

Abstract
In sickle cell disease (SCD), ocular lesions result from stasis and occlusion of small eye vessels by sickled erythrocytes. Vaso-occlusive disease of the retina can be responsible for nonproliferative (NPR) and proliferative retinopathy (PR). Patients are often asymptomatic until serious complications arise as, vitreous hemorrhage and retinal detachment. This work aimed to study the frequency and pattern of ocular manifestations in Egyptian children and young adults with SCD. In this cross-sectional study, 40 steady state patients (80 eyes) aged 2–28 years (30 children and 10 young adults) with established diagnosis of SCD (26 with homozygous SS and 14 with S/β thalassemia underwent complete ophthalmic examination with dilated fundoscopy. Fluorescein angiography was performed for patients ≥12 years old. The overall frequency of retinal lesions was 47.5 % (46.2 and 50 % of SS and S/β patients respectively). PR and NPR were evident in 32.5 and 27.5 % of all enrolled patients respectively (five patients having both). Peripheral retinal occlusion was a frequent ocular finding in both groups; the youngest patient showing PR was 15 years old. Older age, longer disease duration and splenectomy were significantly more prevalent among patients with PR. Despite lack of visual symptoms, children and young adults are at risk of PR. Frequency of retinal lesions was comparable in SS and S/β patients. Periodic ophthalmologic examination starting at the age of 12 years is recommended for timely-identification of retinal lesions thus minimizing the risk of sight threatening retinopathy.
Keywords: Sickle cell disease, Ocular, Retinopathy, Children
Introduction
Sickle cell disease (SCD) is the most common genetic disease worldwide. In Egypt, HbS carrier rates vary from 9 to 22 % [1] with a heterogeneous distribution. Among Egyptians, most of the reported globin gene haplotypes are the African ones and the SCD phenotype is severe [2].
Increase in life expectancy of SCD patients in recent years has led to emergence of more disease-related complications, including ocular manifestations [3]. SCD can affect every vascular bed in the eye and can cause blindness in advanced stages [4]. Ocular lesions result from stasis and occlusion of the small eye vessels by sickled erythrocytes. Vaso-occlusive disease of the retina can be responsible for nonproliferative and proliferative ocular changes (based on the absence or presence of vascular proliferation respectively). This distinction is important because formation of new vessels is the single most important precursor of potentially blinding complication [3].
Lesions of nonproliferative retinopathy (NPR) include “salmon patches”, venous tortuosity, “black sunbursts”, iridescent spots and angioid streaks that characterize hemorrhagic, infarctive and resolving lesions of sickle retinopathy [5]. The classic signs of proliferative retinopathy (PR) stages are: stage I is peripheral arterial occlusion, stage II is peripheral arteriovenous anastomosis, stage III is sea fan neovascularization, stage IV is vitreous hemorrhage and stage V is fibrovascular proliferation and retinal detachment [6]. Conjunctival lesions are isolated dark red vascular anomalies shaped like commas or cork-screws. Although various systemic complications of SCD are more common in patients with (SS) genotype, visual impairment secondary to proliferation is more common in patients with sickle cell hemoglobin C (SC) disease genotype which is quite rare in Egyptian population. Patients are often asymptomatic until complications arise as, vitreous hemorrhage and retinal detachment. Some studies report that the incidence and prevalence rates of all ocular complications in SCD increase with age and that PR progresses rapidly during adolescence, justifying the routine screening of children [7, 8].
The aim of this work was to study the frequency and pattern of ocular manifestations in Egyptian children and young adults with SCD.
Materials and Methods
This was a cross-sectional study conducted at Pediatric Hematology & BMT Unit, New Children Hospital, Cairo University, a reference center attending to SCD patients, between December 2010 and September 2011. A total of 40 patients (80 eyes) aged 2–28 years (30 children and 10 young adults) with established diagnosis of SCD (26 with homozygous SCD (SS) and 14 with sickle-beta thalassemia (S/β) were enrolled in the study. Patients were allowed to participate in the study only after required consents were willingly given by the patients and/or their guardians. Twenty-four patients were female and 16 male. All recruited patients were in a steady state attending for routine follow-up visit. We excluded patients with psychiatric disease, previous ocular trauma, diabetes mellitus, hypertension, decreased visual acuity (<6/60) or retinopathy due to any cause other than SCD. The study protocol was approved by the Ethical Committee of Cairo University, Egypt, according to the Institutional Committee for the Protection of Human Subjects and adopted by the 18th World Medical Assembly, Helsinki, Finland.
