

Abstract
Aims
Variability in responsiveness to clopidogrel is a clinical problem in secondary prevention after cerebral ischaemia which has been suggested to be linked to competitive metabolization of clopidogrel and cytochrome P450 (CYP) 3A4-oxidated statins such as simvastatin. We assessed the hypothesis that simvastatin, in contrast to CYP 2C9-metabolized fluvastatin, reduces clopidogrel-mediated platelet inhibition.
Methods
We performed a randomized, double-blind, double-dummy, two period crossover study in 13 patients with cerebral ischaemia (8F, 5 M), aged 64.1 ± 8.0 years (mean ± SD). After a 14 day period in which all patients received 75 mg clopidogrel day−1, patients additionally received either 20 mg simvastatin day−1 or 80 mg fluvastatin day−1 for 14 days. Regimens were crossed over after a 14 day wash-out period and switched regimens were continued for another 14 days. Platelet aggregation, clopidogrel active metabolite (CAM) plasma concentrations and routine laboratory parameters including prothrombin time (PT) Quick percent value were assessed at baseline and following each treatment phase.
Results
Clopidogrel reduced platelet aggregation in all patients as expected. Platelet aggregation and CAM plasma concentrations were unaltered when simvastatin or fluvastatin was added to clopidogrel. Simvastatin decreased PT Quick percent value (decrease from 109 ± 10.5% to 103 ± 11%, P < 0.05) when combined with clopidogrel but there was no such change following treatment with fluvastatin and clopidogrel.
Conclusions
Our data indicate that treatment with CYP 3A4-metabolized simvastatin does not jeopardize clopidogrel-mediated inhibition of platelet aggregation. After co-administration of simvastatin and clopidogrel we observed a decrease in the PT Quick percent value which could be due to simvastatin-induced reduction of activity of prothrombin fragment 1 + 2.
Keywords: clopidogrel, CYP 3A4, fluvastatin, platelet aggregation, simvastatin
What is already known about this subject
Ex vivo studies demonstrated that both clopidogrel and simvastin are competitively metabolized by cytochrome P450 (CYP) 3A4 suggesting that variability in responsiveness to clopidogrel can be caused by co-treatment with CYP3A4 metabolized statins.
Non-randomized clinical studies and post hoc analyses of CYP3A4-based statin–clopidogrel interactions showed conflicting results due to heterogeneous study designs.
To date, the existence of clinically relevant CYP3A4-based in vivo interaction remains unclear.
What this study adds
This is the first double-blind randomized crossover study demonstrating that CYP3A4 metabolized simvastatin does not alter clopidogrel mediated platelet inhibition in vivo.
This study may form the basis for follow-up randomized controlled trials to investigate whether there are relevant interactions between clopidogrel and other CYP3A4 metabolized statins.
In this study, we observed decrease in PT Quick percent value when simvastatin but not fluvastatin was added to treatment with clopidogrel, possibly due to simvastatin mediated attenuation of activity of prothrombin fragment 1 + 2 and plasminogen activator inhibitor-1. However the clinical relevance of this finding remains unclear.
Introduction
Platelet aggregation inhibitors such as clopidogrel and HMG-CoA reductase inhibitors (statins) are considered gold standard for secondary prevention after ischaemic stroke [1,2]. However, inter-individual differences in responsiveness to clopidogrel can jeopardize the success of this preventive treatment and are considered a clinical problem of emerging relevance [3]. Among numerous mechanisms that have been speculated to play a role in this phenomenon, clopidogrel–statin interactions are extensively discussed. In particular, competitive metabolization of statins, such as simvastatin and clopidogrel, by hepatic cytochrome P450 (CYP) 3A4 has been identified as a possible pharmacokinetic mechanism of interaction [4]. Clopidogrel is a prodrug which is predominantly converted into the active thiol form, clopidogrel active metabolite (CAM), through oxidation by CYP3A4. CAM forms a disulfide bond on the platelet P2Y12 receptor and thus abolishes the ability of platelets to bind adenosine 5′-diphosphate (ADP). In consequence, the affected platelet is not able to undergo the necessary changes in conformation to bind fibrinogen and thus induce aggregation with other platelets. The CYP3A4 mediated conversion of clopidogrel into its active metabolite is therefore a prerequisite to the unfolding of the platelet inhibiting effect of clopidogrel. Similarly to clopidogrel, some statins (such as simvastatin) are also metabolized by CYP3A4, whereas other statins (such as fluvastatin) are predominantly metabolized by CYP2C9 [5]. A decrease in CYP3A4 mediated biotransformation of clopidogrel into its active metabolite through simultaneous administration of CYP3A4-metabolized statins has therefore been previously suggested to reduce the platelet aggregation inhibitory effect of clopidogrel [6]. However, clinical studies on clopidogrel–statin interactions showed conflicting results which may be due to heterogeneous study designs [4]. In this double-blind randomized crossover study we aimed to investigate the hypothesis that combined administration of clopidogrel with simvastatin but not fluvastatin attenuates clopidogrel active metabolite induced platelet inhibition.
