

Abstract
This study aims to test the hypothesis that thiazolidinedione rosiglitazone (RSG), a selective peroxisome proliferator-activated receptor γ (PPARγ) agonist, causes cardiotoxicity independently of PPARγ. Energy metabolism and mitochondrial function were measured in perfused hearts isolated from C57BL/6, cardiomyocyte-specific PPARγ-deficient mice, and their littermates. Cardiac function and mitochondrial oxidative stress were measured in both in vitro and in vivo settings. Treatment of isolated hearts with RSG at the supratherapeutic concentrations of 10 and 30μM caused myocardial energy deficiency as evidenced by the decreases in [PCr], [ATP], ATP/ADP ratio, energy charge with a concomitant cardiac dysfunction as indicated by the decreases in left ventricular systolic pressure, rates of tension development and relaxation, and by an increase in end-diastolic pressure. When incubated with tissue homogenate or isolated mitochondria at these same concentrations, RSG caused mitochondrial dysfunction as evidenced by the decreases in respiration rate, substrate oxidation rates, and activities of complexes I and IV. RSG also increased complexes I- and III-dependent O2− production, decreased glutathione content, inhibited superoxide dismutase, and increased the levels of malondialdehyde, protein carbonyl, and 8-hydroxy-2-deoxyguanosine in mitochondria, consistent with oxidative stress. N-acetyl-L-cysteine (NAC) 20mM prevented RSG-induced above toxicity at those in vitro settings. Cardiomyocyte-specific PPARγ deletion and PPARγ antagonist GW9662 did not prevent the observed cardiotoxicity. Intravenous injection of 10 mg/kg RSG also caused cardiac dysfunction and oxidative stress, 600 mg/kg NAC antagonized these adverse effects. In conclusion, this study demonstrates that RSG at supratherapeutic concentrations causes cardiotoxicity via a PPARγ-independent mechanism involving oxidative stress-induced mitochondrial dysfunction in mouse hearts.
Keywords: Rosiglitazone, PPARγ, energy metabolism, cardiac function, mitochondria, oxidative stress
Peroxisome proliferator-activated receptor γ (PPARγ), a ligand-activated nuclear transcription factor, is highly expressed in metabolic adipose tissue, brain, immune cells, and retina (Dumasia et al., 2005). It plays an important role in adipocyte differentiation, glucose metabolism, and lipid homeostasis, and participates in monocyte/macrophage differentiation. However, it is expressed at lower levels throughout the cardiovascular system (Barger and Kelly, 2000). Despite its lower expression, loss of PPARγ from the heart resulted in cardiac hypertrophy (Duan et al., 2005), suggesting that the presence of PPARγ may be essential for normal cardiac development. Thiazolidinediones (TZDs) including rosiglitazone (RSG), pioglitazone, ciglitazone, and troglitazone are PPARγ agonists used to reduce insulin resistance and hyperglycemia in type 2 diabetic patients. RSG acts more selectively as a PPARγ agonist than other TZDs (Yki-Jarvinen, 2004). It was approved in 1999 for the treatment of hyperglycemia in type 2 diabetes. Its original approval was based on the ability to reduce insulin-resistance, increase peripheral glucose utilization, decrease hepatic glucose output, and as a result, lower blood glucose concentration (Day, 1999). Initial studies were not adequately powered to determine the effects of this agent on the cardiovascular system and no serious cardiovascular adverse events were recognized at the time of approval. Therefore, the potential for unexpected cardiovascular toxic effects when this agent is administered to patients was uncertain. Subsequently, RSG has been reported to increase risks of heart failure (Home et al., 2007; Lago et al., 2007; McMorran and Vu, 2001; Nesto et al., 2003; Ryden et al., 2007; Singh et al., 2007a,b; Taylor and Hobbs, 2009; Winkelmayer et al., 2008) and myocardial infarction (Nissen and Wolski, 2007; Singh et al., 2007a). Therefore, the cardiovascular safety of RSG has become an increasing concern in the treatment of diabetic patients. Furthermore, altered energy metabolism and cardiac dysfunction are common features of heart failure resulted from different causes, including diabetes. We therefore investigated the effects of RSG on energy metabolism, cardiac mitochondrial, and contractile functions in mouse hearts in this study.
In addition to PPARγ-dependent effects, TZDs have also been reported to exert “off-target” PPARγ-independent effects (Boyle et al., 2008; Duan et al., 2005; Gardner et al., 2005; Mughal et al., 2009). The therapeutic effects of TZDs on diabetic patients involve both PPARγ−dependent and PPARγ−independent mechanisms. Overexpression of cardiomyocyte (CM) PPARγ resulted in cardiac dysfunction in mice (Son et al., 2007), indicating PPARγ activation may be involved in cardiovascular adverse effects related to the treatment of TZDs. Therefore, the appropriateness of PPARγ as a therapeutic target to develop new drugs is questioned. To determine whether PPARγ activation is involved in the effects of RSG on myocardial energy metabolism and cardiac function, CM specific PAPRγ-deficient mice and PPARγ specific inhibitor GW9662 were used in the present study.
Mitochondria are both source and target of reactive oxygen species (ROS; Anderson et al., 2012; Figueira et al., 2013; Matamoros et al., 2013). A major source of mitochondrial and cellular ROS is electron leakage from the mitochondrial electron transport chain (Trachootham et al., 2008). Other endogenous sources of ROS include membrane-associated NADPH oxidase, cytochrome c oxidase, and xanthine oxidase (Trachootham et al., 2008). Mitochondria also possess powerful, multileveled high-capacity ROS elimination systems, including superoxide dismutase (SOD), reduced glutathione (GSH), glutathione peroxidase, and catalase (Trachootham et al., 2008). An increase in ROS production and/or a decrease in ROS-scavenging capacity due to exogenous stimuli or endogenous metabolic alterations can disrupt redox homeostasis, leading to an overall increase of intracellular ROS level, or oxidative stress. Numerous studies have reported that mitochondrial dysfunction is accompanied with oxidative stress in aging and neurodegenerative diseases (Albers and Beal, 2000; Tritschler et al., 1994). Moreover, TZDs have been reported to cause mitochondrial dysfunction (Rachek et al., 2009; Scatena et al., 2004). Thus, the effects of RSG on mitochondrial function and redox homeostasis were also studied in the present study.
Overall, this study was performed to test the hypothesis that RSG causes cardiotoxicity independently of PPARγ. To test this hypothesis, energy metabolism, contractile function, mitochondrial function, and redox homeostasis were measured at the levels of perfused heart, tissue homogenate, and isolated mitochondria from C57BL/6, CM-specific PPARγ-deficient mice, and their littermates. Cardiac function and mitochondrial oxidative stress level were also measured in vivo. We found that RSG at supratherapeutic concentrations caused myocardial energy deficiency at the whole heart level. This adverse effect was related to oxidative stress-induced mitochondrial dysfunction, leading to concomitant cardiac contractile dysfunction, and was independent of CM PPARγ.
MATERIALS AND METHODS
Detailed methods are provided in the Supplementary materials.
Materials
RSG maleate and 2-chloro-5-nitrobenzanilide (GW9662) were purchased from Cayman Chemical (Ann Arbor, MI). [1-14C]-glucose and [1-14C]-palmitic acid were from MP Biomedicals (Solon, OH). N-acetyl-L-cysteine (NAC) and other reagents were purchased from Sigma-Aldrich (St Louis, MO), except when indicated otherwise.
Animals
Male, three-month-old C57BL/6 mice were purchased from the Charles River Laboratories (Charles River, MA). CM-specific PPARγ-knockout (PPARγ−) mice (homozygous floxed PPARγ and α-MHC-Cre positive), and their littermates (PPARγ+, homozygous floxed PPARγ and α−MHC-Cre negative) were generated as previously described (Duan et al., 2005). Animals were maintained in accordance with National Institutes of Health guidelines for the care and use of laboratory animals. The investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the U.S. National Institutes of Health (NIH Publication No. 85-23, revised 1996). The experimental protocols were approved by the Standing Committee on Animals of Harvard Medical Area and followed current National Institutes of Health and American Physiologic Society guidelines.
Acute in vivo administration of drugs
NAC was dissolved in physiological saline, the pH of the NAC solution was adjusted by adding NaOH. RSG stock solution was first dissolved in dimethyl sulfoxide (DMSO), its working solution was then diluted in the saline. At 2 h before in vivo measurement of mouse cardiac function with echocardiography, RSG alone, NAC alone, NAC + RSG, or vehicle (DMSO: saline = 1:95) at the dosage of interest was intravenously injected via the tail vein for 5 min.
Echocardiographic studies and heart tissue collection
Transthoracic echocardiography was performed on mice before and 2 h after intravenous injection of vehicle, RSG (1 or 10 mg/kg), NAC (600 mg/kg), and NAC (600 mg/kg) + RSG 10 mg/kg (n = 5 each group) as described previously (Wang et al., 2010). The following parameters were recorded or derived: left ventricular end-diastolic dimension (LVEDD), left ventricular end-systolic dimension (LVESD), heart rate (HR), left ventricular fractional shortening (FS), and ejection fraction (EF). At the end of each experiment, the heart was removed and irrigated with normal saline to flush the blood from the chambers and vessels. The heart was then frozen in liquid nitrogen and stored at −80ºC for further assays of mitochondrial oxidative stress as described below.
