

Abstract
Mismatch-repair (MMR) systems promote genomic stability by correction of DNA replication errors. Thus, MMR proteins—prokaryotic MutS and MutL homodimers or their MutSα and MutLα heterodimer homologs, plus accessory proteins—specifically couple mismatch recognition to nascent-DNA excision. In vivo excision-initiation signals—specific nicks in some prokaryotes, perhaps growing 3′ ends or Okazaki-fragment 5′ ends in eukaryotes—are efficiently mimicked in vitro by nicks or gaps in exogenous DNA substrates. In some models for recognition–excision coupling, MutSα bound to mismatches is induced by ATP hydrolysis, or simply by binding of ATP, to slide along DNA to excision-initiation sites, perhaps in association with MutLα and accessory proteins. In other models, MutSα·MutLα complexes remain fixed at mismatches and contact distant excision sites by DNA looping. To challenge the hypothesis that recognition complexes remain fixed, we placed biotin–streptavidin blockades between mismatches and pre-existing nicks. In human nuclear extracts, mismatch efficiently provoked the initiation of excision despite the intervening barriers, as predicted. However, excision progress and therefore mismatch correction were prevented.
Keywords: DNA looping, mismatch repair, MMR excision, MutSα sliding, streptavidin–biotin
Introduction
Evolutionarily conserved prokaryotic and eukaryotic mismatch-repair (MMR) systems promote genomic stability by correcting DNA replication errors, antagonizing homeologous recombination between diverged DNA sequences, and responding to a variety of DNA lesions (for reviews, see Buermeyer et al, 1999; Kolodner and Marsischky, 1999; Harfe and Jinks-Robertson, 2000; Bellacosa, 2001). The E. coli mismatch-correction pathway, fully reconstituted from purified proteins, provides a mechanistic paradigm (Lahue et al, 1989): MutS homodimers bind to DNA mismatches (Su and Modrich, 1986), and MutH proteins recognize hemi-methylated d(GATC) sequences (Au et al, 1992)—transitory consequences in vivo of a brief post-replication delay in GATC-adenine methylation of nascent DNA. The two recognition events thus provide the basis for an efficient, yet strand-specific, mismatch correction. Interaction of MutL homodimers with MutS-mismatch complexes activates DNA-strand nicking of nascent DNA at the unmethylated d(GATC) sites (Au et al, 1992; Hall and Matson, 1999). DNA helicase II (UvrD protein) loads at nicks if and only if MutS, a DNA mismatch, and MutL are present (Dao and Modrich, 1998); one or more steps in this chain of events require ATP hydrolysis. UvrD presumably separates strands to facilitate excision of the nicked strand toward and beyond the mismatch, by one of several 3′–5′ or 5′–3′ single-stranded-DNA-specific exonucleases, depending on the nick-mismatch orientation (Dao and Modrich, 1998). Gap filling by the replicative DNA polymerase III holoenzyme and DNA ligation follow.
In eukaryotes, mismatches are recognized by heterodimers with different specificities—typically base mismatches and one or two looped-out extra nucleotides by MSH2·MSH6 (MutSα) and a range of extrahelical loopouts by MSH2·MSH3 in most eukaryotes (for reviews, see Buermeyer et al, 1999; Kolodner and Marsischky, 1999; Harfe and Jinks-Robertson, 2000; Bellacosa, 2001), but a subset of base–base mismatches by an additional MSH2·MSH7 heterodimer in plants (Culligan and Hays, 2000; Wu et al, 2003). The high similarity between MutS and its eukaryotic homologs suggests conservation of the biochemistry of MMR initiation. In contrast, although the MutL-homolog heterodimer MutLα (MLH1·PMS2 (MLH1·PMS1 in yeast)) is required for mismatch correction, neither orthologs of UvrD or MutH, nor of any single-stranded-DNA-specific exonucleases, have been implicated in eukaryotic MMR (Buermeyer et al, 1999; Kolodner and Marsischky, 1999; Harfe and Jinks-Robertson, 2000; Bellacosa, 2001). This suggests less evolutionary conservation of post-mismatch-binding mechanisms. In particular, the absence of DNA-adenine methylation and, apparently, of eukaryotic MutH orthologs, indicate that different nascent-DNA features—perhaps leading-strand 3′ ends and lagging-strand 3′ or 5′ ends—are used to strand-specifically initiate excision towards the mismatch (Pavlov et al, 2003). In mammalian cell extracts, mismatches provoke initiation of excision at pre-existing nicks or gaps in exogenous DNA substrates, with high efficiency and specificity (Holmes et al, 1990; Iams et al, 2002).
Despite the apparent divergence of specific mechanisms of MMR excision, numerous partial-reaction studies with purified proteins suggest general conservation of initial steps involving MutS and MutL homologs. However, the mechanism by which mismatch recognition is coupled to site-specific initiation of excision remains controversial (Figure 1). In one class of coupling models (Figure 1 left), mismatch-bound MutS/MSH dimers are posited to bind ATP and then, perhaps when complexed to MutL/MLH dimers, move away along the DNA contour and search for excision-initiation signals—either by ATP-hydrolysis-dependent translocation (Allen et al, 1997) or by ATP-binding-dependent diffusional sliding, with hydrolysis occurring later (Gradia et al, 1997, 1999). Searching might be performed by more elaborate sliding complexes, to which other proteins have been recruited, such as proliferating cell nuclear antigen (PCNA) (Bowers et al, 2001). Alternatively, recognition complexes containing MutS-homolog proteins, MutL-homolog proteins, perhaps other proteins, and ATP are proposed to remain fixed at mismatches (Figure 1, right), where they use DNA looping to search through space for excision-initiation signals (Junop et al, 2001; Schofield et al, 2001).
Figure 1.
