

Abstract
We recently documented p38δ differential expression and function in oesophageal squamous cell carcinoma (OESCC). This study expands upon these findings and investigates whether p38δ status in OESCC can influence response(s) to cytotoxic drugs. The antiproliferative effect of conventional cisplatin and 5-fluorouracil (CF) treatment was compared with the recently reviewed triple regime of cisplatin, 5-fluorouracil and doxorubicin (ACF). p38δ-positive and p38δ-negative cell lines were employed using cell-growth and clonogenic assays. Key regulators of intrinsic and extrinsic apoptotic pathways were measured. Wound-healing assays and a Boyden chamber were used to investigate the effect of drug treatments on cell migration. Functional networks were analysed in terms of changes in MAPK expression. p38δ-negative OESCC is less sensitive to standard CF chemotherapy compared with p38δ-positive cells. However, following ACF treatment p38δ-negative cells showed markedly decreased proliferation and cell migration, and increased apoptosis. ACF induced apoptosis through the extrinsic pathway involving Fas activation, caspase-8 and caspase-3 cleavage and degradation of PARP. Loss of mitochondrial membrane potential (ΔΨm) was observed but downregulation of multidomain proapoptotic proteins, as well as BH3-only proteins, suggests involvement of pathways other than the mitochondrial pathway. Interestingly, induction of p38 and ERK1/2, but not JNK1/2, was observed following ACF treatment. p38δ-negative OESCC is more resistant to traditional CF treatment compared with p38δ-positive OESCC. In light of these results, p38δ phenotyping of tumour tissue may be of considerable value in deciding on an optimal therapeutic strategy for patients with p38δ-negative OESCC.
Keywords: cisplatin, doxorubicin, 5-fluorouracil, oesophageal squamous cell carcinoma, p38δ MAPK
Introduction
Oesophageal cancer is a highly aggressive and fatal malignancy and is the seventh most common cancer worldwide 1. Oesophageal squamous cell carcinoma (OESCC) is an exceptionally drug-resistant tumour. Although surgery is the best modality in terms of local control 2, outcomes following resection for OESCC remain unsatisfactory because of locoregional and distant failure 3. Preoperative chemotherapy or chemoradiotherapy with a fluoropyrimidine/platinum combination – that is, a cisplatin and 5-fluorouracil (CF) regimen – has been the standard treatment for locally advanced disease since the 1980s. At present, multimodal therapy is being investigated for different stages of OESCC, even if the tumour is operable 4. Preoperative chemotherapy with docetaxel plus CF (DCF) has recently been investigated (with or without radiotherapy) with good local control and pathological remission rate being recorded 4,5. More recently doxorubicin, cisplatin and 5-fluorouracil (ACF) have undergone a revival, demonstrating higher response rates than CF treatment, a good safety profile and promising long-term outcomes for patients with highly advanced oesophageal carcinoma 6–8.
The involvement of p38 MAPKs in a variety of pathological conditions is continuing to fuel interest in this particular family of kinases. It consists of four isoforms: p38α (MAPK14), p38β (MAPK11), p38γ (MAPK12) and p38δ (MAPK13), which to date remains the least studied isoform 9. The expression of p38 as a family has previously been outlined in oesophageal cancer, as well as in other cancer types 10–13. We recently outlined for the first time the differential expression of individual p38 isoforms in cancer and in particular OESCC 14,15. We now know that loss of p38δ expression in OESCC affords a more sinister phenotype, with increased proliferation, migration and anchorage-independent growth, thus identifying p38δ as a possible molecular target in OESCC 15.
Advancing our studies a step further we evaluated whether p38δ status could influence cytotoxic responses to drug treatments in OESCC. We used both negative and positive p38δ cell lines isolated from patients with OESCC with no prior treatment, as previously outlined by us 15. Cell viability, wound healing, migration and apoptosis were evaluated following standard CF treatment and ACF treatment. To carry out functional networks expression analysis, we also analysed changes in ERK1/2, JNK1/2 and p38 MAPK expression. In conclusion, our study indicates that p38δ status may be a predictor of response to chemotherapy in OESCC patients.
Materials and methods
Reagents
All chemicals and cell culture reagents were purchased from Sigma Aldrich (Wicklow, Ireland) and primary antibodies from Cell Signalling Technologies (Hertfordshire, UK), unless otherwise stated.
Cell culture
The KE oesophageal squamous cancer cell lines were a kind gift from Professor T. Fujii, Kurume University School of Medicine, Japan 15–18. Cells were cultured in RPMI-1640 supplemented with 10% foetal calf serum, 100 µg/ml streptomycin and 100 U/ml penicillin. KE cell line features have been summarized previously by us 15.
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide assay
Cell viability was assessed using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay, which depends on the ability of viable cells to reduce MTT to a coloured formazan product, as described previously 19.
Boyden chamber cell migration in-vitro assay
Cells were plated in starvation medium at a density of 3×104 cells/well onto a 96-well plate of the upper chamber. The bottom chamber contained 10% foetal calf serum as the chemoattractant. Cells were left to migrate for 24 h through the matrigel filter (8 μm) and stained as previously described 19.
Wound-healing assay
Cell migration was assessed using an in-vitro wound-healing assay as previously described 20. Cells migrating into the wound were photographed under a phase-contrast microscope 48 h after wounding. Migration was determined using the ImageJ (National Institutes of Health, Bethesda, Maryland, USA) program as an average closed area of the wound relative to the initial wound area at 48 h after wounding.