All recruited patients’ medical records were reviewed. Data obtained included age, gender, hemoglobin genotype, frequency of severe vaso-occlusive events (VOE) requiring hospitalization or ER admission and disease-related manifestations as acute chest syndrome (ACS), bone disease, pulmonary hypertension (PHT), and leg ulcers. Physical examination was carried out in addition to evaluations of hemoglobin (Hb), mean corpuscular volume (MCV), reticulocytes, total leucocytic count (TLC), platelets, serum bilirubin and lactate dehydrogenase (LDH) levels. Fetal hemoglobin (HbF) and sickle hemoglobin (HbS) were measured by hemoglobin electrophoresis.
All patients were then sent to the Institute of Ophthalmologic Research for an ophthalmologic examination. Visual acuity was assessed as best corrected vision using Snellen’s chart for patients aged 6 years and over. For children under 6 years of age, visual acuity assessment was based on mother comment, eye fixation, pupillary reaction, and key pictures.
Slit lamp microscopy was performed for anterior segment examination. Intraocular pressure was measured by applanation tenometer. Under mydriasis (with Tropicamide 1 % and phenylephrine 2 %), fundus examination was done with biomicroscopy using +90 volk lens and indirect examination by Keeler ophthalmoscope. Only patients aged 12 years old or more inclusive with a clinical suspicion of PR were evaluated with fluorescein angiography using Topcon fundus camera (n = 25). We avoided angiography in children below 12 years due to difficulties in cooperation, cannulation, and adjusting fluorescein dose to patient’s age and weight. This was further justified by normal fundus examination noted for all children <12 years. Seven cases aged below 6 years were examined under general anesthesia.
Statistical Analysis
Data were expressed as mean ± standard deviation (±SD), frequencies and percentages when appropriate. Numerical variables of two study groups were compared using Student t test for independent samples when normally distributed and Mann–Whitney U test when not normally distributed. For comparing categorical data, Chi square (χ2) test was performed. Exact test was used instead when the expected frequency was <5. Significance level was defined as p < 0.05. Analyses were conducted using SPSS (v. 20, IBM SPSS Statistics, Chicago, IL).
Results
Table 1 shows the mean age and sex distribution of enrolled SS and S/β thalassemia patients. Statistically non-significant differences were found when comparing both groups for age and gender (p = 0.67 and 0.36 respectively).
Table 1.
Age and sex distribution of enrolled SS and S/β thalassemia patients
| Variable | SS (n = 26) | S/β (n = 14) | p value |
|---|---|---|---|
| Age (years)a | 13.69 ± 6.94 | 14.64 ± 6.46 | 0.67 |
| Males:Females | 9:17 | 7:7 | 0.36 |
aValues expressed as mean ± standard deviation
Visual acuity was 6/6 bilaterally in 38 patients; however, two SS patients had impaired visual acuity of 6/36 and 6/18 respectively due to errors of refraction. None of our patients had any visual symptoms. Normal intraocular pressure was observed in all patients. No patients showed conjunctival signs. Normal fundus was present in 21 (52.5 %) patients (14 SS and 7 S/β). Distribution of retinal lesions detected in studied patients is shown in Table 2. The overall frequency of retinal lesions was 47.5 % (46.2 and 50 % of SS and S/β patients respectively). PR and NPR were evident in 32.5 and 27.5 % of all enrolled patients respectively (five patients having both). A frequent ocular finding in both SCD groups (SS and S/β) was peripheral retinal occlusion which is the first stage of PR. None of our patients showed retinal proliferative changes beyond stage I. No retinal changes could be detected before 12 years of age; the youngest patient showing PR was 15 years old. Figure 1 shows fluorescein angiography of a 20 years old female SS patient showing stage I PR.