Methods
Subjects and ethics
Approval from the Dresden University of Technology Institutional Review Board was obtained and full written informed consent was given by each subject. The study was registered in the World Health Organization Clinical Trials Registry Platform (Study ID EudraCT: 2006-000754-40). Fifteen patients (seven males, eight females) aged 61.7 ± 9.8 years (mean ± SD) with transitory ischaemic attack (TIA) or first acute ischaemic stroke were screened at the Carl Gustav University Hospital Stroke Center. Two of these patients were excluded due to complications in the clinical course 1) necessity of operative cranial decompression following progressive oedema due to cerebellar ischaemic stroke and 2) intracerebral bleeding prior to the beginning of the study. Thirteen patients (five males, eight females) aged 64.1 ± 8.0 years were then included in the study (Table 1) Exclusion criteria were neurological deficits which equalled a National Institutes of Health Stroke Scale (NIHSS) score of more than 4 points, pre-treatment with acetylsalicylic acid, oral anticoagulants or lipid-lowering agents within 6 weeks prior to the beginning of the study, or preceding thrombolysis for acute stroke. Furthermore we excluded patients with dementia, malignant systemic disease, known liver damage, persistent elevation of liver enzymes, previous or current abuse of drugs, medications or alcohol, pregnancy or lactation. No female patients on hormonal replacement therapy were included.
Table 1.
Demographic table with basic characteristics
| Patient | Gender | Group | Age (years) | p2y12 34C > T | p2y12 52G > T | Diagnosis |
|---|---|---|---|---|---|---|
| 1 | M | S-F | 59 | mut/wt | wt/wt | Ischaemic infarction in the posterior cerebral artery territory |
| 2 | M | S-F | 58 | mut/wt | wt/wt | Ischaemic infarction in the middle cerebral artery territory |
| 3 | F | S-F | 70 | mut/wt | mut/wt | Ischaemic infarction in the middle cerebral artery territory |
| 4 | M | S-F | 51 | mut/wt | wt/wt | Ischaemic infarction in the middle cerebral artery territory |
| 5 | F | S-F | 66 | wt/wt | wt/wt | Ischaemic infarction in the middle cerebral artery territory |
| 6 | F | S-F | 65 | mut/mut | wt/wt | Ischaemic brain stem infarction |
| 7 | F | F-S | 60 | wt/wt | wt/wt | Transitory ischaemic attack |
| 8 | F | F-S | 77 | mut/wt | wt/wt | Ischaemic infarction in the posterior cerebral artery territory |
| 9 | F | F-S | 63 | wt/wt | wt/wt | Ischaemic infarction in the middle cerebral artery territory |
| 10 | M | F-S | 54 | mut/wt | wt/wt | Ischaemic infarction in the middle cerebral artery territory |
| 11 | M | F-S | 65 | mut/wt | wt/wt | Ischaemic infarction in the middle cerebral artery territory |
| 12 | F | F-S | 67 | mut/mut | wt/wt | Ischaemic infarction in the middle cerebral artery territory |
| 13 | F | F-S | 78 | mut/wt | wt/wt | Ischaemic brain stem infarction |
F, female; F-S, fluvastatin-simvastatin; M, male; mut, mutated; S-F, simvastatin-fluvastatin; wt, wild type.
Testing protocol
Included patients (n = 13) were randomly assigned to two arms of a double-blind crossover study. In the first phase of this study patients of both study arms orally received 75 mg clopidogrel day−1 for a period of 14 days (run-in phase, Figure 1). Following the run-in phase, patients of one study arm (n = 6, three males and three females) orally received 20 mg simvastatin day−1 in addition to 75 mg clopidogrel day−1 for 14 days (active phase 1). During this period, patients of the other study arm (n = 7, two males and five females) received 80 mg fluvastatin day−1 in addition to clopidiogrel. The treatment regimens were crossed over after a 14 day wash-out period during which all patients received 75 mg clopidogrel day−1 alone (wash-out phase). Switched regimens were then continued for another 13 days (active phase 2). In each patient platelet aggregation assessment and laboratory tests were performed at the beginning of the study (baseline, day 1) as well as before each regimen change (days 15, 29 and 43) and following completion of the study (day 57). Research physicians and patients were blinded for the number-coded study medication using a double-dummy design where both treatment arms received placebo during active phases 1 and 2 in addition to clopidogrel and simvastatin, or clopidogrel and fluvastatin respectively. In the clopidogrel and simvastatin phases, patients received additional placebo looking and tasting identical to fluvastatin whereas in the clopidogel and fluvastatin phases, additional placebo looked and tasted identical to simvastatin. Safety was assessed by interviews and physical examinations and additional laboratory testing at each visit. Compliance was assessed through interviews and validation of the returned empty study medication containers at each visit before handing out the medication for each phase of the study.
Figure 1.
