

Abstract
Background and Purpose
Stress exposure produces excitotoxicity and neuroinflammation, contributing to the cellular damage observed in stress-related neuropathologies. The endocannabinoids provide a homeostatic system, present in stress-responsive neural circuits. Here, we have assessed the possible regulatory role of cannabinoid CB2 receptors in stress-induced excitotoxicity and neuroinflammation.
Experimental Approach
We used wild type (WT), transgenic overexpressing CB2 receptors (CB2xP) and CB2 receptor knockout (CB2-KO) mice exposed to immobilization and acoustic stress (2 h·day−1 for 4 days). The CB2 receptor agonist JWH-133 was administered daily (2 mg·kg−1, i.p.) to WT and CB2-KO animals. Glutamate uptake was measured in synaptosomes from frontal cortex; Western blots and RT-PCR were used to measure proinflammatory cytokines, enzymes and mediators in homogenates of frontal cortex.
Key Results
Increased plasma corticosterone induced by stress was not modified by manipulating CB2 receptors. JWH-133 treatment or overexpression of CB2 receptors increased control levels of glutamate uptake, which were reduced by stress back to control levels. JWH-133 prevented the stress-induced increase in proinflammatory cytokines (TNF-α and CCL2), in NF-κB, and in NOS-2 and COX-2 and in the consequent cellular oxidative and nitrosative damage (lipid peroxidation). CB2xP mice exhibited anti-inflammatory or neuroprotective actions similar to those in JWH-133 pretreated animals. Conversely, lack of CB2 receptors (CB2-KO mice) exacerbated stress-induced neuroinflammatory responses and confirmed that effects of JWH-133 were mediated through CB2 receptors.
Conclusions and Implications
Pharmacological manipulation of CB2 receptors is a potential therapeutic strategy for the treatment of stress-related pathologies with a neuroinflammatory component, such as depression.
Keywords: CB2 receptor, stress, excitotoxicity, neuroinflammation, brain frontal cortex, JWH-133, CB2xP mice
Introduction
The relationship between stress and the immune system has been extensively studied over the last few decades (Licinio and Wong, 1999; Sorrells et al., 2009), but the precise mechanisms involved are still a matter of debate, probably due to the complex interactions existing between the periphery and the CNS (Capuron and Miller, 2011). Chronic exposure to stress and stress-related diseases, such as depression or chronic fatigue syndrome, have been classically associated with an inhibition of adaptive immunity, with important negative effects on health (Herbert and Cohen, 1993). However, more recently, a marked activation of innate inflammatory and immune responses has been found, after stress exposure or during certain episodes of depression (García-Bueno et al., 2008; Farooq et al., 2012). The inflammatory response allows the organism to cope with a wide range of threats and challenges but, under pathological and chronic conditions, the maintenance of this response could become deleterious. For instance, long-lasting stress (physical, psychological or mixed) affected synaptic plasticity, dendritic morphology and neurogenesis in animals (Kim and Yoon, 1998), and induces both clinical and anatomical features of neurotoxic damage in humans (Bremner et al., 1995).
Over the past few years, much effort has been made to elucidate the molecular and cellular events responsible for the brain damage produced by exposure to stress. Stress-induced excitotoxicity follows a massive release of the excitatory amino acid, glutamate, in brain areas such as the frontal cortex (Moghaddam, 1993). This over-accumulation induces the release of pro-inflammatory cytokines such as TNF-α (Madrigal et al., 2002). Stress also activates the inflammatory NF-κB pathway through a TNF-α-dependent mechanism (Madrigal et al., 2002; Bierhaus et al., 2003). NF-κB activation elicits the expression and activity of pro-inflammatory enzymes, such as the inducible NOS (NOS-2) and COX-2 (Madrigal et al., 2006). The result of this sequence of events is the accumulation of oxidative and nitrosative mediators, which alter membrane phospholipids and cause cell damage by lipid peroxidation. This has been observed in the brain after exposure to stress (Madrigal et al., 2006). This potentially deleterious neuroinflammatory response is regulated by different anti-inflammatory mechanisms, also activated in the CNS after stress, such as the biosynthesis of 15d-PGJ2, a COX-2-derived lipid mediator and a potent endogenous agonist of the anti-inflammatory transcription factor PPAR-γ (García-Bueno et al., 2008).
Currently, the study of anti-inflammatory pathways has focused on the endocannabinoid system (ECS). The ECS comprises a group of endogenous arachidonate-based lipids (mainly anandamide and 2-arachidonoylglycerol), known as ‘endocannabinoids’; the corresponding GPCRs, CB1 and CB2 receptors (nomenclature follows Alexander et al., 2013a), the two main synthetic enzymes, N-acylphosphatidylethanolamine-phospholipase D and diacylglycerol lipase, and, finally, the degradative enzymes, fatty acid amide hydrolase and monoacylglycerol lipase. The ECS is considered to be an endogenous homeostatic system activated by different immune challenges, restoring brain balance in different experimental settings (Mechoulam and Shohami, 2007; Bambico et al., 2009; Cabral and Griffin-Thomas, 2009). In particular, exposure to stress up-regulated CB1 receptors in the frontal cortex and selective pharmacological activation of these receptors prevented the stress-induced excitotoxic and neuroinflammatory processes (Zoppi et al., 2011).
The other cannabinoid receptor type, the CB2 receptor, has been recognized as a major regulator of the immune system in the periphery, as it is highly expressed in a wide range of immune cells (Arévalo-Martín et al., 2003). However, it is now accepted that CB2 receptors are also expressed in CNS, by microglia, astrocytes and subpopulations of neurons present in brain areas related to the hypothalamic–pituitary–adrenal (HPA) axis (Gong et al., 2006), although the extent of CB2 receptor expression in neurons remains controversial (Atwood and Mackie, 2010). Furthermore, CB2 receptors were inducible in microglia under neuroinflammatory conditions, suggesting that such up-regulation could be a common pattern of response against different types of chronic human neurodegenerative and neurological pathologies (Bisogno and Di Marzo, 2010). Although there is still controversy regarding the role of the CB2 receptor in the brain (Atwood and Mackie, 2010), specific functions for CB2 receptors in neuropsychiatric conditions are currently emerging (Onaivi et al., 2012).
Taking into account all these findings, the aim of the present study was to explore the effect of genetic (overexpression or knockout) or pharmacological (with the selective agonist JWH-133) manipulations of CB2 receptors, on stress-induced excitotoxicity and neuroinflammation in mice.
Methods
Animals
All animal care and experimental protocols adhered to the guidelines of the Animal Welfare Committee of the Universidad Complutense, in accordance with European legislation (2003/65/EC). All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). A total of 75 animals were used in the experiments described here. Transgenic mice overexpressing CB2 receptors (CB2xP) and CB2 receptor knockout mice (CB2-KO) were kindly supplied by Dr. Jorge Manzanares (Instituto de Neurociencias, Universidad Miguel Hernández-CSIC Alicante, Spain), and their corresponding wild-type (WT) littermates (ICR –Swiss strain) by Harlan Iberica (Madrid, Spain). The mice were housed individually with standard temperature and humidity conditions in a 12 h light/dark cycle (lights on at 0800 h) with free access to food and water. All the animals were maintained under constant conditions for 4 days before the exposure to stress.
Stress protocol
Male adult (8-12 weeks old) mice were exposed to sub-chronic immobilization and acoustic stress (2 h from 1300 to 1500 h for 4 days), as previously described (Kiank et al., 2006). Stressed animals were given a lethal injection of sodium pentobarbital (320 mg·kg−1 i.p.) immediately after the last stress session (while still in the restrainer). Control animals were not submitted to stress but were handled at 1300 h for a few seconds, food and water were removed for 2h and then killed as described above. Blood for plasma determinations was collected by cardiac puncture and anticoagulated with trisodium citrate [3.15% (w/v), 1 volume citrate per 9 volumes blood]. The mice were decapitated, the brain was removed from the skull and, after careful removal of the meninges and blood vessels, the frontal cortical areas from both brain hemispheres were excised and frozen at −80°C until assayed. The frontal cortex we used included the prefrontal cortex, cingulate cortex and motor cortex (M1 and M2); the cortical areas that we isolated were from Bregma 3mm to 0.5mm, approximately. The more lateral and ventral cortical structures (primary somatosensory cortex, granular insular cortex) were excluded. This brain area was chosen because of its relatively high levels of CB2 receptors (Gong et al., 2006) and its susceptibility to excitotoxic and neuroinflammatory processes elicited by stress (García-Bueno et al., 2008). The frontal cortex is also an important neural substrate for the regulation of the responses of the HPA axis to stress (Radley et al., 2006).
