

Abstract
Following biosynthesis, both GLUT1 and VSV-G proteins appear rapidly (2–3 h) at the plasma membrane, whereas GLUT4 is retained in intracellular membrane compartments and does not display any significant insulin responsiveness until 6–9 h. Surprisingly, the acquisition of insulin responsiveness did not require plasma membrane endocytosis, as expression of a dominant-interfering dynamin mutant (Dyn/K44A) had no effect on the insulin-stimulated GLUT4 translocation. Furthermore, expression of endocytosis-defective GLUT4 mutants or continuous surface labeling with an exofacial specific antibody demonstrated that GLUT4 did not transit the cell surface prior to the acquisition of insulin responsiveness. The expression of a dominant-interfering GGA mutant (VHS-GAT) had no effect on the trafficking of newly synthesized GLUT1 or VSV-G protein to the plasma membrane, but completely blocked the insulin-stimulated translocation of newly synthesized GLUT4. Furthermore, in vitro budding of GLUT4 vesicles but not GLUT1 or the transferrin receptor was inhibited by VHS-GAT. Together, these data demonstrate that following biosynthesis, GLUT4 directly sorts and traffics to the insulin-responsive storage compartment through a specific GGA-sensitive process.
Keywords: biosynthesis, GGA, GLUT4, insulin, trafficking
Introduction
One unresolved problem in regulated secretion is the mechanism by which cargo is initially sorted into the appropriate responsive compartment. Although tremendous progress has been made in understanding the regulation and formation of synaptic vesicles and secretory granules, little is known about the formation of regulated compartments responsible for integral membrane protein trafficking. In general, two pathways have been suggested to account for the compartmentalization and sorting of membrane proteins in regulated trafficking. For example, the yeast amino-acid permease Gap1p is directly sorted to the plasma membrane following biosynthesis when grown on a reduced nitrogen source. In contrast, on nitrogen-rich media, Gap1p is redirected to the vacuole without ever traversing the cell surface (Roberg et al, 1997; Lemmon and Traub, 2000; Helliwell et al, 2001). This resembles mammalian secretory granule formation in that it results from a selective trafficking of protein from the trans-Golgi network (TGN) to either the plasma membrane (constitutive secretion) or intracellular storage compartments (for regulated secretion). On the other hand, it has been reported that newly synthesized synaptic vesicle proteins such as synaptophysin first traffic from the TGN to the cell surface. Following endocytosis, synaptophysin is then sorted into synaptic vesicles through the endosome system (Régnier-Vigouroux et al, 1991; de Wit et al, 1999). Thus, it appears that membrane proteins can enter their specific regulated secretory compartments either directly following biosynthesis or from plasma membrane internalization.
One of the best examples of hormone-regulated membrane protein trafficking is the insulin-stimulated translocation of the GLUT4 glucose transporter to the plasma membrane (Czech and Corvera, 1999; Watson and Pessin, 2001; Bryant et al, 2002). Under basal conditions, GLUT4 is sequestered in a specialized intracellular insulin-responsive GLUT4 storage compartment (GSC). Following insulin stimulation, approximately 50% of the intracellular GLUT4 molecules redistribute to the plasma membrane, an event that is necessary for the subsequent increase in glucose uptake. Despite extensive research on the intracellular compartmentalization and trafficking of GLUT4 vesicles, little in known about the trafficking itinerary of newly synthesized GLUT4 protein. A key question is the route by which GLUT4 enters the GSC. One possibility is that newly synthesized GLUT4 gains access to the GSC by first trafficking to the plasma membrane and undergoing endocytosis, followed by recycling and sorting through endosomes en route to the GSC. Alternatively, GLUT4 could be routed directly from the TGN into the GSC. To date, no studies have investigated the specific pathway by which newly synthesized GLUT4 protein gains access to the GSC. To address this question, we have taken the approach of analyzing the initial trafficking itinerary of newly synthesized GLUT4 protein during its entry into the GSC. Our data demonstrate that following biosynthesis GLUT4 enters the GSC without first transiting the plasma membrane. Furthermore, this initial sorting decision is specific for GLUT4 and is dependent upon the GGA adaptor protein complex.
Results
Intracellular localization and trafficking of VSV-G, GLUT1 and GLUT4 following initial biosynthesis
VSV-G protein has been used extensively to study membrane transport processes in eucaryotic cells and represents a trafficking marker for the constitutive exocytotic pathway (Orci et al, 1986; Bergmann, 1989). At 2 h post-transfection, VSV-G showed a pronounced perinuclear localization pattern, as well as a small amount of plasma membrane labeling (Figure 1A, panel a). The extent of cell-surface localization increased by 3 h post-transfection and was persistent throughout the time course examined (Figure 1A, panels b–g). GLUT1 is a glucose transporter isoform highly related to GLUT4 but localized primarily to the plasma membrane and undergoes constitutive exocytosis (Rea and James, 1997; Pessin et al, 1999). Similar to the VSV-G protein, an enhanced green fluorescent protein (EGFP) epitope-tagged GLUT1 glucose transporter (GLUT1-EGFP) also rapidly trafficked to the cell surface by 2–3 h post-transfection (Figure 1A, panels h–n). In contrast to the localization of VSV-G and GLUT1, GLUT4 is sequestered in various intracellular compartments in the basal state (Holman and Sandoval, 2001; Bryant et al, 2002). Consistent with these findings, GLUT4-EGFP localized to the perinuclear region with no appreciable accumulation at the plasma membrane at any of the time points examined (Figure 1A, panels o–u). Thus, despite the 65% amino-acid identity between GLUT1 and GLUT4, these two isoforms display dramatically different trafficking and localization patterns within 2 h following biosynthesis.
Figure 1.

Newly synthesized VSV-G and GLUT1 proteins rapidly accumulate at the plasma membrane, whereas GLUT4 is retained in the perinuclear region. (A) Differentiated 3T3L1 adipocytes were electroporated with 50 μg of cDNAs encoding VSV-G (panels a–g), GLUT1-EGFP (panels h–n) or GLUT4-EGFP (panels o–u). Following transfection, the cells were immediately incubated for 2–12 h, fixed and processed for confocal fluorescent microscopy. The VSV-G protein was detected with a monoclonal VSV-G antibody followed by a Texas Red-conjugated secondary antibody. GLUT1-EGFP and GLUT4-EGFP were detected by GFP fluorescence. These are representative experiments independently performed 3–4 times. (B) Differentiated 3T3L1 adipocytes were electroporated with 50 μg GLUT4-EGFP cDNA and incubated for 3–12 h. The cells were then either left untreated (basal, panels a–f) or stimulated with 100 nm insulin (insulin, panels g–l) for 30 min. The cells were then fixed and processed for confocal fluorescent microscopy. These are representative experiments independently performed 3–5 times. (C) Differentiated 3T3L1 adipocytes were electroporated with 50 μg of cDNAs encoding GLUT1-EGFP (open symbols) or GLUT4-EGFP (closed symbols) and either untreated (circles) or incubated with 100 nm insulin for 30 min (squares) at the times indicated. The number of cells displaying a continuous EGFP rim fluorescence is expressed as the average from the counting of 100 cells from four independent experiments.
