

Abstract
Background and Purpose
T-cell infiltration, interstitial fibrosis and cardiac dysfunction have been observed in diabetic patients with cardiovascular diseases. PKC-θ is crucial for the activation of mature T-cells. We hypothesized that inhibition of PKC-θ might protect diabetic hearts through inhibition of T-cell stimulation and maintenance of tight junction integrity.
Experimental Approach
A model of type 1 diabetes was induced by streptozotocin (STZ) (50 mg kg–1 for 5 days) in male C57BL/6J wild-type (WT) mice and Rag1 knockout (KO) mice which lack mature lymphocytes. A cell-permeable selective PKC-θ peptide inhibitor (PI) was administered i.p. (0.2 mg kg–1·day–1) for 4 weeks (first phase) and 2 weeks (second phase). At the end of the 11th week, cardiac contractile force was measured in isolated perfused hearts. Cardiac morphology and fibrosis were determined. Phosphorylation of PKC-θ at Tyr358, infiltrated T-cells and tight junction protein ZO-1 within the hearts were detected, using immunohistochemcial techniques.
Key Results
PI did not affect high blood glucose level in both WT and Rag1 KO diabetic mice. Diabetes induced cardiac fibrosis in WT mice but not in Rag1 KO mice. PI attenuated cardiac fibrosis and improved cardiac contractility of WT diabetic hearts. PI decreased expression of phosphorylated PKC-θ, reduced the infiltration of T-cells and increased ZO-1 expression within WT diabetic hearts.
Conclusion and Implications
Inhibition of PKC-θ improves cardiac function and reduces cardiac fibrosis in WT mice with streptozotocin-induced diabetes. Mature T-cells play a key role in pathophysiology of diabetic cardiomyopathy.
Keywords: diabetic cardiomyopathy, PKC-θ, T-cell, tight junction, ZO-1
Introduction
Diabetic cardiomyopathy, the major cardiovascular complication of diabetes, is a distinct entity independent of coronary artery disease despite earlier beliefs common in the diabetic population (Spector, 1998; Bell, 2003; Poornima et al., 2006). The experimental model of type 1 diabetes mellitus induced by streptozotocin reproduces the structural and cellular abnormalities of diabetic cardiomyopathy including cardiac fibrosis and cardiac inflammation, which lead to left ventricular dysfunction (Tschöpe et al., 2005; Asbun and Villarreal, 2006; Poornima et al., 2006). Cardiac inflammation can be characterized by increased levels of pro-inflammatory cytokines and transendothelial migration of immunocompetent cells including T-cells. Diabetes-associated hyperglycaemia and dyslipidaemia are associated with pro-inflammatory effects affecting the endothelium. Hyperglycaemia induces the appearance of cellular adhesion molecules, leading to increased adherence of monocytes. It also alters the permeability of the endothelial basement membrane, leading to accelerated lipid deposition and macrophage recruitment (King and Wakasaki, 1999; Lamarche et al., 1999; Tschöpe et al., 2005). Tight junctions between endothelial cells form a vascular barrier, which in the diabetic state becomes more vulnerable and permeable as a result of endothelial cell abnormalities. Zona occludens (ZO) proteins are important constituents of the tight junction proteins. Both ZO-1 and ZO-2 are present in tight junctions between endothelial cells and ZO-1 has recently been shown to be reduced in failing human hearts (Jesaitis and Goodenough, 1994; Inoko et al., 2003; Kostin, 2007; Laing et al., 2007). PKC, a member of the cAMP-dependent PK/PKG/PKC family, is a serine/threonine-related PK with 11 isoforms and plays a key role in many signal transduction pathways (Newton, 2001; Geraldes and King, 2010; Alexander et al., 2013). Activation of the diacylglycerol/PKC pathway has been observed in patients with diabetes (Koya and King, 1998). PKC activation directly increases the permeability of albumin and other macromolecules through barriers formed by endothelial cells (Lynch et al., 1990; Wolf et al., 1991). However, it is unclear which isoform is crucial to the modulation of tight junction permeability in the heart. PKC-θ, a member of the novel PKC subfamily is a key player in T-cell activation and survival (Sun et al., 2000; Hayashi and Altman, 2007; Geraldes and King, 2010). Furthermore, inhibition of PKC-θ prevented blood–brain barrier dysfunction through inhibition of ZO-1 phosphorylation (Rigor et al., 2012). Hence, we hypothesized that inhibition of PKC-θ could attenuate myocardial dysfunction and fibrosis by enhancing the integrity of tight junctions, decreasing T-cell infiltration, as well as T-cell activation and survival.
Methods
Animals
All animal care and experimental procedures complied with the Guide for the Care and Use of Laboratory Animals of the United States National Research Council (8th edition, 2011) and were approved by the Institutional Animal Care and Use Committee at South Dakota State University. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). A total of 62 animals were used in the experiments described here. Male C57BL/6J mice (22–25 g) were obtained from the Charles River Laboratories (Wilmington, MA, USA) and two breeding pairs of Rag 1 knockout (KO) mice without mature lymphocytes (20 g) were obtained from the Jackson Laboratory (Bar Harbor, ME, USA) and bred in the Animal Research Wings at South Dakota State University.