Mitochondrial membrane potential (ΔΨm) assay
The decline of mitochondrial membrane potential (ΔΨm) was assessed using a mitochondrial voltage-sensitive dye, 5,5′,6,6′-tetrachloro-1,1′,3,3′- tetraethylbenzimidazole carbocyanide iodide (JC-1), according to the manufacturer’s instructions 21. The dye underwent a reversible change in fluorescence emission from red (i.e. aggregate forms of JC-1) to green (i.e. monomer forms of JC-1) as the mitochondrial membrane potential decreased. Briefly, KE cells were or were not treated with drugs for 24 h. Cells were then washed twice with PBS and loaded with JC-1 for 30 min at 37°C. Images were obtained using an inverted fluorescence microscope (Olympus, Southend-on-Sea, UK). The ratio of red to green fluorescence intensity, which was indicative of a change in ΔΨm, was calculated using the ImageJ software.
Immunoblot analysis
Supernatants used for immunoblotting with specific antibodies, PARP, caspases-3 and 8, Puma, Bak, Bik, Bim, Bid, Bax, p38, phospho-p38, JNK1/2, phospho-JNK1/2, ERK1/2 and phospho-ERK1/2 (New England Biolabs, Hertfordshire, UK), as well as Fas and FasL (Santa Cruz Biotechnology, Santa Cruz, California, USA), have previously been described by us 14,15. Chemiluminescence detection was performed using SuperSignal WestDura Extended Duration Substrate (Pierce Biotechnology, Rockford, Illinois, USA), and bands were visualized using a Syngene G:Box ChemiXR5 Gel Documentation System (Syngene, Cambridge, UK). Images were quantified using ImageJ software.
Colony formation assay
A colony formation assay determines whether cells can recover from treatment. Following treatment, viable cells were reseeded in fresh media (without drug) in a six-well plate (in triplicate) and allowed to grow for 14 days. Colonies were stained with MTT and subsequently counted using ImageJ software.
Statistical analysis
Statistical comparisons were made by analysis of variance with subsequent application of Student’s t-test, as appropriate. The non-parametric Mann–Whitney U-test pairwise comparisons were also performed. As the results obtained using both methods were in agreement, only results for the Student’s t-test are shown.
Results
p38δ MAPK-negative OESCC shows decreased sensitivity to chemotherapeutic drugs
Monotherapy is not beneficial in the treatment of patients with oesophageal cancer 22. Thus, we evaluated the cell viability of KE p38δ-positive and p38δ-negative OESCC cell lines (differential p38δ expression was recently reported by us 15) following double versus triple drug treatments using a range of concentrations of cytotoxic drugs. Interestingly, KE-3 and KE-8 (p38δ-negative cell lines) are significantly less sensitive to CF treatment compared with KE-4, KE-5, KE-6 and KE-10 (p38δ-positive cell lines; Fig. 1a–c). As docetaxel has recently been added to the CF regime 4,5, we investigated the effect of DCF on cell viability. There was a further decrease in KE-3 and KE-8 cell viability (Fig. 1a and b). However, no further reduction in cell viability was observed in the KE p38δ-positive cell lines upon DCF treatment using physiologically relevant drug concentrations (30 μmol/l; Fig. 1c) 23–25. As doxorubicin (together with CF) has recently re-emerged as a promising drug treatment strategy for patients with oesophageal cancer 6–8, we also investigated its effects. Interestingly, the greatest loss in KE-3 and KE-8 cell viability was after triple CF plus doxorubicin (ACF) treatment (Fig. 1a and b). Of note, these results are comparable with the loss in cell viability seen in the p38δ-positive cells upon CF treatment at physiological concentrations (Fig. 1c). Taking the results we obtained with physiological drug concentrations of CF (30 μmol/l each) a step further, we investigated the appropriate triple ACF drug treatment concentrations for KE-3 and KE-8 cell lines. Using a range of doxorubicin drug concentrations, we ascertained that the best triple ACF concentration using physiological concentrations of all three drugs is 30 μmol/l each of CF and 1 μmol/l doxorubicin (Fig. 1d). No significant further decrease in cell viability was observed at a higher doxorubicin drug concentration of 3 μmol/l (Fig. 1d).
Fig. 1.
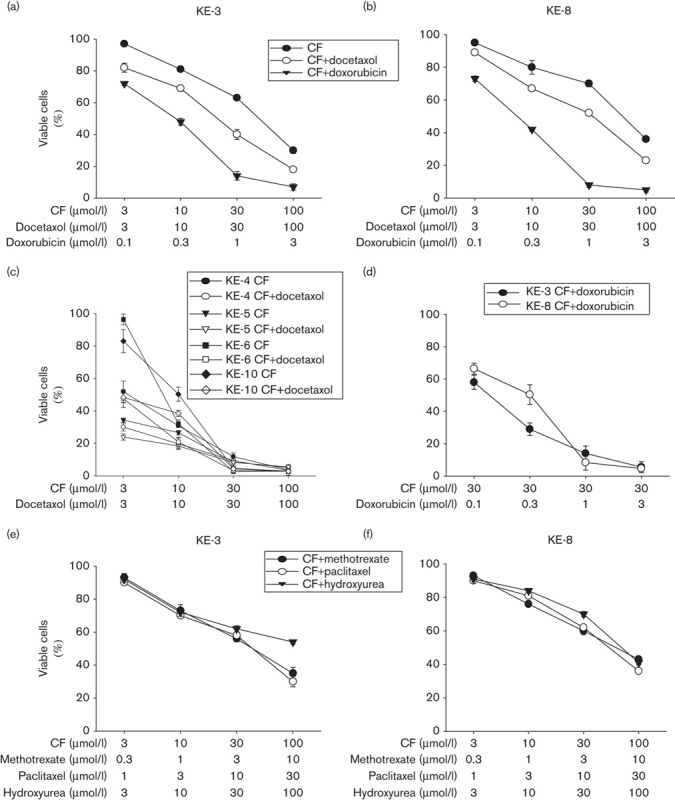
Sensitivity of OESCC to cancer chemotherapeutic drugs is correlated to the presence or absence of p38δ MAPK. (a–f) KE cell lines (KE-3 and KE-8, p38δ negative; KE-4, KE-5, KE-6 and KE-10, p38δ positive) were seeded (2×105) in six-well plates, and cell viability was assessed using the MTT assay (as described in the Materials and methods section) at 48 h to determine the sensitivity of each cell line to different drug combinations. (a, b) KE-3 and KE-8 cells were subjected to double treatment with cisplatin (3–100 μmol/l) and 5-fluoruracil (3–100 μmol/l; CF), or triple treatment with cisplatin (3–100 μmol/l), 5-fluorouracil (3–100 μmol/l; CF) and either docetaxel (3–100 μmol/l) or doxorubicin (0.1–3 μmol/l). (c) KE-4, KE-5, KE-6 and KE-10 cells were treated either with cisplatin (3–100 μmol/l) and 5-fluoruracil (3–100 μmol/l; CF), or with cisplatin (3–100 μmol/l), 5-fluorouracil (3–100 μmol/l; CF) and docetaxel (3–100 μmol/l). (d) KE-3 and KE-8 cells were subjected to triple treatment with constant drug concentrations of cisplatin (30 μmol/l) and 5-fluorouracil (30 μmol/l; CF) plus varying drug concentrations of doxorubicin (0.1–3 μmol/l). (e, f) KE-3 and KE-8 cells were subjected to triple treatment with cisplatin (3–100 μmol/l), 5-fluorouracil (3–100 μmol/l; CF) and either methotrexate (0.3–10 μmol/l), paclitaxel (1–30 nmol/l) or hydroxyurea (3–100 μmol/l). Results shown in (a–f) are mean±SE of three independent experiments. MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide; OESCC, oesophageal squamous cell carcinoma.