Table 2.
Distribution of retinal lesions among enrolled SS and S/β thalassemia patients
| Variable | SS (n = 26) | S/β (n = 14) |
|---|---|---|
| Retinal lesions (n = 19)a | 12 | 7 |
| NPR (n = 11)b | 7 | 4 |
| Sunburst lesion (n = 2) | 2 | – |
| Retinal vascular tortuosity (n = 10) | 6 | 4 |
| PR (n = 13) | 7 | 6 |
All values are expressed as absolute numbers
NPR nonproliferative retinopathy, PR peripheral retinopathy
aFive of 19 patients showed signs of both NPR and PR
bOne of 11 patients had both sunburst lesion and retinal vascular tortuosity
Fig. 1.
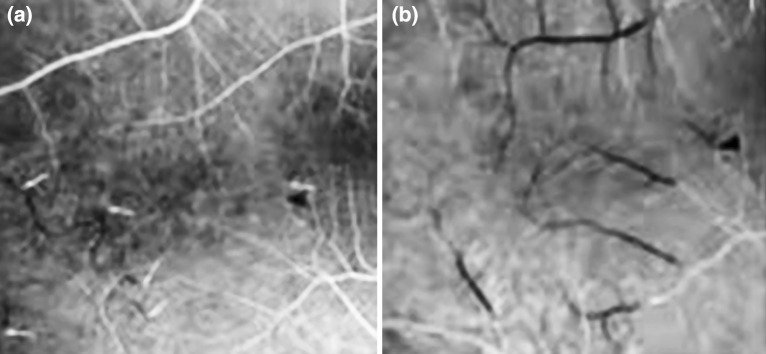
Fluorescein angiography of a 20 years old female SS patient showing stage I PR
Table 3 demonstrates clinical characteristics of patients without and with retinal lesions and PR. Patients showing retinal lesions and PR were significantly older and had longer disease duration. No sex predominance was noted among patients with PR. Comparing frequencies of disease-related events (ACS, PHT, bone disease, and leg ulcers), VOE, hospitalization and transfusion showed no significant differences between patients without and with PR (Table 3). Patients with PR showed significantly higher frequency of splenectomy (p = 0.004).
Table 3.
Clinical characteristics of patients according to retinal lesions and presence or absence of proliferative retinopathy
| Variable | Normal retina (n = 21) | Retinal lesions (n = 19) | p value | PR− (n = 27) | PR+ (n = 13) | p value |
|---|---|---|---|---|---|---|
| Age (years) | 9.17 ± 4.64 | 19.05 ± 4.77 | 0.00** | 10.57 ± 5.12 | 20.69 ± 4.42 | 0.00** |
| Gendera | ||||||
| Male (n = 16) | 11 (52.4 %) | 5 (26.3 %) | 0.09 | 11 (40.7 %) | 5 (38.5 %) | 0.89 |
| Female (n = 24) | 10 (47.6 %) | 14 (73.7 %) | 16 (59.3 %) | 8 (61.5 %) | ||
| Genotypea | ||||||
| SS (n = 26) | 14 (66.7 %) | 12 (63.2 %) | 0.81 | 19 (70.4 %) | 7 (53.8 %) | 0.3 |
| S/β (n = 14) | 7 (33.3 %) | 7 (36.8 %) | 8 (29.6 %) | 6 (46.2 %) | ||
| Disease duration (years) | 6.26 ± 4.77 | 14.47 ± 4.9 | 0.00** | 7.43 ± 5.25 | 15.85 ± 4.3 | 0.00** |
| VOE/year | 1.62 ± 1.6 | 1.89 ± 1.37 | 0.56 | 1.7 ± 1.54 | 1.85 ± 1.41 | 0.78 |
| Hospitalization/year | 1.14 ± 1.6 | 1.0 ± 1.0 | 0.74 | 1.15 ± 1.51 | 0.92 ± 0.95 | 0.62 |
| Transfusion/year | 2.48 ± 2.66 | 2.37 ± 3.7 | 0.91 | 2.48 ± 3.12 | 2.31 ± 3.35 | 0.87 |
| Bone diseasea (n = 4) | 1 (4.8 %) | 3 (15.8 %) | 0.24 | 2 (7.4 %) | 2 (15.4 %) | 0.42 |
| PHTa (n = 3) | 3 (14.3 %) | 0 (0 %) | 0.08 | 3 (11.1 %) | 0 (0 %) | 0.21 |
| ACSa (n = 1) | 0 (0 %) | 1 (5.3 %) | 0.28 | 1 (3.7 %) | 0 (0 %) | 0.48 |
| Gall bladder diseasea (n = 1) | 0 (0 %) | 1 (5.3 %) | 0.28 | 0 (0 %) | 1 (7.7 %) | 0.14 |
| Splenectomya (n = 8) | 2 (9.5 %) | 6 (31.6 %) | 0.09 | 2 (7.4 %) | 6 (46.2 %) | 0.004** |
| Hydroxyureaa (n = 27) | 14 (66.7 %) | 13 (68.4 %) | 0.9 | 18 (66.7 %) | 9 (69.2 %) | 0.87 |
Values expressed as mean ± standard deviation unless mentioned otherwise
