardiac electrophysiological properties and increases the incidence of arrhythmias.35–37
Cardiac electrical instability has been identified to precede the onset of VF.38–40 T-wave alternans are a predictor of spontaneous VF in a canine model of sudden cardiac death.41 T-wave alternans are also associated with sudden death in dialysis patients.3 In this study, we did not identify any apparent abnormality of automaticity but instead, observed EAD, triggered activity, and increased propensity for the development of alternans. Our observation of increased CaT alternans susceptibility in CKD rats is suggestive of abnormal intracellular calcium handling.
Weiss et al.42 reported that functionally altered Cav1.2–ryanodine receptor type 2 couplon could lead to CaT alternans. Cav1.2, one of the major cardiac calcium channels, was downregulated in CKD rats. Furthermore, NCX1 was upregulated, and SERCA2a was downregulated. These abnormalities of intracellular calcium handling may play a role in ventricular arrhythmogenesis. Both PTH and FGF23 have been shown to alter intracellular calcium in cell culture models of cardiomyocytes. Rat cardiomyocytes incubated with PTH have a rapid increase in intracellular calcium, leading to increased beats per minute and apoptosis. Furthermore, incubation of the cells with uremic serum from animals reproduced these findings but not if the serum was from an animal that had undergone parathyroidectomy.43 FGF23 can directly induce LVH and cardiomyocyte hypertrophy in mice through the phospholipase–C-calcineurin–nuclear factor of activated T cells axis.44 In cultured cardiomyocytes, FGF23 can induce a rise in intracellular calcium that can be prevented by pretreatment with verapamil.45 Both PTH and FGF23 progressively rise with loss of kidney function in humans with CKD46 and this animal model, perhaps explaining the increased calcium influx, and might further alter normal calcium recycling within the cytoplasm. This result, together with a downregulation of SERCA2a, may lead to a concomitant increase in arrhythmias.22 However, it is important to point out that we assessed whole-heart CaTs, not single-cell intracellular calcium like in the experiments above. Furthermore, we performed electrophysiology studies ex vivo without circulating PTH or FGF23. Therefore, studies that examine the effects of treatments to lower PTH and FGF23 and resulting electrophysiology are needed to confirm this hypothesis.
Cardiac ion channel remodeling has been reported in various animal models of hypertrophic cardiomyopathy.36 The most common finding is downregulation of transient outward potassium current (Ito), which is composed of Kv1.4 and Kv4.3. It is also the major repolarization mechanism in rodent hearts. Our results are consistent with this finding, showing decreased expression of Kv1.4 and Kv4.3. This finding would lead to reduced repolarizing reserve, which may play a role in the observed increased APD alternans and early depolarization. Future studies will need to focus on pharmacologic blockade of the channels to confirm a causative role of specific channel abnormalities.
In summary, our data indicate the occurrence of cardiac electrical remodeling abnormalities in the rat model of CKD that favors onset and maintenance of VF and thus, predisposes to sudden cardiac death. These remodeling abnormalities include loss of repolarization reserve (EAD and alternans) and altered cellular calcium homeostasis with reduced sarcoplasmic reticulum calcium content. Should these studies be confirmed in other models of CKD, these studies provide new insight in the pathogenesis of arrhythmias and sudden death in CKD.