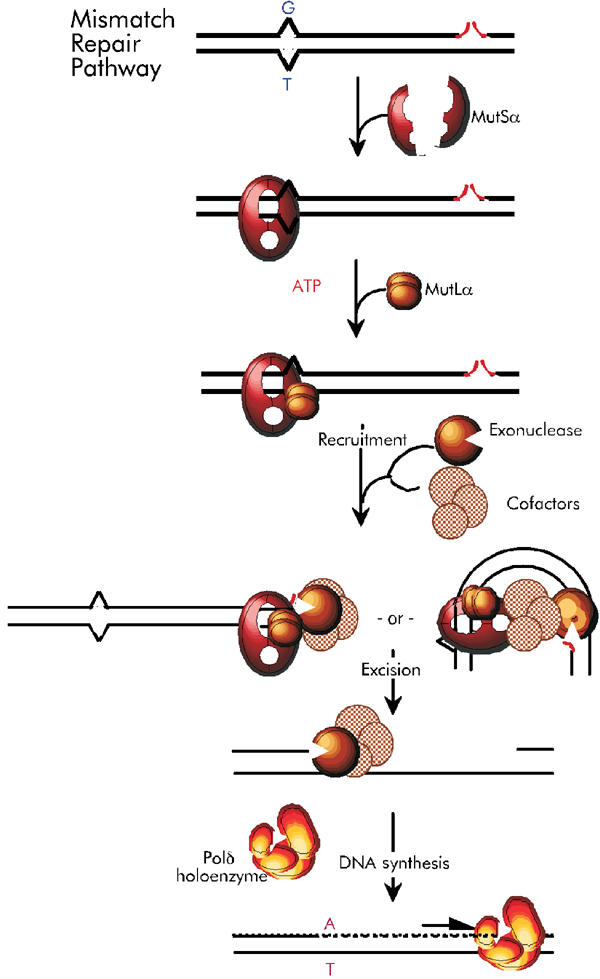
Alternative mismatch-correction pathways. Mismatches (G/T) are bound by MutSα heterodimers (red) and MutLα (bronze), in the presence of ATP, forming recognition complexes. Possible recognition-complex accessory proteins, such as PCNA, are not shown. Mismatch recognition is coupled to identification of the excision-initiation nick, concomitant with recruitment of exonuclease(s) (bronze) and (a) excision co-factor(s), for example, helicases (pink), via recognition-complex sliding (left) or contact of mismatch-bound recognition complexes with nicks or nick-associated proteins, by DNA bending (right). Excision proceeds from nicks to approximately 0.15 kbp past the mismatch, and DNA synthesis restores homoduplex (A/T) DNA.
The important starting points for these models have been numerous ingenious partial-reaction studies, using one or two purified proteins—typically MutS or MutSα, sometimes plus MutL or MutLα—with mismatched DNA substrates, for analysis of protein–DNA interactions and ADP/ATP transactions under a variety of conditions. Among partial-reaction studies that have given rise to the two alternative models, those that employed relatively short synthetic mismatched-DNA oligomers with ends blocked by DNA cruciforms or streptavidin–biotin complexes have been pivotal (Allen et al, 1997; Gradia et al, 1997, 1999; Junop et al, 2001; Schofield et al, 2001; Acharya et al, 2003). Prevention of the rapid ATP-dependent dissociation of MutS or MutSα observed for open-ended linear mismatched DNA by such barriers suggested sliding/translocation by these proteins; modulation of this behavior by MutL or MutLα shed further light on recognition and coupling mechanisms. ATP induced sliding of even ATP-hydrolysis-deficient (but binding-proficient) mutant MutSα proteins (Iaccarino et al, 1998). In other partial-reaction studies, ATP promoted the formation of MutS·MutL (Acharya et al, 2003) or MutSα·MutLα (Raeschle et al, 2002) complexes on mismatched DNA; neither their formation nor their apparent sliding required ATP hydrolysis (or binding) by MutL/MutLα.
Here we analyze MMR recognition–excision coupling in human nuclear extracts, which correct mismatches in nicked plasmids with high efficiency and specificity (Wang and Hays, 2002a,2002b). We challenge fixed-recognition-complex/DNA-looping models by testing a key prediction: that internal barriers, expected to block sliding/translocating of putative recognition complexes from mismatches to nicks, should not prevent efficient initiation of mismatch-dependent excision. We used new techniques (Wang and Hays, 2002a) to construct circular substrate plasmids containing single mismatches and defined nicks, and to generate in both the shorter (0.3-kbp) and longer (1.9-kbp) nick-mismatch paths, streptavidin–biotin complexes similar to those previously placed by others (Blackwell et al, 1998, 2001; Gradia et al, 1999) at DNA ends, to block ATP-activated hMutSα sliding. We assayed excision (gap formation in the absence of exogenous dNTPs) at specific points by a new method (Wang and Hays, 2002a), and also measured mismatch correction in complete reaction mixtures. A (G/T) mismatch efficiently provoked 3′ to 5′ excision of the nicked strand, measured 30–60 nt along the shorter nick–mispair path, despite the intervening streptavidin–biotin blockades. Therefore, fixed-recognition-complex hypotheses were not falsified. The barrier blocked subsequent progress of excision to the mismatch, and thus prevented correction.
Results
Substrate construction and validation
We constructed circular mismatched-DNA substrates containing barriers chemically identical to those used previously to prevent ATP-activated sliding of hMutSα off the ends of linear mismatched-DNA oligoduplexes (Blackwell et al, 1998, 2001; Gradia et al, 1999): streptavidin bound to biotin linked to a thymine nucleotide by a 15-carbon chain. Figure 2A schematically depicts the construction procedure: (i) high-yield production of 30-nt gapped plasmids and ligation of mismatch (T/G)-containing 30-mer oligonucleotides into the gaps, using recently described techniques (Wang and Hays, 2001, 2002a); (ii) production of two additional gaps flanking the mismatch—approximately 160 bp counterclockwise (ccw) and 140 bp clockwise—and ligation of biotinylated 30-mers into them; (iii) introduction of a single excision-initiation nick 0.31 kb ccw from the mismatch (substrate E); and (iv) binding of streptavidin to the biotinylated DNA (substrate F). Thus, streptavidin–biotin blockades would be present in both the shorter (0.31-kbp) and longer (1.87-kbp) paths from the nick to the mismatch.
Figure 2.
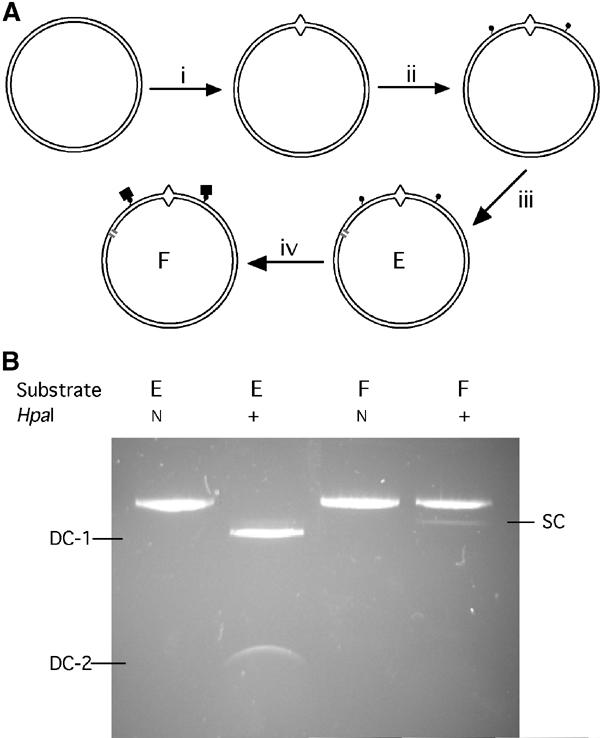
Construction and verification of substrates. (A) Substrate construction. Beginning with plasmid pUC19PPH (Wang and Hays, 2003), intermediates and substrates were constructed via steps (i)–(iv), as described in the text. Biotinylated residues in substrate (E) are indicated by small filled circles attached to DNA, and streptavidin, bound to biotin in substrate (F) by larger filled squares. (B) Verification of substrates. Substrate (E) was mock-incubated (lanes 1, 2) or incubated with streptavidin (lanes 3, 4), then treated with HpaI endonuclease (lanes 2, 4) or mock-treated (lanes 1, 3), and analyzed by agarose-gel electrophoresis. N, not added; band designated SC is the product of single cleavage, at one or the other HpaI site, that is, linearization; bands designated DC are products of double cleavage.
