

Abstract
Background and Purpose
Relaxin family peptide receptor 3 (RXFP3) is expressed in brain areas important for processing sensory information and feeding, suggesting that it may be a target for anti-anxiety and anti-obesity drugs. We examined the effects of H3 relaxin, the biased agonist H2 relaxin and the antagonist, R3(BΔ23–27)R/I5, on RXFP3 signalling to establish their suitability as tools to assess the physiological roles of RXFP3.
Experimental Approach
The signalling profile of the RXFP3 ligands was determined using reporter gene assays, multiplexed signalling assays and direct examination of receptor–G protein and receptor–β-arrestin interactions using BRET.
Key Results
H2 relaxin activated p38MAPK and ERK1/2 with lower efficacy than H3 relaxin, but had similar efficacy for JNK1/2 phosphorylation. H2 or H3 relaxin activation of p38MAPK, JNK1/2 or ERK1/2 involved Pertussis toxin-sensitive G-proteins. R3(BΔ23–27)R/I5 blocked H3 relaxin AP-1 reporter gene activation, but not H2 relaxin AP-1 activation or H3 relaxin NF-κB activation. R3(BΔ23–27)R/I5 activated the SRE reporter, but did not inhibit either H2 or H3 relaxin SRE activation. R3(BΔ23–27)R/I5 blocked H3 relaxin-stimulated p38MAPK and ERK1/2 phosphorylation, but was a weak partial agonist for p38MAPK and ERK1/2 signalling. p38MAPK activation by R3(BΔ23–27)R/I5 was G protein-independent. H3 relaxin-activated RXFP3 interacts with Gαi2, Gαi3, GαoA and GαoB whereas H2 relaxin or R3(BΔ23–27)R/I5 induce interactions only with Gαi2 or GαoB. Only H3 relaxin promoted RXFP3/β-arrestin interactions that were blocked by R3(BΔ23–27)R/I5.
Conclusion and Implications
Understanding signalling profile of drugs acting at RXFP3 is essential for development of therapies targeting this receptor.
Keywords: RXFP3 signalling, relaxin, R3(BΔ23–27)R/I5
Introduction
Relaxin-3 is the most recently discovered neuropeptide of the relaxin/insulin-like family of peptides (Bathgate et al., 2002). Relaxin-3 and its cognate receptor relaxin family peptide receptor 3 (RXFP3; nomenclature follows Alexander et al., 2013) are expressed in brain areas important for processing sensory information and in feeding (Liu et al., 2003b; Sutton et al., 2004). Relaxin-3 modulates behavioural responses to stress (Tanaka et al., 2005) and appetite (McGowan et al., 2007), suggesting key physiological roles in neuroendocrine and sensory processing. Relaxin-3 expression increases in rats in response to physical stress by a corticorelin-dependent mechanism (Tanaka et al., 2005) and injection into the paraventricular nucleus of the hypothalamus or the fourth ventricle increases food intake (McGowan et al., 2007). Recent comprehensive studies of the neuroanatomical distribution of relaxin-3 and RXFP3 in mouse brain confirmed that most relaxin-3/RXFP3–rich brain regions are involved in behavioural activation including septohippocampal, arousal and sensory/visuospatial control circuits or in stress and affective responses including hypothalamic and extrahypothalamic circuits that play crucial roles in corticotrophin-releasing factor-mediated effects and regulation of the hypothalamic–pituitary–adrenal axis (Smith et al., 2010). Thus, the relaxin-3/RXFP3 system is involved in arousal, stress, affective and cognitive circuits.
Some cross reactivity exists between relaxin peptides and their receptors. H3 relaxin also activates RXFP1 (Bathgate et al., 2002; 2006b) and RXFP4 (Liu et al., 2003a). RXFP3-selective agonists and antagonists are useful for the study of the physiological roles of RXFP3 and provide the basis for the development of novel anti-anxiety and anti-obesity drugs.
H3 relaxin contains A- and B-chains linked by two disulphide bonds (Bathgate et al., 2002; 2006a). The chimeric H3 relaxin B-chain/human insulin-like peptide (INSL) 5 A-chain (R3/I5) is selective for RXFP3 over RXFP1 (Liu et al., 2005) as is R3(BΔ23–27)R/I5 (Kuei et al., 2007; Hossain et al., 2009) (Figure 1) making it a useful tool for the study of the role of RXFP3 in behavioural disorders and obesity.
Figure 1.
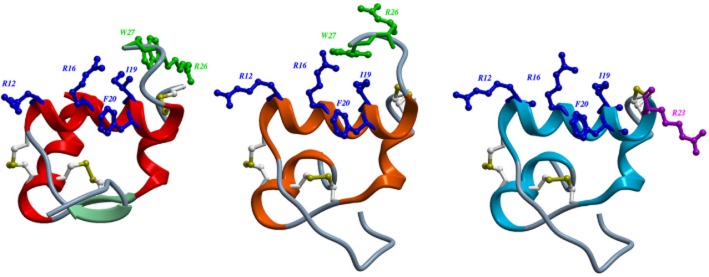
Structures of H3 relaxin (A); chimeric H3 relaxin B-chain/INSL5 A-chain (R3/I5) (B) and R3(BΔ23–27)R/I5 (C). The structures of H3 relaxin (2fhw) and R3/I5) (2k1v) were downloaded from the RSCB protein data bank and displayed using ICM-pro (Molsoft L.L.C., San Diego, CA, USA). The heterodimeric peptides contain an A-chain and B-chain linked by disulphide bonds (Bathgate et al., 2002; 2006a). Residues important for RXFP3 binding are in blue and for activation in green. Arg23 in R3(BΔ23–27)R/I5 is important for antagonist activity and shown in purple (Kuei et al., 2007; Hossain et al., 2009). (A) The NMR structure of H3 relaxin (Rosengren et al., 2006). (B) R3/I5 comprises the H3 relaxin B-chain and the human INSL5 A-chain (Sutton et al., 2004). NMR studies indicate that R3/I5 has a very similar structure to H3 relaxin (Haugaard-Jonsson et al., 2008). (C) The R3/I5 analogue, R3(BΔ23–27)R/I5 has the C-terminus of the B-chain truncated and with a terminal Arg residue (Kuei et al., 2007; Hossain et al., 2009).
RXFP3 is a family A GPCR that upon activation couples to Gαi/o, releases Gβγ and interacts with β-arrestins. RXFP3 agonists cause inhibition of forskolin-stimulated cAMP accumulation and stimulation of ERK1/2 phosphorylation in a Gαi/o-dependent manner (Liu et al., 2003b; van der Westhuizen et al., 2007; 2010). H2 relaxin also binds to RXFP3, but activates a different signalling pattern to H3 relaxin. Whereas stimulation of RXFP3 with H3 relaxin causes activation of AP-1 and NF-κB reporter genes, only AP-1 was stimulated by H2 relaxin. ADP ribosylation of Gαi/o by Pertussis toxin (PTX) blocked all reporter gene activation except for H3 relaxin-stimulated activation of AP-1 (van der Westhuizen et al., 2010). These data provided the first evidence for ligand-directed signalling bias (LDSB) at RXFP3.
LDSB (or stimulus-bias) describes the ability of individual ligands to stabilize different receptor conformations and consequently to display distinct efficacy or potency profiles for different signalling pathways. Thus it is increasingly important to monitor many different signalling pathways in order to determine whether agonists or antagonists display LDSB. In this study, we investigated whether LDSB occurs to H3 relaxin, H2 relaxin and R3(BΔ23–27)R/I5 using reporter gene assays and specific assays for ERK1/2, JNK and p38MAPK. Additionally, we directly examined the interactions between RXFP3, G proteins and β-arrestins. H3 relaxin, H2 relaxin and R3(BΔ23–27)R/I5 couple to distinct patterns of signalling through RXFP3. H3 relaxin activated AP-1, NF-κB and serum response element (SRE) reporter genes whereas H2 relaxin activated AP-1 and SRE, and R3(BΔ23–27)R/I5 only SRE. R3(BΔ23–27)R/I5 antagonized H3 relaxin-mediated AP-1 activation, but not NFkB, SRE or AP-1 or SRE activation by H2 relaxin. All three ligands caused p38MAPK phosphorylation whereas H3 and H2 relaxin caused JNK1/2 phosphorylation and H3 relaxin caused ERK1/2 phosphorylation that was much greater than that to H2 relaxin or R3(BΔ23–27)R/I5. In p38MAPK and ERK1/2 assays, R3(BΔ23–27)R/I5 was a partial agonist with greater efficacy for p38MAPK than ERK1/2 phosphorylation. Finally, we identified G proteins and scaffold proteins that couple to RXFP3 following its activation. RXFP3 coupled to Gαi2 or GαOB when activated by R3(BΔ23–27)R/I5, H2 or H3 relaxin, which also stimulated coupling to Gαi3 or GαOA. H3 relaxin, but not other ligands induced RXFP3/β-arrestin interactions that were inhibited by R3(BΔ23–27)R/I5. In summary, we demonstrated distinct patterns of signalling for H3 and H2 relaxin and R3(BΔ23–27)R/I5 at the RXFP3 receptor.