Concise Methods
Animal Model
Male Cy/+ rats, Han:SPRD rats with autosomal dominant polycystic kidney disease, were used for this study. The Cy/+ rat is a spontaneous cystic kidney disease model with a defect in the samcystin (Cy) gene. However, unlike other models of cystic kidney disease caused by abnormal components of the cilia resulting in increased intracellular calcium,47 the samcystin protein does not affect the cilia but instead, binds to a cytoplasmic RNA binding protein bicc-1.12,48 How that binding leads to cystic disease is unknown. Male heterozygous rats (Cy/+) develop characteristics of CKD (azotemia or elevated BUN) around 10 weeks of age, which progress to terminal uremia by about 40 weeks. This animal model spontaneously develops manifestations of CKD, including biochemical abnormalities and reduced GFR, including hypertension, left ventricular hypertrophy, renal osteodystrophy, and arterial calcification.7–10 Cy/+ rats (called CKD) were 35 weeks old, with the exception of one animal studies at 39 weeks. On the basis of previous 24-hour urine collections,8 the GFR at 35 weeks is approximately 10%–20%. Normal littermates or purchased Han:SPRD rats of similar age were used as NL rats. For cardiac electrophysiological studies, 12 CKD and 9 NL rats were used. Optical mapping data from one NL rat and one CKD rat were discarded because of technical problems. The Langendorff heart preparation requires prolonged (2 hours or more) perfusion and thus, alters histology and PCR. Therefore, tissue for histology was analyzed from sections of CKD and NL rats from a previous study,11 with confirmation from rats that underwent the electrophysiology studies to directly correlate the results to the presence and absence of fibrosis. For real-time PCR and Western blots, we used previously frozen samples from these latter animals11 and additional samples from previously frozen tissue. All tissues were from animals at 35 weeks of age. All procedures were reviewed and approved by the Indiana University School of Medicine Institutional Animal Care and Use Committee.
Serum Biochemical Measurements
Before electrophysiological studies, blood was collected by tail vein for biochemical measurements (n=9 for NL and n=6 for CKD). Plasma was analyzed for BUN, calcium, and phosphorus using colorimetric assays (Point Scientific, Canton, MI). Intact PTH was determined by ELISA (Alpco, Salem, NH). FGF23 was assessed with a two-site assay (Immunotopics, San Clemente, CA).8 Plasma potassium was measured using stored samples from 35-week-old animals because of inadequate volume available in the rats undergoing the electrophysiological studies (because no cardiac puncture could be done) using a clinical autoanalyzer and colorimetric assay.
Echocardiography and ECG
Rats were anesthetized by isoflurane, and an Acuson Cypress Plus Portable Ultrasound Machine (SIEMEMS, Malvern, PA) was used for echocardiography. Then, surface ECG was recorded for 3 minutes. The signals were filtered from 0.05 to 100 Hz and digitized at 1 kHz with AxoScope (Molecular Devices, Downingtown, PA).
Langendorff Heart Preparation
Immediately after euthanasia with isoflurane, the heart was harvested by thoracotomy in ice-cold cardioplegic solution and cannulated through the aorta. Blood was flushed out by injection of a 50-ml cardioplegic solution composed of 129 mM NaCl, 12 mM KCl, 0.9 mM NaH2PO4, 20 mM NaHCO3, 1.8 mM CaCl2, 0.5 mM MgSO4, and 5.5 mM glucose. Subsequently, the heart was connected to a Langendorff apparatus and perfused with oxygenated Tyrode solution heated to 37.5°C. The composition of Tyrode solution was 125 mM NaCl, 7 mM KCl, 0.5 mM MgCl2, 23 mM NaHCO3, 2 mM NaH2PO4, 1.8 mM CaCl2, 5.5 glucose, and 2% BSA equilibrated with 95% O2 and 5% CO2 to maintain a pH of 7.4.
Optical Mapping
The perfusate was maintained at 37.5°C with a constant flow rate around 9 ml/min. After 15 minutes of stabilization, 10 μM voltage-sensitive dye RH237 (Life Technologies, Carlsbad, CA) and 1.5 μM calcium-sensitive dye Rhod-2 (Life Technologies) were added to the perfusate. The heart was then washed for 15 minutes followed by the addition of 15 μM excitation-contraction uncoupler, blebbistatin (Tocris Bioscience, Minneapolis, MN). A laser light at 532 nm was used to excite the stained heart, and fluorescence was collected. One camera acquired calcium fluorescence through a 585±20-nm band-pass filter, whereas the other camera simultaneously recorded voltage fluorescence through a 710-nm long-pass filter. Fluorescence image was captured at a frame rate with 2 ms/frame and 100×100 pixels, with spatial resolution of 0.35×0.35 mm2/pixel for 4 seconds. To determine the rhythm of the Langendorff heart, pseudo-ECG was used. It was obtained with widely spaced bipolar electrodes.