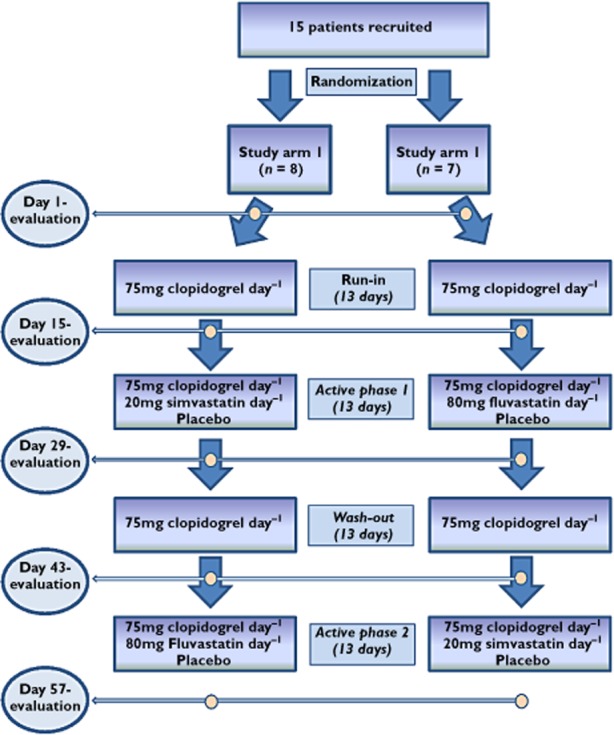
The flow chart illustrates each phase of this randomized, double-blind, double-dummy, two period crossover-study. The yellow circles indicate the time points of evaluation
Blood sampling
Blood samples were obtained on days 1, 15, 29 and 57. Blood samples were anticoagulated with one tenth volume 0.109 mol l−1 buffered trisodium citrate in vacutainer plastic tubes. All tests were performed within 2 h after blood sampling.
Platelet aggregation assessment
Platelet aggregation was the primary outcome parameter in this study. Platelet-rich plasma (PRP, 250 GPt l−1) was obtained by centrifuging trisodium citrate buffered blood samples for 10 min. Following removal of PRP, platelet-poor plasma (PPP) was further centrifuged for 3 min. Aggregation assessment was performed by using an APACT-4 aggregometer (LABiTec, Ahrensburg, Germany) in 250 μl minicuvettes stirred at 1000 rev min–1 at 37°C. The 100% transmission value was determined using platelet-poor plasma. The 0% transmission baseline was established with PRP (adjusted from 200 × 103 μl−1 up to 300 × 103 μl−1). Adenosine diphosphate (ADP, 4 μmol l−1) was used as agonist. After 10 min, the maximal percentage of aggregation was recorded [7]. In order to compare platelet aggregation assessment with an alternative established technique, platelet aggregation was additionally assessed using the PFA-100® device (Siemens, Eschborn, Germany) as a secondary outcome parameter. This device was used to induce a high-shear environment in which the ability of platelets to occlude an aperture in a membrane was measured. Therefore, trisodium citrate buffered blood was aspirated with controlled flow intensity through the aperture in a membrane which was laminated with collagen/ADP. The device measured the duration from the beginning of aspiration to occlusion of the aperture through platelet adhesion and aggregation (clotting time, s). The reference range was 67 to 120 s [8].
Clopidogrel active metabolite assessment
Serum concentrations of CAM were measured to assess quantitatively the effects of simvastatin or fluvastatin co-administration on the CYP-3A4-related biotransformation of clopidogrel as a secondary outcome parameter in this study. Quantification of CAM plasma concentrations was performed using liquid chromatography-tandem mass spectrometry as previously described [9,10]. Briefly, CAM requires stabilization in biological samples due to its thiol group. Therefore an alkylating reagent (2-bromo-3′-methoxyacetophenone) was used to stabilize CAM in blood while an analogue of the derivatived CAM was used as the internal standard (IS). The CAM derivative and IS were then separated on a C18 high-performance liquid chromatography (ODS) column and quantified by tandem mass spectrometry with electrospray ionization.
P2Y12 genotype assessment
The occurrence of the polymorphisms 34C>T and 52G>T of the platelet P2Y12 receptor were assessed as a secondary outcome to investigate whether results of platelet aggregation measures following clopidogrel application might be biased by genetic inter-individual differences [11]. Whole blood samples were used to isolate genomic DNA following standard procedures. The polymorphisms 34C>T and 52G>T of the P2Y12 gene were determined using a primer-introduced restriction analysis-polymerase chain reaction assay (PIRA-PCR assay) as previously described [12]. As the restriction enzyme for 34C>T, MboI was used, whereas SmaI enzyme was used for 52G>T.
Additional laboratory testing
On days 15, 29, 43 and 57 serum concentrations of cholesterol, low density lipoprotein-C (LDL-C), high density lipoprotein-C (HDL-C), triglycerides, C-reactive protein (CRP), creatinine and bilirubin as well as haemogram were assessed. Prothrombin time (PT) was measured on the same evaluation days and the PT Quick percent value was calculated. Also, serum concentrations of liver enzymes alanine transaminase (ALAT), aspartate transaminase (ASAT) and γ-glutamyl transpeptidase (γ-GT) were measured to detect any hepatotoxic adverse events on the same days.
Statistical analysis
All statistical tests were performed using SPSS (IBM, New York, NY, USA).