We verified the stability of the attached biotin and the efficiency of streptavidin binding to it under the same conditions used for the MMR reactions (Figure 2B). Endonuclease HpaI readily cleaved almost all biontinylated substrates (E) and control substrates (not shown) at two sites—each 2 bp away from biotin-linked thymine (Figure 2B, lanes 1 and 2). However, after incubation of substrate E with streptavidin, just 6% of biotin–streptavidin substrates (F) were cut once, and almost none was cut twice (Figure 2B, lanes 3 and 4). Thus, streptavidin–biotin complexes, but not biotinylated thymines, occlude the adjacent HpaI sites, and these complexes are present at both positions in almost the entire DNA. Digestion of streptavidin-bound substrate with HpaI and AlwNI endonucleases generated products consistent with cutting at the unique AlwNI site and at either the proximal or distal HpaI site, to the same extent: 1.75 and 0.44 kbp, and 1.44 and 0.75 kbp, respectively (data not shown). Thus, the rare failures of streptavidin to bind were distributed equally between the two biotin sites (roughly 3% each).
Streptavidin–biotin complexes chemically identical to those previously placed at the ends of linear duplexes were present here in circular DNA. Without repeating previous studies in detail, we wanted to verify whether these interior complexes also possessed the same critical physical property, namely the ability to block sliding of (ATP-bound) hMutSα on DNA, as detected by mobility-shift assays. We first cut out from non-nicked substrate E the 0.5-kb DNA fragments that contain the G/T mismatch flanked at each end by biotin residues, and electrophoretically purified and 32P-endlabeled the fragments. In these fragments, streptavidin–biotin barriers would be approximately 150 and 20 bp from the respective ends. We then used electrophoretic mobility shift assays to measure stable binding to the biotinylated DNA, complexed or not with streptavidin, in the presence or absence of ATP (Figure 3). As expected, free DNA, whether mismatched/biotinylated (E) or homoduplex/unbiotinylated (HM), migrated most rapidly (Figure 3, lanes 1, 3, 4, 6), and bound streptavidin (F) decreased DNA mobility (Figure 3, lanes 2 and 5). In mobility shift assays with fragments of this length, some DNA may not migrate out of the wells (position marked ‘Well'). Disregarding material in wells, 84% of biotinylated-only and 90% of streptavidin-bound mismatched (G/T) DNA were shifted by hMutSα binding in the absence of ATP (Figure 3, lanes 1 and 2); only about 4% of homoduplex DNA was shifted (lane 3), consistent with previous studies by several groups using linear DNA with terminal biotin/streptavidin (Blackwell et al, 1998, 2001; Acharya et al, 2003). ATP strongly reduced (seven-fold) MutSα shifting of free biotinylated mismatched DNA, but only weakly reduced (1.7-fold) shifting of the streptavidin-bound mismatched DNA (Figure 3, lanes 4 and 5). This substantial but incomplete trapping of hMutSα on linear DNA, in which a (G/T) mismatch is flanked by biotin–streptavidin blockades interior to both ends, compares well to previously reported 70% retention on 201-bp mismatched DNA with similar blockades, but at the ends (Blackwell et al, 1998). The low but significant loss of MutSα from this 0.5-kb substrate in the presence of ATP might reflect partial dissociation directly into solution. We performed the mobility-shift assay under conditions permissive or restrictive for ATP hydrolysis—either in the presence of 5 mM Mg2+ at 25°C (Figure 3), or without Mg2+ on ice (data not shown), with similar results. The streptavidin–biotin complexes produced here in circular mismatched DNA thus appear highly similar to those used previously (Blackwell et al, 1998, 2001; Gradia et al, 1999), at least with respect to sliding by (ATP-bound) hMutSα, with some minor caveats (see under ‘Discussion').
Figure 3.
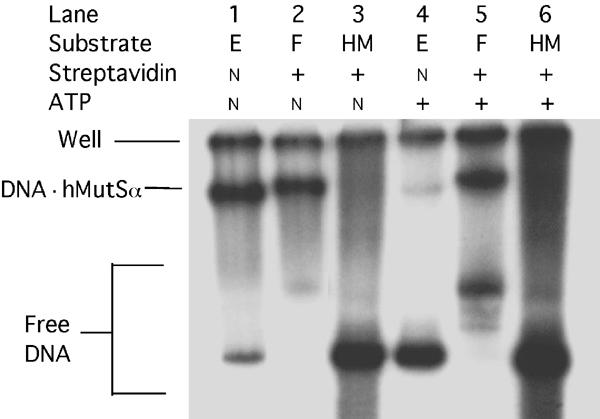
ATP-resistant retention of hMutSα on end-blocked mismatched DNA. The indicated 0.5-kb DNA fragments (6 fmol) cleaved from biotinylated, mismatched-DNA substrate (E) (Figure 1A) or nonderivatized homoduplex DNA (HM) were purified and endlabeled, and incubated first with streptavidin in binding buffer (to convert substrate (E) to substrate (F)), and then with purified hMutSα (0.25 pmol) plus 125 ng nonspecific competitor DNA for 10 min, as described under ‘Materials and methods'. After addition of ATP to 200 μM, where indicated, incubations were continued for an additional 5 min. Mixtures were separated by electrophoresis in 5% nondenaturing polyacrylamide gels (acrylamide to bisacrylamide ratio of 37.5:1), which were dried and analyzed by phosphorimaging. The fastest-moving DNA bands (lanes 1, 3, 4, 6) correspond to free DNA fragments (not bound to MutSα) from substrates (E) or (HM). The slower-moving faint free-DNA band in lane 2 corresponds to the small amount of substrate (F) with both streptavidin complexes but not bound to hMutSα, as do the more intense free DNA bands in lane 5. The two faint bands moving slightly faster in lane 5 presumably correspond to fragments with one complex, either 150 bp from one end or 20 bp from the other end—locations expected to affect mobility differentially. ATP would be expected to cause hMutSα to slide off the free ends of both the latter DNA species. Intensities of the indicated slowest-moving bands, corresponding to DNA hMutSα complexes, were compared to the total of bound plus nonbound free DNA, excluding DNA not fully complexed with streptavidin (faint bands), and disregarding DNA trapped in the wells (‘Well'). Bound fractions were 84 and 90%, respectively, for biotinylated-only (lane 1) and streptavidin-complexed (lane 2) DNA in the absence of ATP, but 12 and 51%, respectively, for these substrates in the presence of ATP (lanes 4 and 5).
