

Abstract
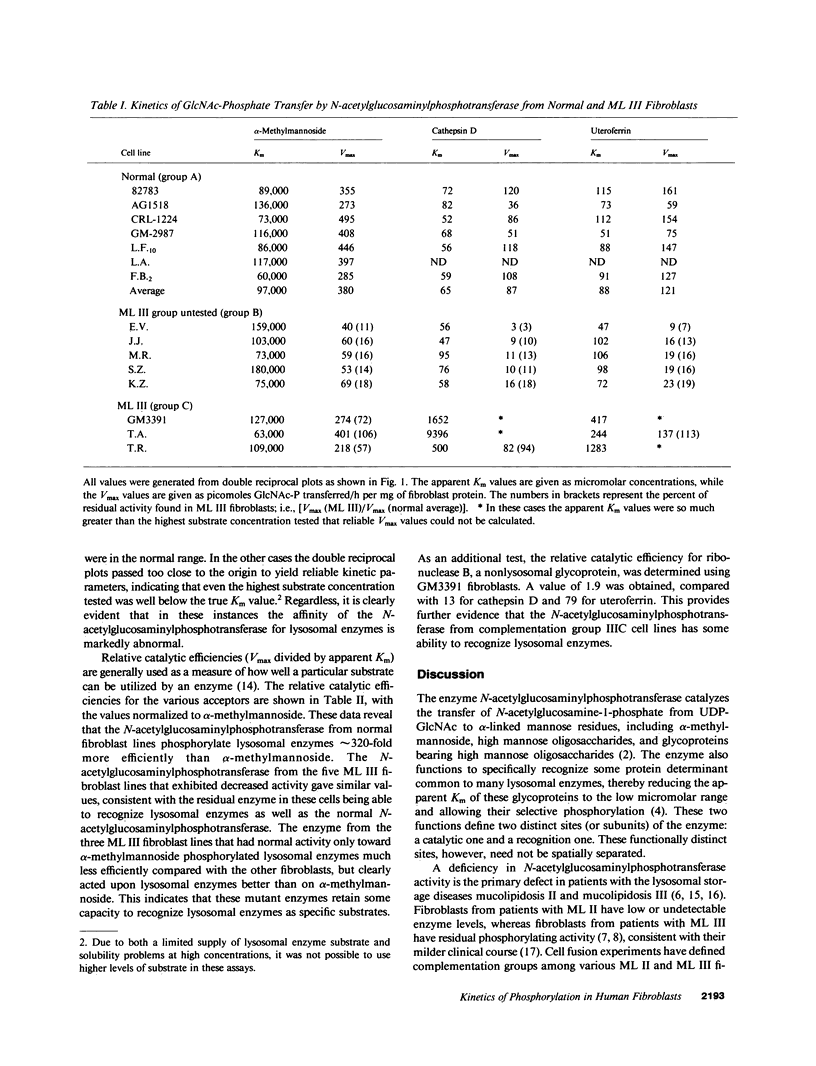
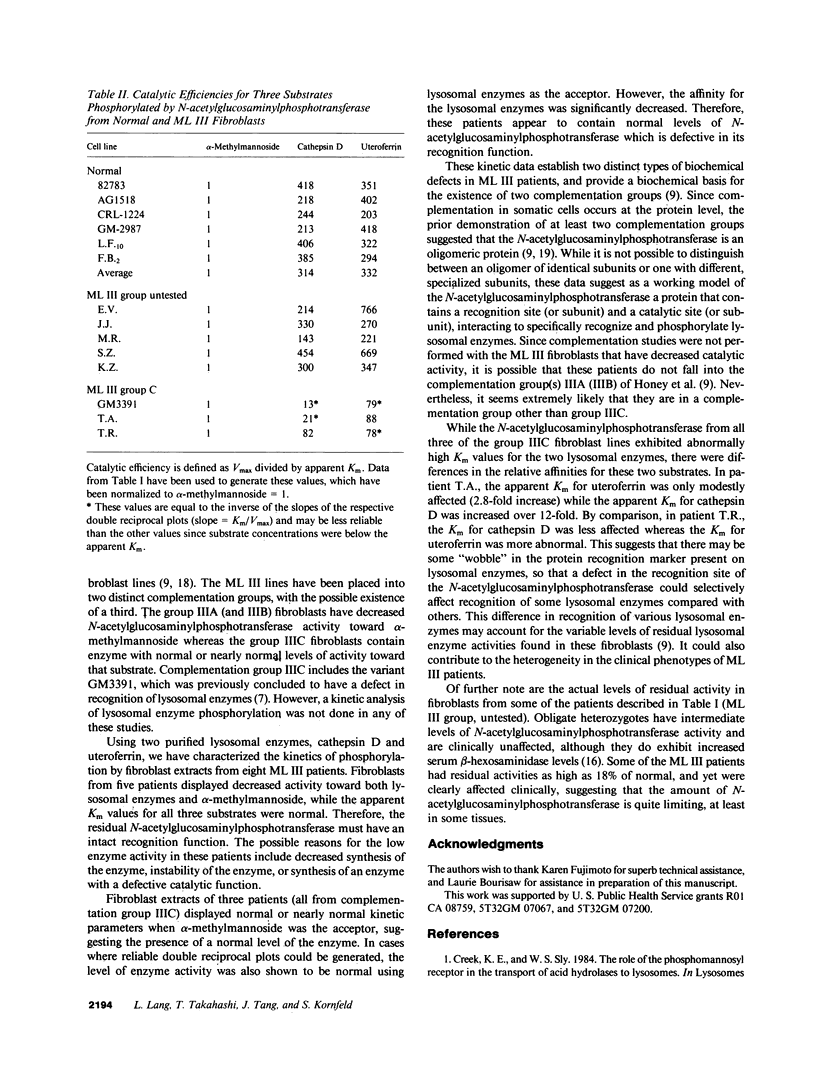
The primary genetic defect in the lysosomal storage disease mucolipidosis III (ML III) is in the enzyme uridine diphospho-N-acetylglucosamine:lysosomal enzyme N-acetylglucosamine-1-phosphotransferase. This enzyme has two well-defined functions: specific recognition of lysosomal enzymes (recognition function) and phosphorylation of their oligosaccharides (catalytic function). Using fibroblasts from patients with ML III as the source of enzyme, and alpha-methylmannoside and two lysosomal enzymes as the substrates, we have identified defects in both of these functions. In one group of fibroblasts, the catalytic activity of the N-acetylglucosaminylphosphotransferase is decreased while the ability to recognize lysosomal enzymes as specific substrates remains intact. In the second group of fibroblasts, the ability to recognize lysosomal enzymes is impaired while the catalytic activity of the enzyme is normal. These data provide a biochemical rationale for the previously described genetic heterogeneity among patients with ML III (Honey, N. K., O. T. Mueller, L. E. Little, A. L. Miller, and T. B. Shows, 1982, Proc. Natl. Acad. Sci. USA., 79:7420-7424).