We next determined the time-dependent acquisition of insulin sensitivity of the newly synthesized GLUT4 protein (Figure 1B). As previously observed, in the absence of insulin, GLUT4 was localized in the perinuclear region of the cells with little if any detectable plasma membrane accumulation (Figure 1B, panels a–f). Insulin stimulation in cells expressing GLUT4 from 3 to 5 h had no significant difference in the distribution of the newly synthesized GLUT4 protein (Figure 1B, panels g–i). However, at 6 h post-transfection, there was a small but readily observable insulin-stimulated cell-surface localization of GLUT4 (Figure 1B, panel j). At 9–12 h after transfection, insulin promoted a robust plasma membrane translocation of GLUT4 (Figure 1B, panels k and l). The insulin-stimulated translocation of the newly synthesized GLUT4 was essentially identical at 12 and 24 h (data not shown). The insulin-responsive aminopeptidase (IRAP) is colocalized with GLUT4 and displays essentially identical sequestration and insulin-stimulated trafficking properties (Kandror and Pilch, 1994; Malide et al, 1997; Martin et al, 1997; Elmendorf et al, 1999; Garza and Birnbaum, 2000; Subtil et al, 2000). Examination of the time dependence of newly synthesized IRAP translocation also demonstrated the acquisition of insulin sensitivity 6–9 h post-transfection (data not shown). Quantification of the time-dependent appearance of GLUT1 and GLUT4 at the plasma membrane is presented in Figure 1C.
To insure that the counting of cells displaying GLUT4 at the plasma membrane accurately reflected translocation, we compared the time-dependent acquisition of insulin-stimulated translocation by both a single-cell quantification and an enzymatic total cell population assay (Supplementary Figure 1D and E). As observed in the plasma membrane surface counting assay, quantification of the total extent of myc-GLUT4 translocation by the horseradish peroxidase enzyme-antibody linked assay demonstrated a similar time-dependent acquisition of insulin-stimulated translocation (Figure 1D). Similarly, single-cell quantification of myc-GLUT4-EGFP translocation also resulted in similar time dependence with a small stimulation observed at 6 h and near maximal stimulation by 12 h (Figure 1E). Together, these data demonstrate that following electroporation the newly synthesized GLUT4 protein requires 6–9 h to display insulin responsiveness.
Electroporation does not impair insulin signaling or translocation of the endogenous GLUT4 protein
Since the adipocytes were transfected by electroporation, it remained formally possible that the time dependence for insulin-stimulated GLUT4 translocation may have resulted from a transient impairment in insulin signaling rather than a real delay in the sorting of GLUT4 into the appropriate regulatory compartment. We therefore compared the insulin stimulation of endogenous GLUT4 translocation with that of the newly synthesized IRAP protein. At 3 h post-transfection, the expressed EGFP-IRAP protein was localized in the perinuclear region and did not undergo any appreciable insulin-stimulated translocation (Figure 2A, panels b and e). In contrast, in the same cells, the endogenous GLUT4 protein was recruited to the plasma membrane following insulin stimulation (Figure 2A, panels a and d). In parallel, we also examined the translocation of the endogenous GLUT4 protein in the plasma membrane sheet assay (Figure 2B). Plasma membranes isolated from adipocytes 3 h post-transfection had a relatively low level of immunoreactive GLUT4 in the basal state (Figure 2B, panel a). In the field shown, one of the plasma membrane sheets displayed a strong level of GLUT1-EGFP labeling (Figure 2B, panel b). Insulin stimulation resulted in the translocation of the endogenous GLUT4 protein as depicted by the increase in plasma membrane immunofluorescence (Figure 2B, panel d). This occurred in several of the plasma membrane sheets including those derived from cells transfected with GLUT1-EGFP (Figure 2B, panel e).
Figure 2.

Electroporation does not disrupt insulin-stimulated translocation of the endogenous glut4 protein. (A) Differentiated 3T3L1 adipocytes were transfected with 50 μg of IRAP-EGFP cDNA and the cells incubated for 2.5 h. The cells were then either left untreated (basal, panels a–c) or incubated with 100 nm insulin (insulin, panels d–f) for 30 min. The samples were then fixed and examined for endogenous GLUT4 translocation using a GLUT4 polyclonal antibody (panels a and d) or GFP fluorescent for EGFP-IRAP translocation (panels b and e). The merged images are presented in panels c and f. This is a representative experiment independently performed three times. (B) 3T3L1 adipocytes were transfected with 50 μg of GLUT1-EGFP cDNA and the cells incubated for 2.5 h. The cells were then either left untreated (basal, panels a–c) or incubated with 100 nM insulin (insulin, panels d–f) for 30 min. Plasma membrane sheets were then prepared and labeled with a GLUT4 polyclonal antibody (panels a and d) or detected by GFP fluorescence for GLUT1-EGFP (panels b and e). The merged images are presented in panels c and f. This is a representative experiment independently performed four times.
Entry of newly synthesized GLUT4 into the insulin-responsive compartments is not affected by inhibition of plasma membrane endocytosis
To determine whether newly synthesized GLUT4 transits the plasma membrane prior to entering the GSC, we employed a dominant-interfering dynamin mutant (Dyn/K44A) that inhibits GLUT4 endocytosis (Al-Hasani et al, 1998; Kao et al, 1998; Volchuk et al, 1998). To ensure that endocytosis is inhibited prior to the secretory trafficking of GLUT4 despite the coexpression of the two proteins, we took advantage of the fungal metabolite Brefeldin A (BFA), which reversibly blocks endoplasmic reticulum to Golgi trafficking (Pelham, 1991; Klausner et al, 1992). Since dynamin is a soluble protein, BFA treatment does not affect dynamin's ability to associate with the plasma membrane (Supplementary Figure 3D). In contrast, integral membrane proteins such as GLUT4 are trapped in the endoplasmic reticulum in the presence of BFA. Since the BFA-induced block of endoplasmic reticulum-to-Golgi trafficking is readily reversible, membrane trafficking processes resume normally following washout of the drug. Thus, cells were co-transfected with wild-type dynamin (Dyn/WT) or the mutant dynamin (Dyn/K44A) plus GLUT4-EGFP and immediately treated with BFA for 3 h. Following BFA washout, we determined the time-dependent localization and acquisition of insulin responsiveness of the newly synthesized GLUT4 protein (Figure 3).
Figure 3.

