

Abstract
2q23.1 deletion syndrome is characterized by intellectual disability, speech impairment, seizures, disturbed sleep pattern, behavioral problems, and hypotonia. Core features of this syndrome are due to haploinsufficiency of MBD5. Deletions that include coding and noncoding exons show reduced MBD5 mRNA expression. We report a patient with a neurological and behavioral phenotype similar to 2q23.1 deletion syndrome with an inherited intronic deletion in the 5-prime untranslated region of MBD5. Our data show that this patient has normal MBD5 mRNA expression; therefore, this deletion is likely not causative for 2q23.1 deletion syndrome. Overall, it is important to validate intronic deletions for pathogenicity.
Keywords: 2q23.1 deletion syndrome, MBD5, 5′UTR, Intronic deletion, Gene expression, Familial variant
The 2q23.1 microdeletion syndrome (OMIM 156200) is a neurodevelopmental disorder characterized by intellectual disability, severe speech impairment, seizures, disturbed sleep pattern, behavioral problems, microcephaly, hypotonia and short stature [1,2] and is caused by deletions of chromosome 2 involving the 2q23.1 band or mutation in the methyl-CpG binding protein 5 gene, MBD5 (OMIM 611472) [2]. MBD5 belongs to the MBD family of proteins, which play critical roles in transcriptional regulation and development. The gene structure contains five non-coding exons at the 5-prime end, followed by 10 coding exons (Figure 1). Two protein isoforms have been described [3]. The first isoform contains 1494 amino acids and is encoded by exons 6–15 (Figure 1). The second isoform is shorter, contains 851 amino acids, and is encoded by exons 6–9. Talkowski et al. [2] showed that deletions that include noncoding exons, which do not alter the MBD5 protein coding sequence, show reduced MBD5 mRNA expression, similar to that observed in individuals with larger 2q23.1 microdeletions encompassing the entire MBD5 gene. The individuals with these MBD5 non-coding exonic deletions exhibit most features of 2q23.1 microdeletion syndrome [2].
Figure 1.
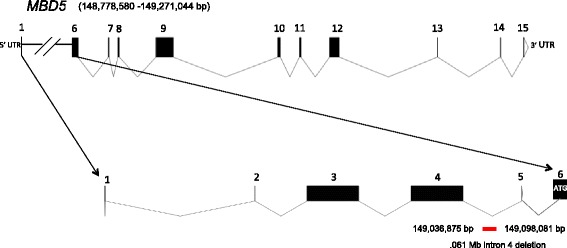
Intragenic deletion involving a portion of MBD5 intron 4 . A schematic representation of MBD5, depicting the 5′-UTR, exons (black boxes), and intronic regions. We depict the .061 Mb intron 4 deletion (in red) in the 5′-noncoding region of MBD5 at chr(2)(q23.1); (chr2.hg19:g.149,036,875-149,098,081).
In this study, a 4-year-old boy (SMS431) was referred for a possible diagnosis of 2q23.1 deletion syndrome based upon array CGH findings using Nimblegen CGX-12 (Roche Technologies, Penzberg, Germany). The DNA microarray analysis of SMS431, his clinically normal parents, and paternal grandparents was performed in a clinical laboratory, revealing a 0.061 Mb paternally inherited deletion at 2q23.1, arr [hg19] 2q23.1 (149,036,875-149,098,081)x1. This region involves a portion of intron 4 of MBD5 (Figure 1). This deletion also was found to be present in the paternal grandfather (Figure 2).
Figure 2.
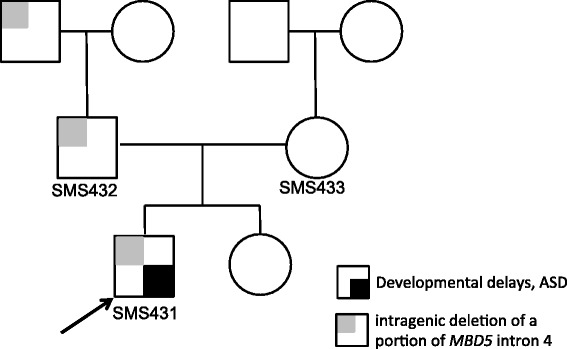
Pedigree of a family with intron 4 deletion of MBD5. The proband (SMS431) is denoted by arrow and small black box. The squares and circles symbolize males and females, respectively. The small light grey boxes denote family members carrying the MBD5 intron 4 deletion.