As other chemotherapeutic drug combinations have also been tested for their efficacy in patients with OESCC, namely CF plus methotrexate 26, CF plus paclitaxel 26,27 and CF plus hydroxyurea 28, we also investigated these three additional drug combinations again using a range of different drug concentrations. Interestingly, KE-3 and KE-8 showed less sensitivity to all three triple-drug combinations compared with ACF treatment (Fig. 1e and f). Thus, as the greatest loss in KE-3 and KE-8 cell viability was observed with triple ACF treatment (Fig. 1a, b and d), all subsequent experiments compared traditional CF treatment with ACF treatment.
ACF treatment significantly delays wound healing and migration compared with CF treatment
A key characteristic of cancer cells is their ability to migrate and progress from primary tumours to metastases in distant organs 15. We examined whether the p38δ status of OESCCs could influence their wound healing and cell migration following drug treatment. Initially, we identified the highest concentration of drug(s) that does not lead to loss of cell viability – that is, 3 μmol/l of each drug for CF and 300 nmol/l doxorubicin (data not shown). Double CF treatment of KE-6 and KE-10 (p38δ-positive cells) for 48 h brought about a significant delay in wound healing, with a 60.0±7 and 66.0±1.2% loss in wound healing, respectively, compared with untreated cells (Fig. 2a and b). In contrast, CF treatment did not impair the ability of KE-3 and KE-8 cells to migrate into the wound (Fig. 2a and b). Triple ACF treatment, however, significantly delayed the wound-healing ability of KE-3 and KE-8 cell lines, with a 72.8±2.5 and 84.0±3.9% loss in wound healing at 48 h, respectively. There was a further 15±5.8% loss in wound healing upon ACF treatment, compared with CF treatment alone, of KE-6 cells but no significant change in KE-10 cells (Fig. 2a and b). Doxorubicin (300 nmol/l) alone did not have any significant effect on the wound-healing ability of either p38δ-positive or p38δ-negative cells (Fig. 2a and b). Further, we compared the migration of p38δ-positive and p38δ-negative cells following CF versus ACF treatment using a Boyden chamber assay. Double CF treatment induced a significant (78.7±5.2 and 78.7±7.8%, respectively) decrease in cell migration of KE-6 and KE-10 cells at 24 h; however, no significant change in cell migration was observed upon CF treatment of KE-3 and KE-8 cells (Fig. 2c). In contrast, triple ACF treatment of KE-3 and KE-8 significantly decreased their cell migration by 75.9±2.4 and 81.3±6.9%, respectively (Fig. 2c).
Fig. 2.
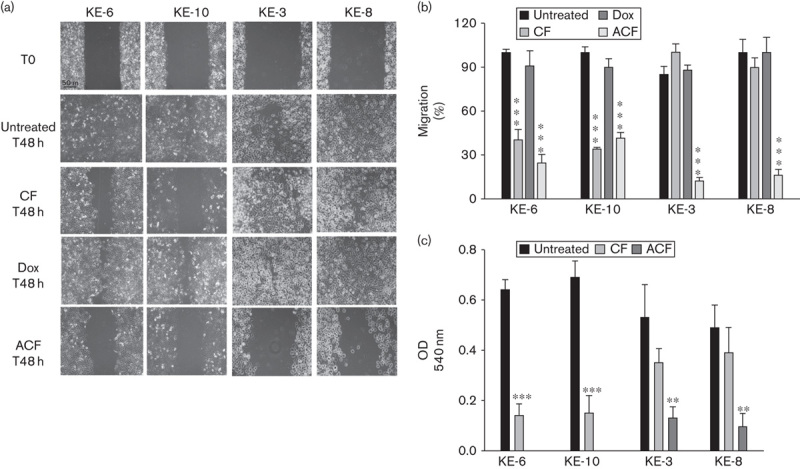
Effect of chemotherapeutic cytotoxic drugs on wound healing and migration of KE p38δ MAPK positive and negative cell lines. KE-6 and KE-10 cells (p38δ positive), as well as KE-3 and KE-8 cells (p38δ negative), were analysed for (a, b) wound healing and (c) cell migration following treatment with cisplatin (3 μmol/l) and 5-fluoruracil (3 μmol/l; CF), doxorubicin (Dox; 300 nmol/l) alone or cisplatin (3 μmol/l), 5-fluorouracil (3 μmol/l) and doxorubicin (300 nmol/l; triple treatment; ACF). (a) Representative wound-healing images at 0 and 48 h with or without drug treatment. The results shown in (a) are representative of three independent experiments, whereas the results shown in (b) and (c) are the mean±SE of three independent experiments. ***P<0.001, **P<0.01, significant changes from control untreated cells.