In vitro treatment of langendorff-perfused hearts with drugs
Mice were heparinized (100 units, IP) 15 min before being sacrificed. Hearts were isolated and perfused in the Langendorff mode as described (He et al., 2007) in the following protocols. Protocol 1: hearts from C57BL/6 (n = 15), PPARγ+ (n = 10), and PPARγ− mice (n = 11) were perfused with regular Krebs-Henseleit (KH) buffer containing (in mM) NaCl 118, KCl 5.3, CaCl2 2.5, MgSO4 1.2, EDTA 0.5, NaHCO3 25, and glucose 10, and pyruvate 0.5. After a 30 min stabilization, the hearts underwent 24 min baseline perfusion, followed by infusion of freshly prepared RSG solution (1.5mM) or vehicle using Genie Plus Infusion Syringe Pump into the KH buffer in a mixing chamber above the heart at 0.067, 0.2, 0.67, and 2% of coronary flow rate to deliver 1, 3 (therapeutic concentration), 10, and 30μM (supratherapeutic concentration) RSG and equivalent vehicle (0.004, 0.011, 0.036, and 0.11% DMSO) for 24 min each. Isovolumic contractile performance and 31P-NMR spectra were collected continuously and simultaneously during the infusion of RSG or vehicle as described below. Protocol 2: hearts from C57BL/6 (n = 8), PPARγ+ (n = 7), and PPARγ− mice (n = 7) were perfused with the regular KH buffer for a 30 min stabilization. Protocol 3: hearts from C57BL/6 mice were perfused with modified KH buffer containing physiological concentrations (0.4mM) of mixed free fatty acid (palmitate, palmitoleic, linoleic, and oleic) carried in 1% bovine serum albumin (BSA), glucose (5.5mM), β-hydroxybutarate (0.19mM), and lactate (1.0mM) and equivalent NaCl, KCl, CaCl2, MgSO4, EDTA, and NaHCO3 as in regular KH buffer. After a 30 min stabilization, the hearts were continually perfused for another 60 min with the modified KH buffer supplemented with vehicle (equivalent DMSO solution for dissolving RSG and GW9662, n = 7), 10μM RSG (n = 6), 30μM RSG (n = 7), 10μM GW9662 (n = 6), 10μM GW9662 +10μM RSG (n = 7), 20mM NAC (n = 6), and 20mM NAC + 10μM RSG (n = 7). Protocol 4: hearts from C57BL/6 (n = 50) were perfused with the modified KH buffer for a 30 min stabilization. At the end of each experiment, the heart was freeze-clamped with Wollenberger tongs precooled in liquid nitrogen and stored at –80ºC for further assays as described below.
Measurement of isovolumic contractile function
Isovolumic contractile performance data including left ventricular systolic pressure (LVSP), end diastolic pressure (EDP), HR, the rate of tension development (+dP/dt), and the rate of relaxation (−dP/dt) were collected online using a PowerLab data acquisition system (ADInstruments, Colorado Springs, CO) after a 30 min stabilization period in the above protocols 1 and 3 as described in the supplemental methods.
31P-NMR spectroscopy
31P-NMR spectra were collected continuously and simultaneously during measurement of isovolumic contractile performance in the isolated hearts in protocol 1 using 31P-NMR spectroscopy (He et al., 2007). The intracellular concentration of ATP, PCr, Pi, and free energy of ATP hydrolysis (ΔGATP) at the whole heart level were calculated as described in the supplemental methods.
HPLC assay
The samples for high-performance liquid chromatography (HPLC) measurement were prepared as described (Lazzarino et al., 2003). Total ATP, ADP, AMP, NAD, and NADH were separated through a Kromasil C-18 reverse phase column (250 × 4.6-mm, 5 μm-particle-size) with tetrabutylammonium hydroxide as the pairing reagent during 100 min at 0.8 ml/min of pump flow rate. The energy charge is defined by ([ATP] + 0.5[ADP])/([ATP] + [ADP] + [AMP]).
Tissue culture
CMs and cardiac fibroblasts (CFs) were derived from the ventricles of eight-week-old C57BL/6 mice. CMs were isolated using Adumyt Kit following the manufacturer's protocol (Cellutron Life Technologies ac-7031) and seeded at 5 ×104 cells per well of a 96-well plate in DMEM containing 10% fetal bovine serum (FBS). CFs were isolated by enzymatic digestion with a collagenase buffer as previously reported (Frangogiannis et al., 2007). Briefly, three hearts were isolated, dissected free of vessels and atria, transferred to 1 ml of collagenase buffer, and quickly minced into small pieces. Digestion with collagenase buffer continued until no visible tissue fragments were left. The isolated cell suspensions from each round were pelleted and washed. All cell suspensions were combined, seeded in 100-mm dishes in full medium supplemented with 10% FBS and antibiotic-antimycotic solution. After overnight incubation, nonadherent cells were removed, and adherent cells were cultivated. On reaching confluence, cells were detached with trypsin/EDTA, split in a 1:2 or 1:4 ratio, and recultured. The second-passage CFs were seeded at 5 ×104 cells per well of a 96-well plate in 10% FBS DMEM.
Bioluminescence assay of intracellular ATP
For intracellular ATP measurement, both CMs and CFs cultured in 96-well plates in 10% FBS DMEM were rinsed with phosphate-buffered saline and the media were replaced with 0.5% FBS high-glucose DMEM supplemented with RSG or/and GW9662 at the concentrations of interest for 60 min. Intracellular ATP content in these cells was measured by luciferase-driven bioluminescence according to the manufacturer's protocol (ATP bioluminescence assay kit HS II; Roche 1699709).
Substrate oxidation rates
Oxidation rate of palmitate and glucose was measured using [1-14C]-glucose and [1-14C]-palmitic acid in fresh tissue homogenates prepared from hearts perfused in protocols 2 and 4 as reported (Benton et al., 2008).
Isolation of mitochondria and nuclei
Myocardial mitochondria and nuclei were isolated and purified from hearts collected in both in vivo and in vitro studies as described in the supplemental methods.
Mitochondrial respiration
Oxygen consumption was measured in isolated mitochondrial in air-saturated buffer (pH 7.2, 25°C) with a Clark-type oxygen electrode as reported (Brunmair et al., 2004) with minor modifications. Briefly, isolated mitochondria (∼40 μg protein) were preincubated for 60 min at 25°C in isolation buffer additionally containing 0.3% BSA (wt/vol) and 1, 3, 10, and 30μM RSG or vehicle, which did not affect the rates of oxygen consumption. For stimulating mitochondrial respiration, 4mM inorganic phosphate was then added together with 5mM glutamate + 5mM malate. After 3 min, mitochondrial respiration was accelerated by the addition of 200μM ADP allowing ATP synthesis, and the rates of oxygen consumption were measured in state 3 (i.e., in the presence of ADP). After the quantitative consumption of added ADP, the rate of oxygen consumption was measured in state 4 (i.e., in the absence of ADP; Gnaiger et al., 2000).
Activities of mitochondrial electron transport chain complexes
Isolated mitochondria were preincubated with drugs of interest and vehicle for 60 min, the activities of mitochondrial electron transport chain complexes I, II, III, IV, and V were then measured spectrophotometrically as described in the supplemental methods.
ROS production
To assess the effects of RSG on ROS production by heart tissues, homogenates of heart tissues perfused in protocol 4 were preincubated with RSG (1–30μM) and vehicle. O2− production by tissue homogenate was measured using lucigenin-enhanced chemiluminescence as described previously (Li et al., 2002). A low lucigenin concentration (5μM) was employed to minimize artifactual O2− production owing to redox cycling. Proteins in homogenate were diluted in modified 4-2-hydroxyethyl-1-piperazineethanesulfonic acid (HEPES) buffer and distributed (100 μg/well) onto a 96-well microplate. To measure NADPH oxidase-dependent O2− production, NADPH (100μM), dark-adapted lucigenin (5μM), nitric oxide synthase inhibitor NG-nitro-l-arginine methyl ester (L-NAME 100μM), mitochondrial electron chain inhibitor rotenone (5μM), and xanthine oxidase inhibitor oxypurinol (100μM) were added to wells just before reading O2−. To measure mitochondrial complex I-dependent O2− production, glutamate (5mM) + malate (5mM), dark-adapted lucigenin (5μM), L-NAME (100μM), oxypurinol (100μM), and NADPH oxidase inhibitor diphenyleneiodonium (DPI, 100μM) were added to wells just before reading. To measure mitochondrial complex III-dependent O2− production, succinate (10mM) + rotenone (5μM), dark-adapted lucigenin (5μM), L-NAME (100μM), DPI (100μM), and oxypurinol (100μM) were added to wells just before reading. To measure xanthine oxidase-dependent O2− production, xanthine (10mM) dark-adapted lucigenin (5μM), rotenone (5μM), L-NAME (100μM), and DPI (100μM) were added to wells just before reading. Light emission was recorded every minute for 60 min, O2− production was expressed as mean arbitrary light units/minute/mg protein (MLU/min/mg Pr). All studies were performed in duplicates.
Mitochondrial antioxidant defense system
To assess the effects of RSG on mitochondrial antioxidant ability, isolated mitochondria from hearts perfused in protocol 4 were preincubated with RSG (1–30μM) and vehicle (0.11% DMSO) for 60 min, commercially available assay kits were used to measure the activities of catalase (Invitrogen A22180), glutathione peroxidase (Trevigen 7512–100-K), and Mn2+-SOD (Trevigen 7500–100-K) in isolated mitochondria following the instructions of manufacturers. Similarly, the content of reduced glutathione (GSH) was measured in isolated mitochondria using Glutathione Assay Kit (Trevigen 7511–100-K).
Oxidative damage
Isolated mitochondria or nuclei from in vitro langendorff hearts perfused in protocol 4 were preincubated with RSG (1–30μM) and vehicle (0.11% DMSO) for 60 min. Oxidative damage to lipids in mitochondria was assessed by measuring malondialdehyde (MDA) content using lipid peroxidation assay kit (Bio Vision K739–100). Oxidative damage to proteins in isolated mitochondria and nuclei was assessed by measuring the content of protein carbonyl using protein carbonyl enzyme immuno-assay (ELISA) kit (Northwest Life Science Specialties NWK-PCK01) according to the instruction of the manufacturer. Oxidative damage to DNAs in mitochondria or nuclei was assessed by measuring the level of 8-hydroxy-2-deoxyguanosine (8-OHdG) using a specific enzyme-linked immunosorbent assay (8-OHdG ELISA; Northwest Life Science Specialties, NWK-8OHDG02). Isolation of nuclear DNA was performed by digestion with proteinase K and then extracted in chloroform/isoamyl alcohol and phenol as previously described. Mitochondrial DNA was obtained by treating isolated mitochondria with proteinase K (10 mg/ml) in sodium dodecyl sulfate (54 mg/ml) followed by chloroform/isoamyl alcohol and phenol extraction and cold ethanol precipitation. To prevent sample oxidation, all mitochondrial and nuclear extracts were normalized to 1–1.5 mg/ml in resuspension buffer with 5mM butylated hydroxyl toluene. Standards were prepared and experimental procedures were performed according to the manufacturer's protocols. In another set of experiments, MDA, protein carbonyl, and 8-OHdG were determined in myocardial mitochondria of mice subjected to acute in vivo treatment with RSG and NAC using the above assay kits.