Induction of diabetes
Type 1 diabetes was induced by 5 days of consecutive injections of streptozotocin (Sigma-Aldrich, St. Louis, MO, USA) at a dose of 50 mg kg–1 of body weight in 0.1 M sodium citrate buffer (pH 4.5). Sodium citrate buffer given alone served as vehicle control. Plasma glucose levels were measured at the beginning, 4 and 11 weeks after streptozotocin injection by using a Contour glucose meter (Bayer, Mishawaka, IN, USA). Mice with fasting plasma glucose over 2 g L-1 were considered diabetic.
Measurements of general characteristics
Mice were randomly divided into three groups: vehicle control (control), untreated diabetes (STZ) and diabetes treated with the PKC-θ inhibitor (PI + STZ). The peptide inhibitor of PKC-θ (PI; Calbiochem, San Diego, CA, USA) was administered once daily (0.2 mg kg–1 i.p.) in two phases: the first 4 weeks (first phase) and the last 2 weeks (second phase). During the treatment period, body weight was assessed twice a week. Plasma glucose levels were evaluated using a glucose meter and serum insulin levels were assessed using an ultrasensitive mouse insulin elisa kit (Crystal Chem Inc., Downers Grove, IL, USA).
Flow cytometry
To detect CD4 and CD8 cells in the blood, antibodies FITC-CD4 or PE-CD8 and isotype control (1 µL each; BD Pharmingen, San Jose, CA, USA) were added to blood samples (100 µL) taken from the tail vein and collected in tubes containing heparin (BD Diagnostics, Franklin Lakes, NJ, USA). After incubation for 30 min at room temperature in the dark, 2 mL of 1 × RBC lysis buffer (BD Pharmingen) was added. The mixture was incubated for 15 min followed by centrifugation at 500× g for 5 min at room temperature. The pellet was isolated and suspended in 200 µL of flow cytometry staining buffer (0.5% BSA in 1 × PBS, pH 7.4). CD4 and CD8 cells were analysed by flow cytometry using a FACScan flow cytometer and CellQuest Pro software (BD Biosciences, San Jose, CA, USA).
Haemodynamic study
At termination (11 weeks after the first injection), mice from each group were weighed and then killed following anaesthesia with phenobarbital (120 mg kg–1, i.p.) and heparin (500 U kg–1, i.p.). Lack of toe pinch reflex indicated that the surgical anaesthesia was sufficient for operation. Blood samples were collected and serum was extracted and stored at −80°C until analysed. Hearts were removed and washed in ice-cold arresting solution (NaCl 120 mmol L–1, KCl 30 mmol L–1), and cannulated via the aorta with a 20 gauge stainless steel blunt needle. Hearts were perfused at 70 mmHg on a modified Langendorff apparatus using Krebs–Henseleit solution (NaCl 118.5 mmol L–1, NaHCO3 25.0 mmol L–1, KCl 4.75 mmol L–1, KH2PO4 1.18 mmol L–1, MgSO4 1.19 mmol L–1, D-glucose 11.0 mmol L–1, CaCl2 1.41 mmol L–1) gassed with 95% O2 and 5% CO2 at 37°C, as previously described (Jin et al., 2004). Ventricular function was measured by a force displacement transducer (model T03; Grass, Warwick, RI, USA) attached to the apex of the heart with a thin thread and metal hook. The resting tension was adjusted to 0.30 g (Xi et al., 2008). Cardiac contractile force (Fc) and the maximal rate of development of contractile force (± dFc/dtmax), coronary flow and heart rate were recorded with Biopac MP 100 data system (Goleta, CA, USA).
Haematoxylin and eosin (H&E) staining and Masson's trichrome staining of cardiac sections
After cardiac contractile force measurement, hearts were perfused with 4% paraformaldehyde in 0.1 M phosphate buffer, pH 7.5 for 10 min for fixation. They were then were removed from the perfusion apparatus and placed in 4% paraformaldehyde overnight at 4°C. Paraffin sections (5 µm) were prepared for standard H&E staining as well as Masson's trichrome staining. Slides were observed under upright microscope (Zeiss, Overkochen, Germany). The area of fibrosis was quantified by using NIH Image J (Bethesda, MA, USA).
Immunohistochemical (IHC) staining of the heart section
IHC staining for CD3, ZO-1 and PKC-θ phosphorylated at Tyr538 (pT538) was performed on paraffin sections (5µm). Briefly, heart sections first underwent deparaffinization and antigen retrieval; and, after blocking with normal goat serum (1/200), antibodies against CD3 (sc20047; Santa Cruz, CA, USA), ZO-1 (617300; Invitrogen, Carlsbad, CA, USA) or pT538-PKC-θ (700043; Invitrogen) was added to the sections. Sections were incubated overnight at 4°C, and an isotype second antibody was used as a background staining control. Sections were rinsed with PBS (three times, 5 min each), and then incubated with a biotin-conjugated IgG secondary antibody (sc-2017; Santa Cruz, CA, USA). Sections were washed and incubated with AB enzyme reagent (sc-2017; Santa Cruz) for 30 min then washed with PBS (three times, 5 min each). After that, heart sections were incubated with one to three drops of peroxide substrate for 2 min following 5 min of washing with running water and then counterstained with haematoxylin. A brown colour indicated positive staining. The intensity of IHC staining was analysed by using computer-assisted morphometry (ImageJ, NIH, MA, USA) as described (Zhao et al., 2010; Wang et al., 2013).
Data analysis
Results were expressed as means ± SEM. One-way anova was used for statistical analysis of data obtained followed by Student–Newman–Keuls test for multiple comparisons of group means. P-values less than 0.05 were considered to indicate statistical significance.
