

Abstract
Aim
A close correlation exists between positron emission tomography (PET)-determined histamine H1-receptor occupancy (H1RO) and the incidence of sedation. Antihistamines with H1RO <20% are classified as non-sedating. The objective was to compare the H1RO of bilastine, a second generation antihistamine, with that of hydroxyzine.
Methods
This randomized, double-blind, crossover study used PET imaging with [11C]-doxepin to evaluate H1RO in 12 healthy males (mean age 26.2 years), after single oral administration of bilastine (20 mg), hydroxyzine (25 mg) or placebo. Binding potentials and H1ROs were calculated in five cerebral cortex regions of interest: frontal, occipital, parietal, temporal, insula. Plasma bilastine concentrations, subjective sedation (visual analogue scale), objective psychomotor performance (digital symbol substitution test), physiological variables and safety (adverse events, AEs), were also evaluated.
Results
The mean binding potential of all five regions of interest (total binding potential) was significantly greater with bilastine than hydroxyzine (mean value 0.26 vs. 0.13, P < 0.01; mean difference and 95% CI −0.130 [−0.155, 0.105]). There was no significant difference between bilastine and placebo. Overall H1RO by bilastine was significantly lower than that by hydroxyzine (mean value −3.92% vs. 53.95%, P < 0.01; mean difference and 95% CI 57.870% [42.664%, 73.075%]). There was no significant linear relationship between individual bilastine plasma concentrations and total binding potential values. No significant between-treatment differences were observed for sedation and psychomotor performance. Twenty-six non-serious AEs were reported. Sleepiness or sedation was not reported with bilastine but appeared in some subjects with hydroxyzine.
Conclusions
A single oral dose of bilastine 20 mg had minimal H1RO, was not associated with subjective sedation or objective impairment of psychomotor performance and was devoid of treatment-related sedative AEs, thus satisfying relevant subjective, objective and PET criteria as a non-sedating antihistamine.
Keywords: antihistamines H-1, bilastine, histamine H-1-receptor occupancy, positron emission tomography, PET
What is already known about this subject
Bilastine, a non-sedating, second generation antihistamine, is approved for the treatment of allergic rhinoconjunctivitis and urticaria.
In clinical trials, the sedative properties of bilastine and placebo were similar. Bilastine has shown no effect on psychomotor performance or driving ability.
Bilastine histamine H1-receptor occupancy (H1RO) has not yet been evaluated by positron emission tomography (PET).
What this study adds
This study is the first to measure cerebral H1RO of bilastine 20 mg by PET. Bilastine showed practically no cerebral H1RO (−3.92 ± 14.39%).
Results are in accordance with the clinical classification of bilastine as a second generation, non-sedating antihistamine.
Introduction
First generation antihistamines (e.g. hydroxyzine) are still widely used clinically, despite undesirable effects such as central nervous system (CNS) impairment and strong sedation [1,2]. Even at recommended doses, first generation agents may cause dangerous situations for individuals who are driving, piloting planes or operating heavy machinery [2–4]. Second generation antihistamines (e.g. cetirizine, loratadine) may be ‘relatively non-sedating’, i.e. they produce minimal cognitive and psychomotor dysfunction at recommended doses, but dose-related dysfunction at higher dosages, or truly non-sedating even at high doses (e.g. fexofenadine). Many physicochemical properties of drugs such as molecular weight, lipophilicity, polar surface area and number of hydrogen bonds, in addition to being a substrate of P-glycoprotein, limit blood–brain barrier penetration [1,3,5].
Studies of the CNS effects of antihistamines are often conducted in healthy volunteers because the results can be extrapolated to patients with allergic disease. Although allergic mediators may alter blood–brain barrier permeability, results from volunteers are good predictors of the effects in patients [6]. The merits of subjective documentation of drowsiness and objective CNS testing may be limited in some situations by an individual's motivation or familiarity with tests [1]. Positron emission tomography (PET) represents a major breakthrough, providing a sensitive reference method for quantifying CNS penetration. The histamine H1-receptor occupancy (H1RO) of antihistamines can relate to psychometric and other tests of CNS function [1,7,8]. Pooled analyses of clinical trial data have confirmed a close correlation between PET-determined H1RO and the incidence of sedation [2,8].
H1RO >50% has been clearly linked with a high rate of sleepiness and cognitive decline. After single dose oral administration of various antihistamines, values of H1RO vary widely up to about 75% with ketotifen 1 mg [3,8]. Overall, three categories of antihistamine can be conventionally defined according to H1RO intervals: non-sedating (<20% occupancy), less sedating (20–50%) and sedating (>50%) [2,3].
Altogether, the non-sedating properties of antihistamines require full characterization according to subjective sleepiness recorded on scales such as the Stanford Sleepiness Scale; objective assessments of cognitive and psychomotor function (e.g. critical flicker fusion, choice reaction time, digit symbol substitution test [DSST]); and PET evaluation of H1RO <20% [1,3]. Subjective sleepiness should be determined in large scale, double-blind, placebo-controlled studies, and assessments of cognitive function (at least two tasks) should also be performed in large study populations. No statistically significant differences should be observed between the test antihistamine and placebo [3]. Various methods of PET imaging with radiolabelled [11C]-doxepin have been used successfully for quantification of H1RO; for example, a simplified reference tissue model approach and Logan graphical analysis with reference tissue or arterial sampling [9–12].
The current crossover trial was designed to determine H1RO in the brain, using [11C]-doxepin and PET, after single dose oral administration of the second generation antihistamine bilastine (20 mg), the first generation agent hydroxyzine (25 mg) and placebo, in healthy male volunteers. Bilastine is approved in several countries for the treatment of allergic rhinoconjunctivitis (seasonal and perennial) and urticaria [13]. It is a non-sedating and long-acting antihistamine, with selective H1-receptor affinity, and no affinity for other aminergic receptors [14,15]. In controlled clinical trials at the recommended therapeutic dose of 20 mg once daily, the CNS effects and safety profile of bilastine were similar to those of placebo; no significant difference in the incidence of drowsiness was noted between bilastine and placebo. Other clinical trials have shown bilastine 40 mg day−1 to have no effect on psychomotor performance or driving ability [16,17]. As is the case for fexofenadine, bilastine is also a P-glycoprotein substrate and its physicochemical properties can limit blood–brain barrier penetration [18]. The current study is the first to assess brain H1RO by bilastine.