Activities of creatine kinase, citrate synthase, and glycolytic enzymes
Fresh tissue homogenates prepared from hearts perfused in protocols 2 and 4 were preincubated with RSG (1–30μM) and vehicle for 60 min. Aliquots were removed for protein assay with Lowry method and for creatine assay with fluorometric method (Kammermeier, 1973). Triton X-100 was then added to the homogenate at a final concentration of 0.1%. Tissue activities of the enzymes citrate synthase (CS), creatine kinase (CK), phosphofructokinase (PFK), lactate dehydrogenase (LDH), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) were measured as described in supplemental methods.
Statistical analysis
The data are presented as the mean ± standard deviation (SD). ANOVA was used to compare measurements among all groups. A post-hoc Bonferoni test was used for comparison of the means. Differences were declared statistically significant at p < 0.05. GraphPad Prism version 5.0 (San Diego, CA) was used for statistical computations and graphs.
RESULTS
RSG Caused Myocardial Energy Deficiency and Mitochondrial Dysfunction
Using 31P-NMR spectroscopy, we measured intracellular phosphocreatine (PCr) and ATP, and calculated free energy of ATP hydrolysis (ΔGATP) in isolated beating hearts perfused in Langendorff mode with regular KH buffer containing 10mM glucose and 0.5mM pyruvate. At baseline, all hearts from C57BL/6 mice showed visible and similar PCr and ATP resonance areas, PCr/ATP ratio was about 1.8 (Fig. 1A). At the human therapeutic concentrations of 1 and 3μM, RSG showed no marked effects on the resonance areas and concentrations of intracellular PCr ([PCr]) and ATP ([ATP]). At the supratherapeutic concentrations of 10 and 30μM, however, RSG decreased [PCr], [ATP], and ΔGATP compared with baseline and vehicle treatment (Figs. 1A–D). To confirm the results from 31P-NMR spectroscopy, we freeze-clamped hearts from those mice at the end of each experiment and then measured total ATP, ADP, and AMP content using HPLC and calculated energy charge. Consistent with the above results, total ATP content, ATP to ADP ratio, and energy charge decreased following acute RSG treatment compared with vehicle control (Supplementary table 1). To determine whether the detrimental action of RSG on myocardial energy metabolism is related to the lack of fatty acid in perfusate, another set of mouse hearts was perfused with modified KH buffer containing 5.5mM glucose, 0.4mM mixed free fatty acid (palmitate, palmitoleic, linoleic, and oleic), 0.19mM β-hydroxybutarate, and 1mM lactate. Using HPLC, we found that RSG at the above supratherapeutic concentrations also decreased total ATP content, ATP to ADP ratio, and energy charge in fatty acid-perfused hearts compared with vehicle control (Figs. 1E–G).
FIG. 1.
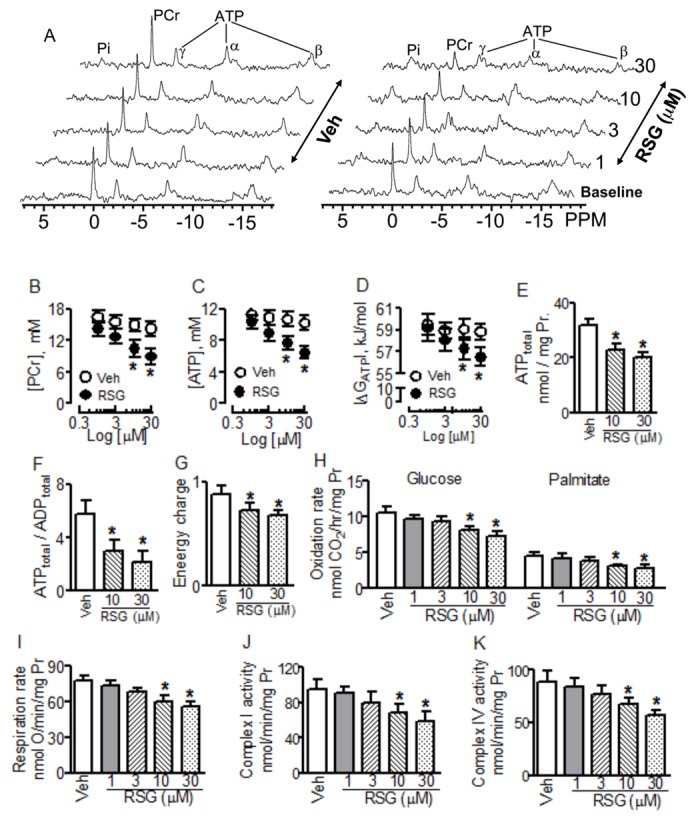
Effects of rosiglitazone (RSG) on myocardial energy metabolism and mitochondrial function in hearts isolated from C57BL/6 mice. Hearts were perfused with regular Krebs-Henseleit buffer containing 10mM glucose and 0.5mM pyruvate from (A) to (D) (n = 7–8) and with modified Krebs-Henseleit buffer containing 5.5mM glucose, 0.4mM mixed free fatty acid from (E) to (K) (n = 6–7). (A) Representatives of 31P-NMR spectra. The data shown from (B) through (K) are mean ± SD. *p < 0.05 versus vehicle (Veh). Pr: protein.
Because mitochondrial oxidation of fatty acid and glucose is a major source of ATP in CMs, we measured glucose and palmitate oxidation rates using [1-14C]-glucose and [1-14C]-palmitic acid, respectively, in fresh tissue homogenates. At the therapeutic concentrations of 1 and 3μM, incubation of RSG with myocardial homogenates for 60 min did not change glucose and palmate oxidation rates. At the supratherapeutic concentrations of 10 and 30μM, however, it decreased oxidation rates of glucose and palmitate compared with vehicle treatment (Fig. 1H). Consistently, RSG decreased mitochondrial respiration rate at these supratherapeutic concentrations (Fig. 1I).
We next determined the effects of RSG on both mitochondrial and cytosolic rate-limiting enzymes controlling ATP synthesis. When incubated with fresh tissue homogenate or isolated mitochondria for 60 min, RSG at 1 and 3μM did not affect the activities of cytosolic and mitochondrial enzymes tested compared with vehicle treatment. At the supratherapeutic concentrations of 10 and 30μM, however, RSG decreased the activities of mitochondrial complexes I and IV (Figs. 1J and K), but did not alter the activities of other mitochondrial enzymes CS, CK, Complexes II, III, V, and cytosolic enzymes PFK, LDH, and GAPDH (Supplementary table 1 and Supplementary fig. 1).
RSG Caused Myocardial Energy Deficiency and Mitochondrial Dysfunction Independently of PPARγ
To determine whether RSG caused myocardial energy deficiency via activation of CM PPARγ, we genetically deleted CM PPARγ as described previously (Duan et al., 2005). At baseline, the resonance areas, [PCr] and [ATP] in PPARγ− hearts were not different from those in PPARγ+ hearts, indicating the loss of regulatory action of CM PPARγ on myocardial energy metabolism can be compensated in vivo. At 1 and 3μM, RSG showed no significant effects on the resonance areas, [PCr] and [ATP], whereas at 10 and 30μM, however, RSG decreased the resonance areas, [PCr] and [ATP], in both PPARγ− and PPARγ+ hearts in parallel compared with their vehicle controls (Figs. 2A–C). Additionally, total ATP content, ATP to ADP ratio, energy charge, oxidation rates of glucose and palmitate, mitochondrial respiration rate, and activities of complexes I and IV were also decreased following acute treatment with RSG at the above supratherapeutic concentrations in both PPARγ− and PPARγ+-hearts comparatively (Figs. 2D-J and Supplementary table 1). These results indicate that the higher concentrations of RSG caused myocardial energy deficiency and mitochondrial dysfunction in the CMs in a PPARγ−independent manner. To rule out the possibility that RSG caused myocardial energy deficiency and mitochondrial dysfunction through activation of PPARγ in other cardiac cells including fibroblast, smooth muscle cells, and endothelial cells, we examined the effects of GW9662, a specific PPARγ antagonist on the detrimental actions of RSG on myocardial energy metabolism and mitochondrial function. As shown in Figure 3 and Supplementary figure 2, perfusion of hearts from C57BL/6 mice with 10μM GW9662 for 60 min affected neither total ATP content, nor ATP/ADP ratio, nor energy charge. This antagonist did not reverse the decreases in total ATP content, ATP/ADP ratio, and energy charge caused by RSG at 10μM in those hearts. Furthermore, 10μM GW9662 showed no effects on the oxidation rates of glucose and palmitate, mitochondrial respiration rate, or the activities of mitochondrial complexes I and IV, it did not antagonize the downregulations of those parameters by RSG at the supratherapeutic concentration of 10μM, either. Additionally, treatments with RSG at the supratherapeutic concentrations of 10 and 30μM for 60 min significantly decreased intracellular ATP content in cultured mouse CMs. In contrast, treatment with 10μM RSG showed no effect on intracellular ATP content in cultured mouse CFs, treatment with 30μM RSG only slightly decreased intracellular ATP content in these fibroblasts. Interestingly, pretreatment with 30μM GW9662 did not prevent the decreases in intracellular ATP content caused by RSG in these cultured CMs or CFs (Supplementary fig. 3).
FIG. 2.