Expression of a dominant-interfering dynamin mutant (Dyn/K44A) does not affect the time-dependent acquisition of insulin responsiveness. Differentiated 3T3L1 adipocytes were electroporated with 100 μg GLUT4-EGFP plus 100 μg HA-Dyn/WT (A) or HA-Dyn/K44A (B) cDNAs. Immediately following transfection, the cells were incubated with 5 μg/ml BFA for 3 h to allow dynamin expression but prevent GLUT4-EGFP exit from the endoplasmic reticulum. BFA was then removed and at the times indicated either untreated (basal, panels a–e) or treated with 100 nM insulin (insulin, panels f–j) for 30 min. Cells were labeled with an HA antibody and a Texas Red-conjugated donkey anti-mouse secondary antibody to detect dynamin expression. These are representative images obtained from 3–6 independent experiments. (C) The data were quantified by determining the number of adipocytes displaying a continuous GLUT4-EGFP rim fluorescence in cells coexpressing Dyn/WT (squares) and Dyn/K44A (circles) in the basal (open symbols) and insulin-stimulated (filled symbols) states. These data represent the average with standard deviation from the counting of 100 cells from four independent experiments.
Treatment with BFA immediately following transfection had no significant effect on the level of expression or localization of Dyn/WT or Dyn/K44A (data not shown). However, GLUT4 displayed a dispersed pattern consistent with localization to the endoplasmic reticulum (Figure 3A and B, panels a and f). Removal of BFA resulted in the re-assembly of the Golgi complex and the normal pattern of intracellular GLUT4 localization (Figure 3A and B, panels b and g). In the presence of noninhibitory Dyn/WT, the distribution of GLUT4 remained primarily in the perinuclear region with no evidence for plasma membrane accumulation in the unstimulated state (Figure 3A, panels a–e). Expression of Dyn/WT had no significant effect on the acquisition of insulin-responsive GLUT4 translocation that began 6 h following BFA washout and increased to maximal levels between 9 and 12 h (Figure 3A, panels h–j). In contrast, in the absence of insulin, the cells expressing Dyn/K44A displayed a slow time-dependent accumulation of GLUT4 at the cell surface that was detectable between 9 and 12 h following BFA removal (Figure 3B, panels a–e).
These data are consistent with previous data demonstrating that in the basal state the rate of exocytosis is relatively slow and that the inhibition of endocytosis results in the slow accumulation of GLUT4 at the cell surface (Kao et al, 1998). However, even in the presence of Dyn/K44A, insulin stimulated the translocation of newly synthesized GLUT4 at 6 h with maximal stimulation between 9 and 12 h (Figure 3B, panels f–j). Quantification of the extent of GLUT4-EGFP translocation to the plasma membrane under these conditions is presented in Figure 3C. As is readily apparent, expression of Dyn/WT had no significant effect on the basal or time-dependent acquisition of insulin-stimulated GLUT4 translocation following BFA removal (Figure 3C, open and filled squares). Similarly, the expression of Dyn/K44A had no significant effect on the time-dependent acquisition of insulin-responsive GLUT4 translocation (Figure 3C, filled circles). The slower accumulation of GLUT4 at the plasma membrane in unstimulated cells expressing Dyn/K44A (Figure 3C, open circles) is consistent with a slow basal rate of plasma membrane recycling that occurs after GLUT4 enters the GSC. It should also be noted that the greater extent of insulin-stimulated GLUT4 translocation in Dyn/K44A-expressing cells was fully accounted for by the increase in basal cell-surface GLUT4 levels. Essentially identical data were obtained when clathrin-dependent endocytosis was inhibited by the dominant-negative A1 fragment of amphiphysin or the epsin amino-terminal homology (ENTH) domain (data not shown).
The newly synthesized GLUT4 protein does not transit the plasma membrane en route to the insulin-responsive storage compartment
The Dyn/K44A construct inhibited endocytosis of the pre-existing GLUT4 protein. However, it remains formally possible that the newly synthesized GLUT4 is initially routed to and retrieved from the plasma membrane through an endocytic mechanism that is insensitive to inhibition by Dyn/K44A. To address this possibility, we utilized an exofacial myc-tagged GLUT4-EGFP construct that contains a myc-epitope tag in the large extracellular loop between transmembrane domains 1 and 2 of GLUT4 (Figure 4). This construct has been used previously in sensitive measurements of the cell-surface exposure of GLUT4 (Bogan and Lodish, 1999; Kanzaki et al, 2001; Shigematsu et al, 2002). Thus, if newly synthesized myc-GLUT4-EGFP is first routed to the plasma membrane, the exofacial myc epitope will be accessible to anti-myc antibodies present in the culture medium. Adipocytes were transfected with either myc-GLUT4-EGFP or with GLUT4-EGFP (which lacks the myc epitope) to control for nonspecific antibody binding. In the absence of dynamin expression, there was a low level of background myc reactivity on the cell surface that increased approximately seven-fold following insulin stimulation at the 9 h time point (Figure 4A, bars 1 and 2). Essentially, an identical extent of insulin-stimulated myc-GLUT4-EGFP translocation was obtained in cells coexpressing Dyn/WT (Figure 4A, bars 3 and 4). Although the maximal extent of myc-GLUT4-EGFP at the plasma membrane in the presence of insulin was the same, coexpression of Dyn/K44A increased the basal cell-surface exposure (Figure 4A, bars 5 and 6).
Figure 4.
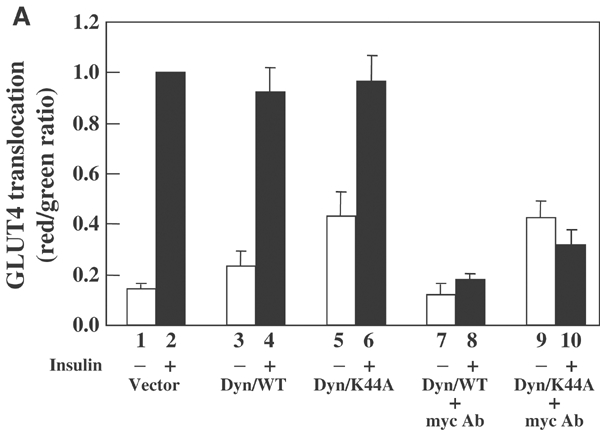
The newly synthesized GLUT4 protein does not transit the plasma membrane prior to becoming insulin responsive. (A) Differentiated 3T3L1 adipocytes were electroporated with 100 μg myc-GLUT4-EGFP with and without 100 μg Dyn/WT or Dyn/K44A cDNAs. Immediately following transfection, the cells were left untreated (bars 1–6) or incubated with 2 μg/ml of the myc monoclonal antibody (bars 7–10) for 6 h at 37°C. The cells were then serum starved in the absence (bars 1–6) or presence of 2 μg/ml of the myc monoclonal antibody (bars 7–10) for 2.5 h. The cells were then either untreated (open bars) or treated with 100 nM insulin (filled bars) for 30 min. Thus, the total duration of time in these experiments was 9 h. The cells in bars 1–6 were washed, fixed, blocked and incubated with the primary myc antibody and then Texas Red-conjugated rabbit anti-mouse secondary antibody. The cells in bars 7–10 were washed to remove the unbound myc antibody, fixed, blocked and then directly incubated with the Texas Red-labeled rabbit anti-mouse secondary antibody. Quantification of myc-GLUT4-EGFP translocation was determined. These data represent the average with standard deviation from the quantification of 30 cells in three independent experiments.