Methods
Study design
This was a phase I, double-blind, placebo-controlled, crossover study, in which subjects were randomized to one of six test-treatment sequences (two subjects per sequence) comprising the three test compounds. A screening visit was conducted within 3 weeks before the first experimental session, and each subject participated in three sessions. In each session, subjects received a single oral dose of bilastine, hydroxyzine or placebo, followed by intravenous [11C]-doxepin (Molecular Imaging Centre, CRC Mar, Barcelona, Spain). To allow for washout of bilastine and hydroxyzine, there was an interval of at least 1 week between sessions. At 3–7 days after completion of all three sessions, a final study visit was conducted which consisted of a physical examination, assessment of laboratory and urinary parameters and final safety evaluation. The total study duration for each subject, including screening, experimental sessions and washout, and the final study visit, was at least 6–8 weeks.
The primary study end point was brain H1RO. Secondary outcome measures comprised plasma bilastine concentrations; subjective evaluation of sedation (drowsiness, absent-mindedness, and sleepiness) assessed by visual analogue scales [VAS 0–100 mm]; objective assessment of psychomotor performance (DSST), physiological variables (systolic and diastolic blood pressure, heart rate [measured using a Carescape™ V100 monitor, GE Healthcare, Milwaukee, WI, USA] and oral temperature); and a safety evaluation which included adverse events (AEs) reported spontaneously by subjects or detected by the study investigator. The DSST, a classical test designed to evaluate recognition and recording of visual information [18], is a subset of the Adult Intelligence Scale-Revised [19] and has been used widely to evaluate sedation induced by drugs [20–22]. The three visual analogue scales selected are commonly used to evaluate clinical sedation in studies in healthy subjects or patients, and have been used in previous studies of bilastine [23,24] or sedatives [20,21].
The study was approved by the Clinical Research Ethical Committee, Parc de Salut MAR, and the Spanish Medicines Agency, and was performed in accordance with the Declaration of Helsinki (2008), Good Clinical Practices of the International Conference on Harmonization (ICH) and applicable laws and regulations (Royal Decree 223/2004, Directive 2001/20/CE).
Subjects
Twelve healthy men, aged 20–39 (mean 26.2 ± 5.7) years and with body mass index 22.4–27.0 (mean 24.7 ± 1.68) kg m−2, participated in and completed the study. None of the subjects smoked regularly (>5 cigarettes day−1). Pre-study alcohol and caffeine consumption were <30 g day−1 and <5 cups day−1, respectively, and all participants had negative pre-study urinary drug abuse screens. At the initial screening visit, serology tests were negative for HIV and hepatitis B and C, vital signs were normal and no subjects had clinically significant laboratory abnormalities. Written informed consent to participate was obtained prior to any study-related procedure.
Test compound administration
The test compounds were single oral doses of bilastine 20 mg (Bilaxten® 20 mg, tablets; FAES FARMA, S.A., Leioa-Bizkaia, Spain), hydroxyzine 25 mg (Atarax® 25 mg, tablets; UCB Pharma, S.A., Barcelona, Spain) and placebo. For all treatments, tablets were encapsulated in opaque capsules to ensure blinding. Each test compound was administered with 250 ml of water.
Experimental sessions (PET measurement)
A schematic overview of the PET study design is shown in Figure 1. Subjects were admitted to the clinical research unit the evening before each experimental session. A magnetic resonance imaging scan was performed that evening. The following morning, subjects were woken at 07.00 h. Baseline and vital signs were recorded, and a sedation questionnaire (VAS) and DSST were completed. The test compound was administered at 08.00 h. One hour later, subjects were moved to the molecular imaging centre for PET examination. PET was performed using a Siemens ECAT EXACT HR+ scanner (Siemens, S.A., Madrid, Spain). The image acquisition protocol was similar to that used in previous studies assessing H1RO by antihistamines [25].
Figure 1.
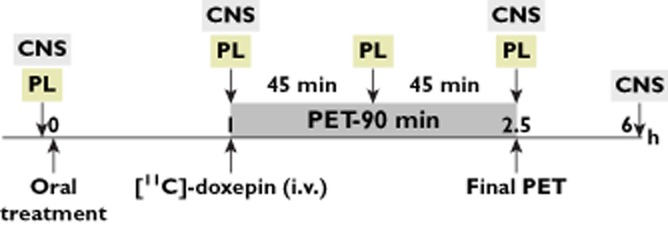
PET study design. PL: Blood sampling for determination of plasma concentrations of bilastine. CNS: central nervous system assessment (subjective evaluation of sedation: drowsiness, absent-mindedness and sleepiness, assessed by visual analogue scales [0–100 mm]; objective assessment of psychomotor performance [digit symbol substitution test])
Before intravenous administration of [11C]-doxepin, each subject was placed on the PET couch and underwent a 10 min transmission scan for subsequent correction of transmission attenuation. After injection of the radioligand, emission acquisition was performed for 90 min (including 26 frames: 8 × 15 s, 3 × 60 s, 5 × 120 s, 5 × 300 s and 5 × 600 s). The radioactive dose did not exceed 8 mCi (296 MBq) per scan. [11C]-doxepin radiochemical purity was ≥95% and its specific radioactivity at the time of injection was 50.12 ± 19.00 GBq μmol−1. The injected dose and cold mass of [11C]-doxepin were 7.11 ± 0.63 mCi and 3.68 ± 2.45 μg, respectively.