Preparation of cytosolic and nuclear extracts
A modified procedure based on the method of Schreiber et al. (1989) was used. Briefly, tissues (30 mg) were homogenized with 300 μL of buffer (composition, in mM; HEPES, 10; EDTA, 1; EGTA, 1; KCl, 10; DTT, 1; phenylmethylsulfonyl fluoride, 0.5; NaF, 5; NaVO4, 1; sucrose, 0.5; and Na2MoO4, 10, with aprotinin, 0.1 μg L-1; leupeptin, 1 μg L-1; and Nα-p-tosyl-L-lysine-chloromethyl ketone, 1 μg mL-1; pH 7.9). After 15 min, Nonidet P-40 (Roche®, Mannheim, Germany) was added to a final concentration of 0.5%. The tubes were gently vortexed for 15 s, and nuclei (in the pellet) were collected by centrifugation at 8000× g for 5 min. Supernatants were taken as a cytosolic fraction. The pellets were resuspended in 100 μL of buffer supplemented with 20% glycerol and 0.4 mol·L−1 KCl, and gently shaken for 30 min at 4°C. Nuclear protein extracts were obtained by centrifugation at 13 000× g for 5 min, and aliquots of the supernatant were stored at −80°C. All steps of the fractionation were carried out at 4°C.
Western blot analysis
To determine the expression levels of the astroglial excitatory amino acid transporter-2 (SLC1A2; EAAT-2; nomenclature follows Alexander et al., 2013b), NOS-2 and COX-2, homogenates of the frontal cortex were used. In the case of the NF-κB subunit p65, the analysis was carried out in nuclear extracts of the homogenates and, for the inhibitory protein of NF-κB IκBα, cytosolic extracts were used (see above). After determining and adjusting protein levels, homogenates of frontal cortex were centrifuged (12 000× g, 20 min at 4°C) and the supernatant mixed with Laemmli sample buffer with β-mercaptoethanol (50 μL·mL−1 of Laemmli; Bio-Rad®, Hercules, CA, USA) and 20 μL of the mixture (containing 2 μg protein·μL−1 ) was loaded into an electrophoresis gel. After separation, proteins from the gels were blotted onto a nitrocellulose membrane (Amersham Ibérica, Madrid, Spain) with a semi-dry transfer system (Bio-Rad) and were incubated with the specific antibodies shown below.
All antibodies were supplied by Santa Cruz Biotechnology (CA, USA). A rabbit polyclonal antibody against EAAT-2, raised against an epitope corresponding to amino acids 1-85 mapping at the N-terminus of EAAT-2 of human origin in a dilution of 1:1000 in 5% BSA in TBS-Tween (sc-15317); a rabbit polyclonal antibody against IκBα (epitope mapping at the C-terminus of IκBα of human origin) in a dilution of 1: 1000 in 5% skimmed milk in BSA;a rabbit polyclonal antibody against NF-κB p65 (epitope mapping within the N-terminus of NF-κB p65 of human origin) in a dilution of 1:500 in BSA 2% (sc-109); a rabbit polyclonal antibody against NOS-2 raised against a peptide mapping at the amino terminus of NOS-2 of human origin in a dilution of 1:1000 in TBS-Tween (sc-651); e) a goat polyclonal antibody against COX-2 raised against a peptide mapping at the C-terminus of COX-2 of human origin in a dilution of 1:750 in 5% BSA in TBS-Tween (sc-1745).
After washing with 10 mM Tris-buffered saline containing 0.1% Tween-20 (Bio-Rad), the membranes were incubated with the respective HRP-conjugated secondary antibodies for 90 min at room temperature. Blots were imaged using an Odyssey® Fc System (Li-Cor Biosciences®, Lincoln, NE, USA) and were quantified by densitometry (NIH ImageJ® software; National Institutes of Health, Bethesda, MD, USA). All densitometries are expressed in arbitrary units of optical density. In all Western blot analyses, the housekeeping gene β-actin was used as loading control except for analysis of NF-κB, in which the loading control was the nuclear factor SP1 (blots shown in the respective figures).
mRNA analysis
Total cytoplasmic RNA was prepared from samples of frontal cortex using TRIzol® reagent (Invitrogen®, Carlsbad, CA, USA); aliquots were converted to cDNA using random hexamer primers. Quantitative changes in mRNA levels were estimated by reverse transcription-PCR using the following cycling conditions: 35 cycles of denaturation at 95°C for 10 s, annealing at 58–61°C for 15 s depending on the specific set of primers and extension at 72°C for 20 s. Reactions were carried out in the presence of SYBR green (1:10 000 dilution of stock solution from Molecular Probes, Eugene, OR, USA), carried out in a 20 L reaction in a Rotor-Gene (Corbett Research, Mortlake, NSW, Australia). Relative mRNA concentrations were obtained by comparing the take-off point of the different samples using the software provided in the unit. It establishes an inverse correlation between the number of cycles before take-off and the concentration of mRNA, while assigning arbitrary units to the results obtained. Tubulin primer levels were used to normalize data. See Supporting Information Appendix S1 for details about the primers used.
Preparation of synaptosomes
Synaptosomes were prepared from anterior cortical structures of the right hemisphere of the forebrain, discarding other brain areas such as the olfactory bulb or basal ganglia. The tissues were dissected on ice and all subsequent steps were performed at 4°C. Tissue was immediately homogenized in 25 volumes of 0.32 M sucrose in a glass homogenizer fitted with a Teflon pestle. The homogenate was centrifuged at 200× g for 10 min, and the supernatant was then collected and centrifuged at 20 000× g for 20 min. The pellet was resuspended in 0.32 M sucrose and centrifuged at 20 000× g for 20 min. The crude synaptosomal pellet was finally resuspended in 1 mL of 0.32 M sucrose.
[3H]Glutamate uptake by synaptosomes
Sodium-dependent glutamate uptake by synaptosomes (see above) was measured, as follows. In brief, 25 μL of aliquots of synaptosomes were added to 250 μL of incubation buffer (5mM Tris, 10mM HEPES, 2.5mM KCl, 1.4M NaCl, 1.2mM CaCl2, 1.2mM MgCl2, 1.2mM KH2PO4, and 10mM dextrose, pH 7.4.) containing L-[3H]glutamic acid 0.125 mM (1 mCi·mL−1; Amersham Biosciences Europe GmbH, Freiburg, Germany) and incubated for 3 min at 37°C in a shaking bath. The reaction was terminated using 1 mL of ice-cold choline buffer (incubation buffer in which an equimolar concentration of choline chloride was substituted for NaCl), and the samples were centrifuged at 10 000× g for 2 min to recover the synaptosomes. The bound 3H-radioactivity was measured using a liquid scintillation counter.
Plasma corticosterone levels
Plasma was obtained from blood samples by centrifuging the sample at 1000× g for 15 min immediately after stress. All plasma samples were stored at −20°C before assay. Corticosterone was measured by the RIA kit Coat-a-Count® (Siemens, Los Angeles, CA, USA) in a γ counter. The values obtained in the control animals (84±12 ng mL−1) matched the kit manufacturer's expected values in adult mice at the time of blood collection (approximately 15:00h).
Levels of nitrite (NO2−)
The stable metabolite of the free radical NO, NO2− was assayed by the Griess method (Green et al., 1982), measuring the optical density at 540nm in a microplate reader (Synergy 2; BioTek, Winooski, VT, USA).
Lipid peroxidation
Lipid peroxidation was measured using the thiobarbituric acid test for malondialdehyde (MDA) following the method described by Das and Ratty (1987) with some modifications. Frontal cortex was sonicated in 10 volumes of 50 mM phosphate buffer (pH 7.4) and deproteinized with 40% trichloroacetic acid and 5 mol·L−1 HCl, followed by the addition of 2% (w/v) thiobarbituric acid in 0.5 mol·L−1 NaOH. The reaction mixture was heated in a water bath at 90°C for 15 min and centrifuged at 12 000× g for 10 min. The pink chromogen was measured at 532 nm in a Beckman DU-7500 spectrophotometer (Beckman® Coulter, Brea, CA, USA).
PGE2 levels in frontal cortex
PGE2 levels were measured by enzyme immunoassay (EIA) using reagents in kit form (PGE2 EIA Kit-Monoclonal, Cayman Chemical®, Tallin, Estonia). Samples of frontal cortex (20 mg) were sonicated in 300 μL of homogenization buffer (0.1 M phosphate buffer, pH 7.4, 1 mM EDTA and 10 μM indomethacin) and purified by incubation with ethanol (4 x sample volume) for 5 min at 4°C and then centrifugation at 3000× g for 10 min. Extracts were acidified with glacial acetic acid to pH 3.5, and PGE2 was extracted using SPE (C-18) columns (Amersham Biosciences, Buckinghamshire, UK) rinsed with methanol and water. After the application of samples, columns were washed with water and hexane. PGE2 was eluted with ethyl acetate. Samples were then evaporated to dryness under nitrogen and resuspended in EIA buffer. Levels of PGE2 were measured at 405 nm following the manufacturer's instructions. The sensitivity of the assay for PGE2 was 15 pg ml−1; intra- and interassay coefficients of variation were 6.6% and 15.5%, respectively, at 62.5 pg ml-1.