PR proliferative retinopathy, VOE vaso-occlusive events, PHT pulmonary hypertension, ACS acute chest syndrome
** Statistically significant (p < 0.05)
aValues expressed as absolute numbers and percentages
Comparing hematological indices of patients without and with PR, failed to reveal significant differences between both groups (Table 4). Patients with PR had higher mean platelets level, however, the difference did not reach level of significance.
Table 4.
Hematological indices among patients without and with peripheral retinopathy
| Variable | PR− (n = 27) | PR+ (n = 13) | p value |
|---|---|---|---|
| Hb (g/dl) | 8.52 ± 1.34 | 7.83 ± 1.27 | 0.13 |
| MCV (fl) | 80.52 ± 10.94 | 84.48 ± 10.92 | 0.29 |
| TLC (×103/ml) | 10.11 ± 5.04 | 12.01 ± 4.81 | 0.26 |
| Platelets (×103/ml) | 369.22 ± 151.48 | 455.46 ± 116.07 | 0.07 |
| Reticulocytes (%) | 7.26 ± 5.99 | 5.51 ± 3.45 | 0.33 |
| Total bilirubin (mg/dl) | 1.73 ± 0.74 | 1.95 ± 0.96 | 0.43 |
| LDH (U/L) | 491.96 ± 141.31 | 531.00 ± 307.97 | 0.58 |
| HbS (%) | 72.65 ± 11.29 | 78.62 ± 9.90 | 0.11 |
| HbF (%) | 16.66 ± 11.04 | 15.22 ± 10.92 | 0.70 |
All values are expressed as mean and standard deviation
PR proliferative retinopathy, Hb hemoglobin, MCV mean corpuscular volume, TLCm total leucocytic count, LDH lactate dehydrogenase
Discussion
Sickle cell disease patients are known to manifest different types of ocular problems; NPR, PR and refractive errors.
Excluding refractive errors, the overall frequency of ocular lesions in our cohort was 47.5 % corroborating previous studies [9–12]. Changes in visual acuity among our patients were attributed to refractive errors. None of previously reported causes of visual impairment in SCD patients as optic atrophy or central retinal occlusion [13] were observed in our patients. MRI and visual evoked potential were not carried out in our patients.
The significance of genotype on overall frequency of ocular lesions was not apparent in our cohort; frequency of retinal lesions being comparable in SS and S/β groups. The most frequent ocular lesions identified in both disease groups in our study were peripheral retinal occlusion and vascular tortuosity. Out of 13 patients showing PR, six had S/β thalassemia confirming a previous observation that S/β thalassemia can lead to serious ischemic retinopathy [14]. Sunbursts lesion, a pigmented chorioretinal scar usually found in the periphery [12] was only detected in two SS patients. This finding is in contrast to previous studies which reported higher frequency of sunbursts lesion [7, 15, 16]. Absence of other non-proliferative ocular signs as salmon patches and iridescent spots among our patients, as previously reported [17], may be attributed to their transient status. Our study failed to detect any proliferative retinal lesions beyond stage I confirming the low frequency of such changes in Egyptian cohort [18].