Methods
Constructs
RXFP3 sequence was amplified from cDNA clone RXFP3-GFP2 (van der Westhuizen et al., 2010) using the forward primer 5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTTCCACCATGCAGATGGCCGAT GCAGCC-3′ and the reverse primer 5′-GGGGACCACTTTGTACA AGAAAGCTGGGTCGTAGGCAGAGCTGCTGGG-3′, purified using the Ultraclean 15 DNA purification kit and cloned into pDONR201 with BP clonase II at 25°C for 16 h. Rluc8 cDNA was amplified, purified and cloned into pEF5/FRT/V5−DEST vector (Kocan et al., 2008). An expression clone of RXFP3-Rluc8 was created using LR clonase at 25°C for 16 h. All construct sequences were confirmed by DNA sequencing at the Australian Genome Research Facility (Melbourne, Australia).
β-Arrestin 1-Venus (arrestin-2) and β-arrestin 2-Venus (arrestin-3) vectors (Venus attached to the C-termini of the β-arrestins) were prepared as described previously (Kocan et al., 2008). G protein cDNA clones Gβ1 and Gα subunits Gαs, Gαi2, Gαi3, GαoA, GαoB, Gαq, Gα11, Gα12 or Gα13 were from Missouri S&T cDNA Resource Center (Rolla, MO, USA; http://www.cdna.org) and Gγ2-Venus was supplied by Michel Bouvier (Department of Biochemistry, Université de Montréal, Montréal, Quebec, Canada; Gales et al., 2005).
Cell culture and transfection
Flp-In CHO parental cells and stable RXFP3-expressing cells (van der Westhuizen et al., 2007), were maintained at 37°C under 5% CO2 in DMEM/Ham's F12 medium containing 5% (v/v) FBS, 2 mM L-glutamine, 100 U·mL–1 penicillin, 100 μg·mL–1 streptomycin. Transient transfections were carried out 24 h after seeding using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA).
Secreted alkaline protease (SEAP)-linked reporter gene assay
Eight cis-acting enhancer elements linked to human placental alkaline phosphatase (SEAP; Mercury Pathway Profiling, Clontech Laboratories, Inc., Mountain View, CA, USA) were used to identify signalling pathways downstream of RXFP3 as described previously (van der Westhuizen et al., 2010). The reporter genes tested were: AP-1, cAMP response element, E-box DNA binding element (Myc), glucocorticoid response element, heat shock response element, nuclear factor of activated T cells, NF-κB and SRE. The SEAP level in the culture medium is directly proportional to intracellular SEAP concentration. A constitutively expressed β-galactosidase reporter gene was cotransfected with the SEAP reporter gene (1:1) to allow for variation in transfection efficiencies between experiments. Flp-In CHO stably expressing human RXFP3 (CHO-RXFP3) cells were plated into 48-well plates (105 cells per well) and grown in DMEM/Ham's F-12 medium for 24 h at 37°C, 5% CO2. Cells were transiently cotransfected with SEAP-linked reporter gene (75 ng per well) and β-galactosidase (75 ng per well) in OptiMEM using Lipofectamine. Subsequently, cells were serum starved for 18 h and then exposed to peptides or 0.001% trifluoroacetic acid, or 10% FBS (positive control). Twenty-four hours following stimulation, samples of medium were taken and frozen at −20°C. Thawed samples (25 μL) were transferred to white 96-well Optiplates, incubated at 65°C for 30 min then cooled on ice. Buffer (1 M diethanolamine and 0.5 mM magnesium chloride, pH 10.3) was added to the SEAP samples and β-galactosidase buffer (0.1 M sodium phosphate dibasic, 1 mM magnesium chloride, and 1.4% (v/v) β-mercaptoethanol, pH 7.0) to the β-galactosidase samples. Subsequently, 4-methylumbelliferyl phosphate (2 mM) for SEAP detection and 4-methylumbelliferyl β-galactopyranoside (2 mM) for β-galactosidase detection were added and plates incubated for 1 h at 22°C in the dark and read on a Packard Fusion-Alpha (excitation 360 nm and emission 440 nm; PerkinElmer Life and Analytical Sciences, Wellesley, MA, USA).
p38MAPK, JNK1/2 or ERK1/2 Phosphorylation Surefire® assay
CHO-RXFP3 cells were plated into 96-well plates (4 × 104 cells per well) and grown overnight as described earlier. Cells were washed twice with PBS and serum starved for 9 h before addition of ligands, serum-free-DMEM/Ham's F-12 medium plus 0.01% w/v BSA (vehicle control) or 10% FBS (positive control) at 37°C. For PTX treatment cells were plated out, allowed to adhere, washed twice with PBS and serum starved in the presence or absence of PTX (100 ng·Ml–1) for 18 h. After experimentation, cells were lysed with 100 μL lysis buffer and frozen at −20°C. p38MAPK, JNK1/2 and ERK1/2 were detected in the same lysate allowing detection of multiple signalling pathways from the same sample. For each kinase detection, 4 μL of cell lysate was transferred to white 384-well microplates (Proxiplates) and 5 μL of AlphaScreen donor beads (40 parts reaction buffer, 10 part activation buffer and 1 part acceptor beads; PerkinElmer Life and Analytical Sciences) added. Plates were incubated for 2 h at 23°C in the dark. Subsequently, 2 μL of dilution buffer with AlphaScreen acceptor beads (20 parts dilution buffer, and 1 part donor beads; PerkinElmer Life and Analytical Sciences) was added and the plates incubated for 2 h at 23°C in the dark. Samples were counted in 384-well microplates (Proxiplates) on an EnVision Multilabel Plate Reader (PerkinElmer Life and Analytical Sciences; excitation 680 nm; emission 520 to 620 nm).
Inhibition of forskolin-stimulated cAMP accumulation
Inhibition of forskolin-stimulated cAMP accumulation was performed (van der Westhuizen et al., 2007) by adding relaxin peptides to the wells for 5 min at 37°C followed by forskolin (3.10−5 M) and incubated at 37°C for 3 min. Cells were lysed as mentioned earlier and 5 μL lysate transferred to an Optiplate (PerkinElmer Life and Analytical Sciences), 5 μL of acceptor beads in detection buffer added to each well under ambient light, and incubated at 22°C in the dark for 30 min. Donor beads and biotinylated cAMP were diluted in detection buffer, and incubated at 22°C in the dark for 30 min. 15 μL of donor bead/biotinylated cAMP were added per well in the 384-well optiplate, and incubated for 16h at 22°C in the dark. Plates were read as described earlier.
Real-time kinetic BRET assays
Flp-InCHO cells were seeded in six-well plates at 600 000 cells per well, transfected with constructs encoding the tagged receptors and signalling proteins then harvested 24 h later in HEPES-buffered phenol-red-free medium containing 5% FBS, plated out in a white 96-well plate (Nunc, Thermo Fisher Scientific Pte. Ltd., Singapore) and incubated at 37°C under 5% CO2. BRET assays were performed 48 h after transfection (Kocan et al., 2008; 2009,). Medium was replaced with phenol-red-free-DMEM containing 5% FBS plus 5 μM coelenterazine h and assays carried out immediately. BRET measurements at 37°C utilized the LUMIstar Omega plate reader and software (BMG LABTECH, Ortenberg, Germany). Emissions were simultaneously measured at 400–475 nm for Rluc8 with coelenterazine h (donor) and at 520–540 nm for Venus (acceptor). Cells were assayed before and after treatment with ligands or 5% FBS phenol-red-free-DMEM medium plus 0.01% w/v BSA (vehicle). Ligand-induced BRET was calculated by subtracting the ratio of emission through the acceptor wavelength window over emission through the donor wavelength window for a vehicle-treated cell sample from the same ratio for a second aliquot of the same cells treated with ligand(s) (Pfleger et al., 2006; Kocan et al., 2008; Mustafa et al., 2012; Ayoub et al., 2013). The final pretreatment reading is presented at the zero time point (time of ligand/vehicle addition).
Data analysis
SEAP activation was expressed as % of β-galactosidase activation and normalized against the positive control. p38MAPK, JNK1/2 and ERK1/2 phosphorylation were calculated against the negative control (vehicle-treated cells). All data are means SEM of n experiments. Reporter gene data were analysed by one-way anova with Dunnett's post hoc test to compare all R3(BΔ23–27)R/I5 + H3 relaxin or R3(BΔ23–27)R/I5 + H2 relaxin samples to the H3 relaxin (10−8 M) or the H2 relaxin (10−8 M) alone using Prism (ver. 5; GraphPad Software, San Diego, CA, USA).
Materials
Flp-InCHO cells, DMEM, DMEM/Ham's F12 media Lipofectamine 2000, OptiMEM, hygromycin B, vectors (pcDNA3.1 +, pDONR201 and pEF5/FRT/V5−DEST), BP and LR clonase proprietary enzyme mix, chemically competent DH5a and XL1−blue Escherichia coli strains and the destination vector conversion kit (Invitrogen); penicillin and streptomycin (ThermoTRACE, Melbourne, Australia); PTX and BSA (Sigma-Aldrich, Castle Hill, Australia); FBS (JRH Biosciences, Lenexa, KS, USA); coelenterazine h (Promega, Madison, WI, USA); tissue culture flasks and plates (Greiner Bio-One, Monroe, NC, USA); white 96- and 384-well Optiplates and Proxiplates (PerkinElmer Life and Analytical Sciences); SEAP reporter gene constructs (Clontech Laboratories, Inc.); Surefire phospho-p38MAPK, phospho-JNK and phospho-ERK kits (TGR Biosciences, Adelaide, Australia and PerkinElmer Life and Analytical Sciences), human gene 3 relaxin (H3 relaxin) (Bathgate et al., 2006b) and R3(BΔ23–27)R/I5 (Kuei et al., 2007) were synthesized at the Florey Institute of Neuroscience & Mental Health (Victoria, Australia) and human gene 2 relaxin (H2 relaxin) was supplied by Corthera Inc. (San Mateo, CA, USA).