Pharmacological tools
The selective CB2 receptor agonist, JWH-133 (6aR,10aR)-6,6,9-trimethyl-3-(2-methylpentan-2-yl)-6a,7,10,10a-tetrahydrobenzo[c]chromene (Tocris Bioscience®, Bristol, UK) was given (i.p.) to groups of WT and CB2-KO mice at the onset of each session of the stress (1300 h). JWH-133 is a potent CB2 receptor agonist (Ki = 3.4 nM), 200-fold selective over CB1 receptors (Pertwee, 1997; Huffman et al., 1999). The dose (2 mg·kg−1) was chosen, based on previous in vivo studies of neuroprotection in mice (Zarruk et al., 2012). The compound was dissolved in DMSO : Tween : PBS (1:1:18), and the total volume injected into each animal was 200 μL. Control (un-stressed) and stressed WT animals, injected with vehicle, were used to allow for the stress produced by the injection and the possible effects of the vehicle used on neuroinflammatory parameters.
Protein assay
Protein levels were measured using Bradford's method (Bradford, 1976).
Data analysis
Data in text and figures are expressed as mean ± SEM. Results from the assays of glutamate uptake by synaptosomes were analysed with one-way anova followed by Dunnett's post hoc test (all groups against control). For multiple comparisons, a two-way anova followed by the Bonferroni post hoc test was used, considering as the first factor the presence or absence of stress and, as the second, the presence or absence of pharmacological or genetic manipulations of the CB2 receptor (receptor overexpression or deletion). To confirm that the anti-inflammatory actions of JWH-133 were dependent on activation of CB2 receptors, a two-way anova followed by the Bonferroni post hoc test was used, considering as first factor the presence or absence of pharmacological treatment, and as second one, the genotype of the mice (WT or CB2-KO). A P-value < 0.05 was considered statistically significant.
Materials
Unless otherwise stated, all chemicals were obtained from Sigma, Madrid, Spain.
Results
CB2 receptor-dependent effects on stress-induced changes in synaptosomal glutamate uptake and on the expression of glutamate transporters
The ECS has been shown to confer neuroprotection by inhibiting glutamatergic excitotoxicity, through a CB1 receptor-related mechanism (Zoppi et al., 2011). We explored the effects of CB2 receptors on glutamate transport in frontal cortical synaptosomes from WT, WT + JWH-133, CB2xP and CB2-KO mice under control and stress conditions.
Treatment of WT mice with JWH-133 (JWH group) or overexpression of CB2 receptors (CB2xP group) increased the control levels of glutamate uptake (Figure 1A,B). As previously described (García-Bueno et al., 2007), exposure to stress markedly decreased glutamate uptake, compared with the control, unstressed, WT group (CWT) (Figure 1A–C). Stress also decreased glutamate uptake in the JWH or CB2xP groups, but only down to the levels observed in the CWT group [JWH F(1, 21) = 7.95, P = 0.014; CB2xP F(1, 21) = 9.87, P = 0.0003].
Figure 1.
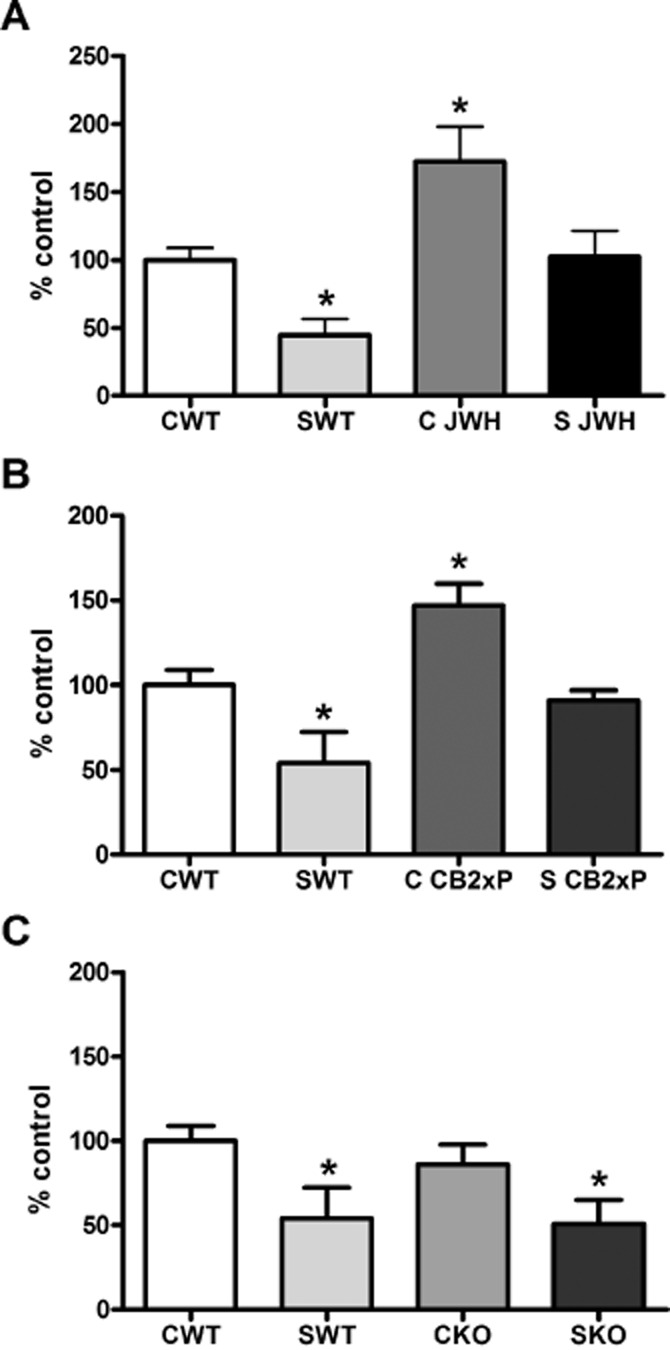
CB2 receptor effects on glutamate uptake mechanisms. (A) Glutamate uptake in frontal cortical synaptosomes from WT mice, with (SWT) or without stress (CWT). The effects of treatment of WT mice with JWH-133, before (C JWH) or after stress (S JWH) is also shown. In (B), the corresponding data from CB2xP mice before (CCB2xP) and after stress (S CB2xP) are shown. In (C), data from CB2-KO mice before (CKO) and after stress (SKO) are shown. The data shown are the means ± SEM of six mice in each group. *P < 0.05 versus CWT; one-way anova with Dunnett's multiple comparison post test.
The absence of CB2 receptors (CB2KO group) did not modify the glutamate uptake by synaptosomes, compared with WT mice, in either control or stress conditions, with stress decreasing uptake in the CB2-KO mice, as it did in the WT mice (Figure 1C) [CB2-KO F(1, 21) = 4.02, P = 0.0236].
We also measured the levels of the major brain glutamate transporter, EAAT-2, in unstressed, WT and CB2xP and CB2-KO mice and found no changes at protein level (data not shown).
CB2 receptor effects on HPA axis activity: plasma corticosterone levels
Corticosterone is the main stress hormone in rodents and is widely known as a classical regulator of the inflammatory and immune response in brain and periphery (Madrigal et al., 2006). In our model, exposure to stress increased plasma corticosterone equally in all groups of animals, compared with their respective controls (Figure 2) [stress F(1, 19) = 120.24, P < 0.0001; JWH treatment F(1, 19) = 1.93, P = 0.184; stress × JWH treatment F(1, 19) = 2.54, P = 0.1305]; [stress F(1, 19) = 36.9, P < 0.0001; CB2xP genotype F(1, 19) = 1.15, P = 0.2963; stress × CB2xP genotype F(1, 19) = 0.62, P = 0.4416]; [stress F(1, 17) = 105.99; CB2-KO genotype F(1, 17) = 1.14, P = 0.3046; stress × CB2-KO genotype F(1, 17) = 1.25, P = 0.2833].
Figure 2.
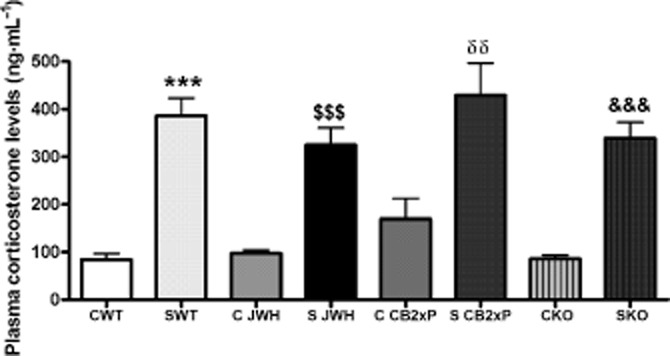
CB2 receptor effects on plasma corticosterone levels. Corticosterone levels in plasma (ng·mL−1) at the time of blood sampling (1500 h) are shown for all experimental groups and conditions (indicated as in Figure 1). Exposure to stress increased plasma corticosterone in all groups, relative to their corresponding control levels. The data represent the mean ± SEM of six mice. ***P < 0.001 versus CWT; $$$P < 0.001 versus C JWH; δδP < 0.01 versus C CB2xP; &&&P < 0.001 versus CKO; two-way anova with Bonferroni post test.