After PET scans, subjects returned to the clinical research unit where they remained until 12 h after study drug administration. Blood samples (5 ml Vacutainer tube containing sodium heparin) for measurement of plasma bilastine concentrations were taken at 0 h (drug administration), 1 h (PET start), 1.75 h (mid-PET) and 2.5 h (PET end). These time points were selected to ensure that PET data were recorded in the interval next to the maximum exposure to bilastine (tmax = 1.3 h) [26]. Bilastine is rapidly absorbed after oral administration, with a tmax of 1–1.5 h and a half-life of 10–12 h [26,27]. Hydroxyzine has a tmax of 2 h and an elimination half-life of 20 ± 4.1 h [28]. Although plasma concentrations of hydroxyzine were not determined, PET data were recorded in the interval around the published tmax (2.1 h) [29]. Vital signs were recorded at 0, 1, 2.5, 6 and 12 h. The sedation questionnaire and DSST were completed at 0, 1, 2.5 and 6 h.
Image and data analysis
PET images were reconstructed by filtered retroprojection and were corrected by attenuation, scatter, random coincidences and dead time, using algorithms from the scanner manufacturer. Dynamic PET images were corrected for motion by realigning each time frame to the averaged frame using a mutual information-based algorithm as implemented in the SPM8 software (Wellcome Trust Centre for Neuroimaging; http://www.fil.ion.ucl.ac.uk/spm) and registered to the subject's MRI. Regions of interest (ROIs) were defined through non-linear registration (using SPM8) of a template MRI and the corresponding atlas (Harvard–Oxford atlas included with FSL 4.1; http://www.fmrib.ox.ac.uk/fsl). ROIs were frontal, occipital, parietal, temporal, insula and cerebellar grey matter. A grey matter map obtained from the subject's MRI scan was applied in order to segment each of the cortical ROIs. Each ROI was then applied to the dynamic PET data to derive regional time–activity curves. Time–activity curves were processed by graphical methods [9], using the cerebellum as the reference region to obtain ‘binding potential’ for the radioligand at baseline and after drug administration [12]. The graphical methods used for image analysis permitted avoidance of arterial blood sampling from subjects; these methods have been described previously and have been validated for H1RO determination [10]. Total binding potential was defined as the mean binding potential value obtained from the five ROIs (frontal, occipital, parietal, temporal, insula).
H1RO was estimated according to equation 1:
 |
1 |
Statistical analyses
All statistical analyses were conducted using IBM SPSS version 18 software (IBM Spain S.A., Barcelona, Spain) and with the significance value (P) set at 0.05.
Repeated measures analysis of variance (anova) with treatment as factor was used to assess the difference between mean total binding potential and binding potential in each ROI. If statistically significant differences were identified, Tukey post hoc tests were applied. The Wilcoxon matched-pairs, signed-rank test was used to determine whether significant differences existed between bilastine and hydroxyzine for H1RO.
Although the protocol employed is not the most appropriate to establish a possible H1RO–plasma concentration relationship, a linear regression between plasma concentrations (AUC(0,2.5h)) vs. binding potential (mean from five ROIs) was calculated as the optimal approach. Bilastine AUC was calculated by the trapezoidal rule.
Values from physiological variables, subjective sedation (drowsiness, absent-mindedness, sleepiness) and DSST (number of correct responses) across the study were transformed to differences from baseline. For each variable, the peak effect in the first 6 h (sedation and psychomotor performance) or 12 h (physiological variable) after administration and the 6 h (sedation and DSST) or 12 h (physiological outcomes) AUC of effects vs. time, calculated by the trapezoidal rule, were determined. These transformations were analyzed by one way repeated measures anova, with test drug as factor. Post hoc multiple comparisons were performed using the Tukey test. Finally, a detailed comparison of the time course of effects was conducted using two way repeated measures anova, with treatment and time as factors. Whenever treatment condition or the treatment condition × time interaction was statistically significant, multiple Tukey post hoc comparisons were performed at each time point using the mean square error term of the treatment condition × time interaction. Any changes in vital signs from screening to the final study visit were evaluated by paired Student's t-test. Descriptive statistics were used to characterize adverse events.
Results
Binding potential and histamine H1-receptor occupancy
Total binding potential was defined as the mean value obtained from the five ROIs. Mean (± SD) total binding potential for [11C]-doxepin was significantly greater with the bilastine than hydroxyzine (0.26 ± 0.07 vs. 0.13 ± 0.07; P < 0.01). No significant difference in mean total binding potential was noted between bilastine and placebo (0.26 ± 0.07 vs. 0.26 ± 0.08, Table 1). The same findings were evident in each of the five ROIs (P < 0.01 for bilastine vs. hydroxyzine, Figure 2). Standard and coloured PET images, obtained after administration of bilastine, hydroxyzine or placebo, are presented in Figure 3.
Table 1.
Binding potential values (mean ± SD) obtained from five regions of interest (ROIs). The total binding potential (mean ± SD) is the average of the five ROIs
| Binding potential (from Logan plot Binding potential = DVR – 1) | |||
|---|---|---|---|
| Region of Interest | Placebo | Bilastine 20 mg | Hydroxyzine 25 mg |
| Frontal | 0.278 ± 0.091 | 0.276 ± 0.075 | 0.122 ± 0.067 |
| Occipital | 0.177 ± 0.070 | 0.193 ± 0.078 | 0.088 ± 0.063 |
| Parietal | 0.273 ± 0.091 | 0.286 ± 0.087 | 0.141 ± 0.063 |
| Temporal | 0.214 ± 0.085 | 0.208 ± 0.068 | 0.094 ± 0.069 |
| Insula | 0.338 ± 0.091 | 0.339 ± 0.070 | 0.205 ± 0.086 |
| Total | 0.256 ± 0.081 | 0.260 ± 0.072 | 0.130 ± 0.067 |
|
Mean difference and 95% CI, placebo vs. bilastine and hydroxyzine Mean difference and 95% CI bilastine vs. hydroxyzine |
0.004 (−0.021, 0.029) (NS) −0.130 (−0.155, −0.105) (P < 0.01) |
−0.126 (−0.151, −0.101) (P < 0.01) | |
Figure 2.
