

Abstract
Aims
This study aimed to assess changes in the plasma concentrationss of 4β-hydroxycholesterol (4βHC) against intravenous (i.v.) and oral midazolam (MDZ) pharmacokinetics (PK) after administration of a potent CYP3A inhibitor [ketoconazole (KETO)] and inducer [rifampicin (RIF)].
Methods
Thirty-two healthy subjects (HS) were allocated into three groups of 12 each in KETO and RIF and 10 in a placebo group (PLB). All HS were randomized to receive oral and i.v. MDZ on day 1 or 2 and on day 15 or 16 after receiving RIF (600 mg once daily), KETO (400 mg once daily) or PLB for 2 weeks. Subjects were followed until day 30. The effect of treatments on 4βHC was assessed by analyzing % change from baseline using a linear spline mixed effects model.
Results
Compared with PLB, KETO decreased 4βHC mean values up to 13% (P = 0.003) and RIF increased 4βHC mean values up to 220% (P < 0.001). Within 14 days of stopping KETO and RIF, 4βHC had either returned to baseline (KETO) or was still returning to baseline (RIF). Compared with baseline, mean oral MDZ AUC increased by 11-fold (90% CI ranging from 9-fold to 13-fold increase) and decreased by 92% (90% CI ranging from 90% to 95% decrease) after KETO and RIF, respectively. Similar trends were observed for 6β-hydroxycortisol : cortisol (6βHCL : CL) urinary ratios.
Conclusions
Changes in plasma 4βHC can be utilized as a surrogate for MDZ PK after multiple doses of potent CYP3A inducers. There is a more limited dynamic range for 4βHC for assessment of potential CYP3A inhibitors. 4βHC is a valuable tool for the assessment of potential CYP3A inducers in early drug development.
Keywords: 4β-hydroxycholesterol, biomarkers, CYP3A, drug–drug interactions, midazolam, saliva
What is already known about this subject
Endogenous plasma 4β-hydroxycholesterol (4βHC) concentrations have been used as a putative CYP3A activity marker. However, placebo controlled studies have not been conducted to assess the dynamic range of potent CYP3A inhibitors and inducers.
What this study adds
This placebo-controlled study assessed the dynamic range of plasma 4βHC concentrations after administration of ketoconazole and rifampicin (for 14 days). A comparison is made with other CYP3A trait measures such as midazolam pharmacokinetics and putative endogenous CYP3A biomarkers. The time course of CYP3A activity before and after administration of CYP3A modulators is also examined. This study validates 4βHC against midazolam pharmacokinetics and also expands the utility of 4βHC to assess CYP3A activity after stopping treatment with CYP3A inducers or inhibitors.
Results from this study show utility of 4βHC to defer or avoid midazolam-based studies, saving cost and avoiding exposing healthy subjects to study medications.
Introduction
At present, the assessment of the propensity of a drug to alter human cytochrome P450 3A (CYP3A) activity through induction or inhibition is generally best assessed through administration of exogenous probes drugs that are relatively specific CYP3A substrates such as midazolam (MDZ) [1,2]. Such assessments add some additional risk to study participants and require additional resources. A validated endogenous marker of CYP3A activity would add significant value to the drug development process by allowing routine monitoring of CYP3A activity in phase 1 and drug–drug interaction studies. Towards this end, 4β-hydroxycholesterol (4βHC) may serve as a promising endogenous biomarker of CYP3A.
Cholesterol is a precursor for the biosynthesis of bile acids and steroid hormones. It is an important element in cells and is readily biotransformed into numerous oxidation products known as oxysterols. Such oxysterols may be formed by different pathways as a consequence of enzymatic cholesterol metabolism [3]. Some of the major oxysterols in human plasma are 27-hydroxycholesterol, 24-hydroxycholesterol, 7α-hydroxycholesterol and 4βHC.
Members of the CYP3A gene subfamily play an important role in the metabolism of many drugs and have been identified as the locus of well-documented drug–drug interactions [4,5]. Other endogenous biomarkers such as 6β-hydroxycortisol : cortisol (6βHCL : CL) ratio and the formation clearance of 6β-hydroxycortisone have been previously assessed in clinical studies [6,7]. It has been demonstrated in vitro that 4βHC is formed primarily by CYP3A, and it has been suggested that 4βHC may be useful as an endogenous biomarker of CYP3A activity [6]. Evidence that 4βHC may correlate with CYP3A4 and CYP3A5 activity has been observed in humans [8]. Patients with epilepsy treated with carbamazepine, phenytoin or phenobarbital, drugs known as strong inducers of CYP3A, have been shown to have highly elevated (7–8-fold higher) concentrations of 4βHC in the plasma relative to the untreated population [9]. However, a preliminary assessment of plasma 4βHC concentrations following 7 days of ketoconazole (KTZ) treatment, a potent inhibitor of CYP3A, were equivocal [10].