Results
R3(BΔ23–27)R/I5 blocks H3 relaxin-stimulated AP-1 reporter gene activation, but not H2 relaxin AP-1 activation or H3 relaxin NF-κB activation. Previously we have shown that H3 relaxin activates both AP-1 and NF-κB reporter genes whereas H2 relaxin activates only the AP-1 reporter (van der Westhuizen et al., 2010). Here, we investigated the effect of the peptide antagonist R3(BΔ23–27)R/I5 on these signalling pathways in the presence and absence of H3 and H2 relaxin. The AP-1 reporter was activated in a concentration-dependent manner (Figure 2A) by H3 relaxin (10−6 M: 113 ± 15; 10−8 M: 44 ± 5), to lesser extent by H2 relaxin (10−6 M: 31 ± 4; 10−8 M: 20 ± 3), but not by R3(BΔ23–27)R/I5 (10−6 M: 10 ± 7; 10−8 M: −2 ± 4) – all expressed as percentage response to 10% FBS (positive control). AP-1 activation by H3 relaxin (10−8 M), but not by H2 relaxin (10−8 M) was blocked by R3(BΔ23–27)R/I5 (10−8 or 10−6 M; Figure 2A). The NF-κB reporter was activated only by H3 relaxin (10−6 M: 113 ± 15; 10−8 M: 54 ± 10), but not by H2 relaxin (10−6 M: 13 ± 6; 10−8 M: −7 ± 4) or R3(BΔ23–27)R/I5 (10−6 M: 9 ± 4; 10−8 M: −10 ± 11). H3 relaxin-stimulated (10−8 M) NF-κB activation was unaffected by R3(BΔ23–27)R/I5 (10−6 M and 10−8 M) (Figure 2B). Thus, H3 relaxin is a full agonist and H2 relaxin a partial agonist for AP-1 signalling and R3(BΔ23–27)R/I5 is an antagonist for H3 relaxin-stimulated AP-1 reporter gene activation, but not H3 relaxin-stimulated NF-κB or H2 relaxin-stimulated AP-1 reporter activation. Interestingly, while having no efficacy for AP-1 activation, R3(BΔ23–27)R/I5 appears to enhance H2 relaxin-induced RXFP3 AP-1 signalling. Experimental controls were parallel treatments of the parent cell line expressing the reporter gene, but not RXFP3. No AP-1 or NF-κB reporter gene activation was detected in Flp-In CHO cells treated with H3 relaxin, H2 relaxin or R3(BΔ23–27)R/I5 (Figure 2A, B).
Figure 2.

Activation of the reporter genes AP-1 (A), NF-κB (B) and SRE (C) and inhibition of the forskolin-stimulated cAMP production (D) in CHO-RXFP3 or Flp-In CHO cells by H3 relaxin, H2 relaxin or R3(BΔ23–27)R/I5. CHO-RXFP3 or Flp-In CHO cells (negative control) were transiently co-transfected with either AP-1-SEAP, NF-κB-SEAP, or SRE-SEAP reporter genes and a constitutively active β-galactosidase reporter. Activation was determined by increased SEAP in the culture medium 24 h after stimulation. H3 relaxin activated AP-1, NF-κB and SRE (A, B, C), but only the AP-1 reporter was blocked by R3(BΔ23–27)R/I5 (A). H2 relaxin stimulated AP-1 and SRE and R3(BΔ23–27)R/I5 had no inhibitory effect (A, C). SRE activation was also detected following R3(BΔ23–27)R/I5 (C). FBS (10%) was used as a positive control in all experiments to demonstrate functional reporter genes. Data are expressed as percentage β-galactosidase activation and normalized to the FBS response. Data are mean ± SEM of six to eight independent experiments, conducted in triplicate. In the cAMP assay (D), forskolin-stimulated cAMP accumulation (30 μM) was inhibited in CHO-RXFP3 cells by H3 relaxin and to a lesser extent by H2 relaxin and R3(BΔ23–27)R/I5. R3(BΔ23–27)R/I5 blocked H3 relaxin, but not H2 relaxin-induced cAMP inhibition. Data are expressed as % response to forskolin. Vehicle represents signalling in the absence of forskolin and ligands. Data are mean ± SEM of three independent experiments, conducted in duplicate. Controls for CHO-RXFP3 cells were parallel treatments of the parent cell line Flp-In CHO. Data were analysed by one-way anova with Dunnett's post hoc test. *P ≤ 0.05 and ***P ≤ 0.001.
R3(BΔ23–27)R/I5 activates the SRE reporter gene but does not inhibit H3 relaxin or H2 relaxin stimulation of SRE
We examined reporter gene responses from eight different cis-acting enhancer elements linked to SEAP reporter genes and of these, R3(BΔ23–27)R/I5 activated the SRE reporter gene, but none of the other signalling pathways. The SRE reporter was activated in a concentration-dependent manner by H3 relaxin (10−6 M: 134 ± 20; 10−8 M: 76 ± 2), H2 relaxin (10−6 M: 101 ± 18; 10−8 M: 74 ± 16) and by R3(BΔ23–27)R/I5 (10−6 M: 99 ± 18; 10−8 M: 54 ± 19) – all expressed as % of the 10% FBS response (positive control). SRE reporter activation by H3 relaxin or H2 relaxin (both 10−8 M) was not blocked by R3(BΔ23–27)R/I5 (10−6 M and 10−8 M; Figure 2C). Thus, R3(BΔ23–27)R/I5 is an agonist at the SRE reporter. In addition, both H3 and H2 relaxin stimulated the SRE reporter gene and these responses were not significantly inhibited by R3(BΔ23–27)R/I5 (10−8 M or 10−6 M). No SRE reporter activation was detected in untransfected Flp-In CHO cells treated with H3 relaxin, H2 relaxin or R3(BΔ23–27)R/I5 (Figure 2C).
R3(BΔ23–27)R/I5 inhibits H3 relaxin-induced but not H2 relaxin-induced inhibition of cAMP accumulation
H3 relaxin, and to a lesser extent H2 relaxin, inhibits forskolin-stimulated cAMP accumulation (van der Westhuizen et al., 2010). Inhibition of forskolin (3.10−5 M) stimulated cAMP accumulation by H3 relaxin (10−8 M) but not H2 relaxin (10−8 M) were inhibited by R3(BΔ23–27)R/I5 (10−6 M) (Figure 2D). No cAMP responses were detected in untransfected Flp-In CHO cells treated with H3 relaxin, H2 relaxin or R3(BΔ23–27)R/I5 (Figure 2D).
Effects of H3 relaxin, H2 relaxin and R3(BΔ23–27)R/I5 on p38MAPK, JNK1/2 and ERK1/2 phosphorylation
The reporter gene assays revealed patterns of activation of AP-1, NF-κB and/or SRE in CHO-RXFP3 cells following addition of H3 relaxin, H2 relaxin or R3(BΔ23–27)R/I5. AP-1 and SRE reporter activation is complex and involves diverse MAPKs. There are three main families of the MAPK: ERKs, JNKs and p38MAPKs and phosphorylation was examined following addition of peptides. H3 and H2 relaxin (10−6 M) activated all three kinases and R3(BΔ23–27)R/I5 (10−6 M) activated p38MAPK and ERK1/2 but not JNK1/2 (Figure 3). Maximal activation of p38MAPK with H3 and H2 relaxin occurred at 5 min and with R3(BΔ23–27)R/I5 at 10 min. JNK1/2 activation by H3 and H2 relaxin occurred at 10 min and pERK1/2 to all three peptides at 5 min (Figure 3). H3 and H2 relaxin produced concentration-dependent activation of p38MAPK and ERK1/2 measured 5 min after treatment (Figure 4A, B). Examination of p38MAPK activation showed that H3 relaxin had much higher efficacy than either H2 relaxin or R3(BΔ23–27)R/I5 (Emax; H3 relaxin, 78.07 ± 6.76; H2 relaxin, 16.87 ± 2.48; R3(BΔ23–27)R/I5, 14.69 ± 1.64) although all three peptides had similar potency (pEC50; H3 relaxin, 8.90 ± 0.27; H2 relaxin, 8.35 ± 0.40; R3(BΔ23–27)R/I5, 9.02 ± 0.33). For ERK1/2 phosphorylation, H2 relaxin and R3(BΔ23–27)R/I5 had poor efficacy compared with H3 relaxin (Emax; H3 relaxin, 93.81 ± 2.31; H2 relaxin, 1.09 ± 0.14; R3(BΔ23–27)R/I5, 1.75 ± 0.16), which also exhibited higher potency (pEC50 H3 relaxin, 9.93 ± 0.09; H2 relaxin, 7.15 ± 0.32; R3(BΔ23–27)R/I5, 8.87 ± 0.29). Interestingly R3(BΔ23–27)R/I5 had higher potency and efficacy than H2 relaxin for ERK1/2 phosphorylation. Regarding JNK1/2 phosphorylation, a response was detected only with highest concentration of H3 or H2 relaxin (10−6 M) suggesting poor coupling to this pathway (data not shown). No p38MAPK, JNK1/2 or ERK1/2 responses were detected in untransfected Flp-In CHO cells treated with H3, H2 relaxin or R3(BΔ23–27)R/I5 (Figure 3D–F). Western blots confirmed that the total p38MAPK, JNK1/2 or ERK1/2 in treated and untreated CHO-RXFP3 cells was not altered (data not shown).