Anti-inflammatory effects elicited by activation of CB2 receptors: mechanisms involved
The ECS has been proposed as an endogenous protective system against excessive inflammatory and immune responses in several CNS pathologies (Wolf et al., 2008). We therefore assayed a number of proinflammatory factors in our model.
Pro-inflammatory cytokines and chemokines in brain
The pro-inflammatory cytokine TNF-α and the chemokine CCL2 (Madrigal et al., 2006; Conductier et al., 2010), are the first pro-inflammatory mediators to be activated in the brain after exposure to stress. We found, by PCR, a consistent increase in mRNA for TNF-α in samples of the frontal cortex from WT mice after stress, an effect that was blocked by JWH and in CB2xP mice (Figure 3A,B) [stress F(1, 14) = 53.64, P < 0.0001; JWH treatment F(1, 14) = 84.45, P < 0.0001; stress × JWH treatment F(1, 14) = 25.93, P = 0.0003]; [stress F(1, 14) = 108.89, P < 0.0001; CB2xP genotype F(1, 14) = 6.05, P = 0.032; stress × CB2xP genotype F(1, 14) = 16.8, P = 0.0018]. However, in CB2-KO mice, stress increased the mRNA for TNF-α mRNA (Figure 3C) [stress F(1, 14) = 33.43, P = 0.0002; CB2-KO genotype F(1, 14) = 11.49, P = 0.0069; stress × CB2-KO genotype F(1, 14) = 0.53, P = 0.4826].
Figure 3.

Anti-inflammatory effects of CB2 receptors in the frontal cortex. Pro-inflammatory cytokines, chemokines and NF-κB in homogenates of the frontal cortex. Quantitative PCR (qPCR) analysis of mRNA for TNF-α. (A) Data are shown for untreated WT mice and WT mice treated with JWH-133, both with and without stress. In (B), results from CB2xP mice, with and without stress and in (C) for CB2-KO mice, with and without stress. Data are normalized by tubulin and are representative of three experiments. *P < 0.05, ***P < 0.001 versus CWT; #P < 0.05, ###P < 0.001 versus SWT; &&P < 0.01 versus CKO; two-way anova with Bonferroni post test. In (D), data for mRNA for CCL2 from untreated WT mice and WT mice treated with JWH-133, both with and without stress. In (E), results from CB2xP mice, with and without stress and in (F) for CB2-KO mice, with and without stress. Data are normalized by tubulin and are representative of three experiments. *P < 0.05, **P < 0.01 versus CWT; #P < 0.05, ##P < 0.001 versus SWT; $$P < 0.01 versus C JWH; &&P < 0.01 versus CKO; two-way anova with Bonferroni post test. (G) Western blot and densitometric analysis of the NF-κB inhibitory protein IκBα in cytosolic extracts of the frontal cortex from untreated WT mice and WT mice treated with JWH-133, both with and without stress. In (H), results from CB2xP mice, with and without stress and in (I) for CB2-KO mice, with and without stress. Data are normalized by β-actin (lower band) and are representative of three experiments. *P < 0.05, ***P < 0.001 versus CWT; ##P < 0.01 versus SWT; $$P < 0.05 versus C JWH; two-way anova with Bonferroni post test. (J) Western blot and densitometric analysis of the NF-κB pro-inflammatory subunit p65 in nuclear extracts of the frontal cortex from untreated WT mice and WT mice treated with JWH-133, both with and without stress. In (K), results from CB2xP mice, with and without stress and in (L) for CB2-KO mice, with and without stress. Data are normalized by β-actin (lower band) and are representative of three experiments. *P < 0.05, ***P < 0.001 versus CWT; ##P < 0.01, ###P < 0.001 versus SWT; two-way anova with Bonferroni post test. AU, arbitrary units.
A slightly different profile was found for the chemokine CCL2. Exposure to stress increased mRNA for CCL2 in the frontal cortex of WT mice and this was blocked only by the overexpression of CB2 receptors, not by JWH-133 treatment (Figure 3D,E) [stress F(1, 18) = 22.53, P = 0.0003; JWH treatment F(1, 18) = 1.45, P = 0.247; stress × JWH treatment F(1, 18) = 0.13, P = 0.7204]; [stress F(1, 18) = 9.97, P = 0.007; CB2xP genotype F(1, 18) = 5.58, P = 0.0331; stress × CB2xP genotype F(1, 18) = 1.16, P = 0.30]. Stressed CB2-KO mice presented higher levels of CCL2 mRNA, compared with SWT group (Figure 3F) [stress F(1, 17) = 19.5, P = 0.0006; CB2-KO genotype F(1, 17) = 11.36, P = 0.0046; stress × CB2-KO genotype F(1, 17) = 1.35, P = 0.2655].
NF-κB
The release of TNF-α after stress activates the NF-κB transcription factor (Madrigal et al., 2002) in the cortex. In our present model, we found that exposure to stress decreased the expression of IκBα in cytosolic extracts of frontal cortex from WT mice. This effect was reversed in CB2xP mice and in WT mice treated with JWH-133 (Figure 3G,H) [stress F(1, 13) = 9.84, P = 0.01; JWH treatment F(1, 13) = 86.39, P < 0.0001; stress × JWH treatment F(1, 13) = 68, P < 0.0001]; [stress F(1, 27) = 24.8, P < 0.0001; CB2xP genotype F(1, 27) = 14.96, P = 0.0007; stress × CB2xP genotype F(1, 27) = 24.8, P < 0.0001]. Levels of the pro-inflammatory NF-κB subunit p65 in nuclear extracts of frontal cortex from different groups studied (Figure 3J,K) mirrored the expression of IκBα, with increased levels in stressed WT mice and blockade in the JWH and CB2xP groups [stress F(1, 30) = 4.73, P = 0.0386; JWH treatment F(1, 30) = 10.7, P = 0.0029; stress × JWH treatment F(1, 30) = 6.96, P = 0.0137]; [stress F(1, 60) = 4.01, P = 0.05; CB2xP genotype F(1, 60) = 5.94, P = 0.018; stress × CB2xP genotype F(1, 60) = 9.04, P = 0.0039].
In CB2-KO mice, the basal IκBα protein levels in frontal cortex were lower than in the WT mice but, after stress, CB2-KO mice presented similar levels to those from the WT group (Figure 3I) [stress F(1, 22) = 7.72, P = 0.013; CB2-KO genotype F(1, 22) = 7.22, P = 0.0156; stress × CB2-KO genotype F(1, 22) = 13.7, P = 0.0018]. In addition, p65 protein expression in nuclear extracts was increased in CB2-KO mice in both control and stress conditions (Figure 3L), suggesting a state of chronic NF-κB activation in these animals [stress F(1, 23) = 6.03, P = 0.024; CB2-KO genotype F(1, 23) = 127.75, P < 0.0001; stress × CB2-KO genotype F(1, 23) = 2.04, P = 0.1705].
Pro-inflammatory enzymes (NOS-2 and COX-2)
NF-κB regulates the expression of genes involved in the production of oxidative, nitrosative and inflammatory mediators after stress exposure (Madrigal et al., 2006). Two major pro-inflammatory, NF-κB dependent, enzymes are NOS-2 and COX-2. Their products (NO and PGE2, respectively) are potent oxidant and pro-inflammatory molecules that are associated with damage and even cell death in many CNS pathologies, including those related to stress (García-Bueno et al., 2008).
We found that NOS-2 and COX-2 expression was increased in the frontal cortex, following stress in WT mice (Figure 4A,D) and treatment of the WT with JWH-133 or the overexpression of CB2 receptors completely blocked this NOS-2 up-regulation (Figure 4A,B). As seen with other pro-inflammatory mediators, the CB2-KO mice, both in control and stress conditions, showed a consistent NOS-2 up-regulation, compared with the WT control groups, and a higher increase after stress (Figure 4C) [stress F(1, 19) = 6.07, P = 0.0255; JWH treatment F(1, 19) = 8.08, P = 0.0118; stress × JWH treatment F(1, 19) = 1.76, P = 0.2027]; [stress F(1, 16) = 4.5, P = 0.0495; CB2xP genotype F(1, 16) = 18.51, P = 0.0013; stress × CB2xP genotype F(1, 16) = 44.25, P < 0.0001]; [stress F(1, 21) = 4.56, P = 0.047; CB2-KO genotype F(1, 21) = 10.77, P < 0.0041; stress × CB2-KO genotype F(1, 21) = 0.91, P = 0.35].
Figure 4.