As 4βHC is formed by metabolism of cholesterol and since cholesterol may change over time due to drug and/or dietary effects, several reports have suggested the utility of normalizing 4βHC concentration with plasma total cholesterol (TC) concentration in order to reduce variability and improve the sensitivity of 4βHC as a marker of CYP3A activity [11,12]. However, no studies have reported a placebo-controlled assessment on the 4βHC response before and after administration of a potent CYP3A inducer and a potent CYP3A inhibitor. An additional candidate for normalization of 4βHC is 4α-hydroxycholesterol (4αHC), an isomer of 4βHC. 4αHC concentrations were not influenced by antiepileptic drugs or KTZ, indicating that 4αHC is unlikely to be a product of CYP3A-catalyzed cholesterol metabolism and suggesting that it would not change in the same direction as TC over time [12,13]. The reported half-life values of 4βHC are wide (ranging from 60 h to 17 days) and its utility as a biomarker of CYP3A activity in short term studies is uncertain due to a hysteresis between CYP3A activity and changes in 4βHC [13,14]. Thus, validation of 4βHC as an endogenous CYP3A biomarker with a commonly accepted exogenous CYP3A probe is required before establishing the full clinical utility for assessment of CYP3A activity. MDZ is a widely accepted and sensitive exogenous probe for CYP3A phenotyping. It has low oral bioavailability and is extensively metabolized by CYP3A, in both the liver and the gut, to its primary, pharmacologically active metabolite, 1-hydroxymidazolam (1-OHMDZ) and to a lesser extent, to 4-hydroxymidazolam and 1,4-dihydroxymidazolam [15]. These metabolites are conjugated and then excreted as glucuronides in the urine. This study was conducted to achieve the following objectives: 1) validate changes in plasma 4βHC concentrations against changes in exposure to MDZ administered orally as a ‘gold standard’ marker of CYP3A activity and intravenously (i.v.) to establish systemic vs. first pass effects by assessing placebo response and time course of plasma 4βHC, 4αHC and TC concentrations during 14 days of dosing with placebo, KTZ or the potent CYP3A inducer, rifampicin (RIF), 2) follow the time course of return towards baseline of plasma 4βHC, 4αHC and TC concentrations for 14 days after discontinuation of these treatments, 3) to compare 4βHC changes over time with and without normalizing by 4αHC and TC, 4) to explore the relationship between saliva MDZ pharmacokinetics and CYP3A biomarkers and 5) to evaluate the utility of other potential endogenous markers of CYP3A activity, namely: urinary 6β-hydroxycortisol : cortisol ratio levels (6βHCL : CL) and formation clearance (CLf) of 6βHCL and 6β-hydroxycortisone (6βHCO).
Methods
Clinical protocol
After signing the written informed consent, 34 healthy subjects in the age range of 18–45 years were enrolled in this open label, parallel group study. A schematic of the study design is presented in Table S1.
The study was approved on August 5 2011 by IntegReview, Ltd, Austin, TX, USA (approval number 3036939A). The study was conducted at PPD, LLC Austin, TX, USA. All healthy subjects were randomized to receive oral (2 mg) as a syrup and intravenous (0.4 mg) MDZ on day 1 or 2, and on day 15 or 16 after receiving once daily RIF 600 mg, KTZ 400 mg or placebo for 2 weeks. Subjects were followed until day 30. Serial blood and saliva samples for MDZ pharmacokinetics were collected at 0, 0.10, 0.25, 0.5, 1, 1.5, 2, 3, 4, 5, 6, 8 and 12 h on days 1, 2, 15 and 16. Serial blood samples were collected for assessment of cortisol, cortisone, 6βHCO and 6βHCL on day 1, 2, 15 and 16. During the same period, urine samples were also collected from 0–12 and 12–24 h ensuring that subjects would void at the end of each collection period. Pre-dose blood samples were collected from days 1 to 30 for assessment of 4βHC, 4αHC and TC. All subjects stayed in-house in the study facility for the entire study duration and were fed with a standard diet.
Study participants were in good general health as assessed by medical history, physical examination, vital signs, 12-lead electrocardiogram and laboratory data. Tobacco smoking and a history of allergy to RIF, MDZ, KTZ and related compounds were also part of the exclusion criteria.
Prior exposure to MDZ, RIF and KTZ and related compounds within 3 months of study drug administration was prohibited. Other prohibited treatments included use of any food or drink containing grapefruit or Seville oranges at least 1 week prior to study start and oral, injectable or implantable hormonal contraceptive agent within 3 months prior to study drug administration. Subjects were asked to abstain from over-the-counter, including St John's wort, and prescription medications from 1 week before the administration of study medication.
Bioanalytical methods
See supplementary methods for detailed information on all bioanalytical methods used in this study.
Analysis of 4βHC and 4αHC
4βHC and 4αHC were measured as previously described with few modifications [8]. Both 4βHC and 4αHC were quantified from calibration curves prepared in plasma using d4-4βHC as a surrogate calibrant at concentrations ranging from 2 to 500 ng ml−1. The limit of quantification was 2 ng ml−1 for all analytes. The intra- and inter-day %CV for 4β- and 4α-hydroxycholesterol were ≤10%.
Analysis of plasma and saliva MDZ and 1-OHMDZ
Plasma and saliva MDZ and 1-OHMDZ were measured using LC-MS assay. Calibration curves for MDZ and 1-OHMDZ were prepared in plasma from 0.1 to 100 ng ml−1 and 0.1 to 50 ng ml−1, respectively. The lower limit of quantification for all analytes in plasma was 0.1 ng ml−1. The lower limit of quantification for all analytes in saliva was 0.025 ng ml−1. The intra- and inter-day %CVs for the plasma assay were ≤4%.
Analysis of plasma and saliva cortisol and cortisone
Plasma cortisol and cortisone were measured using LC-MS assay. Calibration curves for cortisone and cortisol were prepared in surrogate matrix (phosphate buffered saline) from 1 to 250 ng ml−1. The lower limit of quantification for all analytes in urine was 1 ng ml−1. The intra- and inter-day %CV for cortisol and cortisone were ≤8%.