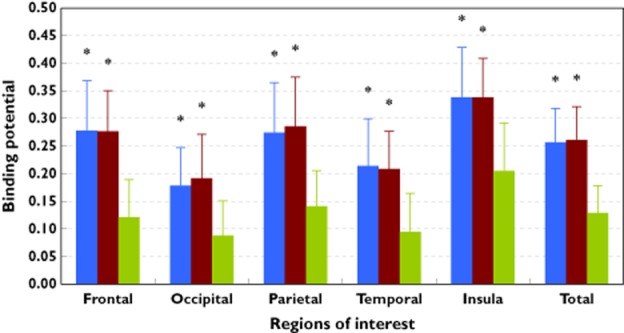
Mean (± SD) binding potential of the radioligand [11C]-doxepin after single dose antihistamine or placebo administration. Data shown are for five individual regions of interest (ROIs), and mean total binding potential for the ROIs combined. *P < 0.01 for all bilastine–hydroxyzine and all hydroxyzine–placebo differences; no significant bilastine–placebo differences.  , placebo;
, placebo;  , bilastine 20 mg;
, bilastine 20 mg;  , hydroxyzine 25 mg
, hydroxyzine 25 mg
Figure 3.
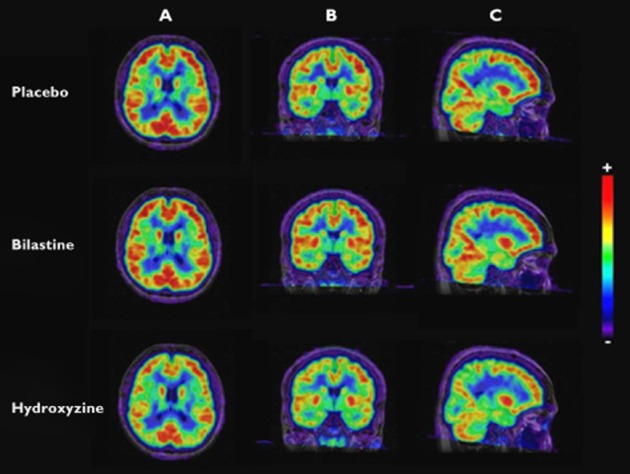
Co-registered magnetic resonance image and [11C]-doxepin positron emission tomography images after administration of placebo, bilastine (20 mg) and hydroxyzine (25 mg). A: horizontal plane. B: coronal plane. C: sagittal plane. Scale + to −: from maximum to minimum binding potential values
Based on binding potential values (see equation 1), mean (± SD) total H1RO by bilastine was significantly lower than that by hydroxyzine (–3.92 ± 14.39% vs. 53.95 ± 14.13%, P < 0.01, Figure 4). The same was true for mean H1RO values in all five ROIs (P < 0.01 for all bilastine–hydroxyzine differences, Table 2).
Figure 4.
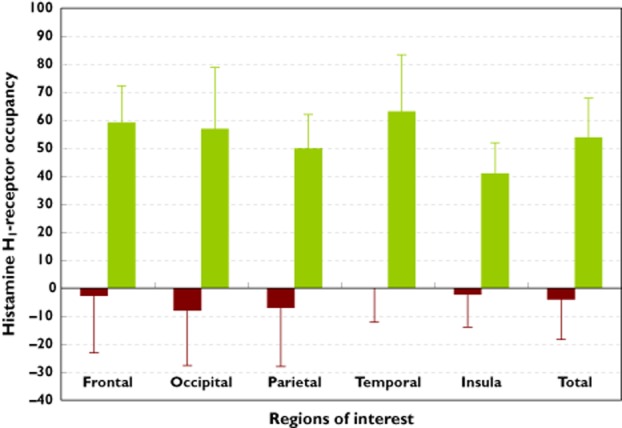
Mean (± SD) brain histamine H1-receptor occupancy (H1RO) after single dose antihistamine administration. Data shown are for five individual regions of interest (ROIs), and mean total H1RO for the ROIs combined. P < 0.01 for all bilastine–hydroxyzine differences.  , bilastine 20 mg;
, bilastine 20 mg;  , hydroxyzine 25 mg
, hydroxyzine 25 mg
Table 2.
Mean (± SD) brain histamine H1-receptor occupancy values (%) for bilastine and hydroxyzine in 12 healthy volunteers
| Receptor occupancy (%) | ||
|---|---|---|
| Region of interest | Bilastine 20 mg (*) | Hydroxyzine 25 mg |
| Frontal | −2.41 ± 20.97 | 58.91 ± 13.37 |
| Occipital | −8.07 ± 19.60 | 56.96 ± 21.95 |
| Parietal | −7.17 ± 20.74 | 49.93 ± 12.29 |
| Temporal | 0.38 ± 12.61 | 62.83 ± 20.60 |
| Insula | −2.32 ± 11.83 | 41.12 ± 11.10 |
| Total | −3.92 ± 14.39 | 53.95 ± 14.13 |
|
Mean difference and 95% CI bilastine vs. hydroxyzine Median difference and 95% CI bilastine vs. hydroxyzine |
57.870% (42.664%, 73.075%) 54.228% (43.713%, 78.819%) |
|
P < 0.01 vs. hydroxyzine.
Plasma bilastine concentrations
Mean plasma bilastine concentrations were 140.4 ± 92.7 ng ml−1 (1 h post-dose), 116.9 ± 73.8 ng ml−1 (1.75 h), and 108.5 ± 55.9 ng ml−1 (2.5 h). Mean bilastine AUC(0,2.5h) was 112.7 ± 64.8 ng ml−1·h, with a range of 2.6–502.3 ng ml−1·h. No significant linear correlation was noted between AUC(0,2.5h) and mean total binding potential for bilastine (r2 = 0.063, P > 0.05, Figure 5).
Figure 5.