Pacing Protocol and Induction of VF
A bipolar pacing lead was placed at the apex of the LV with an output at 2.5 times the diastolic threshold. Optical recording was performed after 40 beats of stable pacing at each PCL. The PCL was progressively shortened from 200 ms in a 20- (for PCL longer than 120 ms) or 10-ms (for PCL shorter than 120 ms) decrement until loss of capture. After 5 minutes of recovery, arrhythmia inducibility was evaluated by a S1-S2 protocol, consisting of continuous 9 beats S1 stimuli (200 ms) followed by an additional premature (S2) beat. A sustained VF longer than 1 minute was regarded as a successful VF induction.
Histology
The heart tissue was fixed in 4% neutral buffered formalin. The hearts were cut transversely, and sections of ventricles were processed routinely and embedded in paraffin. Sections were stained with picrosirius red stain to assess fibrosis using animal hearts from a previous study.11 Heart sections from the animals undergoing electrophysiology studies were stained with a Trichrome/elastin method. The fibrosis score was determined from picrosirius red stain-stained sections by V.H.G., and the percentage of fibrosis was evaluated using a semiquantitative 1+ to 4+ score (1, no fibrosis; 2, minimal interspersed; 3, minimal with focal accumulation; 4, moderate; 5, severe). To confirm the semiquantitative analyses, we obtained an additional opinion by a cardiac pathologist (M.C.F.).
PCR and Western Blotting
PCR and Western blotting were analyzed from hearts of 35-week-old CKD or normal littermate animals at 35 weeks, because it was not feasible to analyze the hearts of the animals that had undergone electrophysiology. Hearts were washed briefly with PBS, and pieces of left ventricular free wall were collected for real-time PCR or Western blotting. Collected tissues were snap-frozen in liquid nitrogen and stored in −80°C until analysis. RNA was extracted by Trizol regent and transformed into cDNA by ImProm-II Reverse Transcription System (Promega, Madison, WI). To access gene expression, 10 ng cDNA was mixed with 300 nM gene-specific primer and SsoFast EvaGreen (Bio-Rad, Hercules, CA). Fluorescence was monitored by MyiQ Single-Color Real-Time PCR Detection System (Bio-Rad). At the end of the PCR reaction, melting point analysis was conducted to verify the specificity of amplicons. PCR efficiency for primers fell in the range from 90% to 110%. The ΔΔ cycle threshold method was used for comparison with glyceraldehyde-3-phosphate dehydrogenase or beta actin as the internal control.
Western blot confirmation was done using standard methods. Because of tissue and antibody availability, we could not confirm all the RNA data, and therefore, we selected three important candidates to confirm protein expression with RNA expression. In brief, protein electrophoresis was done using the Bio-Rad Mini Gel System (Bio-Rad) and transferring to polyvinylidene difluoride membrane (EMD Millipore, Billerica, MA). The membrane was bathed in Tris-buffered saline/Tween 20 with 5% milk for 1 hour and probed with primary antibody overnight. After primary antibody incubation, the membrane was incubated with horseradish peroxidase-conjugated secondary antibodies (Sigma-Aldrich, St. Louis, MO) for 30 minutes. Luminata Crescendo HRP Substrate (EMD Millipore) was added on the membrane according to the manufacturer’s instructions for illumination development. The commercial primary antibodies used in this study included glyceraldehyde-3-phosphate dehydrogenase (Thermo Fisher Scientific, Rockford, IL), Kv1.4 (Alomone Labs, Jerusalem, Israel), and Kv11.1 (Alomone Labs). Antibody against SERCA2 was a gift from Zhenhui Chen as previously described.49
Statistical Analyses
Results were summarized by mean±SEM and compared using ANOVA test, unpaired t test, or Fisher exact test. Bonferroni test was used for post-test comparison after ANOVA. All data were analyzed by Prism 5.0 (GraphPad Software, La Jolla, CA), and figures were processed by Photoshop (San Jose, CA). P value <0.05 is regarded as statistical significant.