Data are presented as mean ± standard deviation (SD). In addition, 95% confidence intervals are given for significant differences in primary outcome parameters. Figures are presented as mean ± standard error of the mean (SEM). Statistical analyses were performed in the intention-to-treat population (n = 13). A linear model with three correlated factors (study arm, study phase and medication) was applied to compare the changes in the primary outcome measure platelet aggregation before and after treatment with simvastatin or fluvastatin and to determine the relative importance of carry-over and order effects on the effect size. Paired t-tests were used to compare changes in secondary outcome measures.
Results
Platelet aggegration assessment
As expected, ADP-induced platelet aggregation was decreased in all patients following clopidogrel administration (72.6 ± 69.2% at baseline vs. 24.5 ± 14.2% after run-in phase, P < 0.05, 95% confidence interval for the difference 0.3, 100.1). When simvastatin or fluvastatin was added to treatment with clopidogrel, ADP-induced platelet aggregation was found unaltered (Figure 2). There was no difference in clotting time as measured by PFA-100® following co-treatment with simvastatin or fluvastatin (104.8 ± 21.9 s pre-simvastatin vs. 102.5 ± 28.4 s post-simvastatin, P = NS and 131 ± 77.1 s pre-fluvastatin vs. 117.0 ± 59.0 s post-fluvastatin, P = NS).
Figure 2.
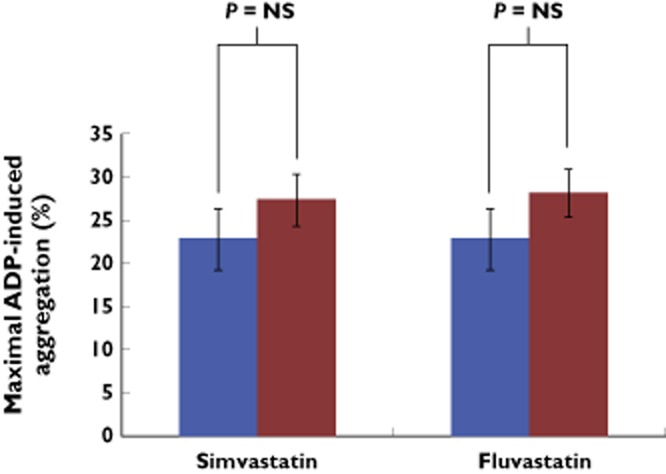
The bar graphs illustrate that aggregation induced by ADP (4 μmol l−1) was unchanged before (blue bars) and after (red bars) administration of simvastatin or fluvastatin in addition to clopidogrel.  , pre-application;
, pre-application;  , post-application
, post-application
Clopidogrel active metabolite
Plasma concentrations of CAM were unaltered when simvastatin or fluvastatin was added to treatment with clopidogrel. (0.56 ± 0.73 ng ml−1 pre-simvastatin vs. 0.36 ± 0.43 ng ml−1 post-simvastatin, P = NS and 0.52 ± 0.84 ng ml−1 pre-fluvastatin vs. 0.36 ± 0.57 ng ml−1 post-fluvastatin, P = NS).
Additional laboratory testing
Cholesterol serum concentrations were decreased (as expected) in all patients following active phases of simvastatin or fluvastatin application compared with baseline: 5.8 ± 1.2 mmol l−1 at baseline vs. 4.1 ± 0.65 mmol l−1 post-active phase 1, P < 0.05 and vs. 4.21 ± 0.74 mmol l−1 post-active phase 2 P = 0.01) whereas there was no such change following the clopidogrel run-in phase (5.69 ± 0.97 mmol l−1, P = NS compared with baseline) and following the wash-out phase (5.94 ± 1 mmol l−1, P = NS compared with baseline). Also LDL-C concentrations were decreased following simvastatin or fluvastatin application compared with baseline (4.09 ± 0.28 mmol l−1 baseline vs. 2.38 ± 0.53 mmol l−1 post-active phase 1, P < 0.001; and vs. post-active phase 2.42 ± 0.5 mmol l−1) whereas LDL-C was unchanged following the run-in phase (3.84 ± 0.86 mmol l−1, P = NS compared with baseline) and following the wash-out phase (4.02 ± 0.87 mmol l−1, P = NS compared with baseline). Haemogram and serum concentration of CRP, cholesterol, LDL-C, HDL-C, triglycerides, creatinine and bilirubin were unaltered when simvastatin or fluvastatin was added to treatment with clopidogrel (data not shown). Simvastatin decreased PT Quick percent value when combined with clopidogrel but there was no such change noted following treatment with fluvastatin and clopidogrel (Figure 3). Liver enzymes (ALAT, ASAT and γ-GT) were neither significantly influenced by co-treatment with simvastatin nor fluvastatin (data not shown).
Figure 3.