Figure 3.

Time course of activation of p38MAPK (A, D), JNK1/2 (B, E) and ERK1/2 (C, F) by H3 relaxin, H2 relaxin or R3(BΔ23–27)R/I5 in CHO-RXFP3 (A, B, C) or Flp-In CHO (D, E, F) cells. Cells were treated with peptides for periods of up to 60 min, and p38MAPK (A, D), JNK1/2 (B, E) and ERK1/2 (C, F) activation was quantified using the appropriate phospho-‘kinase'-specific Surefire AlphaScreen kit. In CHO-RXFP3 cells (A, B, C), H2 relaxin (10−6 M) and H3 relaxin (10−6 M) activated all three kinases, but with different relative efficacies. H2 relaxin had lower efficacy than H3 relaxin for p38MAPK, but equivalent efficacy for JNK1/2 phosphorylation. ERK1/2 phosphorylation was activated by H3 relaxin > H2 relaxin. R3(BΔ23–27)R/I5 (10−6 M) stimulated p38MAPK, had no effect on JNK1/2 and weakly stimulated ERK1/2 with the same level of efficacy as H2 relaxin. Controls for CHO-RXFP3 cells were parallel treatments of the parent cell line Flp-In CHO (D, E, F). Data are mean ± SEM for three to six independent experiments.
Figure 4.
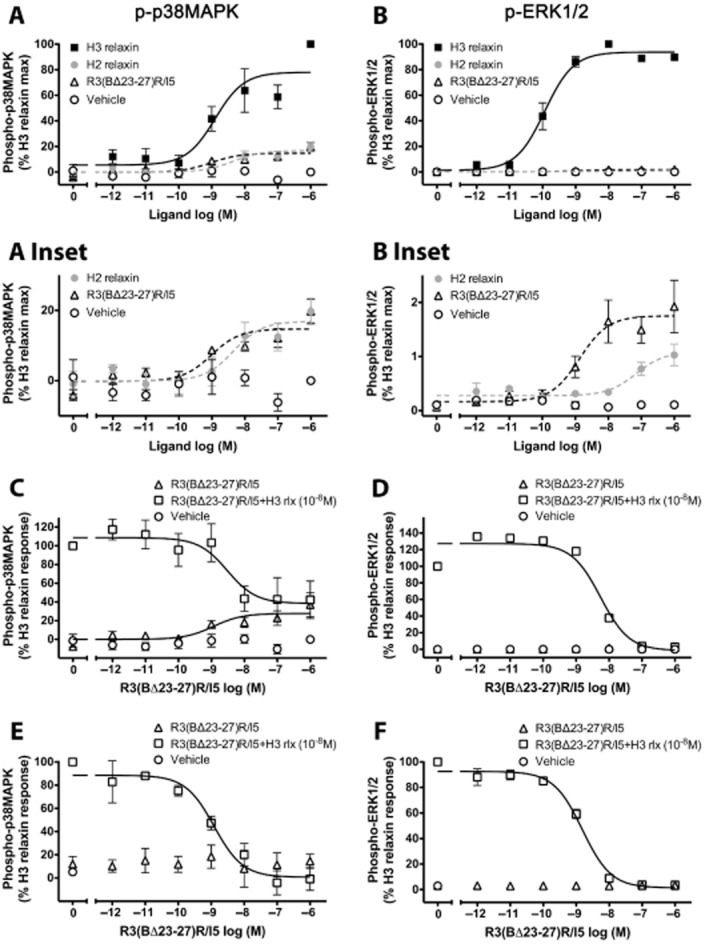
Concentration-dependent activation of p38MAPK (A, C, E) and ERK1/2 (B, D, F) by H3 relaxin, H2 relaxin or R3(BΔ23–27)R/I5 in CHO-RXFP3 cells. H3 relaxin, H2 relaxin or R3(BΔ23–27)R/I5 all activated p38MAPK and ERK1/2 (5 min treatment) with H3 relaxin having the highest efficacy. The effect of R3(BΔ23–27)R/I5 on H3 relaxin-stimulated p38MAPK or ERK1/2 activation was tested either by co-addition of R3(BΔ23–27)R/I5 and H3 relaxin (C, D) or by pre-incubation of cells with R3(BΔ23–27)R/I5 for 1 h prior to H3 relaxin treatment (E, F). R3(BΔ23–27)R/I5 inhibited H3 relaxin-stimulated p38MAPK and ERK1/2 activation in a concentration-dependent manner under both experimental conditions. Data are mean ± SEM for three to four independent experiments.
R3(BΔ23–27)R/I5 inhibits H3 relaxin-stimulated p38MAPK and ERK1/2 phosphorylation
R3(BΔ23–27)R/I5 inhibited H3 relaxin-stimulated AP-1 reporter gene activation. As p38MAPK and ERK kinases are known to contribute to AP-1 activation, we examined the antagonist activity of R3(BΔ23–27)R/I5 on H3 relaxin-stimulated p38MAPK and ERK1/2 phosphorylation under two experimental conditions. Cells were either co-treated with R3(BΔ23–27)R/I5 and H3 relaxin for 5 min or pre-incubated for 1 h with R3(BΔ23–27)R/I5 followed by 5 min treatment with H3 relaxin. R3(BΔ23–27)R/I5 inhibited H3 relaxin-stimulated p38MAPK and ERK1/2 activation in a concentration-dependent manner with similar pIC50 values both when co-added with H3 relaxin (p38MAPK, pIC50 = 8.52 ± 0.46 and ERK1/2, pIC50 = 8.26 ± 0.11; Figure 4C, D) or when pre-incubated for 1 h prior to H3 relaxin treatment (p38MAPK, pIC50 = 8.91 ± 0.20 and ERK1/2, pIC50 = 8.82 ± 0.07; Figure 4E, F). Pre-incubation with R3(BΔ23–27)R/I5 allowed detection of the antagonist activity on H3 relaxin stimulated p38MAPK or ERK1/2 phosphorylation (Figure 4E, F) as after 30 min the response to addition of the antagonist had faded (Figure 3). In contrast, co-addition with H3 relaxin revealed the partial agonist properties of R3(BΔ23–27)R/I5 particularly on p38MAPK phosphorylation (Figure 4C) but not ERK1/2 phosphorylation (Figure 4D). These studies confirmed that R3(BΔ23–27)R/I5 acts as an antagonist or as a partial agonist at RXFP3 depending on experimental conditions.
R3(BΔ23–27)R/I5 activates p38MAPK by a Gi/o independent mechanism whereas H3 relaxin or H2 relaxin-stimulated p38MAPK, JNK1/2 and ERK1/2 responses involve PTX-sensitive G-proteins. To examine whether MAPK activation involved PTX-sensitive G proteins, CHO-RXFP3 cells were treated with PTX (100 ng·mL–1, 18 h) prior to addition of peptides. Activation of p38MAPK, JNK1/2 and ERK1/2 by H3 relaxin (Figure 5A, D, G) or H2 relaxin (Figure 5B, E, H) was blocked by PTX pretreatment. In PTX-pretreated cells, there was >90% inhibition of p38MAPK, JNK1/2 and ERK1/2 phosphorylation compared with cells treated with H3 relaxin or H2 relaxin alone. R3(BΔ23–27)R/I5 activated p38MAPK and ERK1/2 but not JNK1/2 (Figure 5C, F, I), but only ERK1/2 activation was blocked by PTX pretreatment. In PTX-pretreated cells, there was >85% inhibition of ERK1/2 activation compared with cells treated with R3(BΔ23–27)R/I5 alone. These data indicate that H3 and H2 relaxin-stimulated p38MAPK, JNK1/2 and ERK1/2 and R3(BΔ23–27)R/I5-stimulated ERK1/2 involve PTX-sensitive G proteins whereas p38MAPK activation by R3(BΔ23–27)R/I5 is G protein-independent.
Figure 5.

The role of PTX-sensitive G-proteins in p38MAPK (A, B, C), JNK1/2 (D, E, F) and ERK1/2 (G, H, I) activation by H3 relaxin, H2 relaxin or R3(BΔ23–27)R/I5 in CHO-RXFP3 cells. Cells were pretreated with PTX (100 ng·mL–1) for 18 h prior to stimulation with peptides for up to 30 min. Phosphorylation of p38MAPK (A, B, C), JNK1/2 (D, E, F) and ERK1/2 (G, H, I) was quantified using the appropriate phospho-‘kinase'-specific Surefire AlphaScreen kit. Activation of p38MAPK, JNK1/2 and ERK1/2 by H3 relaxin (A, D, G) or H2 relaxin (B, E, H) both at 10−6 M was blocked by PTX. R3(BΔ23–27)R/I5 (10−6 M) activated p38MAPK (C) and ERK1/2 (I) but not JNK1/2 (F); ERK1/2 but not p38MAPK activation was blocked by PTX pretreatment. Data are mean ± SEM for four to seven independent experiments. Data were analysed by Student's t-test. *P < 0.05; **P ≤ 0.05 & P ≥ 0.001; ***P < 0.001.