Analysis of plasma and saliva 6βHCO and 6βHCL
Plasma 6βHCO and 6βHCL were measured using LC-MS assay. Calibration curves for cortisone and cortisol were prepared in surrogate matrix (phosphate buffered saline) from 0.5 to 250 ng ml−1. The lower limit of quantification for all analytes in urine was 0.5 ng ml−1. The intra- and inter-day %CV for cortisol and cortisone were ≤9%.
Statistical methods
The percent change from baseline was calculated for each subject at each time point. The mean percent change from baseline profile for 4βHC, 4αHC and TC were summarized and plotted vs. time for different treatment group.
Trend analysis of plasma 4βHC concentration
In order to determine the days that are associated with a significant difference (α = 0.05) vs. placebo in 4βHC percent change from baseline, a mixed effect model was used. The model included treatment, day and treatment-by-day as fixed effect variables, and subject as a repeated effect. A step-down procedure was used. Starting on day 17, if the P value for the difference of percent change from baseline of 4βHC relative to placebo was smaller than 0.05, the analysis continued to calculation of the P value for the difference on the previous day. Otherwise it stopped.
Time course of plasma 4βHC concentration
The mean percent change from baseline profile for 4βHC, 4αHC and TC was plotted vs. time for the different treatment groups. The effect of multiple doses of KTZ, RIF or placebo on 4βHC was assessed by analyzing the percent change from baseline of 4βHC trough concentration from day 3 to day 30 using the following linear spline mixed effect model.
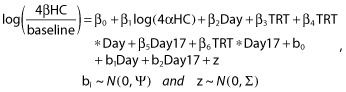 |
where day 17 was defined as a function that is equal to day when day is greater than or equal to 17 and was equal to zero otherwise; TRT was the treatment (KTZ, RIF or placebo); day and day 17 were continuous time variables. β0, …, β6 were fixed effect coefficients, and b0, b1 and b2 were random effect coefficients. The baseline was defined as day 1 predose. Similar analyses using the same model without adjusting covariate or with adjusting covariate log(TC) were performed to understand the impact of the adjusting covariate on the percent change from baseline for 4βHC. [exp(β4) − 1]*100 represented the percent change from baseline in 4βHC (adjusted by 4αHC or TC or no adjustment) per day before day 17 in the treatment arm comparing with the placebo arm. Furthermore, the [exp(β4 + β6) − 1]*100 represented the percent change from baseline in 4βHC (adjusted by 4αHC or TC or no adjustment) per day after day 17 in the interaction drug sequence comparing with the placebo sequence.
Pharmacokinetics of plasma and saliva MDZ and 1-OHMDZ
To assess the effect of 14 day multiple doses of KTZ, RIF or placebo on single dose of MDZ (i.v. and oral), the plasma and saliva pharmacokinetic parameters (AUC(0,∞), AUC(0,t) and Cmax) for MDZ and its metabolite, 1-OHMDZ, following single oral or i.v. doses administration of MDZ with or without co-administration of KTZ, RIF or placebo were analyzed with a mixed effect model. The model included fixed effect for treatment and a repeated effect for subject. Log transformation was applied to AUC(0,∞), AUC(0,t) and Cmax data. Point estimates and 90% CIs for differences on pharmacokinetic parameters between treatments on the natural log scale were exponentiated to obtain estimates for ratios of geometric means on the original scale. In addition, ratio plots (after treatment (F) : before treatment (A)) were generated for MDZ AUC(0,∞) and Cmax after i.v. administration of MDZ.
Cortisol, 6βHCL, cortisone and 6βHCO
CLf of 6βHCO and 6βHCL was calculated as described in Peng et al. [7]. To explore the effect of KTZ, RIF and placebo on the plasma cortisol and cortisone, and urinary cortisol, cortisone, 6βHCO and 6βHCL, the urinary metabolite ratio (UR) at different collection intervals (0–12 h, 12–24 h and 0–24 h) and CLf were analyzed with a similar mixed effect model as the analysis for MDZ pharmacokinetics. Log transformation was applied to CLf and urinary metabolite ratio data and back-transformation of the results was done for reporting purposes.
MDZ pharmacokinetics vs. biomarker relationship
Scatter plots were generated to explore the relationship between the ratio of plasma MDZ AUC(0,∞) and Cmax and 4βHC percent change from baseline, ratio of UR or ratio of CLf before and after 14 days of treatment with KTZ, RIF or placebo.
Results
Safety
Thirty-four healthy subjects (32 males and two females) in the age range of 20–51 years (mean = 32.4 years) were randomized. The small number of female subjects relative to the number of male subjects does not allow for a meaningful assessment of the influence of gender on the study end points. Likewise, quantifying the effect of race was not possible due to the small numbers of subjects and the variability of the end points. The ethnicity of the subjects enrolled in this study was 19 (55.9%) White, 14 (41.2%) African American/Black and 1(2.9%) Asian. There were no deaths or serious AEs in this study. There were a total of 15 subjects (44.1%) with adverse events (AEs) in this study. The most frequently reported AEs were nausea and headache, which were reported in five subjects (14.7%). Three subjects discontinued the study, one due to mild finger swelling and urticaria after the second dose of oral MDZ. None of the clinical laboratory marked abnormalities or ECG abnormalities was considered of clinical significance by the investigator.
4βHC, 4αHC and TC
Figure 1 shows the geometric mean (± SD) percent change from baseline for 4βHC, 4αHC and TC for the entire study period. Table 1 shows the geometric mean percent change from baseline of 4βHC, 4αHC and TC on selected key study days. Administration of placebo treatment for 14 days did not have any significant effect on the percent change from baseline plasma 4βHC concentration–time profiles, which supports the validity of the study.
Figure 1.