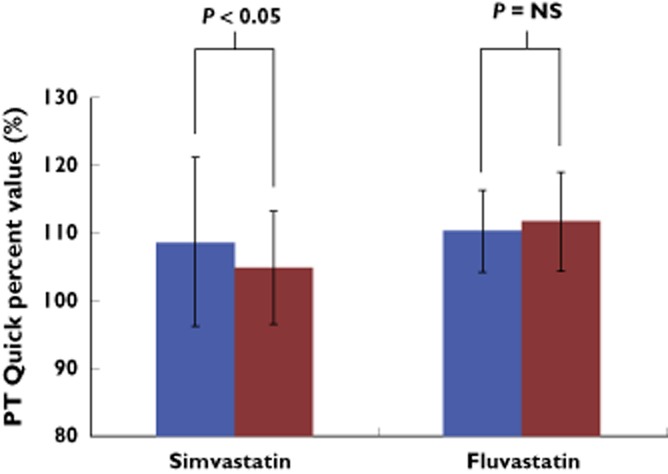
As shown by these bar graphs, PT Quick percent value was decreased post-simvastatin application compared with pre-simvastatin application (left bars) but was unchanged when fluvastatin was added to treatment with clopidogrel (right bars).  , pre-application;
, pre-application;  , post-application
, post-application
DNA analysis
In our study population genotype frequencies for mutated, heterozygous and wildtype alleles for the 34C>T and 52 > G P2Y12 polymorphisms were 15.4% (n = 2), 61.5% (n = 8) and 23.1% (n = 3) and for the 52 > G P2Y12 polymorphism 0% (n = 0), 7.7% (n = 1) and 62.3 % (n = 12), respectively.
Compliance and adverse events
We did not note any malcompliance. Adverse events of mild to moderate severity were noted in both study arms. In study arm 1 (simvastatin-fluvastatin), one patient developed generalized pruritus. In study arm 2 (fluvastatin-simvastatin), one patient reported nose bleeding and one patient reported feeling of abdominal fullness. Another patient in study arm 2 reported general malaise following change of anti-hypertensive medication. All adverse events were transient.
Discussion
The major findings of this study were that 1) neither simvastatin nor fluvastatin altered clopidogrel mediated inhibition of platelet aggregation, 2) serum concentrations of clopidogrel active metabolite were unchanged when either simvastatin or fluvastatin was added to treatment with clopidogrel and 3) PT Quick percent value was decreased when simvastatin was added to treatment with clopidogrel. Taken together, these data suggest that there is no CYP-3A4 mediated interaction between simvastatin and clopidogrel.
The influence of cytochrome P450 isoenzyme activities on the conversion of clopidogrel to its active metabolite has been previously studied with conflicting results. An in vitro study of atorvastatin which is, similarly to simvastatin, a substrate of CYP3A4 found that atorvastatin inhibited the CYP3A4 dependent clopidogrel oxidation and thus decreased the conversion into its active thiol form in a time course and dose dependency manner [13]. The dynamics of this inhibition were characterized by the features of classical competitive inhibition and the results of this study therefore indicated that combined treatment with atorvastatin might reduce the platelet inhibitory effect of clopidogrel through competitive CYP3A4-metabolization. The ex vivo models used in this study were presumed to be applicable to human metabolism but the data were limited by the absence of any in vivo studies. In line with this ex vivo investigation, an open label study by Lau et al. in 44 patients undergoing elective coronary artery stent implantation found that co-administration of 40 mg pravastatin day−1 had no effect on clopidogrel mediated platelet inhibition whereas atorvastatin at doses of 10, 20 and 40 mg day−1 reduced the antiplatelet activity of clopidogrel in a dose-dependent manner [6]. The authors concluded that co-administration with CYP-3A4 metabolized statins, such as atorvastatin, but not pravastatin (which is excreted largely unchanged), reduced the platelet inhibitory effect of clopidogrel. However, in this study no clinical end points were measured and there was no placebo control. Furthermore, the platelet aggregation was measured in this study using plateletworks, a platelet function test which indirectly measures platelet aggregation by quantitative measurement of objects exceeding pre-defined threshold platelet size. This technique is more susceptible to artefacts than platelet aggregometry and therefore potentially overestimates platelet aggregation [14]. In contrast to this study our data showed no influence of CYP 3A4-metabolized simvastatin on platelet aggregation in clopidogrel treated patients. This discrepancy might be explained by differences in design and methodology between both studies. In contrast to the investigation by Lau et al. we applied a randomized, double-blind, double-dummy study design and measured platelet aggregation with two separate validated techniques to minimize methodological bias. Also we excluded the possibility of inter-individual differences by using a crossover design where each patient was subsequently allocated to both treatment regimens (clopidogrel with simvastatin and clopidogrel with CYP2C19-metabolized fluvastatin). In accordance with the results of our study, a retrospective analysis of the Plavix Reduction of New Thrombus Occurrence (PRONTO) study indicated that the administration of statins does not jeopardize the antiplatelet activity of clopidogrel [15]. The results of this investigation were, however, limited by the use of non-randomized data and by the inclusion of both CYP3A4 metabolized statins and statins which have other predominant metabolization pathways. Another post hoc analysis of a multicentre trial in 2116 patients with symptomatic coronary artery disease who were randomly assigned to receive either clopidogrel as a 300 mg loading dose or placebo 3–24 h prior to percutaneous coronary intervention did not find any differences in the occurrence of death, myocardial infarction or stroke between patients receiving statins that are extensively metabolized by CYP34A compared with those that are not [16]. These data indicated that there was no effect on clinical outcome of simultaneous administration of clopidogrel and CYP3A4 metabolized statins. However, in this study patients were not randomized to either receive CYP3A4 metabolized statins or statins with other metabolization pathways, thus leaving open the possibility of selection bias. Similarly, another