Disclosures
None.
Acknowledgments
We thank Dr. Zhenhui Chen for his gift of the antibody to SERCA2.
The study was supported by National Institutes of Health Grants R01-AR058005 (to S.M.M.), R01-HL71140, and P01-HL78931 (to P.-S.C.), a Stuart A. Kleit Professorship (to S.M.M.), a Medtronic-Zipes Endowment (to P.-S.C.), an American Heart Association Midwest Affiliate Winter 2013 Postdoctoral Fellowship (to C.-H.H.), and the Indiana University Health–Indiana University School of Medicine Strategic Research Initiative Grant (to S.M.M.).
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
References
- 1.US Renal Data System : USRDS 2013 Annual Data Report: Atlas of Chronic Kidney Disease and End-Stage Renal Disease in the United States, Bethesda, MD, National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases, 2013 [Google Scholar]
- 2.Herzog CA, Asinger RW, Berger AK, Charytan DM, Díez J, Hart RG, Eckardt KU, Kasiske BL, McCullough PA, Passman RS, DeLoach SS, Pun PH, Ritz E: Cardiovascular disease in chronic kidney disease. A clinical update from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int 80: 572–586, 2011 [DOI] [PubMed] [Google Scholar]
- 3.Green D, Roberts PR, New DI, Kalra PA: Sudden cardiac death in hemodialysis patients: An in-depth review. Am J Kidney Dis 57: 921–929, 2011 [DOI] [PubMed] [Google Scholar]
- 4.Whitman IR, Feldman HI, Deo R: CKD and sudden cardiac death: Epidemiology, mechanisms, and therapeutic approaches. J Am Soc Nephrol 23: 1929–1939, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goldenberg I, Moss AJ, McNitt S, Zareba W, Andrews ML, Hall WJ, Greenberg H, Case RB, Multicenter Automatic Defibrillator Implantation Trial-II Investigators : Relations among renal function, risk of sudden cardiac death, and benefit of the implanted cardiac defibrillator in patients with ischemic left ventricular dysfunction. Am J Cardiol 98: 485–490, 2006 [DOI] [PubMed] [Google Scholar]
- 6.Saxon LA, Bristow MR, Boehmer J, Krueger S, Kass DA, De Marco T, Carson P, DiCarlo L, Feldman AM, Galle E, Ecklund F: Predictors of sudden cardiac death and appropriate shock in the Comparison of Medical Therapy, Pacing, and Defibrillation in Heart Failure (COMPANION) Trial. Circulation 114: 2766–2772, 2006 [DOI] [PubMed] [Google Scholar]
- 7.Moe SM, Chen NX, Seifert MF, Sinders RM, Duan D, Chen X, Liang Y, Radcliff JS, White KE, Gattone VH, 2nd: A rat model of chronic kidney disease-mineral bone disorder. Kidney Int 75: 176–184, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moe SM, Radcliffe JS, White KE, Gattone VH, 2nd, Seifert MF, Chen X, Aldridge B, Chen NX: The pathophysiology of early-stage chronic kidney disease-mineral bone disorder (CKD-MBD) and response to phosphate binders in the rat. J Bone Miner Res 26: 2672–2681, 2011 [DOI] [PubMed] [Google Scholar]
- 9.Cowley BD, Jr., Gudapaty S, Kraybill AL, Barash BD, Harding MA, Calvet JP, Gattone VH, 2nd: Autosomal-dominant polycystic kidney disease in the rat. Kidney Int 43: 522–534, 1993 [DOI] [PubMed] [Google Scholar]