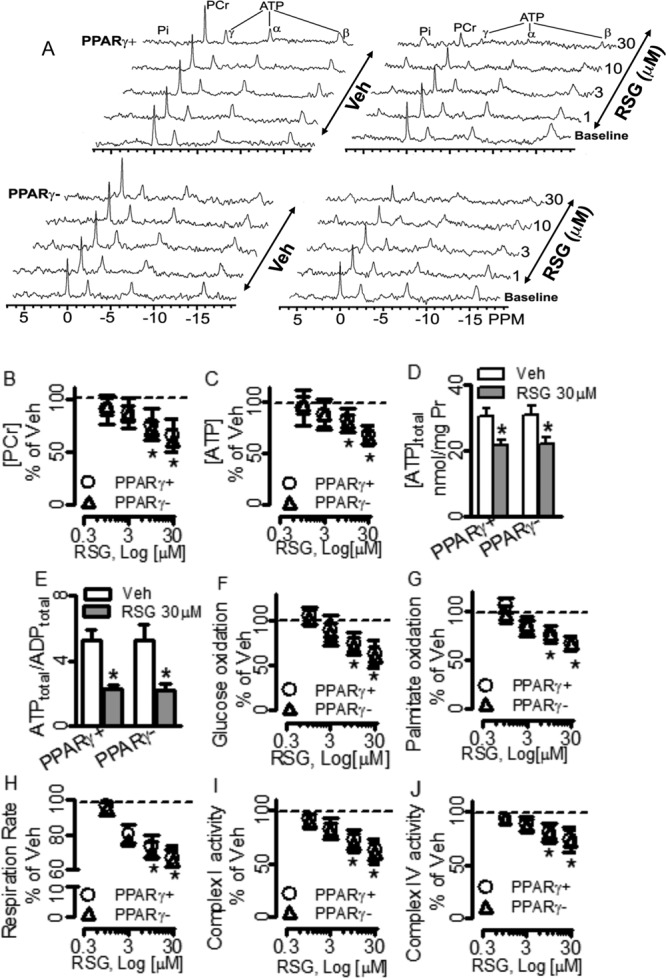
Effects of rosiglitazone (RSG) on myocardial energy metabolism and mitochondrial function in hearts isolated from cardiomyocyte-specific PPARγ-knockout mice (PPARγ−) and littermate controls (PPARγ+). Hearts were perfused with regular Krebs-Henseleit buffer containing 10mM glucose and 0.5mM pyruvate. (A) Representatives of 31P-NMR spectra. The data from (B) through (J) are shown as the mean of percentages ± SD over the vehicle (Veh, indicated by the dashed line), n = 4–8. *p < 0.05 versus Veh. Pr: protein.
FIG. 3.
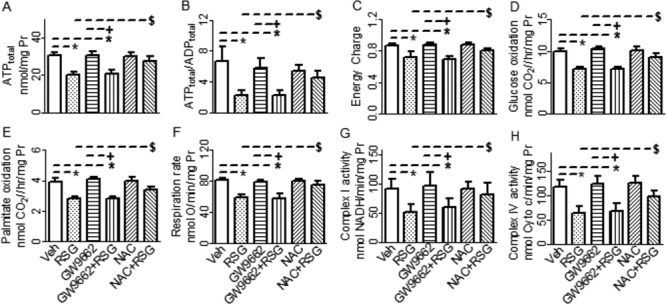
Effects of GW9662 and N-acetyl-L-cysteine (NAC) on rosiglitazone (RSG)-induced down regulations of myocardial energy metabolism and mitochondrial function in hearts isolated from C57BL/6 mice. Hearts were perfused with modified Krebs-Henseleit buffer containing 5.5mM glucose and 0.4mM mixed free fatty acid. The data shown are mean ± SD, n = 5–8. *p < 0.05 versus vehicle (Veh), +p < 0.05 versus GW9662, $p < 0.05 versus RSG alone. Pr : Protein. Cyto c: cytochrome c.
NAC Antagonized RSG-Induced Energy Deficiency and Mitochondrial Dysfunction
To determine whether oxidative stress is involved in RSG-induced myocardial energy deficiency and mitochondrial dysfunction, hearts were perfused with modified KH buffer supplemented with 20mM antioxidant NAC and 10μM RSG for 60 min. As shown in Figure. 3 and Supplementary figure 2, 20mM NAC alone showed no effect on total ATP content, ATP/ADP ratio, energy charge, oxidation rates of glucose and palmitate, mitochondrial respiration rate, or the activities of mitochondrial complexes I and IV compared with vehicle control. NAC, however, at least partially antagonized downregulations of those parameters by RSG at the supratherapeutic concentrations of 10μM.
RSG Caused Mitochondrial Oxidative Stress
To assess the in vitro effects of RSG on redox homeostasis, we determined O2− production by NADPH oxidase (NOX), xanthine oxidase (XO) and mitochondrial electron transport chain (complexes I and III), ROS elimination system and oxidative damages to lipids, proteins and DNAs in isolated mitochondria and nuclei. Here again, at 1 and 3μM, RSG showed no effects on any of the measured parameters. At 10 and 30μM, however, RSG increased mitochondrial complexes I- and III-dependent O2− production (Fig. 4A), decreased the level of mitochondrial GSH and activity of SOD (Figs. 4D and E), and increased the levels of mitochondrial MDA, protein carbonyl and 8-OHdG (Figs. 4F–H). Interestingly, even at the supratherapeutic concentrations of 10 and 30μM, RSG did not affect the activities of catalase and glutathione peroxidase (Figs. 4B and C), and changed neither the level of nuclear protein carbonyl nor the level of nuclear 8-OHdG (Figs. 4G and H).
FIG. 4.
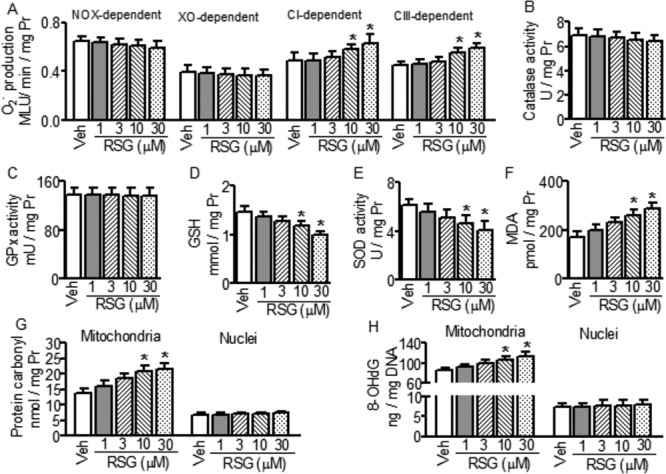
Effects of rosiglitazone (RSG) on O2− production, mitochondrial reactive oxygen species (ROS) elimination and oxidative damages to lipids, proteins and DNAs in mitochondria and nuclei. Data shown are mean ± SD, n = 5–7, *p < 0.05 versus vehicle (Veh). GSH: reduced glutathione; GPx: glutathione peroxidase; SOD: superoxide dismutase; MDA: malondialdehyde; 8-OHdG: 8-hydroxy-2-deoxyguanosine. Pr: protein.
We also assessed the acute effects of RSG on mitochondrial oxidative stress in vivo. At 1 mg/kg, intravenous injection of RSG showed no effect on the levels of mitochondrial MDA, protein carbonyl, and 8-OHdG. At 10 mg/kg, however, RSG increased the levels of these mitochondrial oxidative stress markers. Importantly, NAC 600 mg/kg prevented the above RSG-induced changes of mitochondrial oxidative stress markers in vivo (Figs. 6F–H).
FIG. 6.
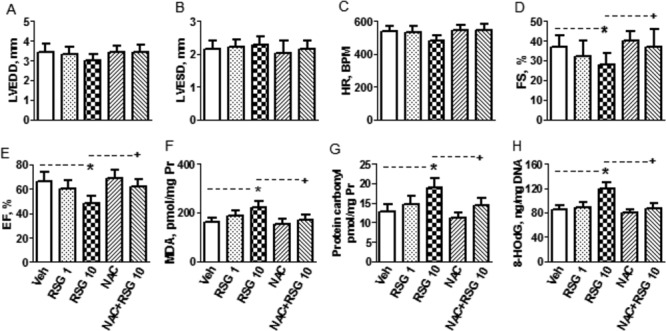
The in vivo effects of rosiglitazone (RSG) on cardiac function and oxidative stress and influence of N-acetyl-L-cysterine (NAC) on RSG-induced changes. From (A) to (E), echocardiographic parameters collected from mice 2 h after intravenous injection of vehicle (Veh), RSG 1 and 10 mg/kg, NAC 600 mg/kg, and NAC 600 mg/kg + RSG 10 mg/kg via the tail vein. LVEDD: left ventricular end-diastolic dimension; LVESD: left ventricular end-systolic dimension; HR: heart rate; FS: fractional shortening; EF: ejection fraction. From (F) to (H), stable markers for oxidative stress (oxidative damages to lipids, proteins and DNAs in mitochondria isolated from hearts subjected to the above in vivo treatments). MDA: malondialdehyde; 8-OHdG: 8-hydroxy-2-deoxyguanosine. Data shown are mean ± SD, n = 5. *p < 0.05 versus Veh, +p < 0.05 versus RSG 10 mg/kg. Pr: protein.
RSG Caused Cardiac Contractile Dysfunction Independently of PPARγ
Energy homeostasis is required for normal cardiac contractile function. As we found that RSG caused energy deficiency, we therefore further tested whether RSG could change cardiac contractile function. In in vitro Langendorff-perfused hearts, cardiac systolic function was assessed by measuring LVSP and +dP/dt. At 1 and 3μM, RSG showed no obvious effects on LVSP and +dP/dt. At 10 and 30μM, however, it decreased LVSP and +dP/dt in hearts from C57BL/6, PPARγ−, and PPARγ+ mice (Figs. 5A and B), indicating acute treatment with RSG at the supratherapeutic concentrations causes cardiac systolic dysfunction. Cardiac diastolic function was assessed by changes in LV EDP and –dP/dt. Similarly, at 1 and 3μM, RSG showed no effect on EDP and –dP/dt. At 10 and 30μM, however, it increased EDP and decreased –dP/dt in all hearts from the above three genotypes (Figs. 5C and D), indicating acute treatment with RSG at the supratherapeutic concentrations also causes cardiac diastolic dysfunction. Interestingly, RSG-induced cardiac dysfunction was not distinguishable among C57BL/6, PPARγ−, and PPARγ+ mice, indicating acute RSG treatment caused cardiac contractile dysfunction independently of CM PPARγ. Additionally, treatment of hearts with 10μM GW9662 for 60 min affected neither cardiac function nor RSG-induced cardiac dysfunction. In contrast, treatment of hearts with 20mM NAC for 60 min did not affect baseline cardiac function, but did prevent cardiac dysfunction caused by RSG at the supratherapeutic concentration of 10μM (Figs. 5E–H).
FIG. 5.