study longitudinally investigated the potential interaction between clopidogrel and CYP3A4-metabolized statins on clinical outcome in 1651 patients with acute coronary syndrome who received 1) neither clopidogrel nor statin therapy, 2) clopidogrel alone, 3) CYP3A4 metabolized statin alone or 4) CYP3A4 metabolized statin and clopidogrel. Rates of cardiovascular events and death at 6 months were unaltered among the four groups and the authors concluded that previous ex vivo observations of CYP3A4 mediated competitive metabolization are not clinically relevant in simultaneous treatment with clopidogrel and statins. The study was limited by non-randomized allocation into the treatment groups based on discharge notes [17]. However the strength of this study was the comparatively large sample size, and its results, viewed in counjuction with our findings, lend support to the conclusion that there is no clinically relevant negative CYP3A4 based interaction between statins and clopidogrel. Another prospective investigation in 75 patients who received acetylsalicylic acid 325 mg day−1 and clopidogrel 300 mg immediately before coronary stent placement found no difference in clopidogrel mediated platelet inhibition between patients pre-treated with atorvastatin, patients pre-treated with any other statins and patients who were not receiving any statins [18]. However, the ‘other statins group’ included both CYP3A4-metabolized statins and non-CYP3A4-metabolized statins, thus diminishing the internal comparability of the data. In addition, this study was limited by the absence of randomization and placebo control. Therefore it remains not fully elucidated to what extent inter-individual differences in responsiveness to clopidogrel have contributed to the results of this study. However, these results are in concordance with our finding of unchanged platelet aggregation following administration of simvastatin or fluvastatin in addition to clopidogrel. Additionaly, in our study, we did not note any differences in serum concentrations of clopidogrel active metabolite between additional simvastatin and additional fluvastatin treatment, suggesting that the conversion of clopidogrel into its active thiol form is not influenced to a quantitatively relevant degree by competitive CYP 3A4-metabolization of simvastatin.
In addition to previously postulated CYP 3A4 based clopidogrel-statin interactions, the 34C>T and 52G>T polymorphisms of the platelet P2Y12 receptor have been linked to inter-individual differences in responsiveness to clopidogrel [19]. In our study population we found frequencies of 34C>T and 52G>T polymorphisms which corresponded with previously described genetic distribution [10]. Even though the presence of P2Y12 receptor polymorphism might contribute to some of the inconsistencies in the findings in the aforementioned previous studies of clopidogrel−statin interactions, in our study the applied crossover design excludes the possibility of P2Y12 receptor polymorphism related group differences as each patient was subsequently allocated to both treatment arms and thus served as his or her own control. Through this crossover design, group differences due to the slight different gender distribution among study arms are also avoided in the comparison of the two applied treatment regimens.
Additional laboratory testing revealed that simvastatin but not fluvastatin attenuated PT Quick percent values. This observation might be explained by simvastatin-induced reduction of activity of prothrombin fragment 1 + 2 and plasminogen activator inhibitor-1 with consecutive attenuation of prothrombinase activity [20]. However, it remains unclear to what extent this influence of simvastatin on haemostatic and fibrinolytic regulation has clinical relevance in secondary prevention of ischaemic cerebrovascular events. This finding should therefore be investigated in further studies which include clinical end points and comparative assessment of haemostatic and fibrinolytic function parameters in different statins.
Limitations
In this study we performed genotyping for the platelet P2Y12 receptor to detect genetic inter-individual differences but we did not assess genetic expression of CYP 2C19 which has previously also been shown to contribute to biotransformation of clopidogrel (beside CYP 3A4) and to be responsible for inter-individual variability in clopidogrel responsiveness [21]. Even though these inter-individual differences might thus have not been detected in our study, the applied crossover design allows for comparison of the crossed over treatment regimens with the exclusion of CYP2C19 induced inter-regimen differences. While internal validity is therefore not jeopardized by absence of data on CYP2C19 gene distribution, generalizability of our findings must be interpreted with caution. A follow-up study in a larger population of patients should therefore include CYP2C19 genotyping and should be powered to detect both inter-group differences and inter-individual distribution of clopidogrel responsiveness in co-treatment with statin. In this study we administered 20 mg of simvastatin which is, based on previous comparative clinical trials, equivalent to a dose of 80 mg of fluvastatin [22]. However, we did not investigate lower or higher doses of both statins so we do not know whether our finding of no clinically relevant interaction between 20 mg of simvastatin and clopidogrel is also applicable to other doses of simvastatin. Our results, however, form the basis for a follow-up study to analyze dose-dependent effects of clopidogrel-simvastatin co-treatment on platelet aggregation.