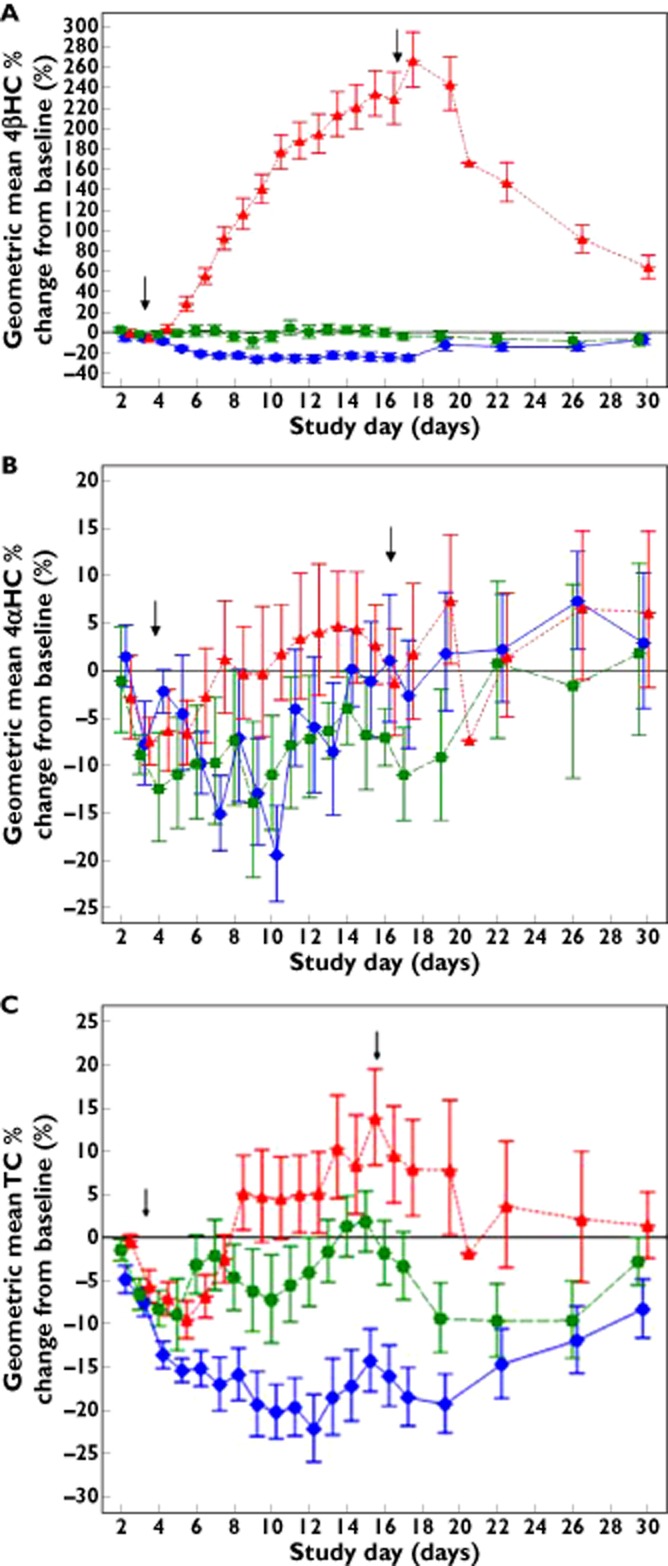
Plot of mean (± SD) plasma concentration–time profiles of 4β-hydroxycholesterol (4βHC) (A), 4α-hydroxycholesterol (4αHC) (B) and total cholesterol (TC) (C) after ketoconazole, rifampicin and placebo treatment (vertical arrows indicate start and stop of treatments).  , ketoconazole;
, ketoconazole;  , rifampicin;
, rifampicin;  , placebo
, placebo
Table 1.
Summary statistics for 4β-hydroxycholesterol, 4α-hydroxycholesterol and total cholesterol
| Analyte | Ketoconazole (% change vs. baseline) | Rifampicin (% change vs. baseline) Time (days)* |
Placebo (% change vs. baseline) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 5 | 7 | 14 | 30 | 5 | 7 | 14 | 30 | 5 | 7 | 14 | 30 | |
| 4β-hydroxycholesterol | −22.9 | −26.6 | −24.8 | −6.8 | 91.6 | 140 | 228.2 | 63.5 | 1.7 | −8.2 | 0.08 | −7.0 |
| 4α-hydroxycholesterol | −15.1 | −12.9 | −1.1 | 2.9 | 1.2 | −0.4 | −1.38 | 6.1 | −9.8 | −13.9 | −7.1 | 1.8 |
| Total cholesterol | −17.0 | −19.4 | −16.1 | −8.3 | −2.6 | 4.8 | 9.5 | 1.4 | −2.1 | −6.2 | −1.8 | −2.8 |
Percent change from baseline after once daily dose (5, 7 or 14 days of dosing) and after stopping treatment for 14 days (day 30).
Temporal variations of 4βHC (trend analysis) were assessed using data from daily plasma 4βHC concentrations from daily samples. 4βHC percent change from baseline showed a statistically significant decrease compared with placebo (P value = 0.0365) after 3 days of treatment with KTZ. Plasma 4βHC concentrations continued to decrease by approximately 23% after 5 days of KTZ administration, and remained decreased throughout the subsequent 9 days of KTZ dosing. Although 4αHC varied during KTZ dosing, there was no clear trend over the 14 days, indicating no major effect of KTZ on 4αHC. There was a modest change in TC concentration after administration of KTZ, with a maximal decrease (20% vs. baseline) observed after 14 days. All analytes either returned to or were approaching baseline by 14 days after stopping KTZ.