We next co-transfected cells with either Dyn/WT or Dyn/K44A and immediately incubated the cells in the continuous presence of the myc antibody. In the basal state, cells expressing Dyn/WT displayed a very low background level of exofacial exposure of the myc antibody (Figure 4A, bar 7). Following insulin stimulation, there was no significant increase in the amount of the pre-bound myc antibody (Figure 4A, bar 8). This was not due to the dissociation of the myc antibody following endocytosis for the following reasons. In control experiments, cells expressing myc-GLUT4-EGFP were incubated with the myc antibody and then treated with or without insulin. The insulin was then removed and endocytosis was allowed to occur, resulting in the retrieval of the antibody-bound myc-GLUT4-EGFP from the cell surface. Cells were then re-stimulated with insulin to allow for a second round of myc-GLUT4-EGFP translocation. This resulted in the re-exposure of the antibody-bound myc epitope to the extracellular environment (data not shown). Having established this assay system, we next coexpressed Dyn/K44A with myc-GLUT4-EGFP in the continuous presence of the myc antibody. This resulted in an increase in the basal exofacial labeling of myc-GLUT4-EGFP, but no increase following insulin stimulation (Figure 4A, bars 9 and 10).
To insure that the myc antibody was fully capable of binding under these conditions and did not dissociate following endocytosis , insulin-stimulated cells were incubated with the myc antibody (Supplementary Figure 4B). These data demonstrated that following insulin removal the GLUT4-bound myc antibody was internalized and subsequently responded to a second round of insulin stimulation. Thus, these data are consistent with the slow basal rate of accumulation of the myc-GLUT4-EGFP at the cell surface in the presence of Dyn/K44A. Together, these data demonstrate that during the 9 h incubation following the release of the BFA block, the exofacial myc epitope of GLUT4 was not accessible to the myc antibody present in the extracellular medium.
To further confirm that GLUT4 is directly transported to its responsive compartment without first transiting the plasma membrane, we also took advantage of mutations in the GLUT4 amino-terminal FQQI and carboxyl-terminal SLL motifs that markedly reduce the rate of plasma membrane endocytosis (Holman and Sandoval, 2001). As previously observed, the newly synthesized wild-type GLUT4 protein was excluded from the plasma membrane in the basal state but acquired insulin responsiveness 6–9 h following expression (Figure 5A). Similarly, expression of the endocytosis-deficient mutant GLUT4/SAA displayed essentially an identical time course of insulin-stimulated translocation (Figure 5B). In parallel, a structurally distinct endocytosis-deficient mutant AQQI also resulted in the same time-dependent acquisition of insulin-stimulated translocation (Figure 5C). It should be noted that similar to Dyn/K44A expression, at the later time points, both the GLUT4/AQQI and the GLUT4/SAA mutants slowly accumulated at the plasma membrane in the basal state. These data are consistent with the slow exit of GLUT4 from the GSC in the basal state and since endocytosis is inhibited, GLUT4 slowly accumulates at the plasma membrane. In any case, taken together these data provide compelling evidence that the newly synthesized GLUT4 protein directly traffics to the insulin-responsive storage compartment and acquires insulin sensitivity independent of plasma membrane endocytosis.
Figure 5.

Expression of an endocytosis-defective GLUT4 mutant does not impair insulin stimulation of the newly synthesized GLUT4 protein. Differentiated 3T3L1 adipocytes were electroporated with 50 μg of the wild-type GLUT4-EGFP fusion protein (A, GLUT4/WT), the carboxyl-terminal endocytosis-defective GLUT4-EGFP mutant (B, GLUT4/SAA) or the amino-terminal endocytosis-defective GLUT4-EGFP mutant (B, GLUT4/AQQI). At the times indicated, the cells were then either left untreated (filled circles) or stimulated with 100 nM insulin (open circles) for 30 min. The cells were then fixed and processed for confocal fluorescent microscopy. The data were quantified by determining the number of adipocytes displaying a continuous myc-GLUT4 rim fluorescence. These data represent the average with standard deviation from the counting of 100–150 cells from 4–6 independent experiments.
Dominant-interfering GGA mutant inhibits entry of newly synthesized GLUT4 into the GSC
Having established that newly synthesized GLUT4 directly traffics to the GSC, we next investigated the requirements for this initial sorting decision. Recent studies have observed that the GGA (Golgi-localized, γ-ear-containing ARF-binding proteins) family of vesicle coat proteins functions in the TGN exit of the cation-independent mannose-6-phosphate receptor (CI-MPR) (Puertollano et al, 2001). Since the CI-MPR appears to partially colocalize with GLUT4 (Kandror and Pilch, 1996; Rea and James, 1997), we examined the effect of a dominant-negative GGA mutant (VHS-GAT) on the acquisition of insulin-stimulated GLUT4 translocation (Figure 6). As expected, expression of VHS-GAT completely blocked the TGN exit of newly synthesized MPR (data not shown). Similarly, expression of VHS-GAT markedly reduced the extent of plasma membrane-localized GLUT4 in the basal state and completely prevented insulin-stimulated GLUT4 translocation (Figure 6A and B). However, expression of VHS-GAT had no significant effect on the appearance of newly synthesized GLUT1 or VSV-G protein at the plasma membrane (Figure 6C and D). As controls, the expression of full-length GGA had no significant effect on insulin-stimulated GLUT4 translocation or constitutive plasma membrane localization of GLUT1 and VSV-G protein (data not shown).
Figure 6.

The acquisition of insulin-stimulated GLUT4 translocation is GGA dependent. (A) Differentiated 3T3L1 adipocytes were electroporated with 100 μg myc-GLUT4 plus 100 μg of the empty vector (pcDNA3). At various times following transfection, the cells were either left untreated (open circles) or stimulated with 100 nM insulin (filled circles) for 30 min. The cells were then fixed, labeled with a myc antibody and a Texas Red-conjugated donkey anti-mouse secondary antibody to detect myc-GLUT4 at the cell surface. The data were quantified by determining the number of adipocytes displaying continuous myc-GLUT4 rim fluorescence. These data represent the average with standard deviation from the counting of 100 cells from four independent experiments. (B) Differentiated 3T3L1 adipocytes were electroporated with 100 μg myc-GLUT4 plus 100 μg of the cDNA encoding the dominant-interfering GGA mutant EGFP fusion protein (VHS-GAT). The cells were either left untreated (open circles) or stimulated with 100 nM insulin (filled circles) and analyzed for GLUT4 translocation as described above. (C) Differentiated 3T3L1 adipocytes were electroporated with 100 μg of the cDNA encoding myc-GLUT1 plus 100 μg of the empty vector (filled squares) or 100 μg myc-GLUT1 cDNA plus 100 μg of the cDNA encoding the dominant-interfering GGA mutant EGFP fusion protein (open squares). At various times following transfection, the cells were fixed, labeled with a myc antibody and a Texas Red-conjugated donkey anti-mouse secondary antibody to detect myc-GLUT1 at the cell surface as described above. (D) Differentiated 3T3L1 adipocytes were electroporated with 100 μg of the cDNA encoding VSV-G protein plus 100 μg of the empty vector (filled squares) or 100 μg VSV-G cDNA plus 100 μg of the cDNA encoding the dominant-interfering GGA mutant EGFP fusion protein (open squares). These data represent the average with standard deviation from the counting of 100 cells from four independent experiments.