Triple ACF treatment decreases mitochondrial membrane potential and activates extrinsic pathways in p38δ negative OESCCs
Initially, we investigated the involvement of the mitochondria in OESCC apoptosis using the JC-1 cationic dye. It selectively enters the mitochondria and reversibly changes in colour from red to green as the membrane potential decreases 21. KE-6 and KE-10 cells show a marked decrease in ΔΨm following CF and ACF treatment (Fig. 3a and b). Similar losses in ΔΨm in KE-3 and KE-8 cells were observed only upon ACF treatment (Fig. 3a and b). Doxorubicin alone did not alter the ΔΨm of KE-6 and KE-10 cells, but it did produce a significant (P<0.01) decrease in the ΔΨm of KE-3 and KE-8, comparable to that on CF treatment (Fig. 3a and b).
Fig. 3.
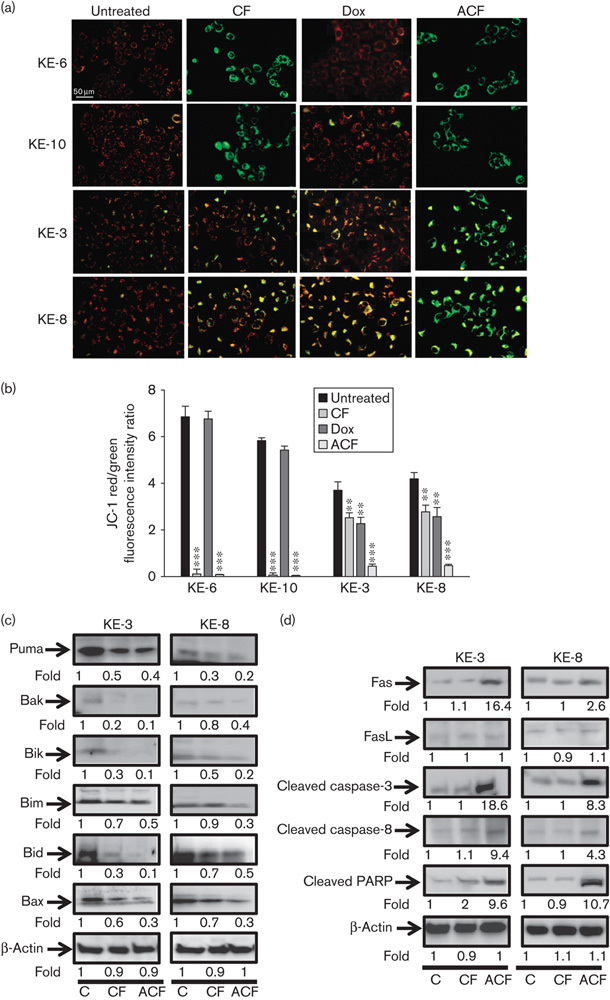
Effect of chemotherapeutic drug treatment on intrinsic and extrinsic mitochondrial pathways in KE-3 and KE-8 cells. (a) The mitochondrial membrane potential (ΔΨm) of KE cells before and after treatment with chemotherapeutic drugs for 24 h. Representative images (red/green mixed channels) show the changes in mitochondrial membrane potential (ΔΨm) in KE cells, detected using JC-1 dye. Red fluorescence indicates high membrane potential and functional capacity in mitochondria, whereas green cytoplasmic fluorescence is indicative of inactive mitochondria. (b) Decreased red/green fluorescence ratio suggests a decrease in ΔΨm. (c) Whole cell lysates were analysed by western blot analysis of proapoptotic Puma, Bak, Bik, Bim, Bid and Bax, as well as (d) Fas and FasL, cleaved caspase-3 and caspase-8 and PARP in KE-3 and KE-8 cells. The results shown in (a), (c) and (d) are representative of four independent experiments, whereas the results shown in (b) are the mean±SE of four independent experiments. ***P<0.001, **P<0.01, significant changes from control untreated cells.
To further investigate the effects of CF-induced versus ACF-induced apoptosis on KE p38δ-negative cells, intrinsic (mitochondrial) and extrinsic (Fas) apoptotic pathways were investigated. Expression of the relevant apoptosis-related proteins was examined by western blot analysis. To investigate the role of the intrinsic mitochondrial pathway we examined the expression levels of Bcl-2 family members including multidomain proapoptotic proteins Bak and Bax, as well as BH3-only proapoptotic proteins Puma, Bik, Bid and Bim. Interestingly CF treatment downregulated all Bcl-2 proapoptotic members examined, with further reductions in expression being observed in the presence of ACF treatment (Fig. 3c). The mitochondrial apoptotic pathway-related caspase-9 was not cleaved (data not shown).
To investigate whether CF and ACF activate the extrinsic apoptotic pathway, we examined the expression levels of death receptor signalling-related proteins including Fas, caspase-3 and caspase-8 and PARP. Alterations in the expression of Fas and FasL, members of the tumour necrosis family, have been reported previously in OESCC with Fas-activated apoptosis being shown to limit the growth of OESCCs 29,30. We observed an increase in Fas expression when both KE-3 and KE-8 cells were treated with ACF, but not on CF treatment, implicating the involvement of the extrinsic death pathway. The expression level of FasL was very low in both cell lines, and there was no appreciable change after treatment with CF or ACF (Fig. 3d). We observed that ACF (but not CF) treatment of KE-3 and KE-8 cell lines activated (cleaved) caspase-3 and caspase-8, as well as activated (cleaved) their substrate PARP, producing the 85 kDa proteolytic fragment indicative of caspase activation and apoptosis (Fig. 3d). The extrinsic apoptotic pathway-related caspases-6 and caspase-7 were not cleaved upon CF or ACF treatment (data not shown).