Terminal streptavidin-biotin blockades were also reported to prevent the loss of ternary complexes containing E. coli MutS and MutL, formed in the presence of ATP, on 0.1-kbp mismatched DNA (Acharya et al, 2003). Substantial reduction in bound protein was observed only in the presence of excess mismatched DNA competitor, suggesting that after ATP-dependent ternary-complex formation at the mismatch, complexes of MutS and MutL rapidly moved off DNA ends, but quickly bound again to mismatched DNA, unless competitor DNA prevented re-binding. However, terminal streptavidin blocked sliding/translocating off ends, making the ternary complexes competitor-resistant. Also, Blackwell et al (2001) demonstrated the dynamic nature of MutSα·MutLα complexed to 0.2-kbp mismatched DNA: only the presence of streptavidin at both ends prevented rapid loss of complexes challenged with excess competitor DNA. Whether or not sliding/translocating MutS(α)–MutL(α) complexes might form in complex nuclear extracts containing PCNA and other proteins that interact with MutSα and MutLα (Bowers et al, 2001) is not known, and is in fact a point of issue here.
MMR excision
We used human nuclear extracts to analyze the effects of streptavidin–biotin blockades on MMR excision, measured in the absence of exogenous dNTPs as the appearance of gapped DNA at various specific positions (Wang and Hays, 2002a), and on mismatch correction (see below). To measure quantitatively and specifically MMR excision gaps in human nuclear extracts, we froze excision gaps by omitting exogenous dNTPs, then bound small radiolabeled oligomer probes to single-stranded DNA at various positions. Previously, this assay was found to be approximately 50:1 specific for mismatched versus homoduplex DNA, over 100:1 specific for excision of nicked versus continuous strands, and 30:1 specific for shorter versus longer nick-mismatch paths (Wang and Hays, 2002a,2002b, 2003). Initiation of excision precedes mismatch correction by about 80 s in these extracts, and both processes show plateau values—typically 40–60% yield—after 6–12 min (Wang and Hays, 2002a,2002b).
Here we analyzed initiation, at a single defined nick, of excision provoked by a mismatch 0.31 kbp away. This mismatch was flanked on each side by a biotinylated nucleotide (0.16 and 0.14 kbp away from the mismatch in the shorter (3′ to 5′) and longer (5′ to 3′) nick-mismatch paths, respectively), to which streptavidin was bound or not. Gapped DNA generated by 3′ to 5′ shorter-path excision of the nicked strand (bottom strand in Figure 4 (lower panel)) was detected by binding of probes complementary to the continuous (top) strand, that is, collinear with the bottom strand, at the positions indicated in Figure 4 (lower panel): (#3), just downstream of the nick (30–60 nt), along the shorter path and thus a measure of excision initiation; (#5), 270 nt further downstream at the mismatch location and thus a measure of excision progress; (#1), 0.8 kbp from the nick, but along the longer nick-mismatch path, and thus a measure of any processive longer-path 5′ to 3′ excision. We did not similarly construct and analyze a substrate in which the shorter nick-mispair path was 5′ to 3′, because nonspecific (mismatch-independent) 5′ to 3′ excision, although quite nonprocessive, is high just downstream of nicks, where mismatch-dependent excision initiation would need to be sensitively measured (Wang and Hays, 2002a).
Figure 4.
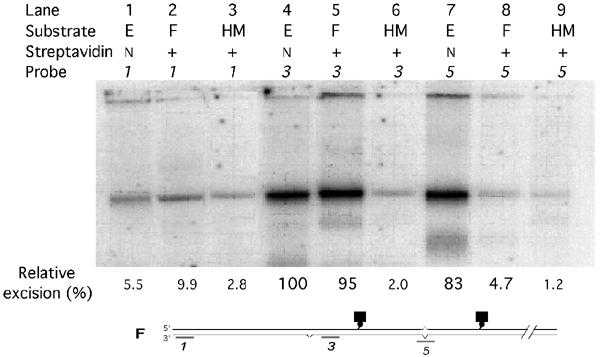
Mismatch-provoked excision in nuclear extracts. Biotinylated circular DNA (shown here as linear duplex), containing a G/T mismatch at 340/340′ bp and a pre-existing nick at position 23 (substrate E), was incubated with streptavidin to produce substrate F where indicated (+). Non-derivatized nicked homoduplex substrate (HM) was similarly treated. DNA substrates were incubated in HeLa-cell extracts for 7 min under MMR conditions, but without exogenous dNTPs. Aliquots of AhdI-endonuclease-cleaved (linearized) reaction products were assayed for excision by annealing to them the indicated radiolabeled 30-mers collinear with the nicked (bottom) strand (probe #1, nt 1351–1381; probe #3, nt 52–80; probe #5, nt 323–352), electrophoresis, and phosphorimaging, as described under ‘Materials and methods'. Indicated biotinylated (streptavidin-bound) nucleotides are at positions 178′ and 487′ in the continuous strand. The apparent uppermost ‘bands' are at the positions of wells. Fastest-moving (but diffuse) minor bands, seen primarily for substrates (E) and (F), have been typical of these assays (Wang and Hays, 2002a,2002b, 2003) and may reflect occasional random cleavage at gaps.
Remarkably, the plateau value for initiation (probe #3) of mismatch-provoked 3′ to 5′ excision in streptavidin-blockade substrate F was 95% of that for biotinylated-only substrate E (Figure 4, Lanes 5 and 4, respectively). To rule out the remote possibility that streptavidin per se, rather than the mismatch, provoked excision of substrate F, we used the techniques described above to construct from plasmid pUC19PA (Wang and Hays, 2003) homoduplex substrates with (single) biotinylated residues or biotin–streptavidin blockades, corresponding to those in the shorter nick-mispair path (see Figure 4, bottom panel). We similarly verified efficient binding of streptavidin to biotin by resistance to HpaI digestion. We compared plateau excision-initiation values (probe #3) among these two homoduplex substrates, nonderivatized homoduplexes, and (again) mismatched-DNA substrates E and F. In this experiment, excision of the streptavidin-blocked substrate (F) was actually initiated slightly more efficiently (107%) than excision of biotinylated-only substrate (E), but apparent excision signals for both biotinylated and biotin–streptavidin homoduplex DNA were at the same background levels as for nonderivatized homoduplex DNA—2.3, 2.2, and 2.0%, respectively (data not shown). In principle, a mismatch might provoke excision at a nick on the other side of the streptavidin barrier in substrate F via some new mechanism—different from that characteristic of biotinylated-only (E) or non-derivatized mismatched-DNA substrates—that nevertheless yielded a similar plateau excision value. Therefore, we also compared the time courses of excision initiation (Figure 5). In two independent experiments, these appeared identical for biotinylated substrates (E) (empty squares and circles) and streptavidin-bound substrates (F) (filled squares and circles). The apparent rates (times to half plateau values) for these experiments agree well with previously observed rates (130 s here versus 122–127 s previously) (Wang and Hays, 2002a,2002b). (Initiation rates in the previous experiments were estimated here by back-extrapolating excision signals measured farther downstream, using the progress rate of 5.1 nt/s.)