While the neurological and behavioral phenotype of SMS431 was remarkably similar to 2q23.1 deletion phenotype (Table 1), it was unclear whether such an intronic deletion in the noncoding region of the MBD5 gene could be deleterious and manifest the phenotypes present in this child. Additionally, both father and paternal grandfather carry this deletion and neither have features associated with 2q23.1 deletion syndrome or other features observed in this child. Reduced MBD5 mRNA expression has been reported in 2q23.1 deletion patients and serves as an additional molecular diagnostic indicator for this disorder [2,4]. Consequently, to determine whether a deletion in the noncoding intron 4 region of MBD5 could result in impaired expression of MBD5 and was, therefore, responsible for the phenotype observed in SMS431, we evaluated MBD5 mRNA gene expression on peripheral blood samples obtained from this family.
Table 1.
Comparison of the phenotype of SMS431 to the prominent features of 2q23.1 deletion syndrome
| 2q23.1 deletion syndrome | SMS431* |
|---|---|
| Developmental delay | + |
| Motor delay | + |
| Language impairment | + |
| Behavioral problems | + |
| Autistic-like symptoms | + |
| Sleep disturbances | + |
| Repetitive behaviors (stereotypies) | + |
| Self-injurious behaviors | + |
| Short attention span | + |
| Aggression | + |
| Seizures | + |
| Infantile hypotonia | + |
| Infantile feeding difficulties | + |
| Eye abnormalities | + |
| Heavy arched eyebrows | - |
| Prominent nose | - |
| Thin upper lip | + |
| Widely spaced teeth | - |
*Intronic deletion of intron 4 in the 5′UTR region of MBD5.
The Institutional Review Board Baylor College of Medicine approved this study. Fresh blood was collected from SMS431 (proband), SMS432 (father), SMS433 (mother), SMS361 (2q23.1 deletion patient) [5], and nine normal controls after informed consent was obtained. Total RNA was isolated according to standard methods (Invitrogen, Carlsbad, CA). RNA was quantified using the NanoDrop ® ND-100 Spectrophotometer and reverse transcribed through qSCRIPT cDNA SuperMix (Quanta Biosciences, Inc., Gaithersburg, MD) according to manufacturer’s instructions. To assess MBD5 mRNA expression, quantitative RT-PCR was performed as previously described [2,6]. Briefly, Taqman minor groove binder probes for MBD5 (OMIM 611472, Hs00289233_m1) and GAPDH (OMIM 138400, Hs9999905_m1) were used. GAPDH was used as the endogenous control. All samples of cDNA were run in triplicate in 10 ul reaction volumes. All samples were run and analyzed according to previously published methods using BioRad CFX Connect™ Real-Time PCR Detection System [2]. Three biological replicates were performed. Results are expressed as fold-change relative to the control sample. Standard error was generated for each sample. A paired t-test was used to determine significance. p <0.01 was considered statistically significant.
As expected, MBD5 mRNA expression for the mother was not statistically different from controls (P = 0.767) (Figure 3), while the 2q23.1 deletion control sample, SMS456, exhibited the typical reduced MBD5 mRNA expression for an individual with an MBD5 deletion (P < 0.0001). The mRNA expression levels for the child (SMS431) and his father (SMS432) were not significantly different from controls (P = 0.060) (Figure 3) or from MBD5 expression for the mother. Further, SMS431 and SMS432 expression patterns were significantly different from a typical 2q23.1 deletion case, SMS456 (P < 0.0001). Since the deletion present in SMS431 and his father did not alter MBD5 mRNA expression, it is highly unlikely that this intronic deletion affects transcription and translation of MBD5, and furthermore, no truncated mRNA, truncated protein or aberrant protein should result.
Figure 3.
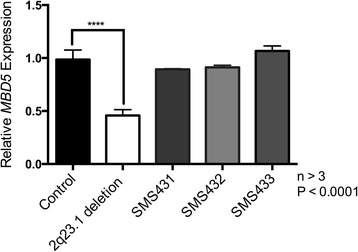
MBD5 mRNA expression is not altered in individuals with MBD5 intronic deletion. Quantitative RT-PCR was used to test MBD5 mRNA expression levels. MBD5 mRNA expression levels are not significantly altered in SMS431 (child) or SMS432 (father) or SMS433 (mother) compared to controls. The expression of a typical 2q23.1 deletion syndrome case shows significantly reduced MBD5 expression compared to controls.
A survey of the Database of Genomic Variants (DGV) contains no deletions that are identical to that observed in the individuals in this family. DVG lists 14 small deletions in the 5′-noncoding region of MBD5, with nine deletions confined to intronic sequences, which were reported to be nonpathogenic or of unknown significance. Five of the nine small deletions were found in intron 4.