The inhibitory effect of VHS-GAT could be a result of a block in GLUT4 entry into the insulin-responsive compartment and/or inhibition of GLUT4 exit from this compartment. To distinguish between these possibilities, we examined the effect of VHS-GAT on the endogenous GLUT4 protein that is already present in the GSC (Figure 7). Microinjection of the empty vector with the plasma membrane marker MBP-Ras demonstrated that in the basal state there was a very low level of GLUT4 in the plasma membrane in both the microinjected and surrounding non-microinjected cells (Figure 7, panels a–c). In contrast, cells stimulated with insulin demonstrated a dramatic increase in GLUT4 at the cell surface (Figure 7, panels d–f). Importantly, cells microinjected with the dominant-interfering VHS-GAT cDNA displayed an identical extent of insulin-stimulated GLUT4 translocation compared to the surrounding non-microinjected cells. Previous studies have demonstrated that overexpression of TC10 markedly inhibits insulin-stimulated GLUT4 translocation from the GSC (Chiang et al, 2001; Watson et al, 2001). As a control, microinjection of the TC10 cDNA markedly inhibited insulin-stimulated GLUT4 translocation compared to the surrounding non-microinjected cells (Figure 7, panels g–i). Quantification of these results is presented in Figure 7B. Together, these data demonstrate that VHS-GAT expression blocks the entry of newly synthesized GLUT4 into its responsive compartment, but does not affect the insulin-stimulated exit from the GSC.
Figure 7.

Insulin-stimulated GLUT4 trafficking from the GSC to the plasma membrane is independent of GGA function. (A) Differentiated 3T3L1 adipocyte nuclei were microinjected with 50 μg of the MBP-Ras fusion cDNA plus 200 μg of either the empty vector (panels a–c), VHS-GAT (panels d–f) or TC10 (panels g–i) cDNAs. The cells were allowed to recover for 24 h and either left untreated (panels a–c) or stimulated with 100 nM insulin (panels d–i) for 30 min. Plasma membrane sheets were then prepared and labeled with an MBP antibody (panels a, d, g) or a GLUT4 antibody (panels b, e, h). The merge images are shown in panels c, f and i. These are representative images obtained from three independent experiments with the visualization of 60 individual MBP-Ras-positive plasma membrane sheets. (B) These data were quantified by counting the number of plasma membrane sheets from microinjected cells that displayed endogenous GLUT4 translocation.
VHS-GAT mutant inhibits the in vitro budding of GLUT4 vesicles
Recently, we have established that isolated heavy (rapidly sedimenting) donor membranes from adipocytes can be used to reconstitute GLUT4 vesicle budding in a temperature-, ATP- and cell extract (cytosol)-dependent manner (Xu and Kandror, 2002). As expected, incubation of donor membranes with GTP and ATP in the absence of cytosol did not result in any significant vesicle protein release (Figure 8A, lane 1). In contrast, the addition of cytosol plus the control GST protein resulted in the release of membrane vesicles containing GLUT4, GLUT1 and the transferrin receptor (Figure 8A, lanes 2 and 3). Incubation of donor membranes with 3 and 30 μg of the VHS-GAT fusion protein resulted in a dose-dependent decrease in the release of vesicles containing the GLUT4 protein (Figure 8A, lanes 4–7). This also occurred with essentially no change in the budding of transferrin receptor-containing vesicles and with a relatively small effect on GLUT1 vesicles. Furthermore, the relative percentage of transferrin receptor in the donor membranes was significantly less than that of GLUT4 and GLUT1 (Figure 8B). Quantification of the vesicle budding data is presented in Supplementary Figure 8C.
Figure 8.
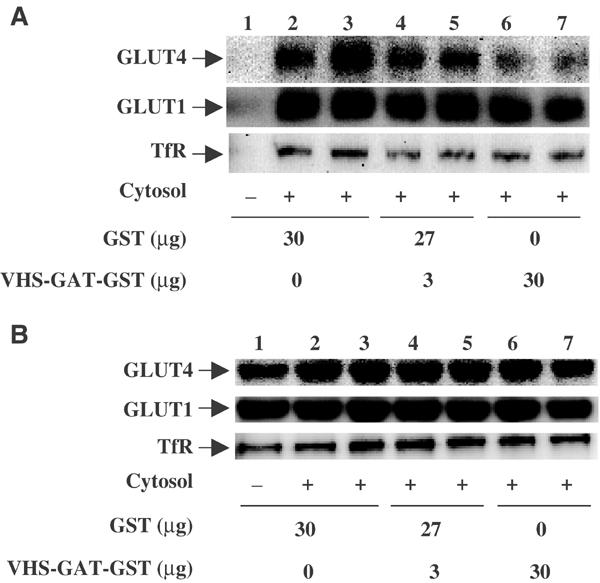
The dominant-interfering GGA mutant inhibits GLUT4 vesicle budding in vitro. (A) Golgi- and endosome-enriched donor membranes were incubated with ATP and GTPγS in the absence (lane 1) or presence (lanes 2–7) of cytosol and as described under Materials and methods. The reactions were also performed in the presence of 30 μg GST (lanes 1–3), 3 μg (lanes 4 and 5) and 30 μg (lanes 6 and 7) of the VHS-GAT-GST fusion protein. The released membrane vesicles (A) and Golgi imput membranes (B) were then subject to immunoblotting using GLUT4, GLUT1 and transferrin receptor (TfR) antibodies. This is a representative immunoblot independently performed four times.
Discussion
One inherent question with any model of regulated membrane trafficking is the source of the cargo protein that enters the responsive compartment. Since GLUT4 is a very stable protein that traffics to the cell surface even in the basal state and subsequently undergo endocytosis, there must be a sorting process within endosomes such that GLUT4 is returned, directly or indirectly, to the GSC (Rea and James, 1997; Holman and Sandoval, 2001). Indeed, following GLUT4 exofacial labeling, the internalized GLUT4 equilibrates with the unlabeled intracellular GLUT4 population and can undergo subsequent rounds of insulin-stimulated translocation (Satoh et al, 1993; Foster et al, 2001; Lampson et al, 2001; Palacios et al, 2001; Shigematsu et al, 2002). However, although numerous studies have examined the steady-state trafficking dynamics of GLUT4, to our knowledge the trafficking route by which newly synthesized GLUT4 initially enters the GCS has not been experimentally studied.