Results
Flow cytometry study of T-cells
Two breeding pairs of Rag1 KO mice which lack mature lymphocytes (Schatz et al., 1989) were purchased from the Jackson Laboratory and bred in our animal facility. To confirm the success of breeding, we performed flow cytometry for both CD4 and CD8 T-cells. Images shown in Figure 1A are representative of flow cytometry results in WT C57BL/6 mice (Figure 1A). Rag1 KO mice did not contain any mature T-cells (Figure 1B).
Figure 1.

Flow cytometry and measurement of general parameters (body weight, plasma glucose and serum insulin) in C57BL/6J WT mice and Rag1 KO mice. (A) Flow cytometry study of CD4 and CD8 T-cell was performed with plasma samples from C57BL/6J WT mice. (B) Flow cytometry study of CD4 and CD8 from Rag1 KO mice. (C) Body weight during the 11 week treatment was measured twice a week for all animals. (D) Blood glucose level was measured before, 4 and 11 weeks after streptozotocin (STZ) injections. (E) Serum insulin level was measured at the end of 11 weeks time point. All values were expressed as means ± SEM. *P < 0.05, compared with the control group. PI + STZ: PKC-θ inhibitor treatment of diabetic mice.
Body weight, glucose and insulin level in wild-type (WT) mice and Rag1 KO mice
During the experiment, the body weight of mice in both untreated diabetes group (STZ, n = 12) and diabetes + PI group (PI + STZ, n = 8) was lower than those in the control group (n = 12). By the end of the treatment period, mice in control group has body weight about 10% higher than the STZ group of mice (P < 0.05, compared with control, Figure 1C). As expected from a model of type 1 diabetes, the STZ group of WT mice displayed hyperglycaemia, as shown by significant increases in blood glucose increase and insulin level decrease (P < 0.05, compared with vehicle control group; Figure 1D, E). As shown in Figure 1, treatment with PI did not have significant effect on animal body weight, blood glucose level or insulin level compared with the untreated diabetes group.
As observed with WT mice, streptozotocin-treated Rag1 KO mice (n = 13) also developed hyperglycaemia, as shown by increased blood glucose levels and decreased insulin, compared with age-matched controls (n = 12, P < 0.05, compared with vehicle control group; Figure 1D, E). The weight of mice in three groups was similar at the beginning of the experiment; however, animal weight started decreasing in both diabetic groups after streptozotocin injection. At the end of 11 weeks of treatment, the body weight of the control mice was about 10% higher than the diabetic mice (P < 0.05, compared with control). As shown in Figure 1, treatment with PI did not have a significant effect on animal body weight, blood glucose level or insulin level, compared with the untreated diabetes group (P > 0.05).
Histology of diabetic hearts
In order to explore the morphological changes of the heart, paraffin sections (5 µm thickness) were prepared and H&E staining was performed. WT hearts from the untreated diabetes group demonstrated a reduction in cardiac muscle cross striations and increased hypereosinophilic compared with control group (Figure 2A). In contrast to WT mice, a significant increase in cardiac tissue integrity was observed in untreated diabetic Rag1 KO mice. The difference between the PI + STZ group and the STZ group in terms of cardiac morphology was largely absent in Rag1 KO mice (Figure 2B).
Figure 2.

Histological examination of hearts from WT mice and Rag1 KO mice in streptozotocin-induced diabetes. (A) H&E staining of cardiac sections from WT mice. Disarrayed myofibres and interstitial oedema were observed in diabetic WT mice. Pretreatment with PI improves the morphology of diabetic hearts. (B) H&E staining of cardiac sections from diabetic Rag1 KO mice. The disturbed structure that was observed in WT mice was largely absent in cardiac tissue from the mutant mice lacking T-cells. Magnification: ×100.
Effect of PI treatment on cardiac fibrosis in diabetic mice
Myocardial fibrosis is commonly observed in diabetic cardiomyopathy and is characterized by excessive production and accumulation of extracellular collagen (Asbun and Villarreal, 2006; Boudina and Abel, 2007). We used Masson's trichrome staining to assess fibrosis in paraffin sections of the heart. In Figure 3A. the blue colour represents collagen staining, indicated with black arrows. The ratio of collagen-stained area to total area was measured (Figure 3B).
Figure 3.

Effect of a selective PKC-θ inhibitor (PI) on cardiac fibrosis in diabetic hearts from WT and Rag1 KO mice. Masson's trichrome staining was used to assess cardiac fibrosis in cardiac tissue samples from all groups. (A) Upper panel: representative images of the whole cross-section of hearts from WT mice. Cardiac sections were observed under low magnification (×16) of stereo microscope and the area of fibrosis (blue staining) was calculated with NIH Image J. Lower panel: representative images, at higher magnification (×400), of trichrome staining of WT hearts. Significant fibrosis was detected in untreated diabetic hearts (STZ) and was attenuated by PI treatment (PI + STZ). M. (B) Quantification of the area of cardiac fibrosis. Data shown are means ± SEM, n = 4–6. *P < 0.05, compared with vehicle control; #P < 0.05, compared with STZ group.
As shown in Figure 3, deposition of collagen was dramatically increased in WT diabetic hearts (Figure 3A, B, P < 0.05 compared with control) and the treatment with PI significantly reduced cardiac fibrosis, compared with the untreated diabetes group (P < 0.05). However, in Rag1 KO mice, streptozotocin treatment did not increase cardiac fibrosis, compared with vehicle control (Figure 3B).