In summary, we found no evidence supporting the hypothesis that combined administration of clopidogrel with CYP3A4-metabolized statins attenuates clopidogrel mediated platelet aggregation inhibition. In contrast, our data, viewed in conjunction with the current literature, suggests that administration of CYP3A4-metabolized statins in clopidogrel treated patients does not induce any changes in the conversion of clopidogrel into its active thiol form and therefore neither has a quantitatively nor clinically relevant influence on clopidogrel efficacy. Furthermore we observed attenuation in PT Quick percent value following simvastatin administration which might be mediated by reduction of activity of prothrombin fragment 1 + 2 and plasminogen activator inhibitor-1.
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare TS, DH, JK, KB, SG, VP, UB, GG had no support from any organization for the submitted work. XG and US report personal fees from Department of Neurology, Faculty of Medicine C. G. Carus, TU Dresden, during the conduct of the study; personal fees from Novartis Pharma, personal fees from Ardey Pharm GmbH, personal fees from One Pharm Research and development GmbH, personal fees from Smith & Nephew, personal fees from Otsuka, grants from DFG/ BMBF program Clinical Trials, several projects, grants from German Ministry of Health, Dementia, personal fees from Schülke & Mayr GmbH, outside the submitted work. HR reports grants and personal fees from Pfizer, personal fees from MSD, personal fees from Novartis, outside the submitted work.
The authors are sincerely thankful to Mr Wildermuth for his technical assistance in conducting the study. Furthermore, the authors would like to express their sincere gratitude to Dr Taubert for performing the assessment of clopidogrel active metabolite concentrations. The authors thank Novartis Pharma for supporting this study financially. Dr Siepmann has made substantial contribution to study design, data analysis and drafting the manuscript. Dr Heinke has made substantial contributions to conducting the study. Dr Kepplinger has made substantial contributions to conducting the study. Dr Barlinn has made substantial contributions to conducting the study. Dr Gehrisch has made substantial contributions to laboratory assessment. Dr Grählert has made substantial contributions to statistical analysis. Dr Schwanebeck has made substantial contributions to statistical analysis. Dr Reichmann has made substantial contributions to manuscript revision. Dr Puetz has made substantial contributions to study design and manuscript revision. Dr Bodechtel has made substantial contributions to study design and manuscript revision. Dr Gahn has made substantial contributions to study design and manuscript revision.
References
- 1.Sudlow CL, Mason G, Maurice JB, Wedderburn CJ, Hankey GJ. Thienopyridine derivatives versus aspirin for preventing stroke and other serious vascular events in high vascular risk patients. Cochrane Database Syst Rev. 2009;(4) doi: 10.1002/14651858.CD001246.pub2. ): CD001246. doi: 10.1002/14651858.CD001246.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Qizilbash N, Lewington S, Duffy S, Peto R. Cholesterol, diastolic blood pressure, and stroke: 13,000 strokes in 450,000 people in 45 prospective cohorts. Lancet. 1995;346:1647–1653. [PubMed] [Google Scholar]
- 3.Angiolillo DJ, Fernandez-Ortiz A, Bernardo E, Alfonso F, Macaya C, Bass TA, Costa MA. Variability in individual responsiveness to clopidogrel: clinical implications, management, and future perspectives. J Am Coll Cardiol. 2007;49:1505–1516. doi: 10.1016/j.jacc.2006.11.044. [DOI] [PubMed] [Google Scholar]
- 4.Tafreshi MJ, Zagnoni LG, Gentry EJ. Combination of clopidogrel and statins: a hypothetical interaction or therapeutic dilemma? Pharmacotherapy. 2006;26:388–394. doi: 10.1592/phco.26.3.388. [DOI] [PubMed] [Google Scholar]
- 5.Neuvonen PJ. Drug interactions with HMG-CoA reductase inhibitors (statins): the importance of CYP enzymes, transporters and pharmacogenetics. Curr Opin Investig Drugs. 2010;11:323–332. [PubMed] [Google Scholar]
- 6.Lau WC, Waskell LA, Watkins PB, Neer CJ, Horowitz K, Hopp AS, Tait AR, Carville DG, Guyer KE, Bates ER. Atorvastatin reduces the ability of clopidogrel to inhibit platelet aggregation: a new drug−drug interaction. Circulation. 2003;107:32–37. doi: 10.1161/01.cir.0000047060.60595.cc. [DOI] [PubMed] [Google Scholar]