Anti-inflammatory effects of CB2 receptors in the frontal cortex; pro-inflammatory enzymes. (A) Western blot and densitometric analysis of NOS-2 in homogenates of the frontal cortex from untreated WT mice and WT mice treated with JWH-133, both with and without stress. In (B), results from CB2xP mice, with and without stress and in (C) for CB2-KO mice, with and without stress. Data are normalized by β-actin (lower band) and are representative of three experiments. *P < 0.05, **P < 0.01 versus CWT; #P < 0.05, ###P < 0.001 versus SWT; two-way anova with Bonferroni post test. (D) Western blot and densitometric analysis of COX-2 in homogenates of the frontal cortex from untreated WT mice and WT mice treated with JWH-133, both with and without stress. In (E), results from CB2xP mice, with and without stress and in (F) for CB2-KO mice, with and without stress. Data are normalized by β-actin (lower band) and are representative of three experiments. *P < 0.05, **P < 0.01 versus CWT; ##P < 0.01 versus SWT; two-way anova with B onferroni post test. (G) Levels of PGE2 in homogenates of the frontal cortex from untreated WT mice and WT mice treated with JWH-133, both with and without stress. In (H), results from CB2xP mice, with and without stress and in (I) for CB2-KO mice, with and without stress. The data represent the mean ± SEM of six mice. *P < 0.05 versus CWT; #P < 0.05, ###P < 0.001 versus SWT; two-way anova with Bonferroni post test. AU, arbitrary units.
The interaction between CB2 receptors and COX-2 was more complex. In the frontal cortex, levels of PGE2, a major product of COX-2 in the brain, were increased after stress exposure in WT mice (Figure 4G). In addition, the pharmacological activation and overexpression of CB2 receptors reduced PGE2 levels, compared with those in the stressed WT animals (Figure 4G,H). However, COX-2 up-regulation produced by stress was only blocked in CB2xP mice (Figure 4E). As it can be observed in Figure 4I, CB2-KO animals exhibited higher levels of PGE2, with and without stress, compared with the corresponding WT groups, but no comparable changes of COX-2 protein were found (Figure 4F). For COX-2, [stress F(1, 16) = 3.9, P = 0.07; JWH treatment F(1, 16) = 0.13, P = 0.722; stress × JWH treatment F(1, 16) = 0.5, P = 0.49]; [stress F(1, 18) = 4.22, P = 0.05; CB2xP genotype F(1, 18) = 6.28, P = 0.0233; stress × CB2xP genotype F(1, 18) = 5.94, P = 0.0278]; [stress F(1, 43) = 5.21, P = 0.028; CB2-KO genotype F(1, 43) = 2.12, P = 0.1534; stress × CB2-KO genotype F(1, 43) = 0.86, P = 0.36]. For PGE2, [stress F(1, 17) = 4.5, P = 0.05; JWH treatment F(1, 17) = 12.4, P = 0.0034; stress × JWH treatment F(1, 17) = 7.71, P = 0.015]; [stress F(1, 16) = 4.96, P = 0.042; CB2xP genotype F(1, 16) = 26.03, P = 0.0002; stress × CB2xP genotype F(1, 16) = 4.74, P = 0.0485]; [stress F(1, 28) = 5.65, P = 0.025; CB2-KO genotype F(1, 28) = 14.13, P = 0.0009; stress × CB2-KO genotype F(1, 28) = 0.02, P = 0.90].
Lipid peroxidation
As a final index of stress-induced cellular damage, we measured the accumulation of the lipid peroxidation marker MDA and of the NO stable metabolite of NO, nitrites (NO2−), in the frontal cortex of the experimental groups. Stress exposure caused a smaller accumulation of MDA in CB2xP and JWH-treated WT mice (Figure 5A,B) [stress F(1, 23) = 4.7, P = 0.042; JWH treatment F(1, 23) = 30.08, P < 0.0001; stress × JWH treatment F(1, 23) = 15.32, P = 0.0009]; [stress F(1, 25) = 4.81, P = 0.039; CB2xP genotype F(1, 25) = 4.33, P = 0.0492; stress × CB2xP genotype F(1, 25) = 4.97, P = 0.036]. Conversely, CB2-KO mice presented higher levels of MDA after stress than the WT group (Figure 5C) [stress F(1, 19) = 15.61, P = 0.011; CB2-KO genotype F(1, 19) = 5.1, P = 0.038; stress × CB2-KO genotype F(1, 19) = 0.71, P = 0.41].
Figure 5.
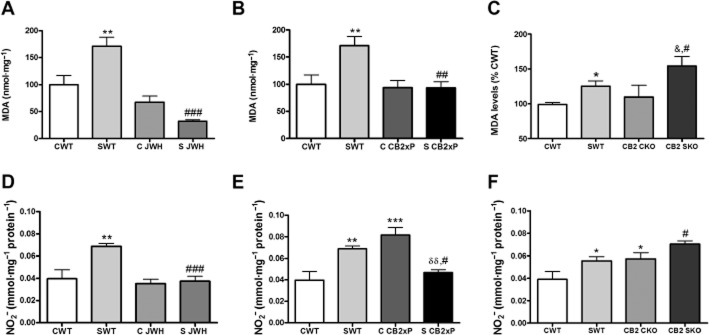
Neuroprotective effects mediated by CB2 receptors against stress-induced oxidative and nitrosative cellular damage. In (A), MDA levels in homogenates of the frontal cortex from untreated WT mice and WT mice treated with JWH-133, both with and without stress. In (B), results from CB2xP mice, with and without stress and in (C) for CB2-KO mice, with and without stress. Data represent the mean ± SEM of six mice. *P < 0.05, **P < 0.01 versus CWT; #P < 0.05, ##P < 0.01, ###P < 0.001 versus SWT; &P < 0.05 versus CKO; two-way anova with Bonferroni post test. In (D), nitrite (NO2−) levels in homogenates of the frontal cortex from untreated WT mice and WT mice treated with JWH-133, both with and without stress. In (E), results from CB2xP mice, with and without stress and in (F) for CB2-KO mice, with and without stress. Data represent the mean ± SEM of six mice. *P < 0.05, **P < 0.01, ***P < 0.001 versus CWT; #P < 0.05, ###P < 0.001 versus SWT; δδP < 0.01 versus C CB2xP; two-way anova with Bonferroni post test.
The results for NO2− (Figure 5D–F) followed a similar pattern in all groups of mice studied, with the exception of the CB2xP mice where there was a significant over-production of NO2−, possibly related to a compensatory mechanism, which may be worth further exploration (Figure 5E) [stress F(1, 18) = 9.79, P = 0.007; JWH treatment F(1, 18) = 12.86, P = 0.0027; stress × JWH treatment F(1, 18) = 7.28, P = 0.0165]; [stress F(1, 19) = 10.91, P = 0.0045; CB2xP genotype F(1, 19) = 14.43, P = 0.0016; stress × CB2xP genotype F(1, 19) = 8.98, P = 0.0085]; [stress F(1, 29) = 11.25, P = 0.0025; CB2-KO genotype F(1, 29) = 14.07, P = 0.0009; stress × CB2-KO genotype F(1, 29) = 0.15, P = 0.70].
Confirmation of the mediation of the effects of JWH-133 by CB2 receptors
Finally, to confirm that the actions of JWH-133 were mediated by CB2 receptors, we examined its effects on some representative inflammatory or oxidative parameters (IκBα and NOS-2 expression, and MDA and NO2− levels) in CB2-KO mice, exposed to stress. As shown in Figure 6A–D, treatment with JWH-133 did not alter any of the responses to stress in CB2-KO mice, suggesting the direct involvement of CB2 receptors in our model. For IκBα Western blot data [JWH treatment F(1, 19) = 15.85, P = 0.0026; CB2-KO genotype F(1, 19) = 71.10, P < 0.0001; JWH treatment × CB2-KO genotype F(1, 19) = 30.83, P = 0.002]. For NOS-2 [JWH treatment F(1, 19) = 9.50, P = 0.0095; CB2-KO genotype F(1, 19) = 69.17, P = 0.001; JWH treatment × CB2-KO genotype F(1, 19) = 4.78, P = 0.049]. For NO2− [JWH treatment F(1, 19) = 9.81, P = 0.0069; CB2-KO genotype F(1, 19) = 29.63, P < 0.0001; JWH treatment × CB2-KO genotype F(1, 19) = 0.63, P = 0.4407]. For MDA [JWH treatment F(1, 19) = 11.12, P = 0.0049; CB2-KO genotype F(1, 19) = 25.21, P = 0.0002; JWH treatment × CB2-KO genotype F(1, 19) = 0.43, P = 0.5216].
Figure 6.

Confirmation that effects of JWH-133 were mediated by CB2 receptors. Western blot and densitometric analysis of the NF-κB inhibitory protein IκBα (A) in extracts of the frontal cortex from stressed WT mice and CB2-KO mice, both with and without JWH-133. In (B), the corresponding data for NOS-2. Data are normalized by β-actin (lower band) and are representative of three experiments. #P < 0.05, ###P < 0.001 versus SWT + VEH; $$$P < 0.001 versus SWT + JW; two-way anova with Bonferroni post test. In (C), results for nitrite levels in homogenates of the frontal cortex from stressed WT mice and CB2-KO mice, both with and without JWH-133. In (D) the corresponding data for MDA, with and without JWH-133. #P < 0.05 versus SWT + VEH; $$P < 0.01, $$$P < 0.001 versus SWT + JW; two-way anova with Bonferroni post test.