Treatment with PI preserves cardiac function in WT diabetic mice
Given the fact that the PKC-θ inhibitor was able to reduce cardiac fibrosis in WT mice, we isolated hearts for Langendorff perfusion and determined cardiac contractile force. The haemodynamic data are shown in Figure 4 and, as expected, diabetes significantly impaired cardiac contractile force (P < 0.05, compared with control group in the WT mice). Treatment with PI preserved cardiac contractility, compared with the untreated diabetes group (P < 0.05).
Figure 4.
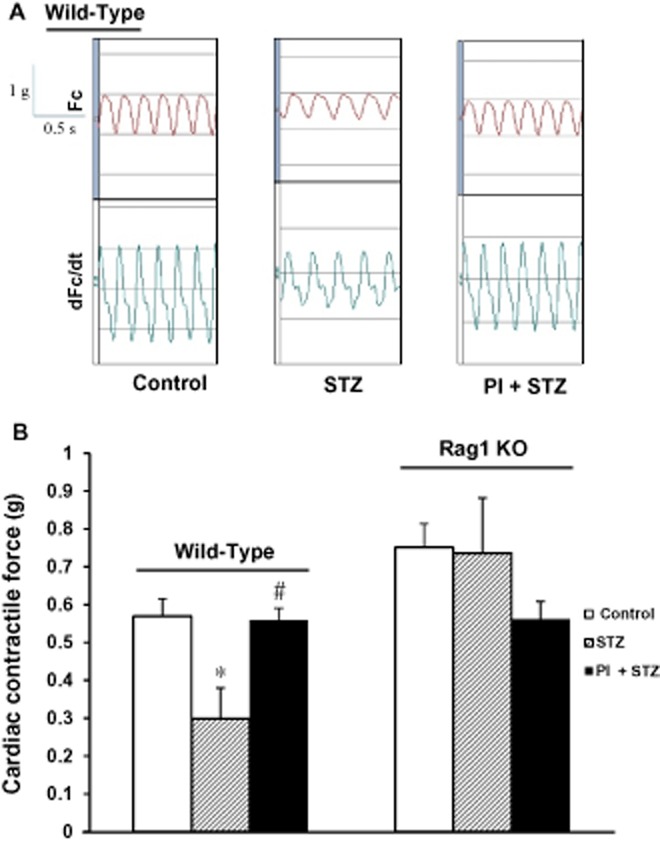
Effect of inhibition of PKC-θ on cardiac contractile force in diabetic hearts. (A) Original recording curve of heart contractility. Untreated diabetic hearts (STZ) show lower contractility in WT mice. Treatment with PI (PI + STZ) preserves the cardiac function of diabetic hearts. (B) Summary data of cardiac contractile force in WT (n = 7 in control; n = 4 in STZ or PI + STZ) and Rag1 KO mice (n = 12 in control, n = 11 in STZ, and 4 in PI + STZ). Data shown are means ± SEM. *P < 0.05 versus control; #P < 0.05 versus STZ. Diabetes significantly reduced cardiac contractility in WT mice whereas cardiac function was maintained in the Rag1 KO mice, lacking mature T-cells.
In the Rag1 KO mice, there was less cardiac fibrosis in the untreated diabetes group (see above) and in the hearts of these mice, streptozotocin also caused less loss of cardiac function. We determined the contractility of heart samples from all three groups as summarised in Figure 4. The cardiac contractile force of the untreated diabetes group was similar to that of the control group. Although the PI group exhibited a lower contractile force, there was no significant difference between PI and control groups.
Expression of phosphorylated PKC-θ (pT538) in diabetic WT and Rag 1 KO mice
Upon activation, PKC-θ is phosphorylated at Tyr538 (pT538). To assess the relevance of activated PKC-θ to cardiomyopathy in the diabetic heart, we measured pT538-PKC-θ expression in heart tissue from all three experimental groups of WT mice and Rag1 KO mice. As shown in Figure 5, both groups of untreated diabetes mice (WT and Rag 1 KO) showed significantly increased pT538-PKC-θ expression (P < 0.05, compared with the corresponding control groups). Treatment with the PKC-θ inhibitor PI decreased expression of pT538-PKC-θ in both strains.
Figure 5.

Phosphorylation of PKC-θ in diabetic hearts, with and without treatment with the PKC-θ inhibitor (PI). Immunohistochemistry was performed on paraffin sections of cardiac tissue from each group. Pictures shown are representative images from each group. (A) Images from WT mice. (B) Images from Rag1 KO mice. The brown colour shows positive staining for phosphorylated PKC-θ (pT538) and was increased in both WT and Rag1 KO diabetic mice. Treatment with PI reduced the expression of pT538-PKC-θ. Magnification: ×400. (C) Computer-assisted morphometric analysis. n = 3 independent studies on each group; *P < 0.05, compared with vehicle control; #P < 0.05, compared with STZ group.
Increased intramyocardial CD3 T-cell infiltrates in WT mice
The infiltration of T-cells in cardiac tissue was determined immunohistochemical staining with anti-CD3 antibody. In WT mice, the myocardium of the diabetic mice, T-cell infiltration was increased in the streptozotocin group compared with control (Figure 6). Intramyocardial infiltrates demonstrated both focal and diffuse patterns of distribution. Infiltrates closely adjacent to cardiomyocytes, suggestive of myocytolysis, were noted, along with T-cell infiltration around blood vessel endothelial cells. Treatment with PI of the diabetic WT mice decreased T-cell infiltration in the myocardium. We also performed immunohistochemistry with the same anti-CD3 antibody in hearts from Rag1 KO mice (images not shown) and as expected, we did not find any positive staining in samples taken from all three groups of Rag1 KO mice.