Our work showed that older age and longer disease duration were significant determinants for development of retinal changes and PR. Several studies reported an increase in incidence and prevalence rates of all ocular manifestations with increasing age [3, 19]. In our cohort, all retinal changes were detected above the age of 12 years and the youngest patient showing PR was 15 years old. Based on these observations, we recommend that children with SS and S/β should be screened for retinopathy using fundoscopic examination beginning at age of 12 years.
Although the mechanism was not clear, few studies [19–21] reported male sex as a risk factor for developing retinopathy. However, our cohort did not show any sex predominance for retinal changes or PR. This may be attributed to our smaller sample size.
Comparing frequencies of disease-related events and VOE between patients with PR and those without in our cohort revealed no significant differences, which support previous reports [4, 21]. However, this conflicts previous observations of ocular complications being attributed directly or indirectly to vaso-occlusion. Our finding may be explained by high prevalence of fetal hemoglobin in our cohort (fetal hemoglobin higher than 12 in 57.5 % of our patients and in 57.7 % of SS group).
In the present study, splenectomy was significantly more common in SCD with PR (p = 0.004). Mean platelet count was higher in patients with PR compared to those without, but the difference did not reach statistical significance (p = 0.07). Both findings suggest a possible role of splenectomy and thrombocytosis, together with the coexistent state of hypercoagulability in SCD, in development of PR.
In our cohort, comparison of hematological indices in patients with and without PR failed to reveal any significant differences. The hematological correlates with retinal lesions are still unclear. Conflicting reports suggest possible effects of total hemoglobin, fetal hemoglobin and HbS [18, 21–23].
In sickle cell retinopathy, treatment strategy is based on closing the sea fan (stage III PR). Patients in stages I and II are not treated because treatment of the ischemic retina does not prevent the formation of sea fan and the majority of patients do not develop sea fan or its complications [24]. The treatment is still controversial because many sea fans regress spontaneously, especially in older SS patients, in whom the incidence of blindness is very small [24]. Photocoagulation has been the most widely used method for treatment of stage III PR [25, 26]. This type of treatment has been effective and safe and prevents subsequent complications [25]. Considering the fact that ocular changes that affect patients with SCD are the consequences of a complex systemic pathophysiological process, prevention of these ocular complications can be achieved through the use of new drugs that focus on the physiopathology of the disease at multiple points, including HbS polymerization, erythrocyte density and cell–cell interactions [24].
Conclusion
Despite lack of visual symptoms, children and young adults are at risk of PR. Frequency of retinal lesions was comparable in SS and S/β patients. Older age, longer disease duration and splenectomy were the only determinants for development of retinal changes and PR in our cohort. Periodic ophthalmologic examination starting at the age of 12 years is recommended for timely-identification of retinal lesions thus minimizing the risk of sight threatening retinopathy.
Acknowledgments
We thank all patients who participated in our study. We would like to express our appreciation to our colleagues and nurses at the Pediatric Hematology and Ophthalmology Departments who facilitated this work.
Conflict of Interest
All authors declare that they have no conflict of interest that may inappropriately influence this work.