Triple ACF treatment induces p38 and ERK1/2 but not JNK1/2 MAPK activation in p38δ-negative OESCC
To gain further insight into CF-induced versus ACF-induced cytotoxic effects, we analysed MAPK expression. We observed p38 phosphorylation upon ACF treatment but not CF treatment (Fig. 4a). CF induced ERK1/2 phosphorylation, which was further enhanced with ACF treatment (Fig. 4a). In contrast, JNK1/2 activation was not observed following either CF or ACF treatment (Fig. 4a).
Fig. 4.
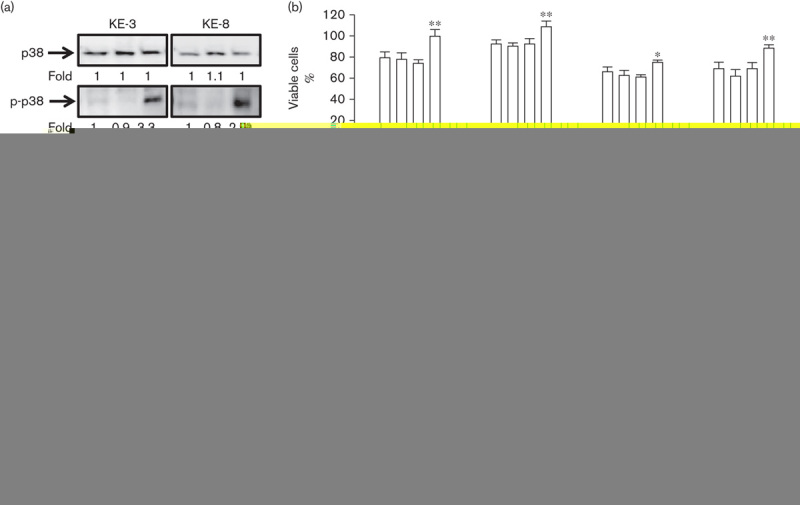
MAPK activation in KE-3 and KE-8 cells following chemotherapeutic drug treatment. Western blot analysis of the three different MAPKs (p38 MAPK, JNKs and ERKs) following treatment with cisplatin (30 μmol/l) and 5-fluoruracil (30 μmol/l; CF), or following triple treatment with cisplatin (30 μmol/l), 5-fluorouracil (30 μmol/l) and doxorubicin (1 μmol/l; ACF) for 24 h. The results shown in (a) are representative of four independent experiments, whereas the results shown in (b) are the mean±SE of four independent experiments. ***P<0.001, **P<0.01, *P<0.05, significant changes from CF-treated or ACF-treated cells.
To assess whether p38 and ERK1/2 activation is causal to the actions of our chemotherapeutics we used specific pharmacological inhibitors of p38 and MEK. Interestingly, both SB203590 (20 micromolar) and BIRB 796 (5 micromolar), inhibitors of p38α and p38β (but not p38γ and p38δ) 24,31 did not prevent the antiproliferative effects of either CF or ACF (Fig. 4b). In fact, in agreement with recent reports (with similar cytotoxic drug combinations), p38 blockade enhanced the cytotoxic effects of ACF treatment significantly (but not CF treatment) at 24 h (Fig. 4b) 24,25. The enhanced antiproliferative effect of p38 inhibition observed at 24 h with ACF was not obvious at 48 h because of the high level of cell death at this time point (Fig. 4b). The specific MEK inhibitor U0126 (20 micromolar) 32 brought about a significant abolition of the effects of both CF and ACF drug treatments in both cell lines, indicating that ERK1/2 may be directly involved in drug-induced cytotoxicity (Fig. 4b).
Recovery of KE-3 and KE-8 cells following drug treatments
One of the most important parameters for the efficacy of chemotherapeutic drug treatments is the long-term effect on cell viability. Thus, we evaluated the effects of CF, doxorubicin alone and ACF treatment on the capacity of KE-3 and KE-8 cells to recover in assays of clonogenic growth. All three different drug treatments had a significant effect on the recovery of both KE-3 and KE-8 cell lines (Fig. 5a and b). However, of note, cell recovery following ACF treatment was never observed, whereas colonies – that is, cell recovery – were observed following treatment with CF or doxorubicin alone (Fig. 5a and b).
Fig. 5.
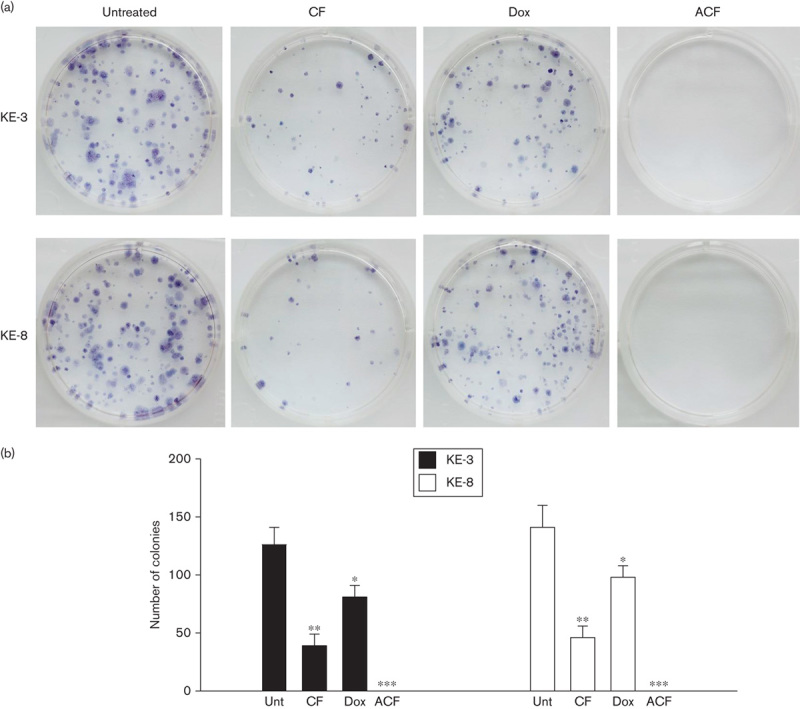
Recovery of KE-3 and KE-8 cells following chemotherapeutic drug withdrawal. The ability of p38δ-negative OESCC cell lines KE-3 and KE-8 to recover after drug withdrawal was assessed with a colony formation assay (clonogenic assay). KE-3 and KE-8 cells were or were not treated (Unt) with cisplatin (30 μmol/l) and 5-fluorouracil (30 µmol/l; CF), doxorubicin (1 μmol/l; Dox) alone or the combination of all three drugs (ACF) for 48 h. Viable, adherent cells were counted and reseeded (1000 cell/well) in a six-well plate (in triplicate) in the absence of drug. (a) Fourteen days later, colonies were stained with MTT. Each well shown is a representative image of nine similar wells (three independent experiments). (b) Colonies were counted using ImageJ software. ***P<0.001, **P<0.01, *P<0.05, significant changes from untreated cells. MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide; OESCC, oesophageal squamous cell carcinoma.