Figure 6.
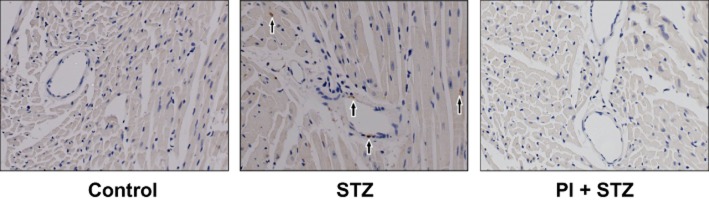
Infiltration of T-lymphocytes in diabetic WT mice. T-cell infiltration, evaluated by immunohistochemical detection of CD3, was increased in diabetic WT mice, compared with control. PI treatment attenuated the infiltration of T-cells into cardiac tissue. Magnification ×400.
Expression of the tight junction protein ZO-1 in WT and Rag1 KO mice
The protein ZO-1 is mainly expressed in tight junctions between endothelial cells including those in blood vessels. In cardiac tissue samples from control WT mice, ZO-1 staining (dark brown colour, Figure 7A) strongly labelled blood vessels and gave rise to a well-defined and homogeneous distribution of label at the border level leading to a well-defined thin line of staining in tissue from both control and PI-treated groups. However, in diabetic WT mice, the staining was not only at a much lower level or absent from blood vessels, but also showed irregular staining in the surrounding tissue, suggesting that diabetes induced degradation and redistribution of this tight junction protein . Treatment of diabetic WT mice with PI restored levels of ZO-1 to almost control values (Figure 7C).
Figure 7.

Effects of PI on expression of the tight junction protein ZO-1 in diabetic WT and Rag1 KO mice. (A) ZO-1 expression was sparsely present in diabetic WT mice and this low level was enhanced by treatment with PI. (B) In diabetic Rag1 KO mice, expression of ZO-1 was decreased by about 40%, compared with control, but this lower level was further decreased after PI treatment. Magnification ×400. (C) Computer-assisted morphometric analysis. n = 3 independent studies on each group; *P < 0.05, compared with vehicle control; #P < 0.05, compared with untreated diabetic (STZ) group.
In cardiac tissue from the control group of Rag1 KO mice, the grade and pattern of immunoreactive ZO-1 in cardiac tissue were similar to those in WT control mice. However, in the untreated diabetic Rag1 KO mice, expression of ZO-1 in blood vessel endothelial cells was moderately decreased (to about 60% control) (Figure 7B, C). Surprisingly, in the group of diabetic Rag1 KO mice treated with PI, ZO-1 expression was further decreased (Figure 7B, C).
Discussion and conclusions
In this report, we identified for the first time a cardiac protective effect of the PKC-θ inhibitor PI in type 1 diabetic cardiomyopathy. Treatment with this PKC-θ pseudosubstrate inhibitor for a long time (11 weeks) was able to preserve tight junction integrity and reduce intramyocardial T-cell infiltration resulting in a significant reduction in cardiac fibrosis as well as improved cardiac function in hearts from mice with diabetes induced by streptozotocin.
Biochemical and genetic approaches have revealed that PKC-θ is required for the activation of mature T-cells as well as for their survival (Sun et al., 2000). Mutation of the PKC-θ gene leads to impaired receptor-induced stimulation of the transcription factors AP-1, NF-κB and NFAT, which results in defective T-cell activation (Hayashi and Altman, 2007). PKC-θ mutation also caused aberrant expression of apoptosis-related proteins, resulting in poor T-cell survival. Recent studies have demonstrated that PKC-θ mutant mice immunized with myelin oligodendrocyte glycoprotein were resistant to the development of clinical experimental autoimmune encephalitis (Salek-Ardakani et al., 2005; Tan et al., 2006; Anderson et al., 2006; Shahabi et al., 2008). In addition to impaired responses in experimental models of autoimmune diseases, PKC-θ mutant mice also display reduced expression of several cytokines (IL-2, IL-4, IFN-γ and TNF-α) at early times (1–3 h) after in vivo i.p. challenge with an anti-CD3 antibody (Tan et al., 2006). The apparently critical role of PKC-θ in Th2- and Th17-mediated responses suggests that pharmacological approaches to inhibit PKC-θ activity may be beneficial in treatment of allergic and autoimmune diseases (Tan et al., 2006).
Intramyocardial inflammation (expression of adhesion molecules, transendothelial immunocompetent infiltrates and cytokine expression) has been noted in streptozotocin-induced diabetes. Some anti-inflammatory agents show protective effect on left ventricular function under hyperglycaemic conditions (Tschöpe et al., 2004; Westermann et al., 2007). The PKC-θ inhibitor that we used in our study is a myristoylated pseudosubstrate inhibitor, which is a cell-permeable, reversible, substrate competitive inhibitor of PKC-θ isozyme. This myristoylated PKC-θ pseudosubstrate inhibitor reduced T-cell chemotaxis (Shahabi et al., 2008). As we showed in our immunohistochemical study of CD3 T-cells in WT mice, the group trteated with the PKC-θ inhibitor PI had reduced intramyocardial infiltration of T-cells. Furthermore, when we used the same experimental protocol with Rag1 KO mice, we found that, although blood glucose levels were still high after streptozotocin, these diabetic mice did not exhibit the increased cardiac fibrosis and impaired cardiac function seen in WT diabetic mice. These results suggest that T-cells are responsible for cardiac dysfunction in this model of type 1 diabetes and that the cardio-protective effect of the PKC-θ inhibitor was related to the presence of T-cells.