- 10.Cowley BD, Jr., Grantham JJ, Muessel MJ, Kraybill AL, Gattone VH, 2nd: Modification of disease progression in rats with inherited polycystic kidney disease. Am J Kidney Dis 27: 865–879, 1996 [DOI] [PubMed] [Google Scholar]
- 11.Moe SM, Chen NX, Newman CL, Gattone VH, 2nd, Organ JM, Chen X, Allen MR: A comparison of calcium to zoledronic acid for improvement of cortical bone in an animal model of CKD. J Bone Miner Res 29: 902–910, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stagner EE, Bouvrette DJ, Cheng J, Bryda EC: The polycystic kidney disease-related proteins Bicc1 and SamCystin interact. Biochem Biophys Res Commun 383: 16–21, 2009 [DOI] [PubMed] [Google Scholar]
- 13.Rensma PL, Allessie MA, Lammers WJ, Bonke FI, Schalij MJ: Length of excitation wave and susceptibility to reentrant atrial arrhythmias in normal conscious dogs. Circ Res 62: 395–410, 1988 [DOI] [PubMed] [Google Scholar]
- 14.Gutierrez OM: The connection between dietary phosphorus, cardiovascular disease, and mortality: Where we stand and what we need to know. Adv Nutr 4: 723–729, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Calvo MS, Moshfegh AJ, Tucker KL: Assessing the health impact of phosphorus in the food supply: Issues and considerations. Adv Nutr 5: 104–113, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weiss JN, Karma A, Shiferaw Y, Chen PS, Garfinkel A, Qu Z: From pulsus to pulseless: The saga of cardiac alternans. Circ Res 98: 1244–1253, 2006 [DOI] [PubMed] [Google Scholar]
- 17.Cao JM, Qu Z, Kim YH, Wu TJ, Garfinkel A, Weiss JN, Karagueuzian HS, Chen PS: Spatiotemporal heterogeneity in the induction of ventricular fibrillation by rapid pacing: Importance of cardiac restitution properties. Circ Res 84: 1318–1331, 1999 [DOI] [PubMed] [Google Scholar]
- 18.Bray MA, Lin SF, Aliev RR, Roth BJ, Wikswo JP, Jr.: Experimental and theoretical analysis of phase singularity dynamics in cardiac tissue. J Cardiovasc Electrophysiol 12: 716–722, 2001 [DOI] [PubMed] [Google Scholar]
- 19.Chen J, Mandapati R, Berenfeld O, Skanes AC, Jalife J: High-frequency periodic sources underlie ventricular fibrillation in the isolated rabbit heart. Circ Res 86: 86–93, 2000 [DOI] [PubMed] [Google Scholar]
- 20.Liu YB, Peter A, Lamp ST, Weiss JN, Chen PS, Lin SF: Spatiotemporal correlation between phase singularities and wavebreaks during ventricular fibrillation. J Cardiovasc Electrophysiol 14: 1103–1109, 2003 [DOI] [PubMed] [Google Scholar]
- 21.Koleganova N, Piecha G, Ritz E, Gross ML: Calcitriol ameliorates capillary deficit and fibrosis of the heart in subtotally nephrectomized rats. Nephrol Dial Transplant 24: 778–787, 2009 [DOI] [PubMed] [Google Scholar]
- 22.Green D, Roberts PR: Ventricular arrhythmias and sudden death in patients with chronic kidney disease. J Ren Care 36[Suppl 1]: 54–60, 2010 [DOI] [PubMed] [Google Scholar]
- 23.Chen NX, Moe SM, Eggleston-Gulyas T, Chen X, Hoffmeyer WD, Bacallao RL, Herbert BS, Gattone VH, 2nd: Calcimimetics inhibit renal pathology in rodent nephronophthisis. Kidney Int 80: 612–619, 2011 [DOI] [PubMed] [Google Scholar]