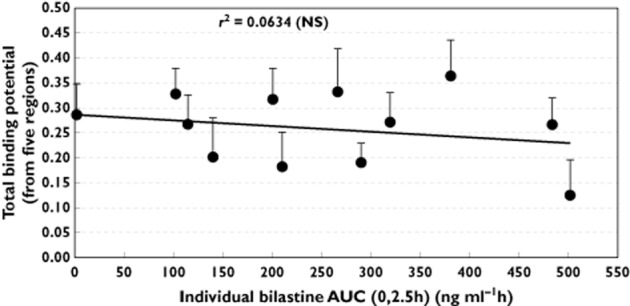
Linear regression between individual bilastine AUC(0,2.5h) (ng ml−1·h) values and total binding potential (average from five regions of interest). The r2 value indicates no significant linear relationship between parameters.  , bilastine 20 mg;
, bilastine 20 mg;  , linear (bilastine 20 mg)
, linear (bilastine 20 mg)
Subjective evaluation of sedation and psychomotor performance
No significant differences were noted between bilastine, hydroxyzine and placebo with regard to mean maximum changes from baseline in VAS scores (Emax) for drowsiness, absent-mindedness and sleepiness or in DSST scores (data not shown). In addition, for all subjective outcomes and DSST, no significant differences were observed between the three study treatments in terms of AUC values. For sedation and DSST, maximum scores were attained at 2.5 h post-dose, but no significant between-treatment differences were noted.
Safety evaluation
Ten study participants (83.3%) reported at least one AE, but none were serious. A total of 26 AEs were reported (Table 3): four subjects had one AE, one had two AEs, two had three AEs, one had four AEs and two subjects had five AEs. Most AEs were mild (84.6%), while the remainder were moderate in intensity. The most frequent AEs were nervous system disorders; headache occurred in four subjects. Overall, nine AEs were reported with bilastine, seven with hydroxyzine and seven with placebo; three other AEs (odynophagia, itchy throat, common cold) occurred between screening and the first dose of study medication. No sleepiness or sedation was reported after bilastine administration, but there were two reports of sedation after hydroxyzine, and one report of sleepiness after placebo. Overall, two AEs (sedation after hydroxyzine) were considered probably related to antihistamine administration, and six AEs (headache, diarrhoea, vomiting) were considered possibly related.
Table 3.
Adverse events (AEs) considered possibly or probably related to study treatment†
| System organ class | AE incidence (%)‡ | Number of AEs | |||
|---|---|---|---|---|---|
| Placebo | Bilastine | Hydroxyzine | Total | ||
| Gastrointestinal disorders | 15.4 | 1 | 3* | 0 | 4 |
| Diarrhoea | 7.7 | 1 | 1* | 0 | 2 |
| Vomiting | 7.7 | 0 | 2* | 0 | 2* |
| Nervous system disorders | 26.9 | 2 | 2* | 3* | 7 |
| Headache | 15.4 | 1 | 2* | 1* | 4 |
| Sleepiness (somnolence) | 3.9 | 1 | 0 | 0 | 1 |
| Sedation | 7.7 | 0 | 0 | 2* | 2* |
Indicated by asterisk (*).
% of all AEs (26).
No differences were observed among treatments with regard to vital signs during the experimental sessions. Also, no clinically significant changes in laboratory parameters and vital signs occurred between selection and final visits.
Discussion
Guidelines from the Consensus Group On New-Generation Antihistamines (CONGA) clearly stipulate that for an antihistamine to be classed as non-sedating, at least three key factors require assessment: self-reporting of sleepiness in antihistamine-treated individuals (it is unscientific to use this measure alone to define the sedative properties of antihistamines [2]); objective evaluation of psychomotor performance and cognitive function through tests such as critical flicker fusion, choice reaction time and DSST; and PET assessment of H1RO [1]. PET assessment provides a sensitive and absolute measure of blood–brain barrier penetration and H1RO [1,7,8]. It is widely acknowledged that H1RO <20% is a requirement for classification of antihistamines as non-sedating [7,30].
The current, placebo-controlled PET study compared brain H1RO by bilastine with that by the first generation antihistamine hydroxyzine in healthy volunteers, using a methodology similar to that adopted in several previous PET studies [7,8,12,25,31–33]. Brain H1RO values after administration of single oral doses of various other antihistamines have been reported previously: bepotastine 10 mg, 15–17% [12,33], cetirizine 20 mg, 26% [31], d-chlorpheniramine 2 mg, 50–77% [7,8,25], diphenhydramine 30–50 mg, 45–56% [12,33], ebastine 10 mg, 10% [25], fexofenadine 120 mg, −0.1% [12,33], hydroxyzine 30 mg, 68% [29], ketotifen 1 mg, 72% [32], loratadine 10 mg, 12% [8], olopatadine 5 mg, 15% [32] and terfenadine 60 mg, 17% [2].
These previous trials have clearly highlighted the rationale for full characterization of the sedative or non-sedative potential of antihistamines. Sedating antihistamines increase the likelihood of accidents while driving, flying planes or operating heavy machinery [2–4]. PET studies have pointed to the possibility of hangover sedation the day after evening administration of diphenhydramine as a sleep aid [33]. PET studies have also facilitated the definition of ‘relatively non-sedating’ antihistamines (e.g. cetirizine, loratadine) which are non-sedating at therapeutic doses, but may cause sedation when used at higher doses (e.g. in ‘over-compliant’ patients who take greater than recommended doses of antihistamines to try to relieve symptoms faster and to a greater degree) [1,3,4].
The current trial represents the first PET study to investigate brain H1RO by bilastine. Results showed statistically significant lower mean binding potential values in all brain ROIs and on average (total binding potential), with hydroxyzine 25 mg vs bilastine 20 mg, and also with hydroxyzine vs placebo. Total binding potential was 0.256 ± 0.081 for placebo, 0.260 ± 0.072 for bilastine 20 mg and 0.130 ± 0.067 for hydroxyzine 25 mg. No significant differences in total binding potential or in each individual ROI were found between bilastine and placebo.