To address this question, we examined the sorting of GLUT4 immediately following its initial biosynthesis thereby avoiding its equilibration with numerous recycling compartments. By examining cells at short time intervals following transient transfection, we have been able to monitor the temporal acquisition of insulin responsiveness for the newly synthesized GLUT4 protein. Whereas VSV-G and GLUT1 localized to the plasma membrane in as little as 2–3 h post-transfection, GLUT4 was retained within the perinuclear region and required 9–12 h to display the full extent of insulin-stimulated plasma membrane translocation. This temporal delay was not due to the inhibition of insulin signaling, as the endogenous GLUT4 translocated normally in cells examined 3 h post-transfection. Thus, our results are consistent with a model wherein newly synthesized GLUT4 undergoes a time-dependent sorting process necessary for the acquisition of insulin responsiveness.
We evaluated whether this temporal delay was due to the additional time required for GLUT4 to undergo plasma membrane trafficking, internalization and subsequent endosomal sorting into the GSC. Using several independent criteria, there was no apparent requirement for the newly synthesized GLUT4 to transit the plasma membrane in order to display insulin-stimulated translocation. This included inhibition of plasma membrane endocytosis by coexpression of a dominant-interfering dynamin mutant and by expression of endocytosis-defective GLUT4 mutants. In addition, continuous incubation with an exofacial myc tag antibody also demonstrated that GLUT4 does not transit the cell surface en route to the GSC. Thus, these data provide compelling evidence that the initial sorting of newly synthesized GLUT4 occurs independently of plasma membrane recycling and therefore must exclusively result from intracellular trafficking events.
Having established this system, we observed that expression of the dominant-interfering VHS-GAT domain of GGA completely inhibited the insulin-stimulated translocation of the newly synthesized GLUT4 protein. Importantly, this mutant had no significant effect on the insulin-stimulated translocation of endogenous GLUT4. Since the transfected GLUT4 behaves identically to endogenous GLUT4 in adipocytes (Dobson et al, 1996; Elmendorf et al, 1999), our data suggest that entry of GLUT4 into the GSC is GGA dependent whereas insulin-stimulated exit from this compartment is GGA independent. This hypothesis is consistent with the inability of VHS-GAT to block the accumulation of constitutive plasma membrane trafficking proteins such as GLUT1 and VSV-G. Furthermore, VHS-GAT strongly inhibited GLUT4 vesicle budding in vitro with relatively little effect on GLUT1 or transferrin receptor vesicle formation. Although these experiments do not distinguish between newly synthesized and recycling GLUT4, they nevertheless show that specific GGA-dependent cargo selection processes operate for GLUT4 vesicle budding events.
Despite these dramatic effects of the dominant-interfering GGA mutant on GLUT4 in vivo and in vitro, GLUT4 does not contain a consensus VHS binding site (acidic cluster-dileucine motif) and we have been unable to detect a direct interaction of GLUT4 with the VHS or the VHS-GAT domains. Nevertheless, the VHS domain can specifically precipitate intracellular GLUT4-containing membrane compartments (unpublished results), suggesting that the GGA-dependent cargo selection of GLUT4 results from the interaction with intermediate adaptor proteins.
The intracellular sorting decision for newly synthesized GLUT4 most likely occurs between the TGN and a post-TGN compartment since the VHS-GAT domain blocks the exit of the MPR from the TGN en route to late endosomes (Puertollano et al, 2001). Although GLUT4 has been suggested to also recycle between endosomes and the TGN (Shewan et al, 2003), it is also possible that the newly synthesized GLUT4 traffics through an intermediate endosome compartment between the TGN and the GSC. In either case, our data are most consistent with the model presented in Figure 9. Following translation and integration into the endoplasmic reticulum, both GLUT1 and GLUT4 are transported to the Golgi and arrive at the TGN. GLUT1 takes a default pathway, similar to constitutive secretion, and is directly transported to the plasma membrane. In contrast, GLUT4 is sorted from GLUT1 through a GGA-dependent coat process and arrives at the GSC, either directly from the TGN or through an intermediate endosome compartment. In the basal state, there is a slow rate of exit from the GSC, but following insulin stimulation exit from this compartment is markedly enhanced. The ability to now functionally distinguish between the initial sorting of GLUT4 into the GSC and the subsequent endocytotic trafficking steps will allow for more detailed analysis of the functional domains and mechanisms responsible for these distinct trafficking decisions.
Figure 9.

Schematic representation of the trafficking of newly synthesized GLUT4 protein in adipocytes. GLUT4 protein undergoes co-translational insertion into the endoplasmic reticulum and traffics through the Golgi into the TGN. GLUT4 exit from the TGN is distinct from GLUT1 protein in that GLUT4 does not initially traffic to the plasma membrane but is targeted directly to the GSC. The exit of GLUT1 from the TGN is GGA independent, whereas the transport of GLUT4 from the TGN to the GSC is GGA dependent. In the basal state, GLUT4 slowly leaves the GSC en route to the plasma membrane where it is recycled back to the GSC through endocytosis and endosome sorting. In the presence of insulin, the transport of GLUT4 out of the GSC is markedly increased and exit from this compartment is GGA independent.
Materials and methods
Culture and transfection of 3T3L1 adipocytes
Murine 3T3L1 adipocytes were cultured and electroporated as previously described (Watson et al, 2001). Following electroporation, cells were allowed to recover for 2–24 h prior to insulin stimulation. Cells that were treated with insulin at the 2 and 3 h time points post-transfection were plated directly into a serum-free medium. For all other time points, cells were first plated in complete media and then switched to the serum-free medium 2–3 h prior to insulin stimulation.
Enhanced green fluorescent protein-tagged constructs
The EGFP-tagged GLUT1, GLUT4 and CI-MPR were prepared as described (Elmendorf et al, 1999; Baumann et al, 2000). The vesicular stomatitis virus G protein (VSV-G) cDNA was obtained from the University of Iowa DNA Core Facility. BFA was from Sigma and used at a final concentration of 5 μg/ml. Adipocytes were processed for confocal microscopy as described previously (Watson et al, 2001). The myc (9E10) and HA monoclonal antibodies were from Santa Cruz Biotechnology and the VSV-G antibody was from Accurate Chemicals. Secondary antibodies were from Jackson Immunoresearch. Images were imported into Adobe Photoshop (Adobe Systems, Inc.) for processing and composite images were generated.