Discussion
In this study, we focused on OESCCs, as this cell phenotype shows differential p38δ expression 15. Loss of this important serine/threonine kinase confers greater tumourgenicity and may be a mechanism by which OESCC promotes carcinogenesis 15. The purpose of this study was to determine whether differences in the OESCC p38δ phenotype could influence the chemosensitivity of OESCC to conventional CF versus ACF drug combinations.
Single-agent response rates of 10–25% remain poor for oesophageal cancer 22. Further, the use of double CF is of limited effectiveness in the treatment of patients with OESCC, with respect to improving overall survival time and patient quality of life 3,8. Triple ACF combination therapy was first reported as far back as 1983 33, but of late it has sparked renewed interest 4,8. Doxorubicin is an effective, widely used chemotherapeutic agent in the treatment of a variety of solid tumours and malignant haematologic diseases 34. There are now reports documenting the usefulness and, importantly, the safety of ACF therapy for the treatment of advanced oesophageal carcinoma 7,8,35. Despite the absence of phase III clinical trials, ACF is currently used in the clinical setting for treatment of patients with OESCC 8.
All three drugs in this study, cisplatin, 5-fluorouracil and doxorubicin, used as monotherapy or double therapy have been reported to induce intrinsic apoptosis in oesophageal cancer 23,36. We examined typical markers of both intrinsic and extrinsic apoptotic cell death in our p38δ-negative cell lines. Although we observed mitochondrial depolarization following ACF treatment, there was no subsequent caspase-9 activation. Interestingly, we also observed downregulation of Bcl-2 multidomain, as well as BH-3 proapoptotic proteins. However, decreased expression of these intrinsic pathway proteins is in agreement with a recent report on oxaliplatin-induced apoptosis in squamous oesophageal cancer cell lines 37. Potentially, the presence of Bcl-2 multidomain and BH3-only proapoptotic molecules may be an important predictor of p38δ-negative OESCC response to combination therapy without being directly involved. Our findings suggest that ACF suppresses cell growth in p38δ-negative OESCC through extrinsic apoptotic pathway activation of the Fas death receptor, with caspase-8 and caspase-3 cleavage and subsequent degradation of PARP.
There are many reports linking MAPK involvement following exposure to mechanistically different chemotherapeutic drugs 38. However, their suppression and activation and indeed the absence of any role in apoptosis have been attributed to all three MAPKs 38–40. Thus, the activation of MAPKs by chemotherapeutic drugs and the subsequent consequences of MAPK activation appear to be very much cell-type specific 39. Although we observed p38 activation upon ACF treatment, our findings with the p38 pharmacological inhibitors do not support a direct role for p38 MAPK activity in drug-induced cytotoxicity. In contrast, ERK1/2 activation may play a more direct role as its inhibition through MEK can partly reverse the antiproliferative effects of both CF and ACF treatments.
The increased antiproliferative and proapoptotic effects of ACF treatment over CF treatment in our study suggest that the former may be considered as mainstay treatment of patients with p38δ-negative OESCC. It remains to be investigated whether patients with p38δ-negative OESCC may benefit more from ACF treatment compared with classical CF treatment, and whether or not ACF treatment can influence overall survival. Further investigations into the mechanistic strategies underpinning p38δ-negativity related loss in drug sensitivity are warranted. The clonogenic assay is a valuable tool in gauging long-term consequences of single, double and triple chemotherapy in p38δ-negative OESCC. This assay closely mirrors the clinical situation in which patients are treated with chemotherapeutic drugs in a pulsed rather than a continuous manner. After 2 weeks of treatment, our ACF-treated p38δ-negative OESCC cells never recovered. Although we observed a statistically significant reduction in colony formation on CF double or doxorubicin treatment, cell recovery was always observed, clearly demonstrating resistance. In general, the MTT assay demonstrated lower cytotoxic activity than the clonogenic assay with these drugs. Of note, a high degree of correlation between both assays is not always apparent, and it may be influenced by both cell type and anticancer drug type 41–43. Thus, unlike ACF treatment, the limited efficacy of CF or doxorubicin alone could provide an opportunity for cancer persistence in patients with p38δ-negative OESCC.
In summary, the present study indicates that p38δ may be a significant predictor of treatment response in patients with p38δ-negative OESCC. p38δ genotyping of pretreatment biopsy may potentially be a useful predictor of response to chemotherapy and ultimately prognosis in OESCC patients. Our data support the value of known p38δ status in the decision process used to inform the optimal treatment of patients with OESCC.