References
- 1.El-Beshlawy A, Youssry I. Prevention of hemoglobinopathies in Egypt. Hemoglobin. 2009;33(Suppl 1):S14–S20. doi: 10.3109/03630260903346395. [DOI] [PubMed] [Google Scholar]
- 2.Soliman AT, El Zalabany M, Amer M, Ansari BM. Growth and pubertal development in transfusion-dependent children and adolescents with thalassemia major and sickle cell disease: a comparative study. J Trop Pediatr. 1999;45:23–30. doi: 10.1093/tropej/45.1.23. [DOI] [PubMed] [Google Scholar]
- 3.Fadugbagbe AO, Gurgel KQ, Mendonca CQ, Cipolotti R, dos Santos AM, Cuevas LE. Ocular manifestations of sickle cell disease. Ann Trop Pediatric. 2010;30(1):19–26. doi: 10.1179/146532810X12637745451870. [DOI] [PubMed] [Google Scholar]
- 4.Gill HS, Lam WC. A screening strategy for the detection of sickle cell retinopathy in pediatric patients. Can J Ophthalmol. 2008;43(2):188–191. doi: 10.3129/i08-003. [DOI] [PubMed] [Google Scholar]
- 5.Ohene-Frempong K, Steinberg MH. Clinical aspects of sickle cell anemia in adults and children. In: Steinberg MH, Forget BG, Higgs DR, Nagel RL, editors. Disorders of hemoglobin: genetics, pathophysiology, and clinical management. Cambridge: Cambridge University Press; 2001. p. 611. [Google Scholar]
- 6.Goldberg MF. Classification and pathogenesis of proliferative sickle cell retinopathy. Am J Ophthalmol. 1971;71(3):649–665. doi: 10.1016/0002-9394(71)90429-6. [DOI] [PubMed] [Google Scholar]
- 7.David RC, Moraes HV, Rodrigues MP. Ocular and electroretinographic changes in sickle cell disease. Arg Bras Oftalmol. 2011;74(3):190–194. doi: 10.1590/S0004-27492011000300009. [DOI] [PubMed] [Google Scholar]
- 8.Babalola OE, Wambebe CO. When should children and young adults with sickle cell disease be referred for eye assessment? Afr J Med Med Sci. 2001;30(4):261–263. [PubMed] [Google Scholar]
- 9.Kaimbo Wa Kaimbo D, Ngiyulu Makuala R, Dralands L, Missotten L. Ocular findings in children with homozygous sickle cell disease in the Democratic Republic of Congo. Bull Soc Belge Ophtalmol. 2000;275:27–30. [PubMed] [Google Scholar]
- 10.Akinsola FB, Kehinde MO. Ocular findings in sickle cell disease patients in Lagos. Niger Postgrad Med J. 2004;11(3):203–206. [PubMed] [Google Scholar]
- 11.dos Santos AM, Faro GB, do Amaral MV, Mendonça Cde Q, Leal BC, Cipolotti R. Retinal impairment in young individuals with sickle cell anemia (hemoglobin SS disease) in university hospital in Northeastern of Brazil. Arq Bras Oftalmol. 2012;75(5):313–315. doi: 10.1590/S0004-27492012000500003. [DOI] [PubMed] [Google Scholar]
- 12.Diallo JW, Sanfo O, Blot I, Meda N, Sawadogo P, Ouedraogo A, Simporé J. Epidemiology and prognostic factors for sickle cell retinopathy in Ouagadougou (Burkina Faso) J Fr Ophtalmol. 2009;32(7):496–500. doi: 10.1016/j.jfo.2009.04.010. [DOI] [PubMed] [Google Scholar]
- 13.Osafo-Kwaako A, Kimani K, Ilako D, Akafo S, Ekem I, Rodrigues O, Enweronu-Laryea C, Nentwich MM. Ocular manifestations of sickle cell disease at the Korle-bu Hospital, Accra. Ghana Eur J Ophthalmol. 2011;21(4):484–489. doi: 10.5301/EJO.2010.5977. [DOI] [PubMed] [Google Scholar]
- 14.Fanny A, Coulibaly F, Gbe K, Meite M, Adjorlolo C, Konan-Toure ML, Berete R, Boni S, Ouattara A, Diallo M. Sickle cell beta-thalassemia leading to serious ischemic retinopathy: a study of 18 patients in Abidjan. J Fr Ophtalmol. 2005;28(4):391–395. doi: 10.1016/S0181-5512(05)81070-8. [DOI] [PubMed] [Google Scholar]