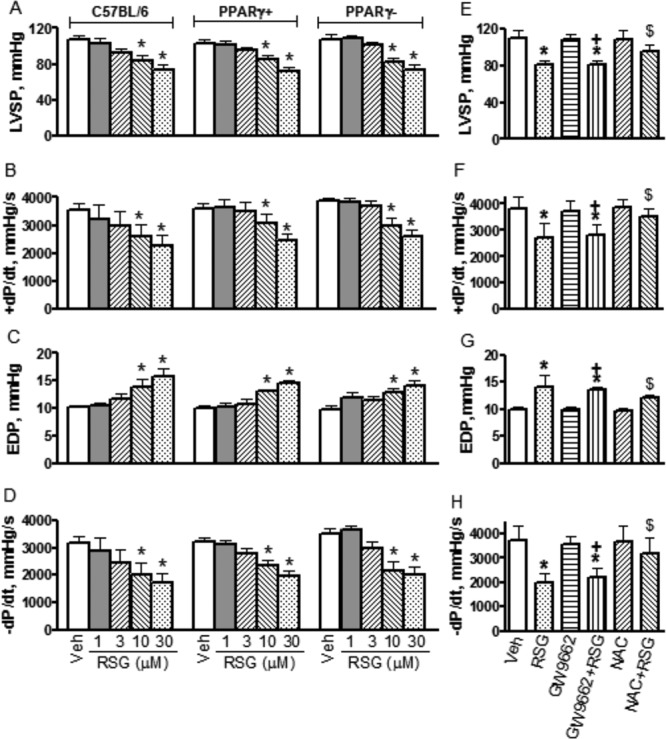
Effects of rosiglitazone (RSG) on cardiac contractile function and influences of GW9622, N-acetyl-L- cysteine (NAC) on RSG-induced changes. From (A) to (D), hearts were isolated from C57BL/6, PPARγ+, and PPARγ− mice and perfused with regular Krebs-Henseleit buffer containing 10mM glucose and 0.5mM pyruvate. From (E) to (H), hearts were isolated from C57BL/6 and perfused with modified Krebs-Hanseleit buffer containing 5.5mM glucose and 0.4mM mixed free fatty acid. Data shown are Mean ± SD, n = 5–8. *p < 0.05 versus vehicle (Veh), +p < 0.05 versus GW9662, $p < 0.05 versus RSG. LVSP: left ventricular systolic pressure; EDP: end diastolic pressure; +dP/dt: rate of tension development; −dP/dt: rate of relaxation.
In vivo study, heart function was assessed by echocardiographic parameters LVEDD, LVESD, FS, and EF. As shown in Figures 6A–6E, at 1 mg/kg, intravenous injection of RSG showed no effect on these parameters. At 10 mg/kg, however, RSG decreased FS and EF, indicating RSG caused cardiac contractile dysfunction at a higher dose. NAC at 600 mg/kg alone showed no effect on cardiac function. In combination with 10 mg/kg RSG, however, it prevented RSG-induced cardiac contractile dysfunction.
DISCUSSION
This study provides evidence that acute treatment with RSG at the supratherapeutic concentrations causes myocardial energy deficiency and mitochondrial dysfunction with concomitant cardiac contractile dysfunction via a PPARγ-independent, mitochondrial oxidative stress related mechanism.
The effects of TZDs on adipose tissue and skeletal muscles are well studied. Their effects on myocardial metabolism and cardiac function, however, are less clear. Only a few studies have systematically assessed the effects of TZDs on cardiac structure and contractile function, but these suffer from important limitations of study design, assessment methods, and execution. In the context of these limited available data, no study has demonstrated pernicious effects of TZDs on myocardial energy metabolism and cardiac function. Only a few randomized clinical trials (Dargie et al., 2007; Ghazzi et al., 1997; St John Sutton et al., 2002) have evaluated the cardiac effects of TZDs, all using echocardiographic parameters in patients with and without heart failure. In these studies, no adverse effects on cardiac structure or function have been observed. The validity of the observations from these studies is notably uncertain due to open-label study design, relatively small sample sizes, and high attrition rates. Because very large numbers of patients with diabetes are being treated with PPARγ agonists TZDs worldwide for long periods of time, uncovering the action of TZDs on myocardial energy metabolism and contractile function has major implications.
In the present study, acute treatment of perfused hearts with RSG at supratherapeutic concentrations caused myocardial energy deficiency as evidenced by the deceases in [PCr], [ATP], total ATP to ADP ratio, and energy charge. To maintain energy homeostasis, the capacities of ATP synthesis by mitochondrial oxidative phosphorylation, glycolysis, and phosphotransferase (i.e., CK) reactions must match the demand for ATP utilization by the sarcomere, ion pumps, etc. (Ingwall, 2009). Therefore, increased ATP utilization and decreased ATP synthesis, singly or in combination, can cause energy deficiency. The free energy of ATP hydrolysis |ΔGATP| decreased following RSG treatment (Fig. 1D and Supplementary fig. 4A). Furthermore, heart mechanical work (assessed by rate pressure product, an indirect index of calcium cycling, metabolic demand, and ATP utilization) also decreased following acute treatment with RSG at the supratherapeutic concentrations (Supplementary fig. 4B). These results suggest that decreased ATP synthesis may be responsible for myocardial energy deficiency caused by RSG. The main pathways for ATP synthesis in hearts are glycolysis, phosphoryltransfer reactions, and substrate oxidative phosphorylation (Ingwall, 2009). RSG even at the supratherapeutic concentrations showed no effect on glycolytic rate-limiting enzymes and the product of CK activity and total creatine content (Supplementary table 1 and Supplementary fig. 1), indicating that neither glycolysis nor phosphoryltransfer reaction is likely to be involved in RSG-induced myocardial energy deficiency. RSG at the supratherapeutic concentrations, however, decreased the oxidation rates of glucose and palmitate (Fig. 1H and Figs. 2F and G).
To explore its underlying mechanism, we tested effects of RSG on major mitochondrial enzymes controlling metabolic flux and ATP production: CS and the electron transport complexes. Acute treatment of isolated mitochondria with RSG showed no effects on CS, complexes II, III and V even at highest concentration tested (Supplementary figs. 1A, C, D, and E). Consistent with the previous report that TZDs inhibited complex I activity in tissue homogenate of rat skeletal muscle and liver, RSG at supratherapeutic concentrations inhibited activities of complexes I and IV in isolated cardiac mitochondria in the present study. The inhibition of complex I can cause impaired oxidation of NADH and in turn decreased NAD content. As a result, NADH/NAD ratio increased due to increased NADH or decreased NAD content following RSG treatment (Supplementary table 1). The impaired oxidation of NADH leads to decreased substrate oxidation and in turn decreased ATP synthesis. Complex IV acts as the terminus of mitochondrial electron transport by accepting four electrons to reduce a single O2 molecule. The reaction is coupled with the transfer of four protons across the mitochondrial membrane, driving ATP synthesis. Thus, the inhibition of both complexes I and IV by RSG reduces ATP synthesis, which manifests as the myocardial energy deficiency induced by RSG in the present study.
Many PPARγ-dependent effects are based upon altered transcription of genes involved in energy metabolism and usually require hours to days to take manifest. The myocardial energy deficiency and cardiac dysfunction were observed after short time incubation with RSG in the present study. Therefore, these adverse effects of RSG at the supratherapeutic concentrations on the heart are unlikely the results of PPARγ-mediated alterations in gene transcription. The PPARγ-independent nature of these adverse effects is also indirectly supported by the ability of RSG to inhibit the activities of complexes I and IV in isolated mitochondria (that presumably lack PPARγ activity). That the adverse effects of RSG are not accounted for the activation of PPARγ in other non-CM cardiac cell types is also supported by the ability of RSG to decrease the intracellular ATP content in cultured CMs and the inability of PPARγ antagonist GW9662 to prevent the decrease in intracellular ATP content in cultured CFs. The inability of CM specific-PPARγ deletion or GW9662 to prevent myocardial high energy deficiency, cardiac mitochondrial, and contractile dysfunction provides the most convincing evidence for PPARγ-independent mechanism responsible for the cardiotoxicity following RSG treatment in the present study. Together with other reports that RSG induced cardiac hypertrophy in CM PPARγ−deficient mice (Duan et al., 2005) and that cardiac hypertrophy associated with the treatment of TZDs was not due to activation of cardiac PPARγ (Sena et al., 2007), our findings indicate that the cardiotoxicity following RSG treatment is not mediated by the activation of PPARγ. These data also suggest that PPARγ remains an appropriate therapeutic target to develop the novel drugs as long as off-target effects are absent.
In this study, we found that RSG at supratherapeutic concentrations caused mitochondrial dysfunction as evidenced by the decreases in ATP level, mitochondrial respiration rate, and oxidation rates of glucose and palmitate. RSG also increased ROS production by mitochondrial electron transport chain. These results can be ascribed to the inhibition of complexes I and IV by RSG as the blockage of electron transport chain increases the ROS generation, but increased ROS may also be the reason that RSG inhibited the activities of complexes I and IV through oxidative modification. On the other hand, RSG decreased SOD activity and free GSH level in our study, these results support that decreased mitochondrial ROS-scavenging capacity may be the important mechanism that RSG caused the oxidative stress. RSG has been reported to bind numerous proteins involved in scavenging of ROS (Hoffmann et al., 2012). Nonspecific binding at supratherapeutic concentrations is the possible mechanism by which RSG decreased free GSH content and SOD activity. RSG-induced oxidative stress is further supported by the fact that it increased the levels of mitochondrial MDA, protein carbonyl, and 8-OHdG in our in vitro and in vivo settings at a supratherapeutic concentration or dose. RSG, however, did not alter the levels of nuclear protein carbonyl and 8-OHdG, indicating that RSG at the concentration tested caused no oxidative damages to nuclear proteins and DNAs, which may be explained by far distance to the major site of ROS generation (electron transport chain) and relatively more potent capacity of nuclear DNA repair compared with mitochondrial DNA repair. Because seven subunits of complex I and three subunits of complex IV are encoded by mitochondrial DNAs, therefore, complexes I and IV are more susceptible to oxidative modification compared with other mitochondrial enzymes encoded mainly by nuclear DNAs.