Identification of interactions between RXFP3 and G proteins following treatment with H3 relaxin, H2 relaxin or R3(BΔ23–27)R/I5. We employed a real-time kinetic BRET assay to investigate ligand-induced interactions between RXFP3 and G-proteins in live cells. Flp-In CHO cells co-expressing RXFP3-Rluc8, Gγ2-Venus, Gβ1 and one of nine Gα subunits (Gs, Gi2, Gi3, GoA, GoB, Gq, G11, G12 or G13) were examined following the treatment with H3 relaxin, H2 relaxin or R3(BΔ23–27)R/I5 (all 10−6 M). All three peptides promoted interactions between RXFP3 and Gi2 or GoB with H3 relaxin producing a markedly higher signal than H2 relaxin or R3(BΔ23–27)R/I5 (Figure 6A, D). In addition, H3 relaxin also promoted RXFP3 interactions with Gi3 and GoA, but this did not occur with H2 relaxin or R3(BΔ23–27)R/I5 (Figure 6B, C). There were no interactions detected between RXFP3 and Gs, Gq (Figure 6E, F) or G11, G12, G13 (data not shown). All of the G-proteins used have been validated and shown to be functional (Jensen et al., 2013) (Kocan et al., unpublished).
Figure 6.
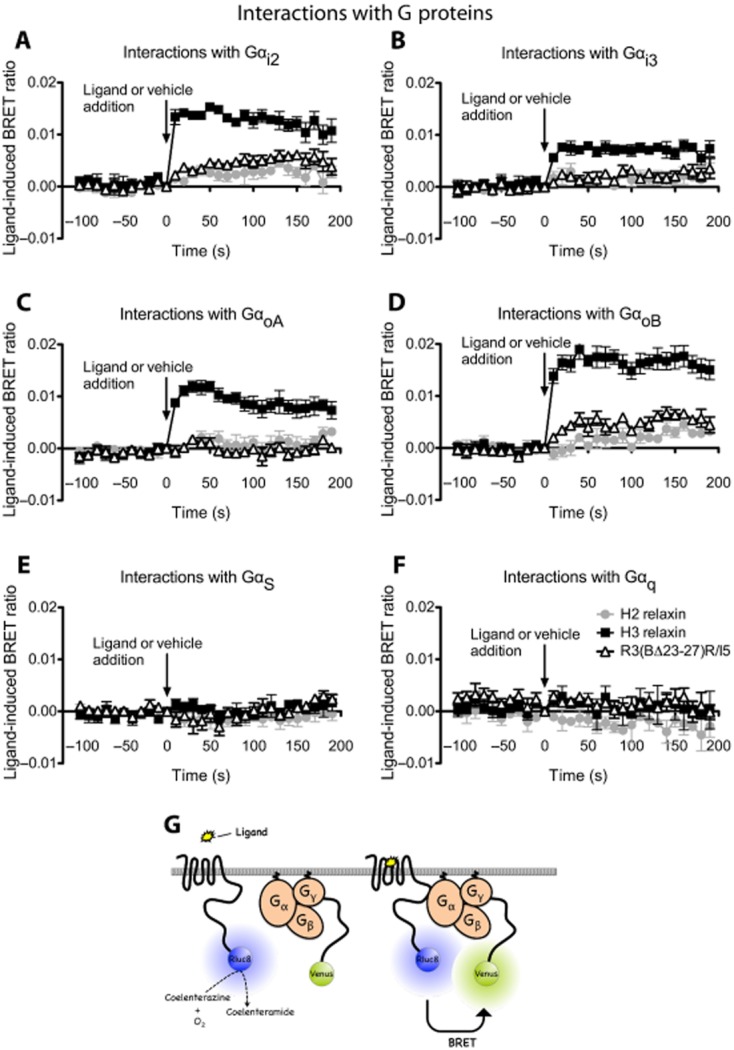
Detection of RXFP3 G protein interactions following treatment with H3 relaxin, H2 relaxin or R3(BΔ23–27)R/I5. Flp-In CHO cells were transiently co-transfected with RXFP3 Rluc8, Gγ2-Venus, Gβ1 and one of Gα subunits (Gαi2, Gαi3, GαoA, GαoB, Gαs, Gαq). Interactions between RXFP3 and G proteins were detected prior to and after treatment with H3 relaxin (10−6 M), H2 relaxin (10−6 M) or R3(BΔ23–27)R/I5 (10−6 M) using real-time BRET. H3 relaxin induced interactions between RXFP3 and Gαi2, Gαi3, GαoA or GαoB (A–D). H2 relaxin or R3(BΔ23–27)R/I5 induced interactions only between RXFP3 and Gαi2 or GαoB proteins and with a smaller signal compared with H3 relaxin (A, D). The ligand-induced BRET ratios were calculated by subtracting the ratio for the vehicle-treated sample from the BRET ratio for each ligand-treated sample as described. Data shown are mean ± SEM of four independent experiments. Illustration of the BRET interaction between receptor and G-protein subunits (G).
H3 relaxin but not H2 relaxin or R3(BΔ23–27)R/I5 promote interactions between RXFP3 and β-arrestin 1 and 2 that are blocked by R3(BΔ23–27)R/I5. Interactions between RXFP3 and β-arrestins 1 and 2 were detected following H3 relaxin, H2 relaxin or R3(BΔ23–27)R/I5 (all 10−6 M) using a real-time kinetic BRET assay. BRET signals were measured from Flp-In CHO cells cotransfected with RXFP3-Rluc8 and one of the β-arrestin fusion proteins (β-arrestin 1-Venus or β-arrestin 2-Venus). Interactions between RXFP3 with β-arrestins were detected following treatment with H3 relaxin but not H2 relaxin or R3(BΔ23–27)R/I5 (Figure 7). No change in BRET ratio was observed following addition of vehicle (see Methods for details). BRET assays were also carried out in cells pre-incubated for 15 min with R3(BΔ23–27)R/I5 (10−8 M or 10−6 M) followed by H3 relaxin (10−8 M) or vehicle (Figure 8). Complete inhibition of the H3 relaxin-induced RXFP3/β-arrestin 1 and RXFP3/β-arrestin 2 signals was observed after R3(BΔ23–27)R/I5 (Figure 8).
Figure 7.

Detection of RXFP3 – β-arrestin interactions following treatment with H3 relaxin, H2 relaxin or R3(BΔ23–27)R/I5 (A, B) and role of β-arrestin in H3 relaxin, H2 relaxin and R3(BΔ23–27)R/I5 – stimulated ERK1/2 signalling (C). Flp-In CHO cells were transiently co-transfected with RXFP3-Rluc8 and β-arrestin 1-Venus or β-arrestin 2-Venus. Ligand-induced interactions between RXFP3 and β-arrestin 1 or β-arrestin 2 were detected using real-time BRET. H3 relaxin (10−6 M) induced RXFP3/β-arrestin 1 and RXFP3/β-arrestin 2 interactions whereas H2 relaxin (10−6 M) or R3(BΔ23–27)R/I5 (10−6 M) had no effect. To examine the role of β-arrestin in ERK1/2 signalling, CHO-RXFP3 cells were transfected with β-arrestin 1 WT (β-arr 1 WT) or dominant negative mutant V53D (β-arr 1 V53D) and tested 48 h later. ERK1/2 activation by H3 relaxin or R3(BΔ23–27)R/I5 (C) was sensitized by dominant negative β-arrestin 1 V53D whereas responses to H2 relaxin were slightly inhibited. Data shown are mean ± SEM of three independent experiments conducted in duplicate. Illustration of BRET between receptor and β-arrestins (D).
Figure 8.

Effect of R3(BΔ23–27)R/I5 and PTX on H3 relaxin-induced RXFP3 – β-arrestin interactions. Flp-In CHO cells were transiently co-transfected with RXFP3-Rluc8 and β-arrestin 1-Venus or β-arrestin 2-Venus. Cells were pretreated with R3(BΔ23–27)R/I5 for 15 min. H3 relaxin(10−6 M)-induced RXFP3 – β-arrestin 1 (A) and RXFP3 – β-arrestin 2 (C) BRET were both completely blocked by R3(BΔ23–27)R/I5. H3 relaxin-induced interactions between RXFP3 and β-arrestin 1 (B) or β-arrestin 2 (D) were partially blocked by PTX suggesting some G protein-independent coupling. Data shown are mean ± SEM of three experiments (β-arrestin) or four to six independent experiments (PTX).
RXFP3/β-arrestin interactions involve Gi/o and G protein-independent mechanisms
To further investigate interactions between RXFP3 and β-arrestins, we examined the involvement of PTX-sensitive G proteins. Flp-In CHO cells transiently co-transfected with RXFP3-Rluc8 and β-arrestin 1-Venus or β-arrestin 2-Venus were pretreated with PTX (100 ng·mL–1, 18 h) prior to stimulation with H3 relaxin (10−6 M). In the pretreated cells, there was ∼50% inhibition of the RXFP3/β-arrestin interactions compared with controls (Figure 8) suggesting that both Gi/o and G protein-independent mechanisms were involved.