Likewise, irrespective of treatment, there was consistency in the binding potential results. Under the conditions used in our study, the occipital region was the region with minimum binding potential values (indicating lower H1-receptor density), and the insula region was the region with the highest binding potential values (indicating higher H1-receptor density). We also observed that the frontal and parietal regions (particularly the frontal region) could possibly represent the total binding potential of the five ROIs, as mean binding potential values obtained in each individual region were close to the total binding potential. Regarding H1RO it can be summarized that lower H1RO in all brain regions and on average (total H1RO) was observed with bilastine compared with hydroxyzine. These differences were statistically significant (P < 0.01). Total H1RO was −3.92 ± 14.39% for bilastine 20 mg compared with 53.95 ± 14.13% for hydroxyzine 25 mg. In this study, it was also noted that the frontal region may be considered to be a representative region of the five brain ROIs assessed, as demonstrated by the similarity in the magnitude of binding potential data vs. mean total binding potential. It is also important to note that total H1RO for bilastine was less than zero. To date, the only other non-sedating antihistamine reported to have negative H1RO is fexofenadine [31]. It is interesting to speculate whether a new category of non-sedating antihistamines, with negative H1RO, can be proposed.
As shown in Figure 5, there was no significant linear relationship between individual values of the AUC(0,2.5h) of bilastine plasma concentrations and the mean total binding potential. A correlation coefficient (r2) of 0.0634 (P > 0.05, NS) was calculated. Nevertheless, a dose–response study would be the best approach to investigate the relationship between bilastine plasma concentrations and total binding potential. The mean total binding potential for placebo (0.2561) fits perfectly with the regression line for bilastine AUC(0,2.5h) values = 0 ng ml−1 h.
The PET assessments were performed between 1–2.5 h post-drug administration in order to obtain measures around the tmax. The results concur in the case of bilastine. A limitation of the study is that hydroxyzine blood concentrations were not determined and the value for tmax was based on previous studies. Bilastine plasma concentrations were lower than expected in one subject and the possibility of excluding his data from the final analysis was considered. Analyses were repeated, again showing low levels of bilastine, and, after considering the absence of correlation between bilastine plasma AUC(0,2.5h) and total binding potential, they were finally included, although we were aware that this would contribute to a greater dispersion of pharmacokinetic data. The reason for such low concentrations of bilastine in the subject is still uncertain, as the record card showed nothing relevant and it could not be explained due to vomiting or other circumstances during the experimental session. Low concentrations in this subject may relate to low bioavailability of bilastine, delayed peak plasma concentration or individual genetic variation in P-glycoprotein.
No significant differences were noted between test compounds with regard to VAS ratings of drowsiness, absent-mindedness and sleepiness or objective assessment of the number of correct responses on the DSST, probably due to inter-subject variability and limited sample size. Although the receptor occupancy of hydroxyzine was >50%, significant CNS effects were not observed which was an unexpected result for a sedating antihistamine with an overall proportional impairment ratio (PIR) of 2.43 [34]. One explanation for this observation is that the assay used in the current study was not the most appropriate for assessing CNS effects after administration of hydroxyzine. Moreover, the 2.5 h assessment was conducted after individuals had been lying down and quiet for a period of 90 min and this may have impacted on the observation of CNS effects. Most subjects sleep during the PET. Indeed, with a mean H1RO of 67.6%, hydroxyzine 30 mg was previously shown to have a significant effect, compared with placebo, on subjective sleepiness only at a single time point (2 h) but not at 2.5 or 3 h [29]. In another study, 12 h after administration, the mean H1RO for diphenhydramine 50 mg was significantly greater than that for bepotastine 10 mg (44.7% and 16.6%, respectively; P < 0.01), yet there were no significant between treatment differences in subjective sleepiness [33]. It is widely acknowledged that assessment of subjective sleepiness is affected by numerous internal and environmental factors and, in isolation, is not the most reliable method for assessing the sedative effect of antihistamines but is useful in real conditions.
The antihistamines in the current trial were well tolerated. Overall, 26 AEs were reported, and all were mild or moderate in intensity. Only eight AEs were considered probably or possibly related to antihistamine administration: headache (two cases after bilastine, one after hydroxyzine), diarrhoea (1, 0), vomiting (2, 0) and sedation (0, 2). No clinically significant changes in laboratory parameters or vital signs occurred during the trial.
Altogether, data indicated that bilastine 20 mg did not cross the blood–brain barrier and did not reach levels sufficient to compete with [11C]-doxepin binding at H1-receptors in any of the five ROIs. These results, highlighting a lack of CNS activity for bilastine, are corroborated by secondary end point safety data, i.e. bilastine was not associated with AEs of sleepiness or sedation considered possibly or probably related to treatment. Data from other studies have also indicated a lack of effect for bilastine on psychomotor performance or driving ability at twice the maximum recommended dosage [16,23].
In summary, brain H1RO by bilastine (after a 20 mg dose) was significantly lower than that by hydroxyzine (25 mg) in all five cerebral cortex ROIs, and overall. Bilastine was not associated with subjective sedation, or objective impairment of psychomotor performance, and was safe and well tolerated and devoid of treatment-related, sedative AEs. Thus, bilastine 20 mg satisfied relevant subjective, objective and PET criteria to be considered as a reliable non-sedating antihistamine.
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author). Research funding for the design and conduct of this study, collection, management, analysis and interpretation of the data was sponsored by FAES FARMA S.A. (Leioa, Bizkaia), Spain. LL and RV are employees of FAES FARMA S.A. (Leioa, Bizkaia), Spain. MF, CPM, EP, EM, MP, SM, SB, SR and JRH have been engaged by FAES FARMA S.A. as experts for various aspects of the studies (PET technique, pharmacokinetic and statistical methods). None of these authors received payments for their contributions to the manuscript.
All authors contributed significantly to the analysis and interpretation of data, were involved in drafting the manuscript or revising it critically for important intellectual content, and gave final approval of the version to be published. CP-M and EP are recipients of a Rio Hortega fellowships (ISCIII-FIS, CM12/00085 and CM13/00016, respectively).
We thank David P. Figgitt PhD, Content Ed Net, for editorial assistance in the preparation of this manuscript. Funding for editorial assistance was provided by FAES FARMA S.A.
Authors' contributions
Conceived and designed the experiments: MF, SB, SR, J-RH, RV.
Performed the experiments: MF, CP-M, EP, EM, MP, SM.
Performed PET scans: SB, SR, CT, J-RH.
Analyzed data: CP-M, SB.
Wrote the paper: MF, CP-M, SB, LL, RV.