In contrast to our findings, some studies have reported that RSG alleviates the oxidative stress (Ceolotto et al., 2007; Hwang et al., 2007; Kavak et al., 2008; Manning et al., 2008; von Bibra et al., 2008), and recent studies reported that acute intravenous administration of RSG did not alter the basic cardiac electrophysiology and hemodynamics parameters both in normal hearts and in hearts subjected to ischemia-reperfusion injury, and that the treatment of rat isolated mitochondria with RSG did not alter cardiac mitochondrial ROS level and membrane potential (Palee et al., 2013; Palee et al., 2011), indicating RSG caused no adverse effects on cardiac contractile function and mitochondrial function. Other studies, however, supported our finding that RSG causes oxidative stress. One reported that in vivo treatment with RSG impaired endothelial function in part through enhanced oxidative stress in mice at high cardiovascular risk (Saraogi et al., 2011). The other reported that in vivo treatment with RSG reduced SOD activity and GSH level, and increased MDA content in cardiac tissues in normal rats (Saraogi et al., 2011). The above discrepancy could be attributable to the differences in dosage or concentrations (therapeutic vs. supratherapeutic) and treatment period (acute vs. chronic) of RSG, experimental setting (in vivo vs. in vitro), animal species (pig/rabbit/rat vs. mouse), animal model (diabetes with/without ischemia-reperfusion vs. nondiabetes), tissue fraction (isolated mitochondria vs. tissue homogenate containing nuclei/PPARγ), parameters to assess mitochondrial function (mitochondrial membrane potential and mitochondrial swelling vs. ATP level, mitochondrial respiration rate, substrate oxidation rates, and enzyme activities), and ROS assay system (isolated mitochondria in the absence of substrate vs. tissue homogenate in the presence of specific substrate). For example, interspecies difference in response to RSG in the heart is supported by a previous study which demonstrated RSG preserved myocardial function, enhanced substrate utilization, and limited the activation of inflammatory signaling pathways in rat hearts subjected to ischemia-reperfusion injury, but this agent had no significant effect on myocardial contractile function, substrate uptake, or expression of proinflammatory cytokines in normal nondiabetic pig hearts subjected to ischemia-reperfusion injury (Xu et al., 2005).
The dose selection rationale for this study was aimed to achieve doses ranging from 1–10 folds over the predicted human efficacious concentration. Clinically, administration of RSG at 8 mg/day results in plasma levels of the drug in the range of 1–3μM. The concentrations of 1, 3, 10, and 30μM used in our in vitro study and dosages of 1 and 10 mg/kg in the in vivo study allowed us to determine the dose response of RSG's actions and to compare our data with others. Consistent with a previous report showing that RSG inhibited complex I activity in tissue homogenate of rat skeletal muscle and liver at 10–100μM (Brunmair et al., 2004), RSG decreased substrate oxidation and mitochondrial respiration, inhibited activities of complexes I and IV, resulting in myocardial energy deficiency and cardiac contractile dysfunction at the supratherapeutic concentrations of 10 and 30μM in this study. Consistent with our study, in vivo administration of RSG at supratherapeutic doses (5–80 mg/kg) has been reported to cause cardiac dysfunction in healthy Sprague Dawley rats (Blasi et al., 2009) and oxidative stress in healthy Wistar rats (Saraogi et al., 2011). On the other hand, recent studies reported that acute intravenous injection of RSG at the therapeutic dose of 1 mg/kg caused no cardiac dysfunction or mitochondrial oxidative stress but facilitated the occurrence of ventricular fibrillation in ischemia-reperfusion hearts of rats or swines (Palee et al., 2013; Palee et al., 2011). In contrast, chronic administration of RSG at the therapeutic concentration or dose has been reported to decrease mitochondrial respiration in human skeletal muscles (Rabol et al., 2010), suppressed activity of mitochondrial complex I, decreased mitochondrial glutathione content, and increased oxidative stress in livers form ob/ob mice (Garcia-Ruiz et al., 2007). The reasons for the discrepancy in the adverse effects at therapeutic level of RSG between our study and the work of others are multiple, including but not limited to the differences in animal species, pathological status, source of the tissues, experiment settings, durations of RSG in system, etc.
There is no evidence that this compound might be concentrated over time in the cell or that the effect is in some other way “cumulative”. However, some patients may benefit from increasing the dose due to the tolerance during a long period of therapeutic time, and diabetic patients with renal dysfunction may have a tendency to accumulate this drug. Therefore, our results from higher concentrations in an acute model may mimic the effects of lower exposure over a longer period of time.
Because beneficial effects of RSG observed in clinical settings are mediated by PPARγ activation following chronic treatment, and PPARγ-dependent actions on the transcription of genes involved in energy metabolism usually take several hours to days (12–24 h) to take place, it is not a surprise that acute in vitro treatment with RSG at the therapeutic concentrations of 1 and 3μM or acute in vivo treatment with RSG at a clinically relevant dose of 1 mg/kg showed no beneficial effects on hearts. The possible reason for RSG caused the cardiotoxicity at supratherapeutic concentrations or dose but not at therapeutic concentrations or dose in our study is that RSG induces mitochondrial oxidative stress due to its nonspecific binding to SOD and GSH at higher concentrations. This is also supported by the ability of NAC to prevent RSG-induced cardiac dysfunction in both in vitro and in vivo settings.
As type 2 diabetes is commonly associated with impaired cardiac function, reduced cardiac reserve, and congestive heart failure in the elderly or in later stages (Barrett-Connor et al., 2004; Kaseta et al., 1999). To avoid the interactive action of diabetic state on the cardiac effects of RSG, we examined the acute effects of RSG on myocardial metabolism and cardiac function in nondiabetic mice with no risk factors for heart failure in this study. On the other hand, as we aimed at the involvement of PPARγ in cardiotoxicity induced by RSG, and other TZDs have some additional PPARα agonist activity (Orasanu et al., 2008) or lower potency to activate PPARγ than RSG (Bishop-Bailey, 2000), we therefore did not comparatively study their actions on myocardial energy metabolism and cardiac function in this study.
Beyond the antidiabetic activity, the activation of PPARγ by TZDs has modulatory effects on growth factor release, cytokine production, cell proliferation and migration, extracellular matrix remodeling, control on cell cycle progression and differentiation (Giannini et al., 2004). RSG has been reported to exert potent anti-inflammatory (Mohanty et al., 2004; Shah et al., 2005) and anticancer effects (Freudlsperger et al., 2006; Han and Roman, 2006; He et al., 2008; Nunez et al., 2006). Therefore, our findings in a nondiabetic model may also have influence upon development of PPARγ as a therapeutic target to develop novel drugs for the treatment of chronic inflammation and cancer.
The precise molecular mechanism for the PPARγ-independent actions of RSG is open to speculation. Two hypotheses are presented. The first is that the initial action of RSG is an inhibition of complexes I and IV, blockage of the electron transport chain at these sites may increase mitochondrial ROS production (especially at complex I), which in addition to direct oxidative mitochondrial damage may cause oxidative modification of SOD and oxidize GSH, thus, leading to additional ROS damage (ROS-induced ROS production), and further loss in the activities of complexes I and IV. The end result is a decrease in oxidative ATP synthesis. The second hypothesis is that the initial RSG action is binding to SOD and GSH, leading to decreases in mitochondrial SOD activity and free GSH level, which in turn increases mitochondrial oxidative stress, leading to oxidative modifications of mitochondrial lipids and proteins that result in the decreases in the activities of complexes I and IV. The end result is of course the same, namely a decrease in oxidative ATP synthesis. Either mechanism manifests similarly on the whole organ level as a decrease in myocardial high-energy phosphates with a concomitant decrease in contractile function.
In conclusion, this study demonstrates that a more selective PPARγ agonist RSG at the supratherapeutic concentrations caused myocardial energy deficiency with concomitant cardiac dysfunction at the whole heart level. These adverse effects are related to oxidative stress-induced mitochondrial dysfunction and independent of cardiac PPARγ activation.
SUPPLEMENTARY DATA
Supplementary data are available online at http://toxsci.oxfordjournals.org/.
FUNDING
National Institutes of Health (R01 HL46033 and HL78634 to J.A.B., P50 HL074734 to F.X.M., and R01 HL083201 to R.M.M.).
Supplementary Material
Acknowledgments
The authors would like to acknowledge Dr Joanne S Ingwall for her helpful comments.
Footnotes
Present Address: Lacrimal Apparatus Center, Department of Ophthalmology, Armed Police General Hospital, 69 Yongding Road, Beijing 100039, China.