- 15.Friberg TK, Young CM, Milner PF. Incidence of ocular abnormalities in patients with sickle hemoglobinopathies. Ann Ophthalmol. 1986;18(4):150–153. [PubMed] [Google Scholar]
- 16.Freitas LG, Isaac DL, Tannure WT, Lima EV, Abud MB, Tavares RS, Freitas CA, Avila MP. Retinal manifestations in patients with sickle cell disease referred to a University Eye Hospital. Arq Bras Oftalmol. 2011;74(5):335–337. doi: 10.1590/S0004-27492011000500005. [DOI] [PubMed] [Google Scholar]
- 17.Cury D, Boa-Sorte N, Lyra IM, Zanette AD, Castro-Lima H, Galvão-Castro B, Goncalves MS. Ocular lesions in sickle cell disease patients from Bahia. Braz Rev Bras Oftalmol. 2010;69(4):259–263. doi: 10.1590/S0034-72802010000400010. [DOI] [Google Scholar]
- 18.Tantawy AA, Andrawes NG, Adly AA, El Kady BA, Shalash AS. Retinal changes in children and adolescents with sickle cell disease attending a paediatric hospital in Cairo, Egypt: risk factors and relation to ophthalmic and cerebral blood flow. Trans R Soc Trop Med Hyg. 2013;107(4):205–211. doi: 10.1093/trstmh/trt008. [DOI] [PubMed] [Google Scholar]
- 19.Leveziel N, Bastuji-Garin S, Lalloum F, Querques G, Benlian P, Binaghi M, Coscas G, Soubrane G, Bachir D, Galactéros F, Souied EH. Clinical and laboratory factors associated with the severity of proliferative sickle cell retinopathy in patients with sickle cell hemoglobin C (SC) and homozygous sickle cell (SS) disease. Medicine (Baltimore) 2011;90(6):372–378. doi: 10.1097/MD.0b013e3182364cba. [DOI] [PubMed] [Google Scholar]
- 20.Rosenberg JB, Hutcheson KA. Pediatric sickle cell retinopathy: correlation with clinical factors. J AAPOS. 2011;15(1):49–53. doi: 10.1016/j.jaapos.2010.11.014. [DOI] [PubMed] [Google Scholar]
- 21.Talbot JF, Bird AC, Maude GH, Acheson RW, Moriarty BJ, Serjeant GR. Sickle cell retinopathy in Jamaican children: further observations from a cohort study. Br J Ophthalmol. 1988;72(10):727–732. doi: 10.1136/bjo.72.10.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hayes RJ, Condon PI, Serjeant GR. Haematological factors associated with proliferative retinopathy in sickle cell-haemoglobin C disease. Br J Ophthalmol. 1981;65(10):712–717. doi: 10.1136/bjo.65.10.712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Al-Hazzaa S, Bird AC, Kulozik A, Serjeant BE, Serjeant GR, Thomas P, Padmos A. Ocular findings in Saudi Arabian patients with sickle cell disease. Br J Ophthalmol. 1995;79(5):457–461. doi: 10.1136/bjo.79.5.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bonanomi MT, Lavezzo MM. Sickle cell retinopathy: diagnosis and treatment. Arq Bras Oftalmol. 2013;76(5):320–327. doi: 10.1590/S0004-27492013000500016. [DOI] [PubMed] [Google Scholar]
- 25.Farber MD, Jampol LM, Fox P, Moriarty BJ, Acheson RW, Rabb MF, et al. A randomized clinical trial of scatter photocoagulation of proliferative sickle cell retinopathy. Arch Ophthalmol. 1991;109(3):363–367. doi: 10.1001/archopht.1991.01080030065040. [DOI] [PubMed] [Google Scholar]
- 26.Condon P, Jampol LM, Farber MD, Rabb M, Serjeant G. A randomized clinical trial of feeder vessel photocoagulation of proliferative sickle cell retinopathy. II. Update and analysis of risk factors. Ophthalmology. 1984;91(12):1496–1498. doi: 10.1016/S0161-6420(84)34121-5. [DOI] [PubMed] [Google Scholar]