Effect of dominant negative β-arrestin on ERK1/2 signalling
H3 relaxin produces the most robust ERK1/2 activation (∼20 fold/basal at 5 min and ∼4 fold/basal at 10 min; Figure 3C) and β-arrestin recruitment to RXFP3 that is inhibited by PTX. To assess the contribution of β-arrestin to ERK1/2 activation we employed a dominant negative mutant of β-arrestin 1 (β-arrestin1 V53D) that disrupts ERK signalling downstream of several GPCRs (Luttrell et al., 1999; Cottrell et al., 2009). ERK1/2 activation in CHO-RXFP3 cells by H3 relaxin, H2 relaxin or R3(BΔ23–27)R/I5 was measured 48 h after transfection with β-arrestin 1 wild type (WT) or β-arrestin 1 V53D. ERK1/2 phosphorylation was expressed as % positive control (10% FBS). The H3 relaxin-induced p-ERK1/2 concentration-response curve was left-shifted in CHO-RXFP3 cells expressing the dominant negative as compared with the β-arrestin 1 WT (pEC50; β-arrestin 1 V53D, 9.04 ± 0.06; β-arrestin 1 WT, 8.16 ± 0.08) with a slight reduction in maximal response (Emax; β-arrestin 1 V53D, 75.55 ± 1.51; β-arrestin 1 WT, 88.30 ± 2.80; Figure 7C). p-ERK1/2 responses to R3(BΔ23–27)R/I5 and H2 relaxin displayed minor changes to the dominant negative but neither was abolished. Western blots confirmed the expression of β-arrestin 1 WT as well as mutant V53D in the same cell sample that was used for ERK1/2 detection (data not shown). β-Arrestin 1 but not β-arrestin 2 is endogenously expressed in CHO cells (confirmed by Western blotting, data not shown) (Baycin-Hizal et al., 2012).
Discussion
Physiological studies suggest that the RXFP3/relaxin-3 system mediates central stress responses and modulates appetite pathways (Bathgate et al., 2002). Recent neuroanatomical studies also suggest roles in arousal, stress, affective and cognitive circuits (Smith et al., 2010). R3(BΔ23–27)R/I5 was synthesized as a RXFP3 antagonist peptide that would be useful for defining the physiological functions of the receptor. In this study, we have identified the specific signalling pathways activated by H3 and H2 relaxin at RXFP3 and examined how R3(BΔ23–27)R/I5 interacts with these signalling cascades.
The R3(BΔ23–27)R/I5 chimeric peptide has been reported to be a selective antagonist at RXFP3 and blocks H3 relaxin-mediated cAMP inhibition (Kuei et al., 2007; Hossain et al., 2009). For the first time, we have identified previously unrecognized signalling properties of R3(BΔ23–27)R/I5 at RXFP3 and shown that it blocks some, but not all responses to relaxin family peptides. Firstly, we showed that R3(BΔ23–27)R/I5 inhibits H3 relaxin-stimulated AP-1 reporter gene activity, but not the H2 relaxin AP-1 response or H3 relaxin NF-κB activation. Secondly, R3(BΔ23–27)R/I5 effectively blocked H3 relaxin-induced, but not H2 relaxin-induced cAMP inhibition, p38MAPK or ERK1/2 phosphorylation. Thus R3(BΔ23–27)R/I5 antagonizes most; however, not all H3 relaxin-stimulated signalling pathways but not responses to H2 relaxin.
Several compounds originally classified as antagonists have the ability to activate their own spectrum of signalling pathways by stabilizing particular conformations of GPCRs. Examples are, ERK1/2 activation by β-adrenoceptor antagonists (Azzi et al., 2003; Baker et al., 2003) and ERK1/2 and p38MAPK activation by the β3-adrenoceptor antagonists SR59230A and L748337 (Sato et al., 2007; 2008). Here we show that R3(BΔ23–27)R/I5 activates several signalling pathways that are a subset of those stimulated by H3 or H2 relaxin such as the SRE reporter. H3 and H2 relaxin also stimulated the SRE reporter and these effects were not inhibited by R3(BΔ23–27)R/I5. SRE signalling involves MAPKs including p38MAPK, JNK1/2 and ERK1/2 (Treisman, 1992). Direct examination of these pathways showed that H3 and H2 relaxin activated maximum ERK1/2 phosphorylation at 5 min (van der Westhuizen et al., 2007) with H3 relaxin having higher efficacy than H2 relaxin. R3(BΔ23–27)R/I5 also activated ERK1/2 with a similar time course and efficacy to H2 relaxin. Activation of ERK1/2 by all peptides was Gαi/o dependent and blocked by PTX pretreatment. Investigation of signalling pathways at RXFP3 revealed that H3 and H2 relaxin, but not R3(BΔ23–27)R/I5 also caused JNK1/2 phosphorylation by a PTX-sensitive mechanism. All three peptides caused p38MAPK phosphorylation with maximum activation observed between 5 and 10 min. However, while responses to H3 and H2 relaxin were PTX-sensitive, those to R3(BΔ23–27)R/I5 were suggesting that were not mediated by Gi/o.
The original paper that describes the synthesis of R3(BΔ23–27)R/I5 (Kuei et al., 2007) measured competition binding in cells expressing RXFP3 where it competed with 125I-H3/INSL5 for binding with a similar affinity to H3 relaxin. It has been suggested that the loss of the terminal tryptophan and shift of arginine in the B-chain results in binding of R3(BΔ23–27)R/I5 to a site on RXFP3 that overlaps with that occupied by H3 relaxin, but is not identical (Hossain et al., 2009). ArgB26 and TrpB27 in the H3 relaxin B-chain appear to be important for RXFP3 activation (Kuei et al., 2007; Hossain et al., 2009) and in R3(BΔ23–27)R/I5 the modified C-terminus with a terminal Arg has an important role in antagonist activity (Hossain et al., 2009). However, R3(BΔ23–27)R/I5 also acts as an agonist with a different signalling profile to H3 relaxin. Like H3 relaxin but with lower efficacy, it activates ERK1/2 through Gαi/o, but unlike H3 relaxin, p38MAPK activation is Gαi/o independent and it fails to cause JNK phosphorylation. Hypothesizing that ArgB26 and TrpB27 in the H3 relaxin B-chain stabilize receptor conformations favourable for coupling to different G-proteins, we tested for interactions between RXFP3 and nine different G-proteins using BRET. H3 relaxin recruited RXFP3 to Gαi2, Gαi3, GαoA and GαoB whereas R3(BΔ23–27)R/I5 and H2 relaxin caused interactions only between RXFP3 and Gαi3 and GαoA. Although the H2 relaxin B-chain lacks ArgB26 it does possess TrpB27, similar to H3 relaxin, and there is evidence that suggests that it binds to a RXFP3 binding site distinct from that identified by H3 relaxin (van der Westhuizen et al., 2010).
In contrast to H3 and H2 relaxin, coupling of R3(BΔ23–27)R/I5 to RXFP3 stabilizes a receptor conformation triggering Gαi/o-independent p38MAPK activation. Not only do GPCRs couple to signalling cascades by both Gα- and Gβγ-subunits, but scaffold proteins such as β-arrestins may also have roles in controlling stress-activated MAPK cascades (Morrison and Davis, 2003; Lefkowitz and Shenoy, 2005). R3(BΔ23–27)R/I5-stimulated Gαi/o-independent p38MAPK phosphorylation had a slower time course, as is often the case with scaffold protein-mediated responses, than the responses to H3 or H2 relaxin. For example, AT1AR ERK phosphorylation via G-proteins, mediated by angiotensin AT1A receptors, peaks at 2 min after angiotensin II whereas β-arrestin-dependent ERK phosphorylation peaks at 10 min (Ahn et al., 2004). Interactions between RXFP3 and β-arrestins using BRET showed that only H3 relaxin induced RXFP3/β-arrestin interactions. Although, β-arrestins are the most commonly studied scaffold proteins, others such as JIP-2 and JIP-4 scaffold stress-activated p38MAPK (see Morrison and Davis, 2003). Thus further experiments are needed to clarify the mechanism involved in Gαi/o-independent activation of p38MAPK by R3(BΔ23–27)R/I5.
The involvement of β-arrestin in ERK1/2 activation downstream of RXFP3 was investigated by comparing cells expressing dominant negative (V53D) versus WT β-arrestin 1. Cells transfected with V53D showed increased sensitivity to H3 relaxin compared with cells transfected with WT and neither showed inhibition suggesting that, unlike several other GPCRs (Luttrell et al., 1999; Cottrell et al., 2009), the RXFP3/β-arrestin 1 interaction contributes little to the ERK1/2 response to H3 relaxin. ERK1/2 responses to H2 relaxin or R3(BΔ23–27)R/I5 although displaying minor changes in sensitivity after WT or V53D transfection were not significantly changed in magnitude in agreement with BRET studies that showed activation of RXFP3 by these peptides produced no measurable interaction between the receptor and β-arrestin.
PTX treatment reduced H3 relaxin-induced interactions of RXFP3 with β-arrestin 1 and β-arrestin 2 by ∼50% (Figure 8B, D). Activated receptors couple to heterotrimeric G-proteins that exchange GDP for GTP and dissociate into Gα and Gβγ subunits. The receptors then recruit and are phosphorylated by the GPCR kinases, GRK2/3, via released Gβγ subunits and bind β-arrestins. Therefore, our data suggest that about half the β-arrestin recruitment follows receptor phosphorylation by GRK2/3, whereas the remainder is either not dependent on receptor phosphorylation, or is facilitated by Gβγ-independent GRKs, such as isoforms 5 and 6. Further studies are required to determine whether β-arrestin recruitment to RXFP3 involves GRK-dependent phosphorylation.