The percent change from baseline plasma 4βHC concentration–time profiles showed an increase by approximately 228% after 14 days of dosing with RIF. 4βHC continued to increase continuously over the daily dosing of RIF for 14 days and it reached statistical significance vs. placebo on day 3 of dosing (P value = 0.001). After administration of RIF was stopped, plasma 4βHC concentrations remained about 60% higher relative to baseline at the end of the 14 days post-treatment observation period. There was no clear trend in 4αHC or TC plasma concentrations observed in the RIF treatment arm.
Time course of plasma 4βHC concentration
To fit the observed effect of 4βHC percent change from baseline after dosing with KTZ, RIF and placebo, a linear spline mixed effect model was developed. To assess the impact of normalizing 4βHC with 4αHC or TC, 4αHC or TC were used as covariates in the model. The estimates of the modelled slope are summarized in Table 2. The modelled trajectory of 4βHC suggested that it would reach baseline concentrations approximately 5 or 18 days after stopping administration of KTZ and RIF, respectively. Use of 4αHC as a covariate improved the estimation of the slope of 4βHC percent change from baseline over time after KTZ treatment (P value for the slope = 0.007) as compared with the slope estimate with TC as a covariate (P value = 0.094) or without these covariates (P value = 0.043). However, the overall conclusions did not change whether or not either 4αHC or TC were used as a covariate in the model. Unlike for KTZ, the slope estimate of 4βHC percent change from baseline over time after administration of RIF did not differ (P < 0.0001) with or without 4αHC or TC in the model.
Table 2.
Summary of modelled mean percent 4β-hydroxycholesterol change from baseline by covariate (4α-hydroxycholesterol, total cholesterol and no covariate) during treatment with ketoconazole and rifampicin on days 3–16 and after stopping treatment (days 17–30)
| Probe | Days 3–16 | Days 17–30 | ||
|---|---|---|---|---|
| Estimate (95% CI) | P value | Estimate (95% CI) | P value | |
| Covariate: 4α-hydroxycholesterol | ||||
| Ketoconazole | −0.014 (−0.023, −0.004) | 0.007 | 0.039 (0.017, 0.060) | 0.001 |
| Rifampicin | 0.094 (0.085, 0.104) | <0.001 | −0.155 (−0.176, −0.133) | <0.001 |
| Covariate: total cholesterol | ||||
| Ketoconazole | −0.007 (−0.016, 0.001) | 0.094 | 0.025 (0.004, 0.047) | 0.022 |
| Rifampicin | 0.091 (0.082, 0.099) | <0.001 | −0.151 (−0.173, −0.130) | <0.001 |
| No covariate | ||||
| Ketoconazole | −0.012 (−0.023, −0.000) | 0.043 | 0.036 (0.014, 0.058) | 0.003 |
| Rifampicin | 0.096 (0.085, 0.108) | <0.001 | −0.160 (−0.182, −0.137) | <0.001 |
Pharmacokinetics of plasma and saliva MDZ and 1-OHMDZ
Table S2 shows the effect of placebo, KTZ and RIF on oral and i.v. MDZ plasma pharmacokinetics.
As expected from the extensive protein binding of MDZ (96% plasma protein binding), the concentrations of MDZ following i.v. dosing were much lower in saliva than in plasma (Table S2) [16]. While the concentrations of 1-OHMDZ were also lower in saliva than in plasma after i.v. administration of MDZ, the difference was not as large as for MDZ due to the lower binding of 1-OHMDZ to serum proteins. MDZ and 1-OHMDZ could be detected in saliva following oral dosing of MDZ. However, the concentrations were high relative to post-i.v. MDZ saliva concentrations and highly variable. This was likely a result of residual MDZ in the buccal cavity from the syrup formulation used in the study and therefore the saliva MDZ and 1-OHMDZ concentrations did not correlate well with plasma concentrations after oral MDZ administration.
The overall exposure (area under the plasma concentration–time curve (AUC)) of 1-OHMDZ decreased after dosing with RIF (Table S3), a finding consistent with the literature [16]. Because the hydroxy-metabolites of MDZ undergo metabolism, their clearance can also be induced by RIF [17,18]. Saliva 1-OHMDZ concentrations following treatment with RIF were highly variable and too low to support reliable calculation of pharmacokinetic parameters.
Results comparing exposure of i.v. MDZ in plasma and saliva are shown in Figure 2. As shown in Table 3 and Table S2, after administration of KTZ, i.v. MDZ AUC(0,∞) geometric mean ratio (GMR) and corresponding 90% CIs were comparable between saliva [5.0 (3.5, 7.1)] and plasma [5.2 (4.1, 6.6)]. After administration of RIF, i.v. MDZ AUC(0,∞) GMR and 90% CI were comparable between saliva [0.6 (0.48, 0.69)] and plasma [0.5 (0.41,0.59)].
Figure 2.