Discussion and conclusions
Our results indicate a general anti-inflammatory role for CB2 receptors in the frontal cortex of mice exposed to sub-chronic restraint and acoustic stress. JWH-133 treatment or overexpression of CB2 receptors resulted in an increase in control levels of glutamate uptake, which was then reduced by stress, back to control levels. These effects are not due to changes in the general response to stress, as the different manipulations of the CB2 receptors did not modify plasma corticosterone levels, in our model.
Although previous results suggested that the excitotoxic process in stress conditions was regulated by CB1 receptors (Zoppi et al., 2011), other authors have demonstrated a role for CB1 and CB2 receptors in the regulation of AMPA excitotoxicity in in vivo and in vitro models of multiple sclerosis, through the up-regulation of EAAT-2 (Docagne et al., 2007; Loría et al., 2010). In our model, we did not find changes in EAAT-2 protein expression. Other approaches such as the determination of glutamate levels in the tissue will help to show whether the effects of CB2 receptor manipulations on glutamate uptake are due to inflammation-related actions on EAAT-2 activity or to a direct effect of the CB2 receptor in neurons.
According to our results, the CB2 receptors were not directly involved in the mechanism(s) controlling the production of plasma corticosterone, using this experimental protocol of stress. In agreement with this, increased plasma corticosterone elicited by systemic endotoxin administration did not change after the pharmacological modulation of CB2 receptors (Roche et al., 2006). However, the expression of these receptors in stress-responsive neural circuits, such as the hippocampus, amygdala and hypothalamus, indirectly suggests that CB2 receptor activation could regulate the neuroendocrine response (García-Gutiérrez et al., 2010). In fact, CB2xP mice, submitted to 30 min of restraint stress, presented lower levels of pro-opiomelanocortin mRNA in the arcuate nuclei than their WT counterparts, as well as a complete block of the stress-induced increase in the mRNA for corticotropin-releasing factor in the paraventricular nucleus of the hypothalamus (García-Gutiérrez and Manzanares, 2011). Thus, more detailed neuroendocrine studies regarding the time course of synthesis and release of corticosterone and other stress hormones in the sub-chronic stress protocol used here are needed to exclude a role of CB2 receptors in the regulation of the activity of the HPA axis.
Classically, because of its high level of expression in diverse types of immune cells and organs (Klein et al., 2003), CB2 receptors mediating anti-inflammatory effects have been described in the periphery. However, anti-inflammatory effects of CB2 receptor agonists in the CNS have been shown in traumatic brain injury, spinal cord injury, stroke and EAE (Arévalo-Martín et al., 2003; Mechoulam and Shohami, 2007; Castillo et al., 2010; Adhikary et al., 2011; Zarruk et al., 2012). Consistent with these findings, CB2 receptors are expressed by glia and neurons in the brain (Gong et al., 2006; Aracil-Fernández et al., 2012), although some controversy still exists, particularly in the case of neurons (Atwood and Mackie, 2010). Here, we have demonstrated that CB2 receptor activation regulated stress-induced neuroinflammation at several levels in the frontal cortex. This anti-inflammatory profile is especially relevant considering that neuroinflammatory processes have been proposed to underlie the pathophysiology of several stress-related neuropsychiatric disorders (Madrigal et al., 2006; Wager-Smith and Markou, 2011).
The regulation by CB2 receptors of the levels of pro-inflammatory cytokines (TNF-α) in the brain has been extensively documented (Jean-Gilles et al., 2010). However, less is known about the inhibitory role of CB2 receptors on production of CCL2 in vivo and only recently an inhibitory effect of CB2 receptor activation on CCL2 mRNA levels in animal models of stroke and multiple sclerosis has been reported (Palazuelos et al., 2008; Zarruk et al., 2012). These results are especially relevant considering that CCL2 is implicated in inflammatory cell migration into inflamed tissues (CNS included) and nociception – processes that have been related to CB2 receptors (Miller and Stella, 2008; Racz et al., 2008; Adhikary et al., 2011)
Our results are in agreement with other authors who demonstrated that CB2 receptor activation inhibited the activity of the major inflammatory mediator NF-κB, in immune cells (macrophages and microglia ) in vitro, exposed to inflammatory and immune stimuli (Jeon et al., 1996; Correa et al., 2010). However, to avoid the oversimplification of the effects of CB2 receptors on TNF-α and NF-κB, it should be noted that some studies have shown the beneficial effects of both inflammatory mediators in neuronal survival (Marchetti et al., 2004). Thus, their precise role in inflammation is still unclear.
The inhibitory effects of CB2 receptor activation on NOS-2 have been studied in animal models of neuropathology in vivo, such as Huntington's disease or stroke (Palazuelos et al., 2009; Zarruk et al., 2012), but the interactions between CB2 receptors and COX may be more complex. Our results suggest possible effects of JWH-133 on the catalytic activity or protein stability of COX-2, on the activity of COX-1 isoform or on tissue-specific PGE2 synthases, all of which remain to be assessed. Indeed, COX-2 inhibition following activation of CB2 receptors has been described in different in vivo and in vitro neuropathological experimental settings (Castillo et al., 2010; Martín-Moreno et al., 2012).
As a result of the over-production of consecutive pro-inflammatory mediators, oxidative and nitrosative cellular damage is produced after stress exposure. Our results suggest that any potential therapeutic use of CB2 receptor activation would utilise its antioxidant profile. Similarly, antioxidant effects mediated by CB2 receptors have also been found in CB2xP mice in an experimental model of Parkinson's disease (Ternianov et al., 2012). The antioxidant effects produced by the pharmacological activation of CB2 receptors have been extensively reviewed for several neurological or neurodegenerative diseases (Fernández-Ruiz, 2009).
CB2xP mice and pretreatment with JWH-133 in WT mice generate different experimental models, because CB2xP mice overexpress CB2 receptors not only in glia but also in neurons, and present endocrine and/or peripheral alterations, such as hyperglycaemia (Romero-Zerbo et al., 2012). Such alterations could limit the conclusions drawn from the use of CB2xP mice. Under the conditions of the present study, the anti-inflammatory profile of both experimental groups was very similar, with the exception of COX-2 protein levels, which were resistant to the effects of treatment with JWH-133, whereas CB2xP mice presented a clear decrease of COX-2 protein. CB2xP mice present an up-regulation of CB2 receptors on frontal cortex neurons, an effect that could affect the characteristic constitutive expression of COX-2 in this brain area (Yamagata et al., 1993).
Although JWH-133 exhibits a higher affinity for CB2 than for CB1 receptors (Huffman et al., 1999), this compound is only selective not specific for CB2 receptors. This is why we tested JWH-133 in CB2-KO animals. Although the anti-inflammatory effects of JWH-133 were absent in CB2-KO animals, we exclude the possibility that this compound elicits CB2 receptor-independent effects, using different doses or routes of administration, or in other models of neuropathology.
In contrast to the anti-inflammatory effects of the CB2 receptor agonist JWH-133, the CB2-KO mice presented an enhanced neuroinflammatory response in the frontal cortex, after stress exposure. Deletion of the CB2 receptors also induced schizophrenia and depression-like behaviours in mice (Ortega-Alvaro et al., 2011), but further investigation is needed to elucidate whether the excessive neuroinflammation present in CB2-KO mice is directly related to the pathophysiology of these major psychiatric diseases or is merely an epiphenomenon.
This study has some limitations. First, although it was not the main goal of this work, assessment of the role(s) of CB2 receptors in the regulation of HPA axis activation requires more detailed neuroendocrine studies, including a time course of the synthesis and release of the main stress hormones. Second, the study of different brain areas involved in the stress response would draw a more comprehensive picture of the regulatory role of CB2 receptors. Third, studies carried out in extended stress exposure models, which induce depressive-like behaviours (e.g. two chronic mild stresses, chronic unpredictable stress) would strengthen translational conclusions.
In conclusion, we have found evidence of anti-inflammatory effects of CB2 receptor activation, which were not related to alterations in plasma corticosterone levels. Activation of CB1 receptors elicits a consistent neuroprotective response in the same stress paradigm (Zoppi et al., 2011), but direct activation of CB2 receptors produces less undesired central effects (e.g. psychoactive effects) (Mechoulam and Parker, 2013). Our previous and current findings open possibilities for the use of dual agonists of CB1 and CB2 receptors or of endocannabinoid reuptake inhibitors, for the management of neurological and neurodegenerative and neuropsychiatric diseases.
Acknowledgments
This research was supported by the Regional Government of Madrid (S2011/BMD-2308. CANNAB), the Spanish Ministries of Science and Innovation (CIBERSAM), the Institute of Salud Carlos III, ISCIII (FIS 10–0123) and Univ. Complutense-Santander (2878–920140). The research was also funded by Red Temática de Investigación Cooperativa en Salud (RETICS, Instituto de Salud Carlos III, MICINN/FEDER): Red de Trastornos Adictivos, RD06/0001/1004 (J. M.). J. R. C. is a Juan de la Cierva postdoctoral fellow (MEC). B. G.-B. is a Ramón y Cajal postdoctoral fellow (MEC).
Glossary
- ECS
endocannabinoid system
- HPA
hypothalamic–pituitary–adrenal
- MDA
malondialdehyde
Conflict of interest
None.