Full text
PDF



Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Honey N. K., Mueller O. T., Little L. E., Miller A. L., Shows T. B. Mucolipidosis III is genetically heterogeneous. Proc Natl Acad Sci U S A. 1982 Dec;79(23):7420–7424. doi: 10.1073/pnas.79.23.7420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LOWRY O. H., ROSEBROUGH N. J., FARR A. L., RANDALL R. J. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951 Nov;193(1):265–275. [PubMed] [Google Scholar]
- Lang L., Kornfeld S. A simplified procedure for synthesizing large quantities of highly purified uridine [beta-32P]diphospho-N-acetylglucosamine. Anal Biochem. 1984 Jul;140(1):264–269. doi: 10.1016/0003-2697(84)90163-5. [DOI] [PubMed] [Google Scholar]
- Lang L., Reitman M., Tang J., Roberts R. M., Kornfeld S. Lysosomal enzyme phosphorylation. Recognition of a protein-dependent determinant allows specific phosphorylation of oligosaccharides present on lysosomal enzymes. J Biol Chem. 1984 Dec 10;259(23):14663–14671. [PubMed] [Google Scholar]
- Mueller O. T., Honey N. K., Little L. E., Miller A. L., Shows T. B. Mucolipidosis II and III. The genetic relationships between two disorders of lysosomal enzyme biosynthesis. J Clin Invest. 1983 Sep;72(3):1016–1023. doi: 10.1172/JCI111025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reitman M. L., Kornfeld S. Lysosomal enzyme targeting. N-Acetylglucosaminylphosphotransferase selectively phosphorylates native lysosomal enzymes. J Biol Chem. 1981 Dec 10;256(23):11977–11980. [PubMed] [Google Scholar]
- Reitman M. L., Lang L., Kornfeld S. UDP-N-acetylglucosamine: lysosomal enzyme N-acetylglucosamine-1-phosphotransferase. Methods Enzymol. 1984;107:163–172. doi: 10.1016/0076-6879(84)07010-5. [DOI] [PubMed] [Google Scholar]
- Reitman M. L., Varki A., Kornfeld S. Fibroblasts from patients with I-cell disease and pseudo-Hurler polydystrophy are deficient in uridine 5'-diphosphate-N-acetylglucosamine: glycoprotein N-acetylglucosaminylphosphotransferase activity. J Clin Invest. 1981 May;67(5):1574–1579. doi: 10.1172/JCI110189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T., Tang J. Cathepsin D from porcine and bovine spleen. Methods Enzymol. 1981;80(Pt 100):565–581. doi: 10.1016/s0076-6879(81)80045-6. [DOI] [PubMed] [Google Scholar]
- Thomas G. H., Taylor H. A., Reynolds L. W., Miller C. S. Mucolipidosis 3 (Pseudo-Hurler polydystrophy): multiple lysosomal enzyme abnormalities in serum and cultured fibroblast cells. Pediatr Res. 1973 Sep;7(9):751–756. doi: 10.1203/00006450-197309000-00004. [DOI] [PubMed] [Google Scholar]
- Varki A. P., Reitman M. L., Kornfeld S. Identification of a variant of mucolipidosis III (pseudo-Hurler polydystrophy): a catalytically active N-acetylglucosaminylphosphotransferase that fails to phosphorylate lysosomal enzymes. Proc Natl Acad Sci U S A. 1981 Dec;78(12):7773–7777. doi: 10.1073/pnas.78.12.7773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varki A., Reitman M. L., Vannier A., Kornfeld S., Grubb J. H., Sly W. S. Demonstration of the heterozygous state for I-cell disease and pseudo-Hurler polydystrophy by assay of N-acetylglucosaminylphosphotransferase in white blood cells and fibroblasts. Am J Hum Genet. 1982 Sep;34(5):717–729. [PMC free article] [PubMed] [Google Scholar]
- Waheed A., Hasilik A., Cantz M., von Figura K. Phosphorylation of lysosomal enzymes in fibroblasts. Marked deficiency of N-acetylglucosamine-1-phosphotransferase in fibroblasts of patients with mucolipidosis III. Hoppe Seylers Z Physiol Chem. 1982 Feb;363(2):169–178. doi: 10.1515/bchm2.1982.363.1.169. [DOI] [PubMed] [Google Scholar]
- Waheed A., Hasilik A., von Figura K. UDP-N-acetylglucosamine:lysosomal enzyme precursor N-acetylglucosamine-1-phosphotransferase. Partial purification and characterization of the rat liver Golgi enzyme. J Biol Chem. 1982 Oct 25;257(20):12322–12331. [PubMed] [Google Scholar]