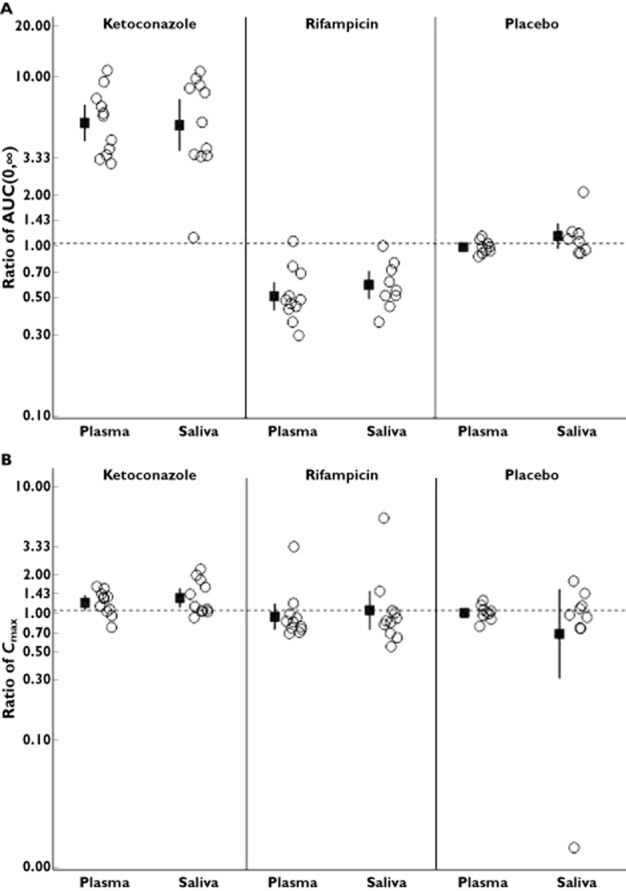
Plot of individual ratios (MDZ + perpetrator : MDZ alone) for i.v. MDZ AUC(0,∞) (A) and Cmax (B).  , GMR (midazolam + DDI : midazolam) with 90% CI; ○, individual ratios (midazolam + DDI : midazolam)
, GMR (midazolam + DDI : midazolam) with 90% CI; ○, individual ratios (midazolam + DDI : midazolam)
Table 3.
Statistical analysis for midazolam pharmacokinetics
| Probe | Comparison | GMR (90% CI) | ||
|---|---|---|---|---|
| AUC(0,∞) | AUC(0,t) | Cmax | ||
| Ketoconazole | MDZi.v. + KETO vs. MDZi.v. | 5.16 (4.063, 6.556) | 3.45 (2.873, 4.145) | 1.16 (1.021, 1.307) |
| MDZoral + KETO vs. MDZoral | 11.53 (9.936, 13.380) | 8.34 (7.243, 9.612) | 3.74 (3.165, 4.423) | |
| Rifampin | MDZi.v. + RIF vs. MDZi.v. | 0.49 (0.406, 0.588) | 0.50 (0.413, 0.594) | 0.90 (0.714, 1.128) |
| MDZoral + RIF vs. MDZoral | 0.08 (0.059, 0.101) | 0.07 (0.054, 0.083) | 0.11 (0.104, 0.126) | |
| Placebo | MDZi.v. + PBO vs. MDZi.v. | 0.95 (0.903, 1.002) | 0.96 (0.915, 1.015) | 0.96 (0.891, 1.041) |
| MDZoral + PBO vs. MDZoral | 1.04 (0.933, 1.167) | 1.04 (0.935, 1.155) | 1.05 (0.878, 1.259) | |
MDZi.v. = 0.4 mg single dose i.v.; MDZoral = MDZ 2 mg single dose orally; KETO = ketoconazole 400 mg once daily for 14 days; RIF = rifampicin 600 mg once daily for 14 days; PBO = placebo once daily for 14 days. CI, confidence interval; GMR, ratio of geometric least-squares means.
Assessment of cortisol, 6β-HCL, cortisone and 6β-HCO
The statistical summary of the urinary 6βHCL ratio data is presented in Table S4. After dosing with KTZ and RIF for 14 days, the mean 6βHCL : CL ratio decreased by approximately 23% and increased by about five-fold, respectively. There were no meaningful changes in the placebo group. There was no apparent diurnal effect (0–12 h vs. 12–24 h collections) either at baseline or after 14 days of any treatment.
The statistical summary of the CLf of 6βHCO and 6βHCL is presented in Table S5. The CLf values of 6βHCO and 6βHCL considerably increased by more than three-fold after 14 days of RIF treatment and decreased by ∼80% after 14 days of KTZ treatment. There was no major difference in CLf values between 6βHCO and 6βHCL alone or in combination.
MDZ–endogenous biomarker relationships
The overall exposure of MDZ before and after administration of RIF, KTZ and placebo were compared to 4βHC concentrations (Figure 3A,B), 6βHCL : CL ratio (Figure S1.) and the CLf values of 6βHCO and 6βHCL (Figure S2). As expected, there was no trend in the data after treatment with placebo. The magnitude of change in MDZ exposure or biomarker response after 14 day treatment with KTZ and RIF are presented on Figure 4.
Figure 3.
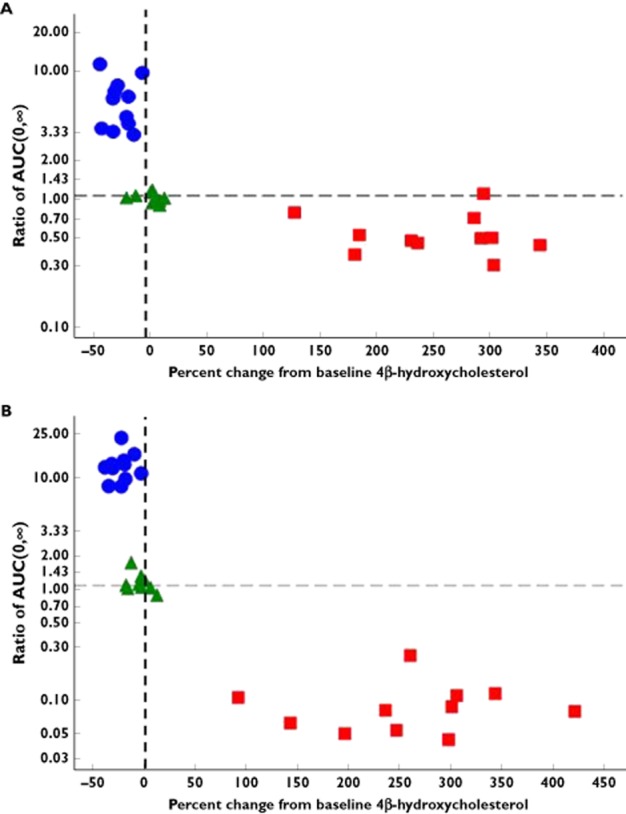
Plasma 4β-hydroxycholesterol vs. MDZ AUC(0,∞) [(MDZ + perpetrator : MDZ alone) ratio] after i.v. (A) and oral (B) MDZ administration. •, ketoconazole; ▀, rifampicin; ▴, placebo
Figure 4.