Supporting Information
Additional Supporting Information may be found in the online version of this article at the publisher’s web-site: http://dx.doi.org/10.1111/bph.12607
Details of experimental procedures.
References
- Adhikary S, Li H, Heller J, Skarica M, Zhang M, Ganea D, et al. Modulation of inflammatory responses by a cannabinoid-2-selective agonist after spinal cord injury. J Neurotrauma. 2011;28:2417–2427. doi: 10.1089/neu.2011.1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: G Protein-Coupled Receptors. Br J Pharmacol. 2013a;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Transporters. Br J Pharmacol. 2013b;170:1706–1796. doi: 10.1111/bph.12450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aracil-Fernández A, Trigo JM, García-Gutiérrez MS, Ortega-Álvaro A, Ternianov A, Navarro D, et al. Decreased cocaine motor sensitization and self-administration in mice overexpressing cannabinoid CB2 receptors. Neuropsychopharmacology. 2012;7:1749–1763. doi: 10.1038/npp.2012.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arévalo-Martín A, Vela JM, Molina-Holgado E, Borrell J, Guaza C. Therapeutic action of cannabinoids in a murine model of multiple sclerosis. J Neurosci. 2003;23:2511–2516. doi: 10.1523/JNEUROSCI.23-07-02511.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwood BK, Mackie K. CB2: a cannabinoid receptor with an identity crisis. Br J Pharmacol. 2010;160:467–479. doi: 10.1111/j.1476-5381.2010.00729.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bambico FR, Duranti A, Tontini A, Tarzia G, Gobbi G. Endocannabinoids in the treatment of mood disorders: evidence from animal models. Curr Pharm Des. 2009;15:1623–1646. doi: 10.2174/138161209788168029. [DOI] [PubMed] [Google Scholar]
- Bierhaus A, Wolf J, Andrassy M, Rohleder N, Humpert PM, Petrov D, et al. A mechanism converting psychosocial stress into mononuclear cell activation. Proc Natl Acad Sci U S A. 2003;100:1920–1925. doi: 10.1073/pnas.0438019100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisogno T, Di Marzo V. Cannabinoid receptors and endocannabinoids: role in neuroinflammatory and neurodegenerative disorders. CNS Neurol Disord Drug Targets. 2010;9:564–573. doi: 10.2174/187152710793361568. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Bremner JD, Randall P, Scott TM, Bronen RA, Seibyl JP, Southwick SM, et al. MRI-based measurement of hippocampal volume in patients with combat-related posttraumatic stress disorder. Am J Psychiatry. 1995;152:973–981. doi: 10.1176/ajp.152.7.973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabral GA, Griffin-Thomas L. Emerging role of the cannabinoid receptor CB2 in immune regulation: therapeutic prospects for neuroinflammation. Expert Rev Mol Med. 2009;11:e3. doi: 10.1017/S1462399409000957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capuron L, Miller AH. Immune system to brain signaling: neuropsychopharmacological implications. Pharmacol Ther. 2011;130:226–238. doi: 10.1016/j.pharmthera.2011.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo A, Tolón MR, Fernández-Ruiz J, Romero J, Martinez-Orgado J. The neuroprotective effect of cannabidiol in an in vitro model of newborn hypoxic-ischemic brain damage in mice is mediated by CB(2) and adenosine receptors. Neurobiol Dis. 2010;37:434–440. doi: 10.1016/j.nbd.2009.10.023. [DOI] [PubMed] [Google Scholar]
- Conductier G, Blondeau N, Guyon A, Nahon JL, Rovère C. The role of monocyte chemoattractant protein MCP1/CCL2 in neuroinflammatory diseases. J Neuroimmunol. 2010;224:93–100. doi: 10.1016/j.jneuroim.2010.05.010. [DOI] [PubMed] [Google Scholar]
- Correa F, Hernangómez M, Mestre L, Loría F, Spagnolo A, Docagne F, et al. Anandamide enhances IL-10 production in activated microglia by targeting CB(2) receptors: roles of ERK1/2, JNK, and NF-kappaB. Glia. 2010;58:135–147. doi: 10.1002/glia.20907. [DOI] [PubMed] [Google Scholar]
- Das NP, Ratty AK. Studies on the effects of the narcotic alkaloids, cocaine, morphine and codeine on nonenzymatic lipid peroxidation in rat brain mitochondria. Biochem Med Metab Biol. 1987;37:256–264. doi: 10.1016/0885-4505(87)90035-1. [DOI] [PubMed] [Google Scholar]
- Docagne F, Muñetón V, Clemente D, Ali C, Loría F, Correa F, et al. Excitotoxicity in a chronic model of multiple sclerosis: neuroprotective effects of cannabinoids through CB1 and CB2 receptor activation. Mol Cell Neurosci. 2007;34:551–561. doi: 10.1016/j.mcn.2006.12.005. [DOI] [PubMed] [Google Scholar]
- Farooq RK, Isingrini E, Tanti A, Le Guisquet AM, Arlicot N, Minier F, et al. Is unpredictable chronic mild stress (UCMS) a reliable model to study depression-induced neuroinflammation? Behav Brain Res. 2012;231:130–137. doi: 10.1016/j.bbr.2012.03.020. [DOI] [PubMed] [Google Scholar]
- Fernández-Ruiz J. The endocannabinoid system as a target for the treatment of motor dysfunction. Br J Pharmacol. 2009;156:1029–1040. doi: 10.1111/j.1476-5381.2008.00088.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Bueno B, Caso JR, Pérez-Nievas BG, Lorenzo P, Leza JC. Effects of peroxisome proliferator-activated receptor gamma agonists on brain glucose and glutamate transporters after stress in rats. Neuropsychopharmacology. 2007;32:1251–1260. doi: 10.1038/sj.npp.1301252. [DOI] [PubMed] [Google Scholar]
- García-Bueno B, Caso JR, Leza JC. Stress as a neuroinflammatory condition in brain: damaging and protective mechanisms. Neurosci Biobehav Rev. 2008;32:1136–1151. doi: 10.1016/j.neubiorev.2008.04.001. [DOI] [PubMed] [Google Scholar]
- García-Gutiérrez MS, Manzanares J. Overexpression of CB2 cannabinoid receptors decreased vulnerability to anxiety and impaired anxiolytic action of alprazolam in mice. Psychopharmacol. 2011;25:111–120. doi: 10.1177/0269881110379507. [DOI] [PubMed] [Google Scholar]
- García-Gutiérrez MS, Pérez-Ortiz JM, Gutiérrez-Adán A, Manzanares J. Depression-resistant endophenotype in mice overexpressing cannabinoid CB(2) receptors. Br J Pharmacol. 2010;160:1773–1784. doi: 10.1111/j.1476-5381.2010.00819.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong JP, Onaivi ES, Ishiguro H, Liu QR, Tagliaferro PA, Brusco A, et al. Cannabinoid CB2 receptors: immunohistochemical localization in rat brain. Brain Res. 2006;1071:10–23. doi: 10.1016/j.brainres.2005.11.035. [DOI] [PubMed] [Google Scholar]
- Green LC, Wagner DA, Glogowski J, Skipper PL, Wishnok JS, Tannenbaum SR. Analysis of nitrate, nitrite, and [15N]nitrate in biological fluids. Anal Biochem. 1982;126:131–138. doi: 10.1016/0003-2697(82)90118-x. [DOI] [PubMed] [Google Scholar]
- Herbert TB, Cohen S. Stress and immunity in humans: a meta-analytic review. Psychosom Med. 1993;55:364–379. doi: 10.1097/00006842-199307000-00004. [DOI] [PubMed] [Google Scholar]
- Huffman JW, Liddle J, Yu S, Aung MM, Abood ME, Wiley JL, et al. 3-(1′,1′-Dimethylbutyl)-1-deoxy-delta8-THC and related compounds: synthesis of selective ligands for the CB2 receptor. Bioorg Med Chem. 1999;7:2905–2914. doi: 10.1016/s0968-0896(99)00219-9. [DOI] [PubMed] [Google Scholar]
- Jean-Gilles L, Gran B, Constantinescu CS. Interaction between cytokines, cannabinoids and the nervous system. Immunobiology. 2010;215:606–610. doi: 10.1016/j.imbio.2009.12.006. [DOI] [PubMed] [Google Scholar]
- Jeon YJ, Yang KH, Pulaski JT, Kaminski NE. Attenuation of inducible nitric oxide synthase gene expression by delta 9-tetrahydrocannabinol is mediated through the inhibition of nuclear factor-kappa B/Rel activation. Mol Pharmacol. 1996;50:334–341. [PubMed] [Google Scholar]
- Kiank C, Holtfreter B, Starke A, Mundt A, Wilke C, Schütt C. Stress susceptibility predicts the severity of immune depression and the failure to combat bacterial infections in chronically stressed mice. Brain Behav Immun. 2006;20:359–368. doi: 10.1016/j.bbi.2005.10.151. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JJ, Yoon KS. Stress: metaplastic effects in the hippocampus. Trends Neurosci. 1998;21:505–509. doi: 10.1016/s0166-2236(98)01322-8. [DOI] [PubMed] [Google Scholar]