The importance of tight junctions in the blood–brain barrier has long been recognized. Many pathological conditions including cerebral hypoxia/reoxygenation injury lead to cell death and tissue injury by modulating tight junction structure and increase blood–brain barrier permeability. It has been reported that PKC-θ and PKC-ζ isozymes are required for dynamic changes in the expression of tight junction proteins and functional integrity of the blood–brain barrier after hypoxia and hypoxia-reoxygenation. The mechanism underlying this biological effect of PKC isozymes appears to be reversible cortical disruption of claudin-5, occludin, and ZO-1 organization and assembly, resulting in a size and time-dependent opening of the cortical blood–brain barrier (Willis et al., 2010). However, the role of tight junctions in the heart is not well understood yet. Our laboratory has previously reported that cardiac ischaemia/reperfusion injury ruptures the structural and functional integrity of coronary endothelial tight junctions and ischaemic preconditioning potently protects the integrity of cardiac tight junctions (Li and Jin, 2012). Myocardial injury is involved in diabetic cardiomyopathy and we therefore investigated changes in tight junctions in the heart, in our model of diabetes.
A particular finding in our present work was the alteration of ZO-1 expression in the diabetic mouse model. We detected ZO-1 expression in both C57BL/6 mice and Rag1 KO mice and compared the expression of ZO-1 in both untreated and treated diabetes groups. Untreated diabetic WT mice exhibited high expression of pT538-PKC-θ and very low expression of ZO-1, whereas expression of ZO-1 in untreated diabetic Rag1 KO mice was only moderately decreased although the expression of pT538-PKC-θ in myocardium was still moderately high. Treatment with PI improved ZO-1 expression in WT mice, presumably by inhibiting T-cell activation, but reduced the expression of ZO-1 in Rag1 KO mice. It has been shown that pro-inflammatory molecules, such as IL-1β and IL-6, increase the disruption of endothelial barriers (Desai et al., 2002; Rigor et al., 2012). IL-1β increases activation of PKC-θ and phosphorylation of PKC-θ at Tyr538 as well as the phosphorylation of ZO-1. Small hairpin RNA silencing of PKC-θ reduces expression of PKC-θ and dysfunction of endothelial barriers (Rigor et al., 2012). In a rat model of global hypoxia and reperfusion injury, hypoxia reduces expression of ZO-1 whereas it enhances the expression of PKC-θ along endothelial cell margins (Willis et al., 2010). The non-selective PKC inhibitor chelerythrine reduced hypoxia-induced vascular permeability and activation of PKC-θ. All these results suggest that high expression and activation of PKC-θ may cause dysfunction and disruption of ZO-1. The mechanism underlying this molecular correlation is not clear and PKC-θ could impair the structure and function of ZO-1 via two mechanisms. Firstly, the activation of PKC-θ induced by hyperglycaemia is critical for the activation of mature T-cells and release of cytokines, including IL-1 and -6. T-cell activation induced by PKC-θ may increase the secretion of pro-inflammatory ILs and reduce the expression of ZO-1 in C57BL/6 mice. In WT mice, the PKC-θ inhibitor PI suppressed activation of T-cells and maintained the structure and function of ZO-1. However, in Rag1 KO mice, diabetes still decreased expression of ZO-1. but to a lesser extent than in WT mice. Secondly, PKC-θ may also mediate the pathological effects of IL-1 or IL-6. In other words, these cytokines reduce the expression of ZO-1 through activation of PKC-θ.
We found that PI treatment reduced the expression of ZO-1 in diabetic Rag1 KO mice, implying that PKC-θ was necessary for the expression of ZO-1. This finding is compatible with the report that the PKC activator diacylglycerol promoted assembly and membrane translocation of ZO-1 (Stuart and Nigam, 1995). Moreover, the PKC activator 12-O-tetradecanoylphorbol-13-acetate enhanced transcription and up-regulation of ZO-1 and function (Balda et al., 1993; Weiler et al., 2005). Although the isoform(s) of PKC involved in the enhancement of transcription remain to be determined, it is plausible that PKC-θ plays a key role in the transcription and synthesis of the ZO-1 protein.
In conclusion, PKC-θ may play a dual role in the regulation of ZO-1. Under physiological conditions, PKC-θ enhanced the transcription and expression of ZO-1 whereas, in pathophysiological situations, overstimulation of PKC-θ activates T-cells, thus increasing secretion of the proinflammatory cytokines IL-1 and IL-6. These pro-inflammatory ILs may cause disruption of ZO-1. We will analyse further the interactions between PKC-θ and ZO-1 with multiple approaches.
Acknowledgments
We would like to thank Dr Xiuqing Wang for the guidance and help in flow cytometry study.
Glossary
- KO
knockout
- PI
peptide inhibitor
- PKC
protein kinase C
- STZ
Streptozotocin
- WT
wild-type
- ZO-1
Zonula occludens-1
Funding sources
This work was supported by the Department of Pharmaceutical Sciences, South Dakota State University, and in part by the Academic and Scholarly Excellence Funds to Z. Q. J. from South Dakota State University.