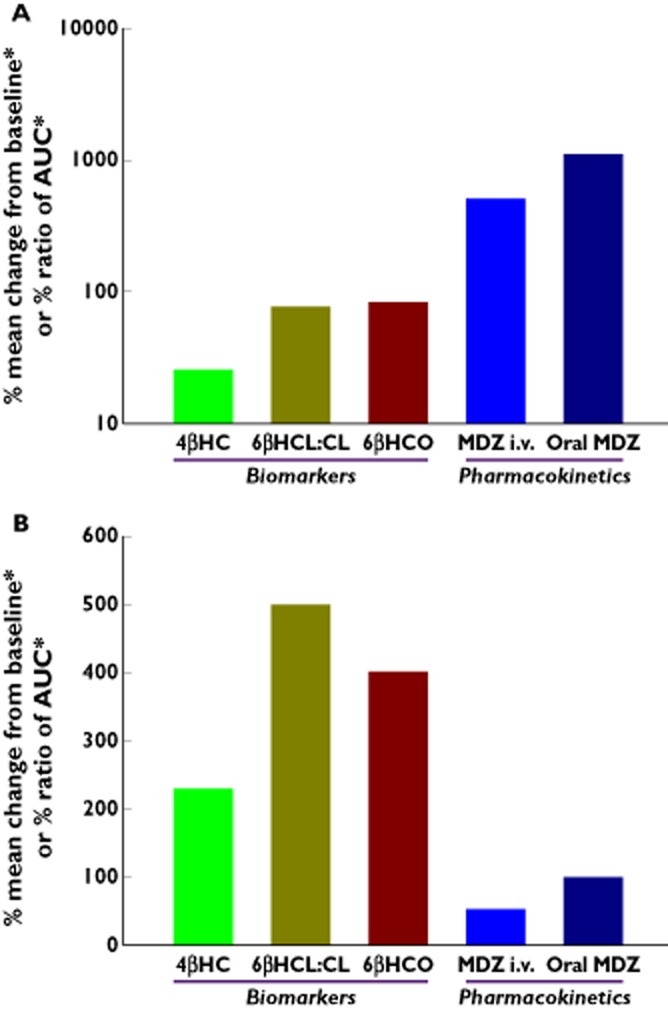
Comparative effect of ketoconazole (A) and rifampicin (B) on plasma 4β-hydroxycholesterol, urinary 6β-hydroxycortisol : cortisol ratio, formation clearance of 6βHC and 6β-hydroxycortisone and MDZ AUC ratio [MDZ + perpetrator : MDZ alone].*Absolute values shown:Ratio (Day 14 AUC(0,∞), Day 1 AUC(0,∞) ) for MDZ i.v. and oral MDZ; Change from baseline for plasma 4βHC, urinary 6βHCL: cortisol ratio and formation clearance of 6βHCO;4βHC = Plasma 4β-hydroxycholesterol;6βHCL:C = Urinary 6β-hydroxycortosol: cortisol ratio;6βHCO = Formation clearance of 6β-hydroxycortisol + 6β-hydroxycortisone;MDZ i.v. = Midazolam i.v. dosing;MDZ Oral = Midazolam oral dosing
After 14 days of KTZ, there is a more limited dynamic range for 4βHC (23% decrease) compared with MDZ exposure (∼11-fold increase after oral dosing), urinary 6βHCL : CL ratio values (∼77% decrease) and the total CLf values of 6βHCO and 6βHCL (∼80% decrease).
However, following 14 days of RIF, all of the endogenous biomarkers studied showed a comparable dynamic range (4βHC 228% increase), urinary 6βHCL : CL ratio ∼500% increase; total CLf of 6βHCO and 6βHCL ∼400% increase) to oral MDZ (>95% decrease).
Discussion
Given the importance of CYP3A, numerous strategies have been employed to facilitate phenotyping and drug–drug interaction assessment in a clinical setting. These have included 1) the utilization of endogenous biomarkers to avoid dosing of a CYP3A probe, 2) assessment of endogenous biomarkers in plasma to avoid collection of urine and 3) the analysis of saliva after probe dosing to obviate the need for blood collection. One or more of these approaches has the potential to facilitate clinical studies with specific subject types (e.g. young, elderly, cancer patients, renal or hepatic impaired, etc) and enable robust assessment of drug interaction time courses. Therefore, an attempt was made to compare plasma 4βHC, urinary 6βHCL : CL ratio, 6βHCO and 6βHCL CLf, i.v. MDZ and oral MDZ as CYP3A trait measures in the same group of subjects following the administration of a potent inducer (RIF) and inhibitor (KTZ).