Figure 5.
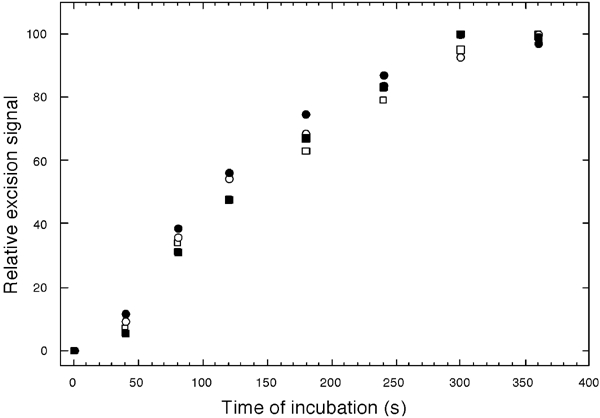
Time courses of mismatch-provoked excision. Preparation of circular substrate (E), incorporating a single biotinylated nucleotide in both paths from the (G/T) mismatch to a pre-existing nick (□, ○), and substrate (F), with streptavidin–biotin complexes at the same positions (▪, •), was as described under ‘Materials and methods'. Substrates were mixed on ice with HeLa nuclear extracts, in MMR buffer lacking exogenous dNTPs, and temperatures were immediately raised to 37°C (time zero). At the times indicated, aliquots were removed and DNA was extracted and analyzed for excision of the nicked strand nt 30–60 along the shorter (3′ to 5′) path towards the mismatch, by annealing of probe #3, gel electrophoresis, and phosphorimaging, as described under ‘Materials and methods'. Excision signals (amounts of probe radioactivity bound) are expressed relative to plateau values at 300 or 360 s. Squares and circles correspond to two independent trials.
Internal biotin did not itself affect MMR reactions: excision-initiation time courses for non-derivatized and biotin-only (and streptavidin–biotin) substrates were similar (see the above paragraph), further progress of excision to the mismatch region (Figure 4, probe #5) was blocked by streptavidin–biotin but not by biotin alone (Figure 4, lanes 7 and 8), and correction of the mismatches in biotinylated substrate (E) was normal (see below and Figure 6).
Figure 6.
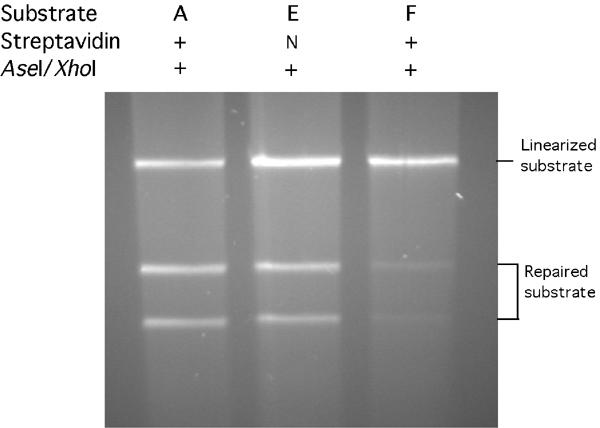
Mismatch correction in nuclear extracts. Circular DNA substrates, all containing G/T mismatches and pre-existing nicks—either unmodified s19ASP, previously designated substrate A (Wang and Hays, 2003), or biotinylated-only substrate E, or substrate E previously incubated with streptavidin to produce substrate F—were incubated in HeLa-cell extracts under standard MMR conditions for 12 min as described under ‘Materials and methods'. Cleavage by XhoI and AseI endonucleases into two bands (‘Repaired substrate') reflects restoration of the XhoI site by correction of G/T to G/C; AseI cleavage alone yields ‘Linearized substrate'.
To estimate strand specificities for mismatch-provoked shorter-path excision in biotin-only (E) and biotin–streptavidin (F) substrates, we measured apparent excision of both the nicked (Figure 4, probe #3) and continuous (probe #3′, complementary to probe #3) strands. These signals were corrected for background values measured using homoduplex substrates. The apparent specificities were 72:1 (E) or 88:1 (F) in favor of the nicked strand (data not shown). To estimate excision-path preferences, we first corrected the apparent efficiencies (Figure 4) of shorter-path (3′ to 5′) excision, measured using probe #3, and of longer-path (5′ to 3′) excision, measured using probe #1, for background probe binding to homoduplex (HM) DNA. For biotinylated substrates (E), the shorter-path efficiency (100–3) was 36-fold higher than that of the longer path (5.5–2.8), and for the blockade-containing substrate the corresponding factor was 13-fold. (Longer-path excision is measured here at a point well beyond the reach of (highly non-processive) nonspecific 5′ to 3′ excision (Wang and Hays, 2002a).) Thus, the strong preference for nick-initiated excision along the shorter nick-mispair path is preserved in the blockade-containing substrates. (Figure 4)
In summary, to the extent that the rate-limiting step in initiation of 3′ to 5′ excision is identification of the excision signal (nick), after recognition of the mismatch and before further assembly of any excision complex, the efficiency (and specificity) of this identification step appears unaffected by intervening biotin–streptavidin barriers (or by biotin alone).
Mismatch correction
We also measured mismatch correction in HeLa-cell nuclear extracts containing added dNTPs, using biotin-free or biotinylated substrates, in the presence or absence of streptavidin (Figure 6). The yields of the two DNA bands diagnostic of accurate correction—corresponding to restriction-endonuclease cleavage at the preexisting AseI site and the restored XhoI site—were 59–65% from a biotin-free substrate, with or without streptavidin present (Figure 6, lane 1, and Wang and Hays, 2003), and 54% from biotinylated substrate (E) with streptavidin absent (Figure 6, lane 2). However, the yield was only 8% from streptavidin-complexed substrate (F) (Figure 6, lane 3), corresponding to the failure of even efficiently initiated excision to progress past the streptavidin–biotin blockades (Figure 4, lanes 7 (E) versus 8 (F)). The minor correction yield (Figure 6, lane 3) may reflect incomplete double binding of streptavidin, as evidenced by the minor HpaI sensitivity (Figure 2B, lane 4). Extended incubation of substrate F, for up to 36 min failed to increase the correction yield (data not shown). Thus, a streptavidin attached to the continuous strand prevents even inefficient correction, by preventing even slow excision of the nicked strand.
Discussion
In human unclear extracts, DNA mismatches in circular substrates provoke initiation of excision at pre-existing nicks dozens to hundreds of nucleotides away, with high specificity and efficiency. To challenge fixed-recognition-complex models for coupling of mismatch recognition to excision initiation, we created internal barriers to protein sliding, by incorporating biotinylated nucleotides in the shorter and longer nick-mispair paths of such substrates, and binding streptavidin to over 90% of them. Mismatch-provoked 3′ to 5′ excision initiated with normal efficiency and specificity, despite these barriers; both initial rates and final plateau values were virtually identical to those observed using biotinylated-only or nonderivatized substrates. Thus, the hypothesis of Hsieh and Yang and co-workers that MutS and MutSα proteins remain at mismatches as a part of recognition complexes that contact excision-initiation signals by DNA bending (Junop et al, 2001; Schofield et al, 2001; Figure 7) was not falsified.