Conflict of interest
The authors declare no conflict of interest.
References
- Alexander SP, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. British Journal of Pharmacology. 2013;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson K, Fitzgerald M, Dupont M, Wang T, Paz N, Dorsch M, et al. Mice deficient in PKC θ demonstrates impaired in vivo T cell activation and protection from T cell mediated inflammatory diseases. Autoimmunity. 2006;39:469–478. doi: 10.1080/08916930600907954. [DOI] [PubMed] [Google Scholar]
- Asbun J, Villarreal FJ. The pathogenesis of myocardial fibrosis in the setting of diabetic cardiomyopathy. J Am Coll Cardiol. 2006;47:693–700. doi: 10.1016/j.jacc.2005.09.050. [DOI] [PubMed] [Google Scholar]
- Balda MS, Gonzalez-Mariscal L, Matter K, Cereijido M, Anderson JM. Assembly of the tight junction: the role of diacyglycerol. J Cell Biol. 1993;123:293–302. doi: 10.1083/jcb.123.2.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell DS. Diabetic cardiomyopathy. Diabetes Care. 2003;26:2949–2951. doi: 10.2337/diacare.26.10.2949. [DOI] [PubMed] [Google Scholar]
- Boudina S, Abel ED. Diabetic cardiomyopathy revisited. Circulation. 2007;115:3213–3223. doi: 10.1161/CIRCULATIONAHA.106.679597. [DOI] [PubMed] [Google Scholar]
- Desai TR, Leeper NJ, Hynes KL, Gewertz BL. Interleukin-6 causes endothelial barrier dysfunction via the protein kinase C pathway. J Surg Res. 2002;104:118–123. doi: 10.1006/jsre.2002.6415. [DOI] [PubMed] [Google Scholar]
- Geraldes P, King GL. Activation of protein kinase C isoforms and its impact on diabetic complications. Circ Res. 2010;106:1319–1331. doi: 10.1161/CIRCRESAHA.110.217117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi K, Altman A. Protein kinase C theta (PKC-θ): a key player in T cell life and death. Pharmacol Res. 2007;55:537–544. doi: 10.1016/j.phrs.2007.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoko A, Itoh M, Tamura A, Matsuda M, Furuse M, Tsukita S. Expression and distribution of ZO-3, a tight junction MAGUK protein, in mouse tissues. Genes Cells. 2003;8:837–845. doi: 10.1046/j.1365-2443.2003.00681.x. [DOI] [PubMed] [Google Scholar]
- Jesaitis LA, Goodenough DA. Molecular characterization and tissue distribution of ZO-2, a tight junction protein homologous to ZO-1 and the Drosophila discs-large tumor suppressor protein. J Cell Biol. 1994;124:949–961. doi: 10.1083/jcb.124.6.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin ZQ, Goetzl EG, Karliner JS. Sphingosine kinase activation mediates ischemic preconditioning in murine. Circulation. 2004;110:1980–1989. doi: 10.1161/01.CIR.0000143632.06471.93. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. NC3Rs Reporting Guidelines Working Group. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King GL, Wakasaki H. Theoretical mechanisms by which hyperglycemia and insulin resistance could cause cardiovascular diseases in diabetes. Diabetes Care. 1999;22(Suppl. 3):C31–C37. [PubMed] [Google Scholar]
- Kostin S. Zonula occludens-1 and connexin 43 expression in the failing human heart. J Cell Mol Med. 2007;11:892–895. doi: 10.1111/j.1582-4934.2007.00063.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koya D, King GL. Protein kinase C activation and the development of diabetic complications. Diabetes. 1998;47:859–866. doi: 10.2337/diabetes.47.6.859. [DOI] [PubMed] [Google Scholar]
- Laing JG, Saffitz JE, Steinberg TH, Yamada KA. Diminished zonula occludens-1 expression in the failing human heart. Cardiovasc Pathol. 2007;16:159–164. doi: 10.1016/j.carpath.2007.01.004. [DOI] [PubMed] [Google Scholar]
- Lamarche B, Lemieux I, Despres JP. The small, dense LDL phenotype and the risk of coronary heart disease: epidemiology, pathophysiology and therapeutic aspects. Diabetes Metab. 1999;25:199–211. [PubMed] [Google Scholar]
- Li Z, Jin ZQ. Ischemic preconditioning enhances integrity of coronary endothelial tight junctions. Biochem Biophys Res Commun. 2012;425:630–635. doi: 10.1016/j.bbrc.2012.07.130. [DOI] [PubMed] [Google Scholar]
- Lynch JJ, Ferro TJ, Blumenstock FA, Brockenauer AM, Malik AB. Increased endothelial albumin permeability mediated by protein kinase C activation. J Clin Invest. 1990;85:1991–1998. doi: 10.1172/JCI114663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J, Drummond G, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton AC. Protein kinase C: structural and spatial regulation by phosphorylation, cofactors, and macromolecular interactions. Chem Rev. 2001;101:2353–2364. doi: 10.1021/cr0002801. [DOI] [PubMed] [Google Scholar]