- 24.Vettori S, Gay S, Distler O: Role of microRNAs in fibrosis. Open Rheumatol J 6: 130–139, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thum T, Gross C, Fiedler J, Fischer T, Kissler S, Bussen M, Galuppo P, Just S, Rottbauer W, Frantz S, Castoldi M, Soutschek J, Koteliansky V, Rosenwald A, Basson MA, Licht JD, Pena JT, Rouhanifard SH, Muckenthaler MU, Tuschl T, Martin GR, Bauersachs J, Engelhardt S: MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature 456: 980–984, 2008 [DOI] [PubMed] [Google Scholar]
- 26.Cutler MJ, Wan X, Plummer BN, Liu H, Deschenes I, Laurita KR, Hajjar RJ, Rosenbaum DS: Targeted sarcoplasmic reticulum Ca2+ ATPase 2a gene delivery to restore electrical stability in the failing heart. Circulation 126: 2095–2104, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guo W, Li H, London B, Nerbonne JM: Functional consequences of elimination of i(to,f) and i(to,s): Early afterdepolarizations, atrioventricular block, and ventricular arrhythmias in mice lacking Kv1.4 and expressing a dominant-negative Kv4 alpha subunit. Circ Res 87: 73–79, 2000 [DOI] [PubMed] [Google Scholar]
- 28.Ataklte F, Erqou S, Laukkanen J, Kaptoge S: Meta-analysis of ventricular premature complexes and their relation to cardiac mortality in general populations. Am J Cardiol 112: 1263–1270, 2013 [DOI] [PubMed] [Google Scholar]
- 29.Foley RN, Parfrey PS, Harnett JD, Kent GM, Martin CJ, Murray DC, Barre PE: Clinical and echocardiographic disease in patients starting end-stage renal disease therapy. Kidney Int 47: 186–192, 1995 [DOI] [PubMed] [Google Scholar]
- 30.Zaslavsky LM, Pinotti AF, Gross JL: Diastolic dysfunction and mortality in diabetic patients on hemodialysis: A 4.25-year controlled prospective study. J Diabetes Complications 19: 194–200, 2005 [DOI] [PubMed] [Google Scholar]
- 31.Amann K, Kronenberg G, Gehlen F, Wessels S, Orth S, Munter K, Ehmke H, Mall G, Ritz E: Cardiac remodelling in experimental renal failure—an immunohistochemical study. Nephrol Dial Transplant 13: 1958–1966, 1998 [DOI] [PubMed] [Google Scholar]
- 32.Ansari A, Kaupke CJ, Vaziri ND, Miller R, Barbari A: Cardiac pathology in patients with end-stage renal disease maintained on hemodialysis. Int J Artif Organs 16: 31–36, 1993 [PubMed] [Google Scholar]
- 33.Mall G, Huther W, Schneider J, Lundin P, Ritz E: Diffuse intermyocardiocytic fibrosis in uraemic patients. Nephrol Dial Transplant 5: 39–44, 1990 [DOI] [PubMed] [Google Scholar]
- 34.Haider AW, Larson MG, Benjamin EJ, Levy D: Increased left ventricular mass and hypertrophy are associated with increased risk for sudden death. J Am Coll Cardiol 32: 1454–1459, 1998 [DOI] [PubMed] [Google Scholar]
- 35.Cooklin M, Wallis WR, Sheridan DJ, Fry CH: Conduction velocity and gap junction resistance in hypertrophied, hypoxic guinea-pig left ventricular myocardium. Exp Physiol 83: 763–770, 1998 [DOI] [PubMed] [Google Scholar]
- 36.Tomaselli GF, Marbán E: Electrophysiological remodeling in hypertrophy and heart failure. Cardiovasc Res 42: 270–283, 1999 [DOI] [PubMed] [Google Scholar]
- 37.Volk T, Nguyen TH, Schultz JH, Faulhaber J, Ehmke H: Regional alterations of repolarizing K+ currents among the left ventricular free wall of rats with ascending aortic stenosis. J Physiol 530: 443–455, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lewis T: Notes upon alternation of the heart. QJM os4: 141–144, 1911 [Google Scholar]