References
- 1.Holgate ST, Canonica GW, Simons FE, Taglialatela M, Tharp M, Timmerman H, Yanai K. Consensus Group on New-Generation Antihistamines (CONGA): present status and recommendations. Clin Exp Allergy. 2003;33:1305–1324. doi: 10.1046/j.1365-2222.2003.01769.x. [DOI] [PubMed] [Google Scholar]
- 2.Yanai K, Zhang D, Tashiro M, Yoshikawa T, Naganuma F, Harada R, Nakamura T, Shibuya K, Okamura N. Positron emission tomography evaluation of sedative properties of antihistamines. Expert Opin Drug Saf. 2011;10:613–622. doi: 10.1517/14740338.2011.562889. [DOI] [PubMed] [Google Scholar]
- 3.Yanai K, Tashiro M. The physiological and pathophysiological roles of neuronal histamine: an insight from human positron emission tomography studies. Pharmacol Ther. 2007;113:1–15. doi: 10.1016/j.pharmthera.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 4.Yanai K, Rogala B, Chugh K, Paraskakis E, Pampura AN, Boev R. Safety considerations in the management of allergic diseases: focus on antihistamines. Curr Med Res Opin. 2012;28:623–642. doi: 10.1185/03007995.2012.672405. [DOI] [PubMed] [Google Scholar]
- 5.Zhao R, Kalvass JC, Yanni SB, Bridges AS, Pollack GM. Fexofenadine brain exposure and the influence of blood–brain barrier P-glycoprotein after fexofenadine and terfenadine administration. Drug Metab Dispos. 2009;37:529–535. doi: 10.1124/dmd.107.019893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rapoport SI. Modulation of blood–brain barrier permeability. J Drug Target. 1996;3:417–425. doi: 10.3109/10611869609015962. [DOI] [PubMed] [Google Scholar]
- 7.Yanai K, Ryu JH, Watanabe T, Iwata R, Ido T, Sawai Y, Ito K, Itoh M. Histamine H1-receptor occupancy in human brains after single oral doses of histamine H1 antagonists measured by positron emission tomography. Br J Pharmacol. 1995;116:1649–1655. doi: 10.1111/j.1476-5381.1995.tb16386.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kubo N, Senda M, Ohsumi Y, Sakamoto S, Matsumoto K, Tashiro M, Okamura N, Yanai K. Brain histamine H1 receptor occupancy of loratadine measured by positron emission topography: comparison of H1 receptor occupancy and proportional impairment ratio. Hum Psychopharmacol. 2011;26:133–139. doi: 10.1002/hup.1184. [DOI] [PubMed] [Google Scholar]
- 9.Logan J, Fowler JS, Volkow ND, Wang GJ, Ding YS, Alexoff DL. Distribution volume ratios without blood sampling from graphical analysis of PET data. J Cereb Blood Flow Metab. 1996;16:834–840. doi: 10.1097/00004647-199609000-00008. [DOI] [PubMed] [Google Scholar]
- 10.Suzuki A, Tashiro M, Kimura Y, Mochizuki H, Ishii K, Watabe H, Yanai K, Ishiwata K. Use of reference tissue models for quantification of histamine H1 receptors in human brain by using positron emission tomography and [11C]doxepin. Ann Nucl Med. 2005;19:425–433. doi: 10.1007/BF02985569. [DOI] [PubMed] [Google Scholar]
- 11.Mochizuki H, Sadato N, Saito DN, Toyoda H, Tashiro M, Okamura N, Yanai K. Neural correlates of perceptual difference between itching and pain: a human fMRI study. Neuroimage. 2007;36:706–717. doi: 10.1016/j.neuroimage.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 12.Tashiro M, Duan X, Kato M, Miyake M, Watanuki S, Ishikawa Y, Funaki Y, Iwata R, Itoh M, Yanai K. Brain histamine H1-receptor occupancy of orally administered antihistamines, bepotastine and diphenhydramine, measured by PET with 11C-doxepin. Br J Clin Pharmacol. 2008;65:811–821. doi: 10.1111/j.1365-2125.2008.03143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Medicines Compendium UK. Summary of Product Characteristics. Ilaxten 20 mg tablets. Available at http://www.medicines.org.uk/EMC/medicine/24461/SPC/Ilaxten+20+mg+tablets/ (last accessed 16 October 2013)
- 14.Corcostegui R, Labeaga L, Innerarity A, Berisa A, Orjales A. Preclinical pharmacology of bilastine, a new selective histamine H1 receptor antagonist: receptor selectivity and in vitro antihistaminic activity. Drugs R D. 2005;6:371–384. doi: 10.2165/00126839-200506060-00005. [DOI] [PubMed] [Google Scholar]
- 15.Corcostegui R, Labeaga L, Innerarity A, Berisa A, Orjales A. In vivo pharmacological characterisation of bilastine, a potent and selective histamine H1 receptor antagonist. Drugs R D. 2006;7:219–231. doi: 10.2165/00126839-200607040-00002. [DOI] [PubMed] [Google Scholar]
- 16.Conen S, Theunissen EL, Van Oers AC, Valiente R, Ramaekers JG. Acute and subchronic effects of bilastine (20 and 40 mg) and hydroxyzine (50 mg) on actual driving performance in healthy volunteers. J Psychopharmacol. 2011;25:1517–1523. doi: 10.1177/0269881110382467. [DOI] [PubMed] [Google Scholar]
- 17.Lucero ML, Gonzalo A, Ganza A, Leal N, Soengas I, Ioja E, Gedey S, Jahic M, Bednarczyk D. Interactions of bilastine, a new oral H1 antihistamine, with human transporter systems. Drug Chem Toxicol. 2012;35(Suppl. 1):8–17. doi: 10.3109/01480545.2012.682653. [DOI] [PubMed] [Google Scholar]
- 18.Hindmarch I. Psychomotor function and psychoactive drugs. Br J Clin Pharmacol. 1980;10:189–209. doi: 10.1111/j.1365-2125.1980.tb01745.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wechsler D. Manual for the Wechsler Adult Intelligence Scale-Revised. New York: The Psychological Corporation; 1981. [Google Scholar]