We have previously shown using reporter gene assays (van der Westhuizen et al., 2010), that H2 relaxin is a biased ligand relative to H3 relaxin at RXFP3. Selective MAPK inhibitors were used to show that H3 relaxin-mediated responses primarily involved P38MAPK with smaller contributions from JNK and ERK1/2 whereas H2 relaxin-mediated responses involved both P38MAPK and JNK with ERK1/2 having a minor role (van der Westhuizen et al., 2010). Our study directly examined MAPKs that contribute to the AP-1 reporter response including p38MAPK (Roux and Blenis, 2004), JNK (Davis, 2000) and ERK (Price et al., 1996; Whitmarsh and Davis, 1996). H2 relaxin activated p38MAPK and ERK1/2 with lower efficacy than H3 relaxin, but had equivalent efficacy for JNK1/2 phosphorylation. Additionally, activation of p38MAPK, JNK1/2 and ERK1/2 signalling by H2 or H3 relaxin involved PTX-sensitive G-proteins. The functional relevance of these findings is supported by several studies. There are dramatic increases in phosphorylated MEK1/2, ERK1/2, JNK1/2/3 following forced swim tests in rats (Shen et al., 2004), and p38MAPK activation is also important in central stress responses (Davis, 2000). As H3 relaxin mRNA is increased in the nucleus incertus after forced swim tests (Tanaka et al., 2005) it is possible that it activates MAPKs through RXFP3, suggesting that H3 relaxin/RXFP3 are involved in central stress responses.
This study identified the signalling cascades activated when H3 relaxin, H2 relaxin and R3(BΔ23–27)R/I5 bind to RXFP3 (Figure 9). H3 relaxin interacts with RXFP3 to cause coupling of the receptor with GαoB, Gαi2, GαoA and Gαi3 whereas H2 relaxin and R3(BΔ23–27)R/I5 signalling involves only GαoB and Gαi2. Only H3 relaxin promotes RXFP3/β-arrestin interaction utilizing both Gi/o-dependent and G-protein-independent mechanisms. H3 relaxin induces strong ERK and weaker JNK and p38MAPK activation and inhibits forskolin-stimulated cAMP accumulation. Both H2 relaxin and R3(BΔ23–27)R/I5 promote much less ERK phosphorylation than H3 relaxin and inhibit forskolin-stimulated cAMP accumulation. H2 relaxin activates JNK and P38MAPK similarly to H3 whereas R3(BΔ23–27)R/I5 does not activate JNK. All of the signalling responses involve Gi/o except the P38MAPK responses to R3(BΔ23–27)R/I5 that are G-protein-independent. R3(BΔ23–27)R/I5 inhibits H3 relaxin-stimulated cAMP inhibition, p38MAPK and ERK phosphorylation and coupling to β-arrestins. These findings provide a better understanding of RXFP3 signalling that is central to the development of novel anti-anxiety and anti-obesity drugs. R3(BΔ23–27)R/I5 stabilizes receptor conformations that couple to a subset of the signalling pathways activated by H3 or H2 relaxin. Thus drugs acting at the RXFP3 receptor can display a variety of profiles for different signalling pathways.
Figure 9.

Signalling pathways activated by H3 relaxin, H2 relaxin and R3(BΔ23–27)R/I5 in CHO cells expressing RXFP3. H3 relaxin interacts with RXFP3 (A) to cause coupling of the receptor with GαoB, Gαi2, GαoA and Gαi3. RXFP3/β-arrestin interactions utilize both Gi/o-dependent and G protein-independent mechanisms. H3 relaxin induces strong ERK and weaker JNK and p38MAPK activation – all through Gi/o. Phosphorylation of MAPKs leads to reporter gene transcription. H3 relaxin also inhibits forskolin-stimulated cAMP accumulation and activates the NF-κB reporter. H2 relaxin (B) promotes coupling of RXFP3 to GαoB and Gαi2 but not GαoA, Gαi3 or β-arrestins. It promotes much less ERK phosphorylation than H3 relaxin, but similar JNK and p38MAPK activation – all through Gi/o proteins. H2 relaxin inhibits forskolin-stimulated cAMP accumulation and activates AP-1 and SRE but not NF-κB. R3(BΔ23–27)R/I5 (C) causes coupling of RXFP3 to GαoB and Gαi2 but not GαoA, Gαi3 or β-arrestins. It also causes ERK but not JNK phosphorylation and activates p38MAPK via Gi/o protein-independent pathways. It also inhibits forskolin-stimulated cAMP accumulation and activates the SRE reporter. R3(BΔ23–27)R/I5 (D) inhibits (red crosses) H3 relaxin-stimulated cAMP inhibition, p38MAPK and ERK phosphorylation and AP-1 reporter transcription. R3(BΔ23–27)R/I5 completely inhibits RXFP3 coupling to β-arrestins induced by H3 relaxin.
Acknowledgments
We thank Dr Nathan Hall for images of peptide structures and Dr Caroline Hick for expert technical advice. We are grateful to A/Prof Ross Bathgate (Florey Institute of Neuroscience and Mental Health), A/Prof Kevin Pfleger (Western Australian Institute for Medical Research and Centre for Medical Research, University of Western Australia) and Dr Bronwyn Evans for useful scientific discussions and advices. Studies carried out at the Florey Institute of Neuroscience and Mental Health were supported by the Victorian Government's Operational Infrastructure Support Program. We are grateful to Andreas Loening and Sanjiv Gambhir (Stanford University, Stanford, CA), Atsushi Miyawaki (RIKEN Brain Science Institute, Wako City, Japan) and Michel Bouvier (Department of Biochemistry, Université de Montréal, Montréal, Quebec, Canada) for providing cDNA constructs. This research was supported by the National Health and Medical Research Council (NH&MRC Australia Project Grants 436713, 454375 and Program Grant 519461).
Glossary
- AP-1
activator protein 1
- CHO-RXFP3
Flp-In CHO cells expressing RXFP3
- GRK
GPCR kinase
- H2 relaxin
human gene 2 relaxin
- H3 relaxin
human gene 3 relaxin
- HEK-RXFP3
HEK293 cells expressing RXFP3
- INSL
insulin-like peptide
- LDSB
ligand-directed signalling bias
- PTX
Pertussis toxin
- R3(BΔ23–27)R/I5
B-chain truncated variant of the chimeric peptide H3 relaxin B-chain/human INSL5 A-chain with a terminal Arg residue
- RXFP
relaxin family peptide receptor
- SEAP
secreted alkaline protease
- SRE
serum response element
Conflict of interest
None.
References
- Ahn S, Shenoy SK, Wei H, Lefkowitz RJ. Differential kinetic and spatial patterns of beta-arrestin and G protein-mediated ERK activation by the angiotensin II receptor. J Biol Chem. 2004;279:35518–35525. doi: 10.1074/jbc.M405878200. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL. Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: G Protein-Coupled Receptors. Br J Pharmacol. 2013;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayoub MA, See HB, Seeber RM, Armstrong SP, Pfleger KD. Profiling epidermal growth factor receptor and heregulin receptor 3 heteromerization using receptor tyrosine kinase heteromer investigation technology. PLoS ONE. 2013;8:e64672. doi: 10.1371/journal.pone.0064672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azzi M, Charest PG, Angers S, Rousseau G, Kohout T, Bouvier M, et al. Beta-arrestin-mediated activation of MAPK by inverse agonists reveals distinct active conformations for G protein-coupled receptors. Proc Natl Acad Sci U S A. 2003;100:11406–11411. doi: 10.1073/pnas.1936664100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker JG, Hall IP, Hill SJ. Agonist and inverse agonist actions of beta-blockers at the human beta 2-adrenoceptor provide evidence for agonist-directed signaling. Mol Pharmacol. 2003;64:1357–1369. doi: 10.1124/mol.64.6.1357. [DOI] [PubMed] [Google Scholar]
- Bathgate RA, Samuel CS, Burazin TC, Layfield S, Claasz AA, Reytomas IG, et al. Human relaxin gene 3 (H3) and the equivalent mouse relaxin (M3) gene. Novel members of the relaxin peptide family. J Biol Chem. 2002;277:1148–1157. doi: 10.1074/jbc.M107882200. [DOI] [PubMed] [Google Scholar]
- Bathgate RA, Ivell R, Sanborn BM, Sherwood OD, Summers RJ. International Union of Pharmacology LVII: recommendations for the nomenclature of receptors for relaxin family peptides. Pharmacol Rev. 2006a;58:7–31. doi: 10.1124/pr.58.1.9. [DOI] [PubMed] [Google Scholar]
- Bathgate RA, Lin F, Hanson NF, Otvos L, Jr, Guidolin A, Giannakis C, et al. Relaxin-3: improved synthesis strategy and demonstration of its high-affinity interaction with the relaxin receptor LGR7 both in vitro and in vivo. Biochemistry. 2006b;45:1043–1053. doi: 10.1021/bi052233e. [DOI] [PubMed] [Google Scholar]