- Klein TW, Newton C, Larsen K, Lu L, Perkins I, Nong L, et al. The cannabinoid system and immune modulation. Leukoc Biol. 2003;74:486–496. doi: 10.1189/jlb.0303101. [DOI] [PubMed] [Google Scholar]
- Licinio J, Wong ML. The role of inflammatory mediators in the biology of major depression: central nervous system cytokines modulate the biological substrate of depressive symptoms, regulate stress-responsive systems, and contribute to neurotoxicity and neuroprotection. Mol Psychiatry. 1999;4:317–327. doi: 10.1038/sj.mp.4000586. [DOI] [PubMed] [Google Scholar]
- Loría F, Petrosino S, Hernangómez M, Mestre L, Spagnolo A, Correa F, et al. An endocannabinoid tone limits excitotoxicity in vitro and in a model of multiple sclerosis. Neurobiol Dis. 2010;37:166–176. doi: 10.1016/j.nbd.2009.09.020. [DOI] [PubMed] [Google Scholar]
- Madrigal JL, Hurtado O, Moro MA, Lizasoain I, Lorenzo P, Castrillo A, et al. The increase in TNF-alpha levels is involved in NF-kappaB activation and inducible nitric oxide synthase expression in brain cortex after immobilization stress. Neuropsychopharmacology. 2002;26:155–163. doi: 10.1016/S0893-133X(01)00292-5. [DOI] [PubMed] [Google Scholar]
- Madrigal JL, García-Bueno B, Caso JR, Pérez-Nievas BG, Leza JC. Stress-induced oxidative changes in brain. CNS Neurol Disord Drug Targets. 2006;5:561–568. doi: 10.2174/187152706778559327. [DOI] [PubMed] [Google Scholar]
- Marchetti L, Klein M, Schlett K, Pfizenmaier K, Eisel UL. Tumor necrosis factor (TNF)-mediated neuroprotection against glutamate-induced excitotoxicity is enhanced by N-methyl-D-aspartate receptor activation. Essential role of a TNF receptor 2-mediated phosphatidylinositol 3-kinase-dependent NF-kappa B pathway. J Biol Chem. 2004;279:32869–32881. doi: 10.1074/jbc.M311766200. [DOI] [PubMed] [Google Scholar]
- Martín-Moreno AM, Brera B, Spuch C, Carro E, García-García L, Delgado M, et al. Prolonged oral cannabinoid administration prevents neuroinflammation, lowers β-amyloid levels and improves cognitive performance in Tg APP 2576 mice. J Neuroinflammation. 2012;9:8. doi: 10.1186/1742-2094-9-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mechoulam R, Parker LA. The endocannabinoid system and the brain. Annu Rev Psychol. 2013;64:21–47. doi: 10.1146/annurev-psych-113011-143739. [DOI] [PubMed] [Google Scholar]
- Mechoulam R, Shohami E. Endocannabinoids and traumatic brain injury. Mol Neurobiol. 2007;36:68–74. doi: 10.1007/s12035-007-8008-6. [DOI] [PubMed] [Google Scholar]
- Miller AM, Stella N. CB2 receptor-mediated migration of immune cells: it can go either way. Br J Pharmacol. 2008;153:299–308. doi: 10.1038/sj.bjp.0707523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moghaddam B. Stress preferentially increases extraneuronal levels of excitatory amino acids in the prefrontal cortex: comparison to hippocampus and basal ganglia. J Neurochem. 1993;60:1650–1657. doi: 10.1111/j.1471-4159.1993.tb13387.x. [DOI] [PubMed] [Google Scholar]
- Onaivi ES, Ishiguro H, Gu S, Liu QR. CNS effects of CB2 cannabinoid receptors: beyond neuro-immuno-cannabinoid activity. J Psychopharmacol. 2012;26:92–103. doi: 10.1177/0269881111400652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega-Alvaro A, Aracil-Fernández A, García-Gutiérrez MS, Navarrete F, Manzanares J. Deletion of CB2 cannabinoid receptor induces schizophrenia-related behaviors in mice. Neuropsychopharmacology. 2011;36:1489–1504. doi: 10.1038/npp.2011.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palazuelos J, Davoust N, Julien B, Hatterer E, Aguado T, Mechoulam R, et al. The CB(2) cannabinoid receptor controls myeloid progenitor trafficking: involvement in the pathogenesis of an animal model of multiple sclerosis. J Biol Chem. 2008;83:13320–13329. doi: 10.1074/jbc.M707960200. [DOI] [PubMed] [Google Scholar]
- Palazuelos J, Aguado T, Pazos MR, Julien B, Carrasco C, Resel E, et al. Microglial CB2 cannabinoid receptors are neuroprotective in Huntington's disease excitotoxicity. Brain. 2009;132:3152–3164. doi: 10.1093/brain/awp239. (Pt 11) [DOI] [PubMed] [Google Scholar]
- Pertwee RG. Pharmacology of cannabinoid CB1 and CB2 receptors. Pharmacol Ther. 1997;74:129–180. doi: 10.1016/s0163-7258(97)82001-3. [DOI] [PubMed] [Google Scholar]
- Racz I, Nadal X, Alferink J, Baños JE, Rehnelt J, Martín M, et al. Crucial role of CB(2) cannabinoid receptor in the regulation of central immune responses during neuropathic pain. J Neurosci. 2008;28:12125–12135. doi: 10.1523/JNEUROSCI.3400-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radley JJ, Arias CM, Sawchenko PE. Regional differentiation of the medial prefrontal cortex in regulating adaptive responses to acute emotional stress. J Neurosci. 2006;26:12967–12976. doi: 10.1523/JNEUROSCI.4297-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche M, Diamond M, Kelly JP, Finn DP. In vivo modulation of LPS-induced alterations in brain and peripheral cytokines and HPA axis activity by cannabinoids. J Neuroimmunol. 2006;181:57–67. doi: 10.1016/j.jneuroim.2006.08.001. [DOI] [PubMed] [Google Scholar]
- Romero-Zerbo SY, Garcia-Gutierrez MS, Suárez J, Rivera P, Ruz-Maldonado I, Vida M, et al. Overexpression of cannabinoid CB2 receptor in the brain induces hyperglycaemia and a lean phenotype in adult mice. J Neuroendocrinol. 2012;24:1106–1119. doi: 10.1111/j.1365-2826.2012.02325.x. [DOI] [PubMed] [Google Scholar]
- Schreiber E, Matthias P, Müller MM, Schaffner W. Rapid detection of octamer binding proteins with ‘mini-extracts’, prepared from a small number of cells. Nucleic Acid Res. 1989;17:6419. doi: 10.1093/nar/17.15.6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorrells SF, Caso JR, Munhoz CD, Sapolsky RM. The stressed CNS: when glucocorticoids aggravate inflammation. Neuron. 2009;64:33–39. doi: 10.1016/j.neuron.2009.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ternianov A, Pérez-Ortiz JM, Solesio ME, García-Gutiérrez MS, Ortega-Álvaro A, Navarrete F, et al. Overexpression of CB2 cannabinoid receptors results in neuroprotection against behavioral and neurochemical alterations induced by intracaudate administration of 6-hydroxydopamine. Neurobiol Aging. 2012;33:421.e1–421.e16. doi: 10.1016/j.neurobiolaging.2010.09.012. [DOI] [PubMed] [Google Scholar]
- Wager-Smith K, Markou A. Depression: a repair response to stress-induced neuronal microdamage that can grade into a chronic neuroinflammatory condition? Neurosci Biobehav Rev. 2011;35:742–764. doi: 10.1016/j.neubiorev.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf SA, Tauber S, Ullrich O. CNS immune surveillance and neuroinflammation: endocannabinoids keep control. Curr Pharm Des. 2008;14:2266–2278. doi: 10.2174/138161208785740090. [DOI] [PubMed] [Google Scholar]
- Yamagata K, Andreasson KI, Kaufmann WE, Barnes CA, Worley PF. Expression of a mitogen-inducible cyclooxygenase in brain neurons: regulation by synaptic activity and glucocorticoids. Neuron. 1993;11:371–386. doi: 10.1016/0896-6273(93)90192-t. [DOI] [PubMed] [Google Scholar]
- Zarruk JG, Fernández-López D, García-Yébenes I, García-Gutiérrez MS, Vivancos J, Nombela F, et al. Cannabinoid type 2 receptor activation downregulates stroke-induced classic and alternative brain macrophage/microglial activation concomitant to neuroprotection. Stroke. 2012;43:211–219. doi: 10.1161/STROKEAHA.111.631044. [DOI] [PubMed] [Google Scholar]
- Zoppi S, Pérez Nievas BG, Madrigal JL, Manzanares J, Leza JC, García-Bueno B. Regulatory role of cannabinoid receptor 1 in stress-induced excitotoxicity and neuroinflammation. Neuropsychopharmacology. 2011;236:805–818. doi: 10.1038/npp.2010.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Details of experimental procedures.