- 20.Abanades S, Farré M, Barral D, Torrens M, Closas N, Langohr K, Pastor A, de la Torre R. Relative abuse liability of gamma-hydroxybutyric acid, flunitrazepam and ethanol in club drug users. J Clin Psychopharmacol. 2007;27:625–638. doi: 10.1097/jcp.0b013e31815a2542. [DOI] [PubMed] [Google Scholar]
- 21.Farré M, Terán MT, Camí J. A comparison of the acute behavioral effects of flunitrazepam and triazolam in healthy volunteers. Psychopharmacology (Berl) 1996;125:1–12. doi: 10.1007/BF02247387. [DOI] [PubMed] [Google Scholar]
- 22.Verster JC, Roth T. Predicting psychopharmacological drug effects on actual driving performance (SDLP) from psychometric tests measuring driving-related skills. Psychopharmacology (Berl) 2012;220:293–301. doi: 10.1007/s00213-011-2484-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garcia-Gea C, Martinez-Colomer J, Antonijoan RM, Valiente R, Barbanoj MJ. Comparison of peripheral and central effects of single and repeated oral dose administrations of bilastine, a new H1 antihistamine: a dose-range study in healthy volunteers with hydroxyzine and placebo as control treatments. J Clin Psychopharmacol. 2008;28:675–685. doi: 10.1097/JCP.0b013e31818b2091. [DOI] [PubMed] [Google Scholar]
- 24.García-Gea C, Martínez J, Ballester MR, Gich I, Valiente R, Antonijoan RM. Psychomotor and subjective effects of bilastine, hydroxyzine, and cetirizine, in combination with alcohol: a randomized, double-blind, crossover, and positive-controlled and placebo-controlled Phase I clinical trials. Hum Psychopharmacol. 2014;29:120–132. doi: 10.1002/hup.2378. [DOI] [PubMed] [Google Scholar]
- 25.Tagawa M, Kano M, Okamura N, Higuchi M, Matsuda M, Mizuki Y, Arai H, Iwata R, Fujii T, Komemushi S, Ido T, Itoh M, Sasaki H, Watanabe T, Yanai K. Neuroimaging of histamine H1-receptor occupancy in human brain by positron emission tomography (PET): a comparative study of ebastine, a second generation antihistamine, and (+)-chlorpheniramine, a classical antihistamine. Br J Clin Pharmacol. 2001;52:501–509. doi: 10.1046/j.1365-2125.2001.01471.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bachert C, Kuna P, Sanquer F, Ivan P, Dimitrov V, Gorina MM, van de Heyning P, Loureiro A Bilastine International Working Group. Comparison of the efficacy and safety of bilastine 20 mg vs. desloratadine 5 mg in seasonal allergic rhinitis patients. Allergy. 2009;64:158–165. doi: 10.1111/j.1398-9995.2008.01813.x. [DOI] [PubMed] [Google Scholar]
- 27.Jauregizar N, de la Fuente L, Lucero ML, Sologuren A, Leal N, Rodríguez M. Pharmacokinetic–pharmacodynamic modelling of the antihistaminic (H1) effect of bilastine. Clin Pharmacokinet. 2009;48:543–554. doi: 10.2165/11317180-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 28.Bartra J, Valero AL, del Cuvillo A, Dávila I, Jáuregui I, Montoro J, Mullol J, Sastre J. Interactions of the H1 antihistamines. J Investig Allergol Clin Immunol. 2006;16(Suppl. 1):29–36. [PubMed] [Google Scholar]
- 29.Tashiro M, Kato M, Miyake M, Watanuki S, Funaki Y, Ishikawa Y, Iwata R, Yanai K. Dose dependency of brain histamine H(1) receptor occupancy following oral administration of cetirizine hydrochloride measured using PET with [11C]doxepin. Hum Psychopharmacol. 2009;24:540–548. doi: 10.1002/hup.1051. [DOI] [PubMed] [Google Scholar]
- 30.Okamura N, Yanai K, Higuchi M, Sakai J, Iwata R, Ido T, Sasaki H, Watanabe T, Itoh M. Functional neuroimaging of cognition impaired by a classical antihistamine, d-chlorpheniramine. Br J Pharmacol. 2000;129:115–123. doi: 10.1038/sj.bjp.0702994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tashiro M, Sakurada Y, Iwabuchi K, Mochizuki H, Kato M, Aoki M, Funaki Y, Itoh M, Iwata R, Wong DF, Yanai K. Central effects of fexofenadine and cetirizine: measurement of psychomotor performance, subjective sleepiness, and brain histamine H1-receptor occupancy using 11C-doxepin positron emission tomography. J Clin Pharmacol. 2004;44:890–900. doi: 10.1177/0091270004267590. [DOI] [PubMed] [Google Scholar]
- 32.Tashiro M, Mochizuki H, Sakurada Y, Ishii K, Oda K, Kimura Y, Sasaki T, Ishiwata K, Yanai K. Brain histamine H1-receptor occupancy of orally administered antihistamines measured by positron emission tomography with [11C]-doxepin in a placebo-controlled crossover study design in healthy subjects: a comparison of olopatadine and ketotifen. Br J Clin Pharmacol. 2006;61:16–26. doi: 10.1111/j.1365-2125.2005.02514.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang D, Tashiro M, Shibuya K, Okamura N, Funaki Y, Yoshikawa T, Kato M, Yanai K. Next-day residual sedative effect after night time administration of an over-the-counter antihistamine sleep aid, diphenhydramine, measured by positron emission tomography. J Clin Psychopharmacol. 2010;30:694–701. doi: 10.1097/jcp.0b013e3181fa8526. [DOI] [PubMed] [Google Scholar]
- 34.Shamsi Z, Hindmarch I. Sedation and antihistamines: a review of inter-drug differences using proportional impairment ratios. Hum Psychopharmacol. 2000;15(Suppl. 1):S3–S30. doi: 10.1002/1099-1077(200010)15:1+<::AID-HUP247>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]