Acknowledgements
This work was supported by the Health Research Board, Ireland (Grant HRA/2009/17 to Barry).
Conflicts of interest
There are no conflicts of interest.
References
- 1.Kayani B, Zacharakis E, Ahmed K, Hanna GB. Lymph node metastases and prognosis in oesophageal carcinoma – a systematic review. Eur J Surg Oncol 2011; 37:747–753. [DOI] [PubMed] [Google Scholar]
- 2.Kuwano H, Fukuchi M, Kato H. Thoracoscopic surgery for esophageal cancer. Ann Thorac Cardiovasc Surg 2006; 12:305–307. [PubMed] [Google Scholar]
- 3.Mariette C, Piessen G, Triboulet JP. Therapeutic strategies in oesophageal carcinoma: role of surgery and other modalities. Lancet Oncol 2007; 8:545–553. [DOI] [PubMed] [Google Scholar]
- 4.Nakajima M, Kato H. Treatment options for esophageal squamous cell carcinoma. Expert Opin Pharmacother 2013; 14:1345–1354. [DOI] [PubMed] [Google Scholar]
- 5.Nakamura K, Kato K, Igaki H, Ito Y, Mizusawa J, Ando N, et al. Japan Esophageal Oncology Group/Japan Clinical Oncology Group. Three-arm phase III trial comparing cisplatin plus 5-FU (CF) versus docetaxel, cisplatin plus 5-FU (DCF) versus radiotherapy with CF (CF-RT) as preoperative therapy for locally advanced esophageal cancer (JCOG1109, NExT study). Jpn J Clin Oncol 2013; 43:752–755. [DOI] [PubMed] [Google Scholar]
- 6.Kosugi S, Kanda T, Nakagawa S, Ohashi M, Nishimaki T, Hatakeyama K. Efficacy and toxicity of fluorouracil, doxorubicin and cisplatin/nedaplatin treatment as neoadjuvant chemotherapy for advanced esophageal carcinoma. Scand J Gastroenterol 2005; 40:886–892. [DOI] [PubMed] [Google Scholar]
- 7.Honda M, Miura A, Izumi Y, Kato T, Ryotokuji T, Monma K, et al. Doxorubicin, cisplatin, and fluorouracil combination therapy for metastatic esophageal squamous cell carcinoma. Dis Esophagus 2010; 23:641–645. [DOI] [PubMed] [Google Scholar]
- 8.Yamasaki M, Miyata H, Fujiwara Y, Takiguchi S, Nakajima K, Nishida T, et al. p53 genotype predicts response to chemotherapy in patients with squamous cell carcinoma of the esophagus. Ann Surg Oncol 2010; 17:634–642. [DOI] [PubMed] [Google Scholar]
- 9.Raman M, Chen W, Cobb MH. Differential regulation and properties of MAPKs. Oncogene 2007; 26:3100–3112. [DOI] [PubMed] [Google Scholar]
- 10.Ono K, Han J. The p38 signal transduction pathway: activation and function. Cell Signal 2000; 12:1–13. [DOI] [PubMed] [Google Scholar]
- 11.Cuenda A, Rousseau S. p38 MAP-kinases pathway regulation, function and role in human diseases. Biochim Biophys Acta 2007; 1773:1358–1375. [DOI] [PubMed] [Google Scholar]
- 12.Cargnello M, Roux PP. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol Mol Biol Rev 2011; 75:50–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wagner EF, Nebreda AR. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer 2009; 9:537–549. [DOI] [PubMed] [Google Scholar]
- 14.Ambrose M, Ryan A, O’Sullivan GC, Dunne C, Barry OP. Induction of apoptosis in renal cell carcinoma by reactive oxygen species: involvement of extracellular signal-regulated kinase 1/2, p38delta/gamma, cyclooxygenase-2 down-regulation, and translocation of apoptosis-inducing factor. Mol Pharmacol 2006; 69:1879–1890. [DOI] [PubMed] [Google Scholar]
- 15.O’Callaghan C, Fanning LJ, Houston A, Barry OP. Loss of p38δ mitogen-activated protein kinase expression promotes oesophageal squamous cell carcinoma proliferation, migration and anchorage-independent growth. Int J Oncol 2013; 43:405–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yamana H, Kakegawa T, Tanaka T, Higaki K, Fujii T, Tou U. Experimental studies on immunotargeting therapy for esophageal carcinoma. Gan To Kagaku Ryoho 1994; 21:755–760. [PubMed] [Google Scholar]
- 17.Tsukahara T, Nabeta Y, Kawaguchi S, Ikeda H, Sato Y, Shimozawa K, et al. Identification of human autologous cytotoxic T-lymphocyte-defined osteosarcoma gene that encodes a transcriptional regulator, papillomavirus binding factor. Cancer Res 2004; 64:5442–5448. [DOI] [PubMed] [Google Scholar]
- 18.Nakao M, Yamana H, Imai Y, Toh Y, Toh U, Kimura A, et al. HLA A2601-restricted CTLs recognize a peptide antigen expressed on squamous cell carcinoma. Cancer Res 1995; 55:4248–4252. [PubMed] [Google Scholar]
- 19.O’Sullivan GC, Tangney M, Casey G, Ambrose M, Houston A, Barry OP. Modulation of p21-activated kinase 1 alters the behavior of renal cell carcinoma. Int J Cancer 2007; 121:1930–1940. [DOI] [PubMed] [Google Scholar]
- 20.Chen L, Zhang JJ, Huang XY. cAMP inhibits cell migration by interfering with Rac-induced lamellipodium formation. J Biol Chem 2008; 283:13799–13805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bernardi P, Scorrano L, Colonna R, Petronilli V, Di Lisa F. Mitochondria and cell death. Mechanistic aspects and methodological issues. Eur J Biochem 1999; 264:687–701. [DOI] [PubMed] [Google Scholar]
- 22.Ku GY, Ilson DH. Chemotherapeutic options for gastroesophageal junction tumors. Semin Radiat Oncol 2013; 23:24–30. [DOI] [PubMed] [Google Scholar]