REFERENCES
- Albers D. S., Beal M. F. Mitochondrial dysfunction and oxidative stress in aging and neurodegenerative disease. J. Neural. Transm. Suppl. 2000;59:133–154. doi: 10.1007/978-3-7091-6781-6_16. [DOI] [PubMed] [Google Scholar]
- Anderson E. J., Katunga L. A., Willis M. S. Mitochondria as a source and target of lipid peroxidation products in healthy and diseased heart. Clin. Exp. Pharmacol. Physiol. 2012;39:179–193. doi: 10.1111/j.1440-1681.2011.05641.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barger P. M., Kelly D. P. PPAR signaling in the control of cardiac energy metabolism. Trends Cardiovasc. Med. 2000;10:238–245. doi: 10.1016/s1050-1738(00)00077-3. [DOI] [PubMed] [Google Scholar]
- Barrett-Connor E., Giardina E. G., Gitt A. K., Gudat U., Steinberg H. O., Tschoepe D. Women and heart disease: The role of diabetes and hyperglycemia. Arch. Intern. Med. 2004;164:934–942. doi: 10.1001/archinte.164.9.934. [DOI] [PubMed] [Google Scholar]
- Benton C. R., Holloway G. P., Campbell S. E., Yoshida Y., Tandon N. N., Glatz J. F., Luiken J. J., Spriet L. L., Bonen A. Rosiglitazone increases fatty acid oxidation and fatty acid translocase (FAT/CD36) but not carnitine palmitoyltransferase I in rat muscle mitochondria. J. Physiol. 2008;586:1755–1766. doi: 10.1113/jphysiol.2007.146563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop-Bailey D. Peroxisome proliferator-activated receptors in the cardiovascular system. Br. J. Pharmacol. 2000;129:823–834. doi: 10.1038/sj.bjp.0703149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasi E. R., Heyen J., Hemkens M., McHarg A., Ecelbarger C. M., Tiwari S. Effects of chronic PPAR-agonist treatment on cardiac structure and function, blood pressure, and kidney in healthy sprague-dawley rats. PPAR Res. 2009;2009:237865. doi: 10.1155/2009/237865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle J. G., Logan P. J., Ewart M. A., Reihill J. A., Ritchie S. A., Connell J. M., Cleland S. J., Salt I. P. Rosiglitazone stimulates nitric oxide synthesis in human aortic endothelial cells via AMP-activated protein kinase. J. Biol. Chem. 2008;283:11210–11217. doi: 10.1074/jbc.M710048200. [DOI] [PubMed] [Google Scholar]
- Brunmair B., Staniek K., Gras F., Scharf N., Althaym A., Clara R., Roden M., Gnaiger E., Nohl H., Waldhausl W. Thiazolidinediones, like metformin, inhibit respiratory complex I: A common mechanism contributing to their antidiabetic actions? Diabetes. 2004;53:1052–1059. doi: 10.2337/diabetes.53.4.1052. [DOI] [PubMed] [Google Scholar]
- Ceolotto G., Gallo A., Papparella I., Franco L., Murphy E., Iori E., Pagnin E., Fadini G. P., Albiero M., Semplicini A. Rosiglitazone reduces glucose-induced oxidative stress mediated by NAD(P)H oxidase via AMPK-dependent mechanism. Arterioscler. Thromb. Vasc. Biol. 2007;27:2627–2633. doi: 10.1161/ATVBAHA.107.155762. [DOI] [PubMed] [Google Scholar]
- Dargie H. J., Hildebrandt P. R., Riegger G. A., McMurray J. J., McMorn S. O., Roberts J. N., Zambanini A., Wilding J. P. A randomized, placebo-controlled trial assessing the effects of rosiglitazone on echocardiographic function and cardiac status in type 2 diabetic patients with New York Heart Association Functional Class I or II Heart Failure. J. Am. Coll. Cardiol. 2007;49:1696–1704. doi: 10.1016/j.jacc.2006.10.077. [DOI] [PubMed] [Google Scholar]
- Day C. Thiazolidinediones: A new class of antidiabetic drugs. Diabet. Med. 1999;16:179–192. doi: 10.1046/j.1464-5491.1999.00023.x. [DOI] [PubMed] [Google Scholar]
- Duan S. Z., Ivashchenko C. Y., Russell M. W., Milstone D. S., Mortensen R. M. Cardiomyocyte-specific knockout and agonist of peroxisome proliferator-activated receptor-gamma both induce cardiac hypertrophy in mice. Circ. Res. 2005;97:372–379. doi: 10.1161/01.RES.0000179226.34112.6d. [DOI] [PubMed] [Google Scholar]
- Dumasia R., Eagle K. A., Kline-Rogers E., May N., Cho L., Mukherjee D. Role of PPAR- gamma agonist thiazolidinediones in treatment of pre-diabetic and diabetic individuals: A cardiovascular perspective. Curr. Drug Targets Cardiovasc. Haematol. Disord. 2005;5:377–386. doi: 10.2174/156800605774370362. [DOI] [PubMed] [Google Scholar]
- Figueira T. R., Barros M. H., Camargo A. A., Castilho R. F., Ferreira J. C., Kowaltowski A. J., Sluse F. E., Souza-Pinto N. C., Vercesi A. E. Mitochondria as a source of reactive oxygen and nitrogen species: From molecular mechanisms to human health. Antioxid. Redox Signal. 2013;18:2029–2074. doi: 10.1089/ars.2012.4729. [DOI] [PubMed] [Google Scholar]
- Frangogiannis N. G., Dewald O., Xia Y., Ren G., Haudek S., Leucker T., Kraemer D., Taffet G., Rollins B. J., Entman M. L. Critical role of monocyte chemoattractant protein-1/CC chemokine ligand 2 in the pathogenesis of ischemic cardiomyopathy. Circulation. 2007;115:584–592. doi: 10.1161/CIRCULATIONAHA.106.646091. [DOI] [PubMed] [Google Scholar]
- Freudlsperger C., Moll I., Schumacher U., Thies A. Anti-proliferative effect of peroxisome proliferator-activated receptor gamma agonists on human malignant melanoma cells in vitro. Anticancer Drugs. 2006;17:325–332. doi: 10.1097/00001813-200603000-00011. [DOI] [PubMed] [Google Scholar]
- Garcia-Ruiz I., Rodriguez-Juan C., Diaz-Sanjuan T., Martinez M. A., Munoz-Yague T., Solis-Herruzo J. A. Effects of rosiglitazone on the liver histology and mitochondrial function in ob/ob mice. Hepatology. 2007;46:414–423. doi: 10.1002/hep.21687. [DOI] [PubMed] [Google Scholar]
- Gardner O. S., Shiau C. W., Chen C. S., Graves L. M. Peroxisome proliferator-activated receptor gamma-independent activation of p38 MAPK by thiazolidinediones involves calcium/calmodulin-dependent protein kinase II and protein kinase R: Correlation with endoplasmic reticulum stress. J. Biol. Chem. 2005;280:10109–10118. doi: 10.1074/jbc.M410445200. [DOI] [PubMed] [Google Scholar]
- Ghazzi M. N., Perez J. E., Antonucci T. K., Driscoll J. H., Huang S. M., Faja B. W., Whitcomb R. W. Cardiac and glycemic benefits of troglitazone treatment in NIDDM. The Troglitazone Study Group. Diabetes. 1997;46:433–439. doi: 10.2337/diab.46.3.433. [DOI] [PubMed] [Google Scholar]
- Giannini S., Serio M., Galli A. Pleiotropic effects of thiazolidinediones: Taking a look beyond antidiabetic activity. J. Endocrinol. Invest. 2004;27:982–991. doi: 10.1007/BF03347546. [DOI] [PubMed] [Google Scholar]
- Gnaiger E., Mendez G., Hand S. C. High phosphorylation efficiency and depression of uncoupled respiration in mitochondria under hypoxia. Proc. Natl. Acad. Sci. U.S.A. 2000;97:11080–11085. doi: 10.1073/pnas.97.20.11080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S., Roman J. Rosiglitazone suppresses human lung carcinoma cell growth through PPARgamma-dependent and PPARgamma-independent signal pathways. Mol. Cancer. Ther. 2006;5:430–437. doi: 10.1158/1535-7163.MCT-05-0347. [DOI] [PubMed] [Google Scholar]
- He H., Javadpour M. M., Latif F., Tardiff J. C., Ingwall J. S. R-92L and R-92W mutations in cardiac troponin T lead to distinct energetic phenotypes in intact mouse hearts. Biophys. J. 2007;93:1834–1844. doi: 10.1529/biophysj.107.107557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Q., Pang R., Song X., Chen J., Chen H., Chen B., Hu P., Chen M. Rosiglitazone suppresses the growth and invasiveness of SGC-7901 gastric cancer cells and angiogenesis in vitro via PPARgamma dependent and independent mechanisms. PPAR Res. 2008;2008:649808. doi: 10.1155/2008/649808. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Hoffmann B. R., El-Mansy M. F., Sem D. S., Greene A. S. Chemical proteomics-based analysis of off-target binding profiles for rosiglitazone and pioglitazone: Clues for assessing potential for cardiotoxicity. J. Med. Chem. 2012;55:8260–8271. doi: 10.1021/jm301204r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Home P. D., Pocock S. J., Beck-Nielsen H., Gomis R., Hanefeld M., Jones N. P., Komajda M., McMurray J. J. Rosiglitazone evaluated for cardiovascular outcomes—an interim analysis. N. Engl. J. Med. 2007;357:28–38. doi: 10.1056/NEJMoa073394. [DOI] [PubMed] [Google Scholar]
- Hwang J., Kleinhenz D. J., Rupnow H. L., Campbell A. G., Thule P. M., Sutliff R. L., Hart C. M. The PPARgamma ligand, rosiglitazone, reduces vascular oxidative stress and NADPH oxidase expression in diabetic mice. Vascul. Pharmacol. 2007;46:456–462. doi: 10.1016/j.vph.2007.01.007. [DOI] [PubMed] [Google Scholar]
- Ingwall J. S. Energy metabolism in heart failure and remodelling. Cardiovasc. Res. 2009;81:412–419. doi: 10.1093/cvr/cvn301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kammermeier H. Microassay of free and total creatine from tissue extracts by combination of chromatographic and fluorometric methods. Anal. biochem. 1973;56:341–345. doi: 10.1016/0003-2697(73)90199-1. [DOI] [PubMed] [Google Scholar]
- Kaseta J. R., Skafar D. F., Ram J. L., Jacober S. J., Sowers J. R. Cardiovascular disease in the diabetic woman. J. Clin. Endocrinol. Metab. 1999;84:1835–1838. doi: 10.1210/jcem.84.6.5735. [DOI] [PubMed] [Google Scholar]
- Kavak S., Ayaz L., Emre M., Inal T., Tamer L., Gunay I. The effects of rosiglitazone on oxidative stress and lipid profile in left ventricular muscles of diabetic rats. Cell Biochem. Funct. 2008;26:478–485. doi: 10.1002/cbf.1469. [DOI] [PubMed] [Google Scholar]
- Lago R. M., Singh P. P., Nesto R. W. Congestive heart failure and cardiovascular death in patients with prediabetes and type 2 diabetes given thiazolidinediones: A meta-analysis of randomised clinical trials. Lancet. 2007;370:1129–1136. doi: 10.1016/S0140-6736(07)61514-1. [DOI] [PubMed] [Google Scholar]