GLUT4 translocation
The quantification of plasma membrane translocation was determined by counting the number of cells displaying a continuous cell-surface fluorescence as described previously (Watson et al, 2001). This was validated by comparison with a single-cell myc-GLUT4-EGFP quantification assay that determines the cell-surface exposure of the myc epitopte normalized for the amount of EGFP expression (Shigematsu et al, 2002). In turn, this was compared with a total transfected cell population GLUT4 translocation assay. In brief, adipocytes were electroporated with either the empty vector or myc-GLUT4 cDNAs (200 μg) and at various times incubated with and without 100 nM insulin for 30 min. The cells were washed with ice-cold PBS and treated with 2 mM KCN for 5 min. Cells were then incubated with 5 μg/ml of the myc monoclonal antibody followed by a second incubation with an HRP-conjugated goat anti-mouse antibody at 1:1000 dilution. The samples were then incubated with 0.4 mg/ml o-phenylenediamine dihydrochloride and 0.4 mg/ml urea hydrogen for 2 min and the reactions were terminated with 3 N HCl. The resultant products were determined by absorbance at 492 nm.
Quantitative exofacial labeling of myc-GLUT4-EGFP
The cell-surface levels of the exofacial myc-tagged GLUT4-EGFP (myc-GLUT4-EGFP) were assayed as described (Shigematsu et al, 2002). To determine the transit of myc-GLUT4-EGFP to the plasma membrane, cells were incubated in the continuous presence of 2 μg/ml myc antibody. Cells were then stimulated with 100 nM insulin for 30 min, extensively washed with ice-cold PBS, fixed in 4% paraformaldehyde, and labeled with the Texas Red-conjugated anti-mouse secondary antibody. The Texas Red/EGFP intensity ratios were measured in 10 cells/experiment for each condition as described (Shigematsu et al, 2002). Under these conditions, there is an approximately 400-fold molar excess of the myc antibody compared to the expressed myc-GLUT4-EGFP protein.
Microinjection and plasma membrane sheet assays
The microinjection and visualization of single 3T3L1 adipocytes was performed as described previously (Baumann et al, 2000). Briefly, the cells were grown on coverslips and the medium was changed to Lebovitz's L-15 medium containing 0.1% bovine serum albumin (BSA) prior to microinjection. Differentiated 3T3L1 adipocytes were impaled using Eppendorf model 5171 micromanipulator and nuclei were injected with 50 μg/ml of the MBP-Ras fusion cDNA plus 200 μg/ml of cDNAs in 100 mM KCl and 5 mM Na2PO4 (pH 7.2) with an Eppendorf model 5246 transinjector. The cells were allowed to recover for 24 h and then stimulated with 100 nM insulin for 30 min followed by the preparation of plasma membrane sheets as described previously (Elmendorf et al, 1998).
In vitro GLUT4 vesicle budding assay
Differentiated 3T3-L1 adipocytes were incubated in serum-free media for 2 h, homogenized and then centrifuged at 16 000 g for 20 min. The supernatant was centrifuged again at 200 000 g for 60 min. The budding reaction mixture consisted of the pellet of the 16 000 g centrifugation (250 μg), the supernatant of the 200 000 g centrifugation (500 μg) and an ATP regeneration system (1 mM ATP, 8 mM creatine phosphate, 1.5 U/ml creatine phosphokinase) as described previously (Xu and Kandror, 2002). This mixture (total volume 250 μl) was incubated with 100 μM GTPγS and the indicated amounts of GST or GST-VHS-GAT fusion protein at 4°C for 20 min. The samples were incubated at 37°C for 20 min and centrifuged again at 16 000 g and then at 200 000 g. The pellet of the 200 000 g centrifugation that contained de novo formed vesicles was then subjected to Western blot analysis.
Supplementary Material
Supplementary Figure 1
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 8
Acknowledgments
This study was supported by National Institutes of Health research grants to JEP (DK55811 and DK33823) and to KVK (DK52057 and DK56736).
References
- Al-Hasani H, Hinck CS, Cushman SW (1998) Endocytosis of the glucose transporter GLUT4 is mediated by the GTPase dynamin. J Biol Chem 273: 17504–17510 [DOI] [PubMed] [Google Scholar]
- Baumann CA, Ribon V, Kanzaki K, Thurmond DC, Mora S, Shigematsu S, Bickel PE, Pessin JE, Saltiel AR (2000) CAP defines a second signaling pathway required for insulin-stimulated glucose transport. Nature 407: 202–207 [DOI] [PubMed] [Google Scholar]
- Bergmann JE (1989) Using temperature-sensitive mutants of VSV to study membrane protein biogenesis. Methods Cell Biol 32: 85–110 [DOI] [PubMed] [Google Scholar]
- Bogan JS, Lodish HF (1999) Two compartments for insulin-stimulated exocytosis in 3T3-L1 adipocytes defined by endogenous ACRP30 and GLUT4. J Cell Biol 146: 609–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant NJ, Govers R, James DE (2002) Regulated transport of the glucose transporter GLUT4. Nat Rev Mol Cell Biol 3: 267–277 [DOI] [PubMed] [Google Scholar]
- Chiang SH, Baumann CA, Kanzaki M, Thurmond DC, Watson RT, Neudauer CL, Macara IG, Pessin JE, Salteil AR (2001) Insulin-stimulated GLUT4 translocation requires the CAP-dependent activation of TC10. Nature 410: 944–948 [DOI] [PubMed] [Google Scholar]
- Czech MP, Corvera S (1999) Signaling mechanisms that regulate glucose transport. J Biol Chem 274: 1865–1868 [DOI] [PubMed] [Google Scholar]
- de Wit H, Lichtenstein Y, Geuze HJ, Kelly RB, van der Sluijs P, Klumperman J (1999) Synaptic vesicles form by budding from tubular extensions of sorting endosomes in PC12 cells. Mol Biol Cell 10: 4163–4176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobson SP, Livingstone C, Gould GW, Tavare JM (1996) Dynamics of insulin-stimulated translocation of GLUT4 in single living cells visualised using green fluorescent protein. FEBS Lett 393: 179–184 [DOI] [PubMed] [Google Scholar]
- Elmendorf JS, Boeglin DJ, Pessin JE (1999) Temporal separation of insulin-stimulated GLUT4/IRAP vesicle plasma membrane docking and fusion in 3T3L1 adipocytes. J Biol Chem 274: 37357–37361 [DOI] [PubMed] [Google Scholar]
- Elmendorf JS, Chen D, Pessin JE (1998) Guanosine 5′-O-(3-thiotriphosphate) (GTPgammaS) stimulation of GLUT4 translocation is tyrosine kinase-dependent. J Biol Chem 273: 13289–13296 [DOI] [PubMed] [Google Scholar]
- Foster LJ, Li D, Randhawa VK, Klip A (2001) Insulin accelerates inter-endosomal GLUT4 traffic via phosphatidylinositol 3-kinase and protein kinase B. J Biol Chem 276: 44212–44221 [DOI] [PubMed] [Google Scholar]