Figure 7.
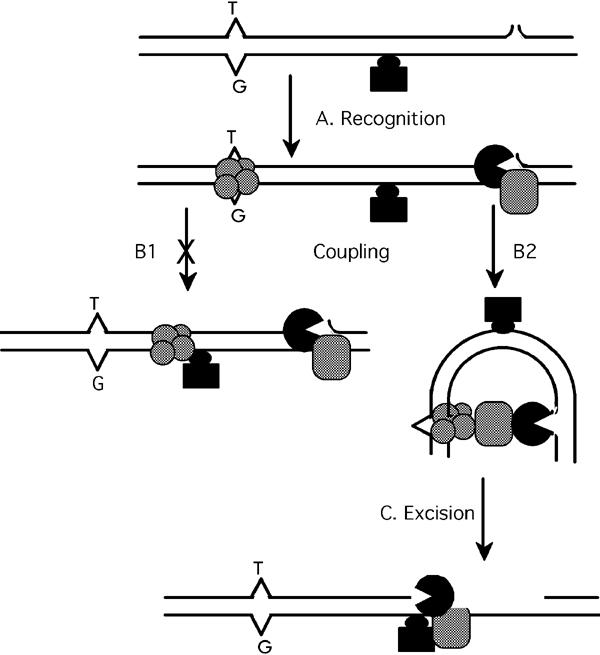
Schematic representation of MMR processing of substrates with internal barriers. Representation of specific proteins and unspecified accessory proteins is less elaborate than in Figure 1. (A) Circular substrate (shown linear) containing a single defined nick and a streptavidin (black rectangle)–biotin (black oval) complex in the mispair-nick path is recognized by a complex of MutSα plus MutLα (hatched circles) and perhaps other proteins (not shown). Recognition of the nick by (a) exonuclease(s) (black) plus accessory protein(s) (hatched rectangle) is depicted, but some or all of these proteins could be recruited during the subsequent coupling process (B) instead. (B) Coupling of mismatch recognition to excision-initiation via (B1) sliding of recognition complexes, not supported by observations here (Figures 4 and 5) or via DNA bending (B2), consistent with these observations. (C) Blockage of excision progress by the barrier attached to the continuous (not excised) strand.
The internal DNA-attached streptavidin–biotin complexes, chemically identical to those previously placed at the ends of linear mismatched DNA, showed similar interference with ATP-induced sliding of purified MutSα (Blackwell et al, 1998, 2001; Gradia et al, 1999). In the highly MMR-competent cell-free extracts employed here, the barrier complex would most likely also block any sliding by more elaborate assemblies containing MutSα plus MutLα, and perhaps MMR accessory factors such as PCNA (Bowers et al, 2001) and/or other proteins. This demonstration of high similarity between terminal and interior streptavidin–biotin complexes by band-shift analysis (Figure 3) is subject to three minor caveats. First, we cannot readily explain the trapping of some DNA in wells. To our knowledge, there have been few, if any, mobility-shift experiments in which MutS-homolog dimers bound to single-mismatch linear DNA as long as 0.5 kbp, especially to DNA incorporating biotin or biotin–streptavidin. Binding to the single mismatch was not readily measurable in agarose gels (data not shown), necessitating polyacrylamide gels. The 0.5-kbp DNA fragments were trapped to a similar degree in wells, regardless of whether or not streptavidin and/or hMutSα were present in the samples. A 100-fold molar excess of (unlabeled) competitor DNA—51-bp linear mismatched (G/T) or homoduplex DNA—reduced radiolabeled well material from a mixture of substrate F and hMutSα only by 26 and 20%, respectively (data not shown). The 0.5-kbp size may be unfortunate—too large for optimal polyacrylamide electrophoresis, but showing a minimal mobility shift in agarose when bound by hMutSα. Second, the minor but significant fraction of hMutSα released from 0.5-kb mismatched DNA containing streptavidin blocks just interior to the ends contrasts with the very low releases from shorter mismatched DNA with streptavidin at the ends (Gradia et al, 1999) (but agrees with the results seen with 0.2-kb end-blocked mismatched DNA (Blackwell et al, 1998)). Although purified hMutSα might sometimes simply dissociate from the 0.5-kbp mismatched DNA directly into solution, we cannot rule out the occasional sliding/translocation of hMutSα past the internal blocks. Finally, the apparent hMutSα retention in the absence of Mg2+ appears to differ from previous reports that complexes formed on (much shorter) linear DNA with streptavidin–biotin end blocks were not retained in the presence of ATP without Mg2+ (Gradia et al, 1999; Raeschle et al, 2002). Thus these hMutSα band-shift experiments do not unequivocally show interior streptavidin blockades to be quantitatively identical to streptavidin blocks at the ends of shorter DNA in every respect. However, the purpose of these experiments was simply to show that interior biotin–streptavidin complexes were not only chemically identical to such complexes at the ends of linear DNA but also similar with respect to retention of hMutSα in the presence of ATP. This appears to be the case, subject to the three caveats above. Furthermore, more elaborate recognition complexes in MMR-competent nuclear extracts might have properties different from purified hMutSα—never sliding/translocating away from mismatches under any circumstances, for example.
Although the identical excision-initiation time courses and similar plateau excision efficiencies observed argue against different coupling mechanisms for biotin-only versus streptavidin–biotin substrates (Figures 4 and 5), coupling by either mechanism might be very fast relative to some other rate-limiting steps, such as recruitment of (a) excision protein(s) to the nick, before and/or after activation by the mismatch. Therefore, we cannot unequivocally rule out sliding of recognition complexes up to barriers, followed by activation of excision from that position through space, or inefficient but not rate-limiting occasional sliding of putative MutSα/MutLα/accessory-protein complexes past the barriers. With respect to the latter possibility, the escape in the presence of ATP of a small fraction of hMutSα molecules from 0.5-kbp mismatched DNA containing streptavidin–biotin complexes near but not at the ends, and the retention of most hMutSα in the presence of ATP, even without Mg2+, might be interpreted as evidence for differences between internal and terminal complexes.
We previously incorporated 20-bp hairpins in the continuous (non-nicked) DNA strand, in the shorter- and longer nick-mismatch paths of circular substrates, otherwise identical to those used here and similarly measured initiation of excision and error correction (Wang and Hays, 2003). Similar to our streptavidin–biotin complexes, internal hairpins had no effect on the efficiency initiation of mismatch-provoked 3′ to 5′ excision along the shorter nick-mispair path, so fixed-recognition-complex models were not falsified.