- Poornima IG, Parikh P, Shannon RP. Diabetic cardiomyopathy: the search for a unifying hypothesis. Circ Res. 2006;98:596–605. doi: 10.1161/01.RES.0000207406.94146.c2. [DOI] [PubMed] [Google Scholar]
- Rigor RR, Beard RS, Jr, Litovka OP, Yuan SY. Interleukin-1β-induced barrier dysfunction is signaled through PKC-θ in human brain microvascular endothelium. Am J Physiol Cell Physiol. 2012;302:C1513–C1522. doi: 10.1152/ajpcell.00371.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salek-Ardakani S, So T, Halteman BS, Altman A, Croft M. Protein kinase C θ controls Th1 cells in experimental autoimmune encephalomyelitis. J Immunol. 2005;175:7635–7641. doi: 10.4049/jimmunol.175.11.7635. [DOI] [PubMed] [Google Scholar]
- Schatz DG, Oettinger MX, Baltimore D. The V (D) J recombination activating gene, Rag-1. Cell. 1989;59:1035–1048. doi: 10.1016/0092-8674(89)90760-5. [DOI] [PubMed] [Google Scholar]
- Shahabi NA, McAllen K, Sharp BM. Stromal cell-derived factor 1-α (SDF)-induced human T cell chemotaxis becomes phosphoinositide 3-kinase (PI3K)-independent: role of PKC-θ. J Leukoc Biol. 2008;83:663–671. doi: 10.1189/jlb.0607420. [DOI] [PubMed] [Google Scholar]
- Spector KS. Diabetic cardiomyopathy. Clin Cardiol. 1998;21:885–887. doi: 10.1002/clc.4960211205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart RO, Nigam SK. Regulated assembly of tight junctions by protein kinase C. Proc Natl Acad Sci USA. 1995;92:6072–6076. doi: 10.1073/pnas.92.13.6072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z, Arendt CW, Ellmeier W, Schaeffer EM, Sunshine MJ, Gandhi L, et al. PKC-θ is required for TCR-induced NF-kB activation in mature but not immature T lymphocytes. Nature. 2000;404:402–407. doi: 10.1038/35006090. [DOI] [PubMed] [Google Scholar]
- Tan SL, Zhao J, Bi C, Chen XC, Hepburn DC, Wang L, et al. Resistance to experimental autoimmune encephalomyelitis and impaired IL-17 production in protein kinase C θ-deficient mice. J Immunol. 2006;176:2872–2879. doi: 10.4049/jimmunol.176.5.2872. [DOI] [PubMed] [Google Scholar]
- Tschöpe C, Spillmann F, Rehfeld U, Koch M, Westermann D, Altmann C, et al. Improvement of defective sarcoplasmic reticulum Ca2+ transport in diabetic heart of transgenic rats expressing the human kallikrein-1 gene. FASEB J. 2004;18:1967–1969. doi: 10.1096/fj.04-1614fje. [DOI] [PubMed] [Google Scholar]
- Tschöpe C, Walther T, Escher F, Spillmann F, Du J, Altmann C, et al. Transgenic activation of the kallikrein–kinin system inhibits intramyocardial inflammation, endothelial dysfunction, and oxidative stress in experimental diabetic cardiomyopathy. FASEB J. 2005;14:2057–2059. doi: 10.1096/fj.05-4095fje. [DOI] [PubMed] [Google Scholar]
- Wang ZF, Wang NP, Harmouche S, Philip T, Pang XF, Bai F, et al. Postconditioning promotes the cardiac repair through balancing collagen degradation and synthesis after myocardial infarction in rats. Basic Res Cardiol. 2013;108:318. doi: 10.1007/s00395-012-0318-9. [DOI] [PubMed] [Google Scholar]
- Weiler F, Marbe T, Scheppach W, Schauber J. Influence of protein kinase C on transcription of the tight junction elements ZO-1 and occludin. J Cell Physiol. 2005;204:83–86. doi: 10.1002/jcp.20268. [DOI] [PubMed] [Google Scholar]
- Westermann D, Van Linthout S, Dhayat S, Dhayat N, Escher F, Bucker-Gartner C, et al. Cardioprotective and anti-inflammatory effects of interleukin converting enzyme inhibition in experimental diabetic cardiomyopathy. Diabetes. 2007;56:1834–1841. doi: 10.2337/db06-1662. [DOI] [PubMed] [Google Scholar]
- Willis CL, Meske DS, Davis TP. Protein kinase C activation modulates reversible increase in cortical blood–brain barrier permeability and tight junction protein expression during hypoxia and posthypoxic reoxygenation. J Cereb Blood Flow Metab. 2010;30:1847–1859. doi: 10.1038/jcbfm.2010.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf BA, Williamson JR, Easom RA, Chang K, Sherman WR, Turk J. Diacylglycerol accumulation and microvascular abnormalities induced by elevated glucose levels. J Clin Invest. 1991;87:31–38. doi: 10.1172/JCI114988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi L, Das A, Zhao ZQ, Merino VF, Bader M, Kukreja RC. Loss of myocardial ischemic preconditioning in adenosine A1 and bradykinin B2 receptors gene knockout mice. Circulation. 2008;118:S32–S37. doi: 10.1161/CIRCULATIONAHA.107.752865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao ZQ, Puskas JD, Xu D, Wang NP, Mosunjac M, Guyton RA, et al. Improvement in cardiac function with small intestine extracellular matrix is associated with recruitment of C-kit cells, myofibroblasts, and macrophages after myocardial infarction. J Am Coll Cardiol. 2010;55:1250–1261. doi: 10.1016/j.jacc.2009.10.049. [DOI] [PubMed] [Google Scholar]