Overall, it is apparent that the intron 4 deletion does not affect expression of MBD5 and likely does not give rise to the features associated with the MBD5 haploinsufficiency that is observed in 2q23.1 deletion syndrome.
Introns in the noncoding region of a gene can play a major role in the transcriptional regulation of a gene and consequently, gene expression. An intron can enhance gene expression through the presence of transcriptional regulatory elements or through structural modulation and splicing [7]. Copy number variants within splice site sequences at the intron-exon junction cause approximately 10% of disease-causing mutations [8]. There are several cases in the medical literature of pathogenic intronic deletions such as NRXN1 deletions (Autism Spectrum Disorder) [9], SLC34A3 deletions (hereditary hypophosphatemic rickets with hypercalciuria) [10,11], PKD1 deletions (Rothmund-Thomson syndrome) [12], and NASE deletions (5-fluorouracil toxicity) [13].
Since the intronic deletion identified in SMS431 does not delete the splice junctions between exon 4 and exon 5 or create new splice junctions, proper mRNA splicing likely still occurs, without impacting expression. Further, deletions in 5-prime UTR introns could lead to altered expression if transcription factor binding is affected. Whether there are transcription factors, enhancers, or silencers that bind to that specific intronic region of MBD5 remains unclear.
In summary, SMS431 exhibited developmental delay, motor delay, severe language impairment, sleep disturbances and behavioral problems that mimicked 2q23.1 deletion syndrome (Table 1). However, these neurological and behavioral phenotypes are observed in other neurodevelopmental disorders. The 2q23.1 deletion syndrome phenotype overlaps with other neurodevelopmental disorders, including Angelman syndrome, Smith-Magenis syndrome, Pitt-Hopkins syndrome, and Kleefstra syndrome [2,6]. SMS431 likely has a different neurodevelopmental disorder that overlaps phenotypically with 2q23.1 deletion syndrome. Since no other copy number variants were identified for this patient, additional testing will be required to determine a cause for the phenotype. The possibility exists that a gene that is involved in a pathway common to MBD5 may be affected, including a gene downstream of MBD5 or binding partner of MBD5. Finally, whole exome sequencing (WES) may be the best approach toward diagnosis for this child.
In conclusion, we highlight the importance of validating intronic deletions for pathogenicity so that accurate and informed diagnosis can be provided to the patient.
Consent
Written informed consent was obtained from the patient’s parents for publication of this paper. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Acknowledgements
We are thankful to the family for their willingness to participate in this study. We thank the Fondation Jerome Lejuene for funding portions of this study. This work was supported in part by resources from the Baylor College of Medicine and the Jan and Dan Duncan Neurological Research Institute.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
Both authors have made substantial contributions to conception and design, acquisition of data, analysis and interpretation of data. SVM performed the qPCR studies and drafted the manuscript. Both authors read and approved the final manuscript.
Contributor Information
Sureni V Mullegama, Email: sureni.mullegama@bcm.edu.
Sarah H Elsea, Email: elsea@bcm.edu.
References
- 1.Shichiji M, Ito Y, Shimojima K, Nakamu H, Oguni H, Osawa M, Yamamoto T. A cryptic microdeletion including MBD5 occurring within the breakpoint of a reciprocal translocation between chromosomes 2 and 5 in a patient with developmental delay and obesity. Am J Med Genet A. 2013;161A:850–855. doi: 10.1002/ajmg.a.35768. [DOI] [PubMed] [Google Scholar]