- 23.O’Donovan TR, O’Sullivan GC, McKenna SL. Induction of autophagy by drug-resistant esophageal cancer cells promotes their survival and recovery following treatment with chemotherapeutics. Autophagy 2011; 7:509–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pereira L, Igea A, Canovas B, Dolado I, Nebreda AR. Inhibition of p38 MAPK sensitizes tumour cells to cisplatin-induced apoptosis mediated by reactive oxygen species and JNK. EMBO Mol Med 2013; 5:1759–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Elsea CR, Roberts DA, Druker BJ, Wood LJ. Inhibition of p38 MAPK suppresses inflammatory cytokine induction by etoposide, 5-fluorouracil, and doxorubicin without affecting tumoricidal activity. PLoS One 2008; 3:e2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen WW, Lin CC, Huang TC, Cheng AL, Yeh KH, Hsu CH. Prognostic factors of metastatic or recurrent esophageal squamous cell carcinoma in patients receiving three-drug combination chemotherapy. Anticancer Res 2013; 33:4123–4128. [PubMed] [Google Scholar]
- 27.Chen J, Pan J, Liu J, Li J, Zhu K, Zheng X, et al. Postoperative radiation therapy with or without concurrent chemotherapy for node-positive thoracic esophageal squamous cell carcinoma. Int J Radiat Oncol Biol Phys 2013; 86:671–677. [DOI] [PubMed] [Google Scholar]
- 28.Taïeb J, Artru P, Baujat B, Mabro M, Carola E, Maindrault F, et al. Optimisation of 5-fluorouracil (5-FU)/cisplatin combination chemotherapy with a new schedule of hydroxyurea, leucovorin, 5-FU and cisplatin (HLFP regimen) for metastatic oesophageal cancer. Eur J Cancer 2002; 38:661–666. [DOI] [PubMed] [Google Scholar]
- 29.Chan KW, Lee PY, Lam AK, Law S, Wong J, Srivastava G. Clinical relevance of Fas expression in oesophageal squamous cell carcinoma. J Clin Pathol 2006; 59:101–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matsuzaki I, Suzuki H, Kitamura M, Minamiya Y, Kawai H, Ogawa J. Cisplatin induces fas expression in esophageal cancer cell lines and enhanced cytotoxicity in combination with LAK cells. Oncology 2000; 59:336–343. [DOI] [PubMed] [Google Scholar]
- 31.He D, Zhao XQ, Chen XG, Fang Y, Singh S, Talele TT, et al. BIRB796, the inhibitor of p38 mitogen-activated protein kinase, enhances the efficacy of chemotherapeutic agents in ABCB1 overexpression cells. PLoS One 2013; 8:e54181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alessi DR, Cuenda A, Cohen P, Dudley DT, Saltiel AR. PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J Biol Chem 1995; 270:27489–27494. [DOI] [PubMed] [Google Scholar]
- 33.Gisselbrecht C, Calvo F, Mignot L, Pujade E, Bouvry M, Danne O, et al. Fluorouracil (F), adriamycin (A), and cisplatin (P) (FAP): combination chemotherapy of advanced esophageal carcinoma. Cancer 1983; 15:974–977. [DOI] [PubMed] [Google Scholar]
- 34.Weiss RB. The anthracyclines: will we ever find a better doxorubicin? Semin Oncol 1992; 19:670–686. [PubMed] [Google Scholar]
- 35.Shimakawa T, Naritaka Y, Asaka S, Isohata N, Murayama M, Konno S, et al. Neoadjuvant chemotherapy (FAP) for advanced esophageal cancer. Anticancer Res 2008; 28:2321–2326. [PubMed] [Google Scholar]
- 36.Juan HC, Tsai HT, Chang PH, Huang CY, Hu CP, Wong FH. Insulin-like growth factor 1 mediates 5-fluorouracil chemoresistance in esophageal carcinoma cells through increasing survivin stability. Apoptosis 2011; 16:174–183. [DOI] [PubMed] [Google Scholar]
- 37.Ngan CY, Yamamoto H, Takagi A, Fujie Y, Takemasa I, Ikeda M, et al. Oxaliplatin induces mitotic catastrophe and apoptosis in esophageal cancer cells. Cancer Sci 2008; 99:129–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boldt S, Weidle UH, Kolch W. The role of MAPK pathways in the action of chemotherapeutic drugs. Carcinogenesis 2002; 23:1831–1838. [DOI] [PubMed] [Google Scholar]
- 39.Brozovic A, Osmak M. Activation of mitogen-activated protein kinases by cisplatin and their role in cisplatin-resistance. Cancer Lett 2007; 251:1–16. [DOI] [PubMed] [Google Scholar]
- 40.De la Cruz-Morcillo MA, Valero ML, Callejas-Valera JL, Arias-González L, Melgar-Rojas P, Galán-Moya EM, et al. p38MAPK is a major determinant of the balance between apoptosis and autophagy triggered by 5-fluorouracil: implication in resistance. Oncogene 2012; 31:1073–1085. [DOI] [PubMed] [Google Scholar]
- 41.Buch K, Peters T, Nawroth T, Sänger M, Schmidberger H, Langguth P. Determination of cell survival after irradiation via clonogenic assay versus multiple MTT assay – a comparative study. Radiat Oncol 2012; 7:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sumantran VN. Cellular chemosensitivity assays: an overview. Methods Mol Biol 2011; 731:219–236. [DOI] [PubMed] [Google Scholar]
- 43.Kawada K, Yonei T, Ueoka H, Kiura K, Tabata M, Takigawa N, et al. Comparison of chemosensitivity tests: clonogenic assay versus MTT assay. Acta Med Okayama 2002; 56:129–134. [DOI] [PubMed] [Google Scholar]