- Lazzarino G., Amorini A. M., Fazzina G., Vagnozzi R., Signoretti S., Donzelli S., Di Stasio E., Giardina B., Tavazzi B. Single-sample preparation for simultaneous cellular redox and energy state determination. Anal. Biochem. 2003;322:51–59. doi: 10.1016/j.ab.2003.07.013. [DOI] [PubMed] [Google Scholar]
- Li J. M., Mullen A. M., Yun S., Wientjes F., Brouns G. Y., Thrasher A. J., Shah A. M. Essential role of the NADPH oxidase subunit p47(phox) in endothelial cell superoxide production in response to phorbol ester and tumor necrosis factor-alpha. Circ. Res. 2002;90:143–150. doi: 10.1161/hh0202.103615. [DOI] [PubMed] [Google Scholar]
- Manning P. J., Sutherland W. H., Walker R. J., Williams S. M., de Jong S. A., Berry E. A. The effect of rosiglitazone on oxidative stress and insulin resistance in overweight individuals. Diabetes Res. Clin. Pract. 2008;81:209–215. doi: 10.1016/j.diabres.2008.04.015. [DOI] [PubMed] [Google Scholar]
- Matamoros M. A., Fernandez-Garcia N., Wienkoop S., Loscos J., Saiz A., Becana M. Mitochondria are an early target of oxidative modifications in senescing legume nodules. New Phytol. 2013;197:873–885. doi: 10.1111/nph.12049. [DOI] [PubMed] [Google Scholar]
- McMorran M., Vu D. Rosiglitazone (Avandia): Hepatic, cardiac and hematological reactions. CMAJ. 2001;165:82–83. 86–87. [PubMed] [Google Scholar]
- Mohanty P., Aljada A., Ghanim H., Hofmeyer D., Tripathy D., Syed T., Al-Haddad W., Dhindsa S., Dandona P. Evidence for a potent antiinflammatory effect of rosiglitazone. J. Clin. Endocrinol. Metab. 2004;89:2728–2735. doi: 10.1210/jc.2003-032103. [DOI] [PubMed] [Google Scholar]
- Mughal R. S., Warburton P., O’Regan D. J., Ball S. G., Turner N. A., Porter K. E. Peroxisome proliferator-activated receptor gamma-independent effects of thiazolidinediones on human cardiac myofibroblast function. Clin. Exp. Pharmacol. Physiol. 2009;36:478–486. doi: 10.1111/j.1440-1681.2008.05088.x. [DOI] [PubMed] [Google Scholar]
- Nesto R. W., Bell D., Bonow R. O., Fonseca V., Grundy S. M., Horton E. S., Le Winter M., Porte D., Semenkovich C. F., Smith S. Thiazolidinedione use, fluid retention, and congestive heart failure: A consensus statement from the American Heart Association and American Diabetes Association. October 7, 2003. Circulation. 2003;108:2941–2948. doi: 10.1161/01.CIR.0000103683.99399.7E. [DOI] [PubMed] [Google Scholar]
- Nissen S. E., Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N. Engl. J. Med. 2007;356:2457–2471. doi: 10.1056/NEJMoa072761. [DOI] [PubMed] [Google Scholar]
- Nunez M., Martin G., Cocca C., Mohamad N., Gutierrez A., Cricco G., Medina V., Rivera E., Croci M., Crescenti E. Effect of rosiglitazone on N-nitroso-N-methylurea-induced mammary tumors in rat. Anticancer Res. 2006;26:2113–2122. [PubMed] [Google Scholar]
- Orasanu G., Ziouzenkova O., Devchand P. R., Nehra V., Hamdy O., Horton E. S., Plutzky J. The peroxisome proliferator-activated receptor-gamma agonist pioglitazone represses inflammation in a peroxisome proliferator-activated receptor-alpha-dependent manner in vitro and in vivo in mice. J. Am. Coll. Cardiol. 2008;52:869–881. doi: 10.1016/j.jacc.2008.04.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palee S., Weerateerangkul P., Chinda K., Chattipakorn S. C., Chattipakorn N. Mechanisms responsible for beneficial and adverse effects of rosiglitazone in a rat model of acute cardiac ischaemia-reperfusion. Exp. Physiol. 2013;98:1028–1037. doi: 10.1113/expphysiol.2012.070433. [DOI] [PubMed] [Google Scholar]
- Palee S., Weerateerangkul P., Surinkeaw S., Chattipakorn S., Chattipakorn N. Effect of rosiglitazone on cardiac electrophysiology, infarct size and mitochondrial function in ischaemia and reperfusion of swine and rat heart. Exp. Physiol. 2011;96:778–789. doi: 10.1113/expphysiol.2011.057885. [DOI] [PubMed] [Google Scholar]
- Rabol R., Boushel R., Almdal T., Hansen C. N., Ploug T., Haugaard S. B., Prats C., Madsbad S., Dela F. Opposite effects of pioglitazone and rosiglitazone on mitochondrial respiration in skeletal muscle of patients with type 2 diabetes. Diabetes Obes. Metab. 2010;12:806–814. doi: 10.1111/j.1463-1326.2010.01237.x. [DOI] [PubMed] [Google Scholar]
- Rachek L. I., Yuzefovych L. V., Ledoux S. P., Julie N. L., Wilson G. L. Troglitazone, but not rosiglitazone, damages mitochondrial DNA and induces mitochondrial dysfunction and cell death in human hepatocytes. Toxicol. Appl. Pharmacol. 2009;240:348–354. doi: 10.1016/j.taap.2009.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryden L., Thrainsdottir I., Swedberg K. Adjudication of serious heart failure in patients from PROactive. Lancet. 2007;369:189–190. doi: 10.1016/S0140-6736(07)60106-8. [DOI] [PubMed] [Google Scholar]
- Saraogi P., Pillai K. K., Singh B. K., Dubey K. Rosiglitazone and pioglitazone aggravate doxorubicin-induced cardiomyopathy in Wistar rats. Biomed. Aging Pathol. 2011;1:65–71. doi: 10.1016/j.biopha.2010.12.012. [DOI] [PubMed] [Google Scholar]
- Savoia C., Ebrahimian T., Lemarie C. A., Paradis P., Iglarz M., Amiri F., Javeshgani D., Schiffrin E. L. Countervailing vascular effects of rosiglitazone in high cardiovascular risk mice: Role of oxidative stress and PRMT-1. Clin. Sci. (Lond) 2010;118:583–592. doi: 10.1042/CS20090289. [DOI] [PubMed] [Google Scholar]
- Scatena R., Bottoni P., Martorana G. E., Ferrari F., De Sole P., Rossi C., Giardina B. Mitochondrial respiratory chain dysfunction, a non-receptor-mediated effect of synthetic PPAR-ligands: Biochemical and pharmacological implications. Biochem. Biophys. Res. Commun. 2004;319:967–973. doi: 10.1016/j.bbrc.2004.05.072. [DOI] [PubMed] [Google Scholar]
- Sena S., Rasmussen I. R., Wende A. R., McQueen A. P., Theobald H. A., Wilde N., Pereira R. O., Litwin S. E., Berger J. P., Abel E. D. Cardiac hypertrophy caused by peroxisome proliferator-activated receptor-gamma agonist treatment occurs independently of changes in myocardial insulin signaling. Endocrinology. 2007;148:6047–6053. doi: 10.1210/en.2006-1559. [DOI] [PubMed] [Google Scholar]
- Shah R. D., Gonzales F., Golez E., Augustin D., Caudillo S., Abbott A., Morello J., McDonough P. M., Paolini P. J., Shubeita H. E. The antidiabetic agent rosiglitazone upregulates SERCA2 and enhances TNF-alpha- and LPS-induced NF-kappaB-dependent transcription and TNF-alpha-induced IL-6 secretion in ventricular myocytes. Cell. Physiol. Biochem. 2005;15:41–50. doi: 10.1159/000083637. [DOI] [PubMed] [Google Scholar]
- Singh S., Loke Y. K., Furberg C. D. Long-term risk of cardiovascular events with rosiglitazone: A meta-analysis. JAMA. 2007a;298:1189–1195. doi: 10.1001/jama.298.10.1189. [DOI] [PubMed] [Google Scholar]
- Singh S., Loke Y. K., Furberg C. D. Thiazolidinediones and heart failure: A teleo-analysis. Diabetes Care. 2007b;30:2148–2153. doi: 10.2337/dc07-0141. [DOI] [PubMed] [Google Scholar]
- Son N. H., Park T. S., Yamashita H., Yokoyama M., Huggins L. A., Okajima K., Homma S., Szabolcs M. J., Huang L. S., Goldberg I. J. Cardiomyocyte expression of PPARgamma leads to cardiac dysfunction in mice. J. Clin. Invest. 2007;117:2791–2801. doi: 10.1172/JCI30335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John Sutton M., St, Rendell M., Dandona P., Dole J. F., Murphy K., Patwardhan R., Patel J., Freed M. A comparison of the effects of rosiglitazone and glyburide on cardiovascular function and glycemic control in patients with type 2 diabetes. Diabetes Care. 2002;25:2058–2064. doi: 10.2337/diacare.25.11.2058. [DOI] [PubMed] [Google Scholar]
- Taylor C., Hobbs F. D. Type 2 diabetes, thiazolidinediones, and cardiovascular risk. Br. J. Gen. Pract. 2009;59:520–524. doi: 10.3399/bjgp09X453440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trachootham D., Lu W., Ogasawara M. A., Nilsa R. D., Huang P. Redox regulation of cell survival. Antioxid. Redox Signal. 2008;10:1343–1374. doi: 10.1089/ars.2007.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tritschler H. J., Packer L., Medori R. Oxidative stress and mitochondrial dysfunction in neurodegeneration. Biochem. Mol. Biol. Int. 1994;34:169–181. [PubMed] [Google Scholar]
- von Bibra H., Diamant M., Scheffer P. G., Siegmund T., Schumm-Draeger P. M. Rosiglitazone, but not glimepiride, improves myocardial diastolic function in association with reduction in oxidative stress in type 2 diabetic patients without overt heart disease. Diab. Vasc. Dis. Res. 2008;5:310–318. doi: 10.3132/dvdr.2008.045. [DOI] [PubMed] [Google Scholar]
- Wang P., Liu J., Li Y., Wu S., Luo J., Yang H., Subbiah R., Chatham J., Zhelyabovska O., Yang Q. Peroxisome proliferator-activated receptor {delta} is an essential transcriptional regulator for mitochondrial protection and biogenesis in adult heart. Circ. Res. 2010;106:911–919. doi: 10.1161/CIRCRESAHA.109.206185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkelmayer W. C., Setoguchi S., Levin R., Solomon D. H. Comparison of cardiovascular outcomes in elderly patients with diabetes who initiated rosiglitazone vs pioglitazone therapy. Arch. Intern. Med. 2008;168:2368–2375. doi: 10.1001/archinte.168.21.2368. [DOI] [PubMed] [Google Scholar]
- Xu Y., Gen M., Lu L., Fox J., Weiss S. O., Brown R. D., Perlov D., Ahmad H., Zhu P., Greyson C., et al. PPAR-gamma activation fails to provide myocardial protection in ischemia and reperfusion in pigs. Am. J. Physiol. Heart Circ. Physiol. 2005;288:H1314–H1323. doi: 10.1152/ajpheart.00618.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yki-Jarvinen H. Thiazolidinediones. N. Engl. J. Med. 2004;351:1106–1118. doi: 10.1056/NEJMra041001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.