Subsequent to initiation, excision proceeds at approximately 5.1 nt/s (Wang and Hays, 2002b). We previously found a 20-bp hairpin in the continuous strand to block excision progress strongly (Wang and Hays, 2003), and here a biotin–streptavidin complex attached to the continuous strand similarly blocks excision per se. Although streptavidin might simply impede the progress of an exonuclease along the nicked strand, the equally strong effect of the continuous-strand hairpin suggests that (a) protein(s) might track along the continuous strand to facilitate excision of the nicked strand (Wang and Hays, 2003).
The mechanism by which excision is directed preferentially down the shorter nick-mispair path—a bias of 20–30-fold in our experiments—remains an enigma. The strong preference is retained here even when streptavidin blocks the shorter (and longer) path(s), suggesting that putative sliding recognition complexes are not needed to direct excision paths. On the other hand, the DNA-looping model provides no obvious mechanism for path preference. Nicking at unmethylated d(GATC) sites in short linear oligoduplexes, by purified E. coli MutH plus MutL proteins, was substantially stimulated in trans by a 56-bp duplex containing a mismatch, in a MutS-dependent manner (Schofield et al, 2001). However, these experiments mimicked only the initial generation of the E. coli strand-specific excision-initiation signal and did not address the efficiency or directionality of excision itself. Some property of the DNA contour between mismatch and excision-initiation site (here the pre-existing nick) might dictate the efficiency of excision back towards the mismatch. This could be intrinsic to the DNA itself—bending strain, for example, or excision direction could be directed by proteins associated with the DNA—perhaps bound between mismatch and nick so as to make a continuous chain of protein–protein contacts that here bypassed the biotin–streptavidin blockade, perhaps by looping out a small portion of DNA containing the blockade. In this respect, the abilities of yeast MutLα protein to bind cooperatively to extended stretches of DNA and to bring two separate regions of DNA together, apparently via dual binding sites in MutLα (Hall et al, 2001), may be relevant.
Materials and methods
DNA substrates and proteins
Plasmid pUC19PPH and numbering of its bla sense-strand (1, 2, 3…) or antisense-strand (1′, 2′, 3′…) nucleotides have been described (Wang and Hays, 2003). The use of site-specific nicking endonuclease N.AlwI to produce plasmids containing a single 30-nt gap at positions 331–361, ligation into them of 30-nt oligomers—creating a (G/T) mispair that inactivates the XhoI site at nt 340 or maintaining the sequence (G/C)—and isolation of supercoiled product were as described (Wang and Hays, 2003). Subsequently, we similarly used nicking endonuclease N.BstNBI to produce in the (G/T) derivative two 30-nt gaps (positions 160′–190′, and nt 469′–499′) of identical sequence and ligated into them 30-mers containing single thymine analogs (nt 178′ and 487′) linked at C-5 to biotin by 15-carbon chains. A third single nick was introduced at position 23 by nicking endonuclease N.Bpu10I, yielding the (G/T) substrate E. Streptavidin (Rockland, Gilbertsville, PA, USA) was routinely bound to substrate E, yielding substrate F, by incubation of 100 ng DNA with an equal volume of 1.0 mg/ml streptavidin solution for 20 min at room temperature, in buffers appropriate to subsequent binding, excision or correction experiments (see below). Streptavidin–biotin complexes at positions 178′ and 487′ were identified by resistance to cleavage by endonuclease HpaI at its sites (nt 171′–176′ and 480′–485′), which were not blocked by biotin alone. Purification of hMutSα protein from HeLa cells was as described (Wang and Hays, 2003).
Bandshift assay for hMutSα binding
As described previously (Wang and Hays, 2003), 0.5-kb fragments were cleaved from non-nicked substrates E or its homoduplex homolog with endonuclease BglII and AflIII, electrophoretically purified, and endlabeled by filling in the recessed 3′ ends using 32P-α-dCTP and three other non-radioactive dNTPs in the presence of DNA polI Klenow fragment (exo−) (New England Biolab). After standard incubation with streptavidin in Binding Buffer, where indicated, these fragments, plus 125 ng of 1-kb ladder DNA (Invitrogen, catalog number 15615-016), were incubated with purified hMutSα (10 min), then with ATP (5 min), where indicated. After addition of 1/5 volume of 50% sucrose, products were separated by electrophoresis through 5% nondenaturing polyacrylamide gels (acrylamide:bis-acrylamide ratio at 37.5:1) as described (Wang and Hays, 2003). Bands were measured by phosphorimaging and used to calculate fractions of retarded DNA.
Measurement of mismatch-provoked excision
Oligomer probes (30-nt) corresponding to substrate sequences 1351–1381 (#1), 52–80 (#3), or 323–352 (#5) were radiolabeled using T4 polynucleotide kinase (Invitrogen) and 32P-γ-ATP, and then adjusted to equal specific activities. After standard incubation with streptavidin in MMR buffer, where indicated, nicked (G/T) substrate E, F, or the homoduplex non-biotinylated homolog (HM) was incubated with HeLa extracts for 7 min, as described above under ‘Mismatch-correction assay', except that exogenous dNTPs were omitted. DNA purified from reaction mixtures was annealed with excess of probes #1, #3, or #5 after cleavage with endonuclease AhdI. Mixtures were separated by electrophoresis and analyzed by phosphorimaging, as described (Wang and Hays, 2003).
Mismatch-correction assay
To assay correction of the G/T mismatch in (nicked) substrate E, F, or the nonbiotinylated (nicked) homolog s19ASP (substrate A), about 75 fmol (100 ng) DNA was incubated at 37°C for 12 min with 100 μg of HeLa nuclear extracts—prepared as described (Wang and Hays, 2003)—in MMR buffer (20 mM Tris–HCl, pH 7.6; 1.5 mM ATP; 5.0 mM MgCl2; 110 mM KCl; 50 μg/ml bovine serum albumin), plus all four dNTPs at 100 μM. DNA purified from reaction mixtures was simultaneously cleaved with endonuclease AseI at its pre-existing site (nt 1569) and challenged with endonuclease XhoI, to assay restoration of the XhoI site (G/T to G/C at 340 bp); mixtures were analyzed by electrophoresis in 1% agarose for 120 min, and DNA band intensities were measured as described (Wang and Hays, 2003). Upon construction, mismatch (G/T)-containing substrates A and E (100 ng) (see under ‘DNA substrates and proteins' above) were routinely tested for resistance to XhoI cleavage, which was undetectable (<2–3%).
Acknowledgments
We thank Drs Andrew Buermeyer, Niels DeWind, Richard Fishel, and Meindert Lamers for careful reading of the manuscript and helpful suggestions. This work was supported by NIH grant ES09848 to JBH.