- 39.Smith JM, Clancy EA, Valeri CR, Ruskin JN, Cohen RJ: Electrical alternans and cardiac electrical instability. Circulation 77: 110–121, 1988 [DOI] [PubMed] [Google Scholar]
- 40.Raeder EA, Rosenbaum DS, Bhasin R, Cohen RJ: Alternating morphology of the QRST complex preceding sudden death. N Engl J Med 326: 271–272, 1992 [DOI] [PubMed] [Google Scholar]
- 41.Tsai J, Cao JM, Zhou S, Swissa M, Cates AW, Kenknight BH, Chen LS, Karagueuzian HS, Chen PS: T wave alternans as a predictor of spontaneous ventricular tachycardia in a canine model of sudden cardiac death. J Cardiovasc Electrophysiol 13: 51–55, 2002 [DOI] [PubMed] [Google Scholar]
- 42.Weiss JN, Nivala M, Garfinkel A, Qu Z: Alternans and arrhythmias: From cell to heart. Circ Res 108: 98–112, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bogin E, Massry SG, Harary I: Effect of parathyroid hormone on rat heart cells. J Clin Invest 67: 1215–1227, 1981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Faul C, Amaral AP, Oskouei B, Hu MC, Sloan A, Isakova T, Gutiérrez OM, Aguillon-Prada R, Lincoln J, Hare JM, Mundel P, Morales A, Scialla J, Fischer M, Soliman EZ, Chen J, Go AS, Rosas SE, Nessel L, Townsend RR, Feldman HI, St John Sutton M, Ojo A, Gadegbeku C, Di Marco GS, Reuter S, Kentrup D, Tiemann K, Brand M, Hill JA, Moe OW, Kuro-O M, Kusek JW, Keane MG, Wolf M: FGF23 induces left ventricular hypertrophy. J Clin Invest 121: 4393–4408, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Touchberry CD, Green TM, Tchikrizov V, Mannix JE, Mao TF, Carney BW, Girgis M, Vincent RJ, Wetmore LA, Dawn B, Bonewald LF, Stubbs JR, Wacker MJ: FGF23 is a novel regulator of intracellular calcium and cardiac contractility in addition to cardiac hypertrophy. Am J Physiol Endocrinol Metab 304: E863–E873, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Isakova T, Wahl P, Vargas GS, Gutiérrez OM, Scialla J, Xie H, Appleby D, Nessel L, Bellovich K, Chen J, Hamm L, Gadegbeku C, Horwitz E, Townsend RR, Anderson CA, Lash JP, Hsu CY, Leonard MB, Wolf M: Fibroblast growth factor 23 is elevated before parathyroid hormone and phosphate in chronic kidney disease. Kidney Int 79: 1370–1378, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Torres VE, Harris PC: Mechanisms of disease: Autosomal dominant and recessive polycystic kidney diseases. Nat Clin Pract Nephrol 2: 40–55, 2006 [DOI] [PubMed] [Google Scholar]
- 48.Brown JH, Bihoreau MT, Hoffmann S, Kränzlin B, Tychinskaya I, Obermüller N, Podlich D, Boehn SN, Kaisaki PJ, Megel N, Danoy P, Copley RR, Broxholme J, Witzgall R, Lathrop M, Gretz N, Gauguier D: Missense mutation in sterile alpha motif of novel protein SamCystin is associated with polycystic kidney disease in (cy/+) rat. J Am Soc Nephrol 16: 3517–3526, 2005 [DOI] [PubMed] [Google Scholar]
- 49.Chen Z, Stokes DL, Rice WJ, Jones LR: Spatial and dynamic interactions between phospholamban and the canine cardiac Ca2+ pump revealed with use of heterobifunctional cross-linking agents. J Biol Chem 278: 48348–48356, 2003 [DOI] [PubMed] [Google Scholar]