- Baycin-Hizal D, Tabb DL, Chaerkady R, Chen L, Lewis NE, Nagarajan H, et al. Proteomic analysis of Chinese hamster ovary cells. J Proteome Res. 2012;11:5265–5276. doi: 10.1021/pr300476w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cottrell GS, Padilla BE, Amadesi S, Poole DP, Murphy JE, Hardt M, et al. Endosomal endothelin-converting enzyme-1: a regulator of beta-arrestin-dependent ERK signaling. J Biol Chem. 2009;284:22411–22425. doi: 10.1074/jbc.M109.026674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–252. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- Gales C, Rebois RV, Hogue M, Trieu P, Breit A, Hebert TE, et al. Real-time monitoring of receptor and G-protein interactions in living cells. Nat Methods. 2005;2:177–184. doi: 10.1038/nmeth743. [DOI] [PubMed] [Google Scholar]
- Haugaard-Jonsson LM, Hossain MA, Daly NL, Bathgate RA, Wade JD, Craik DJ, et al. Structure of the R3/I5 chimeric relaxin peptide, a selective GPCR135 and GPCR142 agonist. J Biol Chem. 2008;283:23811–23818. doi: 10.1074/jbc.M800489200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hossain MA, Bathgate RA, Rosengren KJ, Shabanpoor F, Zhang S, Lin F, et al. The structural and functional role of the B-chain C-terminal arginine in the relaxin-3 peptide antagonist, R3(BDelta23–27)R/I5. Chem Biol Drug Des. 2009;73:46–52. doi: 10.1111/j.1747-0285.2008.00756.x. [DOI] [PubMed] [Google Scholar]
- Jensen DD, Godfrey CB, Niklas C, Canals M, Kocan M, Poole DP, et al. The bile acid receptor TGR5 does not interact with beta-arrestins or traffic to endosomes but transmits sustained signals from plasma membrane rafts. J Biol Chem. 2013;288:22942–22960. doi: 10.1074/jbc.M113.455774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocan M, See HB, Seeber RM, Eidne KA, Pfleger KD. Demonstration of improvements to the bioluminescence resonance energy transfer (BRET) technology for the monitoring of G protein-coupled receptors in live cells. J Biomol Screen. 2008;13:888–898. doi: 10.1177/1087057108324032. [DOI] [PubMed] [Google Scholar]
- Kocan M, See HB, Sampaio NG, Eidne KA, Feldman BJ, Pfleger KD. Agonist-independent interactions between beta-arrestins and mutant vasopressin type II receptors associated with nephrogenic syndrome of inappropriate antidiuresis. Mol Endocrinol. 2009;23:559–571. doi: 10.1210/me.2008-0321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuei C, Sutton S, Bonaventure P, Pudiak C, Shelton J, Zhu J, et al. R3(BDelta23 27)R/I5 chimeric peptide, a selective antagonist for GPCR135 and GPCR142 over relaxin receptor LGR7: in vitro and in vivo characterization. J Biol Chem. 2007;282:25425–25435. doi: 10.1074/jbc.M701416200. [DOI] [PubMed] [Google Scholar]
- Lefkowitz RJ, Shenoy SK. Transduction of receptor signals by beta-arrestins. Science. 2005;308:512–517. doi: 10.1126/science.1109237. [DOI] [PubMed] [Google Scholar]
- Liu C, Chen J, Sutton S, Roland B, Kuei C, Farmer N, et al. Identification of relaxin-3/INSL7 as a ligand for GPCR142. J Biol Chem. 2003a;278:50765–50770. doi: 10.1074/jbc.M308996200. [DOI] [PubMed] [Google Scholar]
- Liu C, Eriste E, Sutton S, Chen J, Roland B, Kuei C, et al. Identification of relaxin-3/INSL7 as an endogenous ligand for the orphan G-protein-coupled receptor GPCR135. J Biol Chem. 2003b;278:50754–50764. doi: 10.1074/jbc.M308995200. [DOI] [PubMed] [Google Scholar]
- Liu C, Chen J, Kuei C, Sutton S, Nepomuceno D, Bonaventure P, et al. Relaxin-3/insulin-like peptide 5 chimeric peptide, a selective ligand for G protein-coupled receptor (GPCR)135 and GPCR142 over leucine-rich repeat-containing G protein-coupled receptor 7. Mol Pharmacol. 2005;67:231–240. doi: 10.1124/mol.104.006700. [DOI] [PubMed] [Google Scholar]
- Luttrell LM, Ferguson SS, Daaka Y, Miller WE, Maudsley S, Della Rocca GJ, et al. Beta-arrestin-dependent formation of beta2 adrenergic receptor-Src protein kinase complexes. Science. 1999;283:655–661. doi: 10.1126/science.283.5402.655. [DOI] [PubMed] [Google Scholar]
- McGowan BM, Stanley SA, White NE, Spangeus A, Patterson M, Thompson EL, et al. Hypothalamic mapping of orexigenic action and Fos-like immunoreactivity following relaxin-3 administration in male Wistar rats. Am J Physiol Endocrinol Metab. 2007;292:E913–E919. doi: 10.1152/ajpendo.00346.2006. [DOI] [PubMed] [Google Scholar]
- Morrison DK, Davis RJ. Regulation of MAP kinase signaling modules by scaffold proteins in mammals. Annu Rev Cell Dev Biol. 2003;19:91–118. doi: 10.1146/annurev.cellbio.19.111401.091942. [DOI] [PubMed] [Google Scholar]
- Mustafa S, See HB, Seeber RM, Armstrong SP, White CW, Ventura S, et al. Identification and profiling of novel alpha1A-adrenoceptor-CXC chemokine receptor 2 heteromer. J Biol Chem. 2012;287:12952–12965. doi: 10.1074/jbc.M111.322834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfleger KD, Seeber RM, Eidne KA. Bioluminescence resonance energy transfer (BRET) for the real-time detection of protein-protein interactions. Nat Protoc. 2006;1:337–345. doi: 10.1038/nprot.2006.52. [DOI] [PubMed] [Google Scholar]
- Price MA, Cruzalegui FH, Treisman R. The p38 and ERK MAP kinase pathways cooperate to activate ternary complex factors and c-fos transcription in response to UV light. EMBO J. 1996;15:6552–6563. [PMC free article] [PubMed] [Google Scholar]
- Rosengren KJ, Lin F, Bathgate RA, Tregear GW, Daly NL, Wade JD, et al. Solution structure and novel insights into the determinants of the receptor specificity of human relaxin-3. J Biol Chem. 2006;281:5845–5851. doi: 10.1074/jbc.M511210200. [DOI] [PubMed] [Google Scholar]
- Roux PP, Blenis J. ERK and p38 MAPK-activated protein kinases: a family of protein kinases with diverse biological functions. Microbiol Mol Biol Rev. 2004;68:320–344. doi: 10.1128/MMBR.68.2.320-344.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M, Horinouchi T, Hutchinson DS, Evans BA, Summers RJ. Ligand-directed signaling at the beta3-adrenoceptor produced by 3-(2-ethylphenoxy)-1-[(1,S)-1,2,3,4-tetrahydronapth-1-ylamino]-2S-2-propan ol oxalate (Sr59230A) relative to receptor agonists. Mol Pharmacol. 2007;72:1359–1368. doi: 10.1124/mol.107.035337. [DOI] [PubMed] [Google Scholar]
- Sato M, Hutchinson DS, Evans BA, Summers RJ. The beta3-adrenoceptor agonist 4-[ [ (hexylamino)carbonyl]amino]-N-[4-[2-[ [ (2S)-2-hydroxy-3-(4-hydroxyphenoxy)prop yl]amino]ethyl]-phenyl]-benzenesulfonamide (L755507) and antagonist (S)-N-[4-[2-[[3-[3-(acetamidomethyl)phenoxy]-2-hydroxypropyl]amino]-ethyl]phenyl] benzenesulfonamide (L748337) activate different signaling pathways in Chinese hamster ovary-K1 cells stably expressing the human beta3-adrenoceptor. Mol Pharmacol. 2008;74:1417–1428. doi: 10.1124/mol.108.046979. [DOI] [PubMed] [Google Scholar]
- Shen CP, Tsimberg Y, Salvadore C, Meller E. Activation of Erk and JNK MAPK pathways by acute swim stress in rat brain regions. BMC Neurosci. 2004;5:36. doi: 10.1186/1471-2202-5-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CM, Shen PJ, Banerjee A, Bonaventure P, Ma S, Bathgate RA, et al. Distribution of relaxin-3 and RXFP3 within arousal, stress, affective, and cognitive circuits of mouse brain. J Comp Neurol. 2010;518:4016–4045. doi: 10.1002/cne.22442. [DOI] [PubMed] [Google Scholar]
- Sutton SW, Bonaventure P, Kuei C, Roland B, Chen J, Nepomuceno D, et al. Distribution of G-protein-coupled receptor (GPCR)135 binding sites and receptor mRNA in the rat brain suggests a role for relaxin-3 in neuroendocrine and sensory processing. Neuroendocrinology. 2004;80:298–307. doi: 10.1159/000083656. [DOI] [PubMed] [Google Scholar]
- Tanaka M, Iijima N, Miyamoto Y, Fukusumi S, Itoh Y, Ozawa H, et al. Neurons expressing relaxin 3/INSL 7 in the nucleus incertus respond to stress. Eur J Neurosci. 2005;21:1659–1670. doi: 10.1111/j.1460-9568.2005.03980.x. [DOI] [PubMed] [Google Scholar]
- Treisman R. The serum response element. Trends Biochem Sci. 1992;17:423–426. doi: 10.1016/0968-0004(92)90013-y. [DOI] [PubMed] [Google Scholar]
- van der Westhuizen ET, Werry TD, Sexton PM, Summers RJ. The relaxin family peptide receptor 3 (RXFP3) activates ERK1/2 through a PKC dependent mechanism. Mol Pharmacol. 2007;71:1618–1629. doi: 10.1124/mol.106.032763. [DOI] [PubMed] [Google Scholar]
- van der Westhuizen ET, Christopoulos A, Sexton PM, Wade JD, Summers RJ. H2 relaxin is a biased ligand relative to H3 relaxin at the relaxin family peptide receptor 3 (RXFP3) Mol Pharmacol. 2010;77:759–772. doi: 10.1124/mol.109.061432. [DOI] [PubMed] [Google Scholar]
- Whitmarsh AJ, Davis RJ. Transcription factor AP-1 regulation by mitogen-activated protein kinase signal transduction pathways. J Mol Med. 1996;74:589–607. doi: 10.1007/s001090050063. [DOI] [PubMed] [Google Scholar]