References
- Acharya S, Foster PL, Brooks P, Fishel R (2003) The coordinated functions of the E. coli MutS and MutL proteins in mismatch repair. Mol Cell 12: 233–246 [DOI] [PubMed] [Google Scholar]
- Allen DJ, Makhov A, Grilley M, Taylor J, Thresher R, Modrich M, Griffith JD (1997) MutS mediates heteroduplex loop formation by a translocation mechanism. EMBO J 14: 4467–4470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Au KG, Welsh K, Modrich R (1992) Initiation of methyl-directed mismatch repair. J Biol Chem 267: 12142–12148 [PubMed] [Google Scholar]
- Bellacosa A (2001) Functional interactions and signaling properties of mammalian DNA mismatch repair proteins. Cell Death Differ 8: 1076–1092 [DOI] [PubMed] [Google Scholar]
- Blackwell LJ, Martik D, Bjornson KP, Bjornson ES, Modrich P (1998) Nucleotide-promoted release of hMutSalpha from heteroduplex DNA is consistent with an ATP-dependent translocation mechanism. J Biol Chem 273: 32055–32062 [DOI] [PubMed] [Google Scholar]
- Blackwell LJ, Wang S, Modrich P (2001) DNA chain length dependence of formation and dynamics of hMutSalpha.hMutLalpha.heteroduplex complexes. J Biol Chem 276: 33233–33240 [DOI] [PubMed] [Google Scholar]
- Bowers J, Tran PT, Joshi A, Liskay RM, Alani E (2001) MSH-MLH complexes formed at a DNA mismatch are disrupted by the PCNA sliding clamp. J Mol Biol 306: 957–968 [DOI] [PubMed] [Google Scholar]
- Buermeyer AB, Deschenes SM, Baker SM, Liskay RM (1999) Mammalian DNA mismatch repair. Annu Rev Genet 33: 533–564 [DOI] [PubMed] [Google Scholar]
- Culligan K, Hays JB (2000) Arabidopsis MutS homologs-AtMSH2, AtMSH3, AtMSH6, and a novel AtMSH7-form three distinct protein heterodimers with different specificities for mismatched DNA. Plant cell 12: 991–1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dao V, Modrich P (1998) Mismatch-, MutS-, MutL-, and helicase II-dependent unwinding from the single-strand break of an incised heteroduplex. J Biol Chem 273: 9202–9207 [DOI] [PubMed] [Google Scholar]
- Gradia S, Subramanian D, Acharya S, Malchov A, Griffith J, Fishel R (1999) hMSH2–hMSH6 forms a hydrolysis-independent sliding clamp on mismatched DNA. Mol Cell 3: 255–261 [DOI] [PubMed] [Google Scholar]
- Gradia S, Acharya S, Fishel R (1997) The human mismatch recognition complex hMSH2–hMSH6 functions as a novel molecular switch. Cell 91: 995–1005 [DOI] [PubMed] [Google Scholar]
- Hall MC, Matson SW (1999) The Escherichia coli MutL protein physically interacts with MutH and stimulates the MutH-associated endonuclease activity. J Biol Chem 274: 1306–1312 [DOI] [PubMed] [Google Scholar]
- Hall MC, Wang H, Erie DA, Kunkel TA (2001) High affinity cooperative DNA binding by the yeast Mlh1–Pms1 heterodimer. J Mol Biol 312: 637–647 [DOI] [PubMed] [Google Scholar]
- Harfe BD, Jinks-Robertson S (2000) DNA mismatch repair and genetic stability. Annu Rev Genet 34: 359–399 [DOI] [PubMed] [Google Scholar]
- Holmes J, Clark S, Modrich P (1990) Strand-specific mismatch correction in nuclear extracts of human and Drosophila melanogaster cell lines. Proc Natl Acad Sci USA 87: 5837–5841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iaccarino I, Marra G, Palombo F, Jiricny J (1998) hMSH2 and hMSH6 play distinct roles in mismatch binding and contribute differently to the ATPase activity of hMutSalpha. EMBO J 17: 2677–2686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iams K, Larson ED, Drummond JT (2002) DNA template requirements for human mismatch repair in vitro. J Biol Chem 277: 30805–30814 [DOI] [PubMed] [Google Scholar]
- Junop MS, Obmolova G, Rausch K, Hsieh P, Yang W (2001) Composite active site of an ABC ATPase: MutS uses ATP to verify mismatch recognition and authorize DNA repair. Mol Cell 7: 1–12 [DOI] [PubMed] [Google Scholar]
- Kolodner RD, Marsischky GT (1999) Eukaryotic DNA mismatch repair. Curr Opin Genet Dev 9: 89–96 [DOI] [PubMed] [Google Scholar]
- Lahue RS, Au KG, Modrich P (1989) DNA mismatch correction in a defined system. Science 245: 160–164 [DOI] [PubMed] [Google Scholar]
- Pavlov YI, Mian IM, Kunkel TA (2003) Evidence for preferential mismatch repair of lagging strand DNA replication errors in yeast. Curr Biol 13: 744–748 [DOI] [PubMed] [Google Scholar]
- Raeschle M, Dufner P, Marra G, Jiricny J (2002) Mutations within the hMLH1 and hPMS2 subunits of the human MutLalpha mismatch repair factor affect its ATPase activity, but not its ability to interact with hMutSalpha. J Biol Chem 277: 21810–21820 [DOI] [PubMed] [Google Scholar]
- Schofield MJ, Nayak S, Scott TH, Du C, Hsieh P (2001) Interaction of Escherichia coli MutS and MutL at a DNA mismatch. J Biol Chem 276: 28291–28299 [DOI] [PubMed] [Google Scholar]
- Su S-S, Modrich P (1986) Escherichia coli mutS-encoded protein binds to mismatched DNA base pairs. Proc Natl Acad Sci USA 83: 5057–5061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Hays JB (2001) Simple and rapid preparation of gapped plasmid DNA for incorporation of oligomers containing specific DNA lesions. Mol Biotechnol 19: 133–140 [DOI] [PubMed] [Google Scholar]
- Wang H, Hays JB (2002a) Mismatch repair in human nuclear extracts. Quantitative analyses of excision of nicked circular mismatched DNA substrates, constructed by a new technique employing synthetic oligonucleotides. J Biol Chem 277: 26136–26142 [DOI] [PubMed] [Google Scholar]
- Wang H, Hays JB (2002b) Mismatch repair in human nuclear extracts. Time courses and ATP requirements for kinetically distinguishable steps leading to tightly controlled 5′ to 3′ and aphidicolin-sensitive 3′ to 5′ mispair-provoked excision. J Biol Chem 277: 26143–26148 [DOI] [PubMed] [Google Scholar]
- Wang H, Hays JB (2003) Mismatch repair in human nuclear extracts: effects of internal DNA hairpin structures between mismatches and excision-initiation nicks on mismatch correction and mismatch-provoked excision. J Biol Chem 278: 28686–28693 [DOI] [PubMed] [Google Scholar]
- Wu S-Y, Culligan KM, Lamers MH, Hays JB (2003) Dissimilar mispair-recognition spectra of Arabidopsis DNA-mismatch-repair proteins MSH2·MSH6 (AtMutSα) and MSH2·MSH7 (AtMutSγ). Nucleic Acid Res 19: 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]