- Garza LA, Birnbaum MJ (2000) Insulin-responsive aminopeptidase trafficking in 3T3-L1 adipocytes. J Biol Chem 275: 2560–2567 [DOI] [PubMed] [Google Scholar]
- Helliwell SB, Losko S, Kaiser CA (2001) Components of a ubiquitin ligase complex specify polyubiquitination and intracellular trafficking of the general amino acid permease. J Cell Biol 153: 649–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holman GD, Sandoval IV (2001) Moving the insulin-regulated glucose transporter GLUT4 into and out of storage. Trends Cell Biol 11: 173–179 [DOI] [PubMed] [Google Scholar]
- Kandror KV, Pilch PF (1994) gp160, a tissue-specific marker for insulin-activated glucose transport. Proc Natl Acad Sci USA 91: 8017–8021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandror KV, Pilch PF (1996) Compartmentalization of protein traffic in insulin-sensitive cells. Am J Physiol 271: E1–E14 [DOI] [PubMed] [Google Scholar]
- Kanzaki M, Watson RT, Khan AH, Pessin JE (2001) Insulin stimulates actin comet tails on intracellular GLUT4 containing compartments in differentiated 3T3L1 adipocytes. J Biol Chem 276: 49331–49336 [DOI] [PubMed] [Google Scholar]
- Kao AW, Ceresa BP, Santeler SR, Pessin JE (1998) Expression of a dominant interfering dynamin mutant in 3T3L1 adipocytes inhibits GLUT4 endocytosis without affecting insulin signaling. J Biol Chem 273: 25450–25457 [DOI] [PubMed] [Google Scholar]
- Klausner RD, Donaldson JG, Lippincott-Schwartz J (1992) Brefeldin A: insights into the control of membrane traffic and organelle structure. J Cell Biol 116: 1071–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampson MA, Schmoranzer J, Zeigerer A, Simon SM, McGraw TE (2001) Insulin-regulated release from the endosomal recycling compartment is regulated by budding of specialized vesicles. Mol Biol Cell 12: 3489–3501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmon SK, Traub LM (2000) Sorting in the endosomal system in yeast and animal cells. Curr Opin Cell Biol 12: 457–466 [DOI] [PubMed] [Google Scholar]
- Malide D, St-Denis JF, Keller SR, Cushman SW (1997) Vp165 and GLUT4 share similar vesicle pools along their trafficking pathways in rat adipose cells. FEBS Lett 409: 461–468 [DOI] [PubMed] [Google Scholar]
- Martin S, Rice JE, Gould GW, Keller SR, Slot JW, James DE (1997) The glucose transporter GLUT4 and the aminopeptidase vp165 colocalise in tubulo-vesicular elements in adipocytes and cardiomyocytes. J Cell Sci 110: 2281–2291 [DOI] [PubMed] [Google Scholar]
- Orci L, Glick BS, Rothman JE (1986) A new type of coated vesicular carrier that appears not to contain clathrin: its possible role in protein transport within the Golgi stack. Cell 46: 171–184 [DOI] [PubMed] [Google Scholar]
- Palacios S, Lalioti V, Martinez-Arca S, Chattopadhyay S, Sandoval IV (2001) Recycling of the insulin-sensitive glucose transporter GLUT4. Access of surface internalized GLUT4 molecules to the perinuclear storage compartment is mediated by the Phe5-Gln6-Gln7-Ile8 motif. J Biol Chem 276: 3371–3383 [DOI] [PubMed] [Google Scholar]
- Pelham HR (1991) Recycling of proteins between the endoplasmic reticulum and Golgi complex. Curr Opin Cell Biol 3: 585–591 [DOI] [PubMed] [Google Scholar]
- Pessin J, Thurmond D, Elmendorf J, Coker K, Okada S (1999) Molecular basis of insulin-stimulated GLUT4 vesicle trafficking: location! location! location!. J Biol Chem 274: 2593–2596 [DOI] [PubMed] [Google Scholar]
- Puertollano R, Aguilar RC, Gorshkova I, Crouch RJ, Bonifacino JS (2001) Sorting of mannose 6-phosphate receptors mediated by the GGAs. Science 292: 1712–1716 [DOI] [PubMed] [Google Scholar]
- Rea S, James D (1997) Moving GLUT4: the biogenesis and trafficking of GLUT4 storage vesicles. Diabetes 46: 1667–1677 [DOI] [PubMed] [Google Scholar]
- Régnier-Vigouroux A, Tooze SA, Huttner WB (1991) Newly synthesized synaptophysin is transported to synaptic-like microvesicles via constitutive secretory vesicles and the plasma membrane. EMBO J 10: 3589–3601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberg KJ, Rowley N, Kaiser CA (1997) Physiological regulation of membrane protein sorting late in the secretory pathway of Saccharomyces cerevisiae. J Cell Biol 137: 1469–1482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh S, Nishimura H, Clark AE, Kozka IJ, Vannucci SJ, Simpson IA, Quon MJ, Cushman SW, Holman GD (1993) Use of bismannose photolabel to elucidate insulin-regulated GLUT4 subcellular trafficking kinetics in rat adipose cells. Evidence that exocytosis is a critical site of hormone action. J Biol Chem 268: 17820–17829 [PubMed] [Google Scholar]
- Shewan AM, Van Dam EM, Martin S, Luen TB, Hong W, Bryant NJ, James DE (2003) GLUT4 recycles via a trans-Golgi network (TGN) subdomain enriched in syntaxins 6 and 16 but not TGN38: involvement of an acidic targeting motif. Mol Biol Cell 14: 973–986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigematsu S, Khan AH, Kanzaki M, Pessin JE (2002) Intracellular insulin-responsive glucose transporter (GLUT4) distribution but not insulin-stimulated GLUT4 exocytosis and recycling are microtubule dependent. Mol Endocrinol 16: 1060–1068 [DOI] [PubMed] [Google Scholar]
- Subtil A, Lampson MA, Keller SR, McGraw TE (2000) Characterization of the insulin-regulated endocytic recycling mechanism in 3T3-L1 adipocytes using a novel reporter molecule. J Biol Chem 275: 4787–4795 [DOI] [PubMed] [Google Scholar]
- Volchuk A, Narine S, Foster LJ, Grabs D, De Camilli P, Klip A (1998) Perturbation of dynamin II with an amphiphysin SH3 domain increases GLUT4 glucose transporters at the plasma membrane in 3T3-L1 adipocytes. Dynamin II participates in GLUT4 endocytosis. J Biol Chem 273: 8169–8176 [DOI] [PubMed] [Google Scholar]
- Watson RT, Pessin JE (2001) Intracellular organization of insulin signaling and GLUT4 translocation. Recent Prog Horm Res 56: 175–193 [DOI] [PubMed] [Google Scholar]
- Watson RT, Shigematsu S, Chiang SH, Mora S, Kanzaki M, Macara IG, Saltiel AR, Pessin JE (2001) Lipid raft microdomain compartmentalization of TC10 is required for insulin signaling and GLUT4 translocation. J Cell Biol 154: 829–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Kandror KV (2002) Translocation of small preformed vesicles is responsible for the insulin activation of glucose transport in adipose cells. Evidence from the in vitro reconstitution assay. J Biol Chem 277: 47972–47975 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 8