- 2.Talkowski ME, Mullegama SV, Rosenfeld JA, van Bon BW, Shen Y, Repnikova EA, Gastier-Foster J, Thrush DL, Kathiresan S, Ruderfer DM, Chiang C, Hanscom C, Ernst C, Lindgren AM, Morton CC, An Y, Astbury C, Brueton LA, Lichtenbelt KD, Ades LC, Fichera M, Romano C, Innis JW, Williams CA, Bartholomew D, Van Allen MI, Parikh A, Zhang L, Wu BL, Pyatt RE, et al. Assessment of 2q23.1 microdeletion syndrome implicates MBD5 as a single causal locus of intellectual disability, epilepsy, and autism spectrum disorder. Am J Hum Genet. 2011;89:551–563. doi: 10.1016/j.ajhg.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Laget S, Joulie M, Le Masson F, Sasai N, Christians E, Pradhan S, Roberts RJ, Defossez PA. The human proteins MBD5 and MBD6 associate with heterochromatin but they do not bind methylated DNA. PLoS One. 2010;5:e11982. doi: 10.1371/journal.pone.0011982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bonnet C, Ali Khan A, Bresso E, Vigouroux C, Beri M, Lejczak S, Deemer B, Andrieux J, Philippe C, Moncla A, Giurgea I, Devignes MD, Leheup B, Jonveaux P. Extended spectrum of MBD5 mutations in neurodevelopmental disorders. Eur J Hum Genet. 2013;21:1457–1461. doi: 10.1038/ejhg.2013.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Williams SR, Mullegama SV, Rosenfeld JA, Dagli AI, Hatchwell E, Allen WP, Williams CA, Elsea SH. Haploinsufficiency of MBD5 associated with a syndrome involving microcephaly, intellectual disabilities, severe speech impairment, and seizures. Eur J Hum Genet. 2010;18:436–441. doi: 10.1038/ejhg.2009.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hodge JC, Mitchell E, Pillalamarri V, Toler TL, Bartel F, Kearney HM, Zou YS, Tan WH, Hanscom C, Kirmani S, Hanson RR, Skinner SA, Rogers RC, Everman DB, Boyd E, Tapp C, Mullegama SV, Keelean-Fuller D, Powell CM, Elsea SH, Morton CC, Gusella JF, DuPont B, Chaubey A, Lin AE, Talkowski ME. Disruption of MBD5 contributes to a spectrum of psychopathology and neurodevelopmental abnormalities. Mol Psychiatry. 2014;19:368–379. doi: 10.1038/mp.2013.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cenik C, Derti A, Mellor JC, Berriz GF, Roth FP. Genome-wide functional analysis of human 5' untranslated region introns. Genome Biol. 2010;11:R29. doi: 10.1186/gb-2010-11-3-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Krawczak M, Thomas NS, Hundrieser B, Mort M, Wittig M, Hampe J, Cooper DN. Single base-pair substitutions in exon-intron junctions of human genes: nature, distribution, and consequences for mRNA splicing. Hum Mutat. 2007;28:150–158. doi: 10.1002/humu.20400. [DOI] [PubMed] [Google Scholar]
- 9.Dabell MP, Rosenfeld JA, Bader P, Escobar LF, El-Khechen D, Vallee SE, Dinulos MB, Curry C, Fisher J, Tervo R, Hannibal MC, Siefkas K, Wyatt PR, Hughes L, Smith R, Ellingwood S, Lacassie Y, Stroud T, Farrell SA, Sanchez-Lara PA, Randolph LM, Niyazov D, Stevens CA, Schoonveld C, Skidmore D, MacKay S, Miles JH, Moodley M, Huillet A, Neill NJ, et al. Investigation of NRXN1 deletions: clinical and molecular characterization. Am J Med Genet A. 2013;161A:717–731. doi: 10.1002/ajmg.a.35780. [DOI] [PubMed] [Google Scholar]
- 10.Ichikawa S, Sorenson AH, Imel EA, Friedman NE, Gertner JM, Econs MJ. Intronic deletions in the SLC34A3 gene cause hereditary hypophosphatemic rickets with hypercalciuria. J Clin Endocrinol Metab. 2006;91:4022–4027. doi: 10.1210/jc.2005-2840. [DOI] [PubMed] [Google Scholar]
- 11.Ichikawa S, Tuchman S, Padgett LR, Gray AK, Baluarte HJ, Econs MJ. Intronic deletions in the SLC34A3 gene: a cautionary tale for mutation analysis of hereditary hypophosphatemic rickets with hypercalciuria. Bone. 2014;59:53–56. doi: 10.1016/j.bone.2013.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang LL, Worley K, Gannavarapu A, Chintagumpala MM, Levy ML, Plon SE. Intron-size constraint as a mutational mechanism in Rothmund-Thomson syndrome. Am J Hum Genet. 2002;71:165–167. doi: 10.1086/341234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van Kuilenburg AB, Meijer J, Mul AN, Meinsma R, Schmid V, Dobritzsch D, Hennekam RC, Mannens MM, Kiechle M, Etienne-Grimaldi MC, Klumpen HJ, Maring JG, Derleyn VA, Maartense E, Milano G, Vijzelaar R, Gross E. Intragenic deletions and a deep intronic mutation affecting pre-mRNA splicing in the dihydropyrimidine dehydrogenase gene as novel mechanisms causing 5-fluorouracil toxicity. Hum Genet. 2010;128:529–538. doi: 10.1007/s00439-010-0879-3. [DOI] [PMC free article] [PubMed] [Google Scholar]