- 7.Born GV. Aggregation of blood platelets by adenosine diphosphate and its reversal. Nature. 1962;194:927–929. doi: 10.1038/194927b0. [DOI] [PubMed] [Google Scholar]
- 8.Mammen EF, Comp PC, Gosselin R, Greenberg C, Hoots WK, Kessler CM, Larkin EC, Liles D, Nugent DJ. PFA-100 system: a new method for assessment of platelet dysfunction. Semin Thromb Hemost. 1998;24:195–202. doi: 10.1055/s-2007-995840. [DOI] [PubMed] [Google Scholar]
- 9.Takahashi M, Pang H, Kawabata K, Farid NA, Kurihara A. Quantitative determination of clopidogrel active metabolite in human plasma by LC-MS/MS. J Pharm Biomed Anal. 2008;48:1219–1224. doi: 10.1016/j.jpba.2008.08.020. [DOI] [PubMed] [Google Scholar]
- 10.Taubert D, Kastrati A, Harlfinger S, Gorchakova O, Lazar A, von Beckerath N, Schömig A, Schömig E. Pharmacokinetics of clopidogrel after administration of a high loading dose. Thromb Haemost. 2004;92:311–316. doi: 10.1160/TH04-02-0105. [DOI] [PubMed] [Google Scholar]
- 11.Fontana P, Dupont A, Gandrille S, Bachelot-Loza C, Reny JL, Aiach M, Gaussem P. Adenosine diphosphate-induced platelet aggregation is associated with P2Y12 gene sequence variations in healthy subjects. Circulation. 2003;108:989–995. doi: 10.1161/01.CIR.0000085073.69189.88. [DOI] [PubMed] [Google Scholar]
- 12.Ke X, Collins A, Ye S. PIRA PCR designer for restriction analysis of single nucleotide polymorphisms. Bioinformatics. 2001;17:9838–9839. doi: 10.1093/bioinformatics/17.9.838. [DOI] [PubMed] [Google Scholar]
- 13.Clarke TA, Waskell LA. The metabolism of clopidogrel is catalyzed by human cytochrome P450 3A and inhibited by atorvastatin. Drug Metab Dispos. 2003;31:53–59. doi: 10.1124/dmd.31.1.53. [DOI] [PubMed] [Google Scholar]
- 14.Lennon MJ, Gibbs NM, Weightman WM, McGuire D, Michalopoulos N. A comparison of Plateletworks and platelet aggregometry for the assessment of aspirin-related platelet dysfunction in cardiac surgical patients. J Cardiothorac Vasc Anesth. 2004;18:136–140. doi: 10.1053/j.jvca.2004.01.015. [DOI] [PubMed] [Google Scholar]
- 15.Serebruany VL, Malinin AI, Callahan KP, Gurbel PA, Steinhubl SR. Statins do not affect platelet inhibition with clopidogrel during coronary stenting. Atherosclerosis. 2001;159:239–241. doi: 10.1016/s0021-9150(01)00606-2. [DOI] [PubMed] [Google Scholar]
- 16.Saw J, Steinhubl SR, Berger PB, Kereiakes DJ, Serebruany VL, Brennan D, Topol EJ. Clopidogrel for the Reduction of Events During Observation Investigators. Lack of adverse clopidogrel-atorvastatin clinical interaction from secondary analysis of a randomized, placebo-controlled clopidogrel trial. Circulation. 2003;108:921–924. doi: 10.1161/01.CIR.0000088780.57432.43. [DOI] [PubMed] [Google Scholar]
- 17.Mukherjee D, Kline-Rogers E, Fang J, Munir K, Eagle KA. Lack of clopidogrel-CYP3A4 statin interaction in patients with acute coronary syndrome. Heart. 2005;91:23–26. doi: 10.1136/hrt.2004.035014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Serebruany VL, Midei MG, Malinin AI, Oshrine BR, Lowry DR, Sane DC, Tanguay JF, Steinhubl SR, Berger PB, O'Connor CM, Hennekens CH. Absence of interaction between atorvastatin or other statins and clopidogrel. Arch Intern Med. 2004;164:2051–2057. doi: 10.1001/archinte.164.18.2051. [DOI] [PubMed] [Google Scholar]
- 19.Ziegler S, Schillinger M, Funk M, Felber K, Exner M, Mlekusch W, Sabeti S, Amighi J, Minar E, Brunner M, Muller M, Mannhalter C. Association of a functional polymorphism in the clopidogrel target receptor gene, P2Y12, and the risk for ischemic cerebrovascular events in patients with peripheral artery disease. Stroke. 2005;36:1394–1399. doi: 10.1161/01.STR.0000169922.79281.a5. [DOI] [PubMed] [Google Scholar]
- 20.Ludwig S, Dharmalingam S, Erickson-Nesmith S, Ren S, Zhu F, Ma GM, Zhao R, Fenton JW, 2nd, Ofosu FA, Velthuis HT, van Mierlo G, Shen GX. Impact of simvastatin on hemostatic and fibrinolytic regulators in type 2 diabetes mellitus. Diabetes Res Clin Pract. 2005;70:110–118. doi: 10.1016/j.diabres.2005.03.025. [DOI] [PubMed] [Google Scholar]
- 21.Scott SA, Sangkuhl K, Stein CM, Hulot JS, Mega JL, Roden DM, Klein TE, Sabatine MS, Johnson JA, Shuldiner AR. Clinical pharmacogenetics implementation consortium guidelines for CYP2C19 genotype and clopidogrel therapy: 2013 update. Clin Pharmacol Ther. 2013;3:317–323. doi: 10.1038/clpt.2013.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Illingworth DR, Tobert JA. A review of clinical comparing HMG-CoA reductase inhibitors. Clin Ther. 1994;16:366–385. [PubMed] [Google Scholar]