Although 4βHC has been suggested as a putative endogenous marker of CYP3A activity, no studies to date have reported a comprehensive assessment comparing oral and i.v. MDZ pharmacokinetics and plasma concentrations of 4βHC after administration of a potent CYP3A inducer or inhibitor. Tomalik-Scharte et al. assessed the suitability of 4βHC as a CYP3A marker and used five cocktail phenotyping studies to compare MDZ and 4βHC parameters [19]. The authors showed a weak correlation with 4βHC and MDZ clearance in healthy subjects and in HIV-positive patients with most of the assessments conducted within less than 2 weeks of treatment. In addition, it is not entirely clear if the background medications had an effect on the 4βHC concentrations in this HIV patient population.
Results from this placebo-controlled study show a validated assessment of CYP3A activity using plasma 4βHC concentrations, urinary 6βHCL : CL ratios and the CLf values of 6βHCO and 6βHCL before and after administration of KTZ and RIF relative to the well-established exogenous probe, MDZ. Additionally, the time course of 4βHC plasma concentrations during and after treatment show the temporal changes in this biomarker and allow an assessment of the utility of this marker in studies of various duration and also guides the length of time required to assess for a return to baseline of CYP3A activity without administration of endogenous probes. Furthermore, as shown in this study, trend analysis of 4βHC return to baseline after cessation of a potent CYP3A inducer and inhibitor provides an opportunity to measure CYP3A activity using a minimally invasive tool instead of one or more MDZ assessments. This is especially significant in the light of the current draft guidance by FDA which states the importance to measure the enzyme activity and the time of return back to baseline when induction or time-dependent inhibition is involved [20].
Although it is known that MDZ systemic exposure is significantly decreased when administered with RIF, the time course of 4βHC plasma concentrations as a result of daily administration of a potent inducer was not previously known. Compared with placebo, 14 days of RIF administration substantially increased the 4βHC concentrations in plasma, reaching ∼230% of baseline concentrations. Following RIF dosing, 4βHC increased in the first 3 days of dosing, the difference from baseline in 4βHC concentrations reaching statistical significance compared with placebo after 3 days (P value = 0.0001). This indicates that the effect of a potent inducer on 4βHC is acute and can also be observed in relatively short term studies with a potent inducer. Increases in 4βHC did not reach a plateau after 14 days of RIF and once the dosing with rifampicin was stopped, 4βHC concentrations declined to concentrations that were ∼60% higher than baseline at the end of the observation period (14 days after stopping treatment). The 4βHC placebo-corrected concentrations did not return to baseline in this period presumably due the time it takes for CYP3A to return to steady-state following induction and due to the relatively long half-life of 4βHC, and the time post-induction to re-establish steady-state 4βHC concentrations [13]. This finding is consistent with the previously published modelled data where 4 weeks after termination of dosing, the average 4βHC concentration was still 30% higher than pretreatment concentrations, and even after 8 weeks slightly increased concentrations of 4βHC were observed [11].
While there was a very clear increase in 4βHC after RIF treatment, KTZ dosing did not reduce 4βHC with a similar amplitude in the opposite direction. Plasma 4βHC concentrations were reduced at statistically significant levels as early as 3 days post KTZ dosing. However, after 14 days of KTZ treatment, compared with placebo there was very little additional decrease in 4βHC concentrations which remained ∼23% (P = 0.0003) lower than baseline. This finding is consistent with a literature report showing that when itraconazole was administered for 8 days in patients with onychomycosis 4βHC was reduced by 20% [21]. The result is consistent also with modelled data showing a ∼40% decrease in plasma 4βHC (vs. baseline) after 14 days of KTZ [9]. While the 4βHC dynamic range was more limited, MDZ pharmacokinetics were consistent with previously reported oral and i.v. AUC increases of 11-fold and four-fold, respectively [22]. This suggests that the utility of 4βHC may be more limited for assessing the in vivo impact of mild and moderate CYP3A inhibitors. Of note, antifungal drugs, such as KTZ, are known to reduce TC likely due to inhibition of lanosterol metabolism [23].
KTZ is a competitive inhibitor and has a short half-life with minimal accumulation after multiple dosing. Zhao et al. reported that for substrates with half-life values much longer than that of KTZ (e.g. 4βHC), a sustained inhibition covering the entire kinetic profile may be required, which can only be achieved by more frequent dosing with KTZ. Thus, dosing with KTZ 200 mg twice daily may have shown a greater magnitude in 4βHC reduction as compared with the values observed after 400 mg once daily dosing in this study [24].
While there may be differential effects of inhibitor or inducers on liver and intestinal CYP3A metabolism, we have shown that both i.v. and oral MDZ pharmacokinetics change as expected (for example, as 4βHC concentrations increased with RIF there was a decrease in MDZ exposure). These findings suggest that evaluating CYP3A enzyme induction during early drug development is possible using an endogenous probe instead of conducting another clinical pharmacology study using an external probe such as MDZ thereby saving time and cost associated with the assessment.
Diczfalusy et al. postulated that the plasma concentration of 4βHC reflects both formation as well as elimination [13]. Furthermore, the reported half-life of 4βHC is relatively long but not yet well defined, ranging from 60 h to 17 days. Interestingly, in this study, the change in 4βHC concentration reached a maximal effect 3 days following dosing with KTZ which is indicative of a relatively short half-life for 4βHC. The kinetics of this biomarker therefore remain to be assessed comprehensively [10]. This study showed not only the impact of KTZ and RIF on plasma 4βHC formation but it also compared the results with a placebo group showing the return to baseline of 4βHC after KTZ dosing is stopped.
Recently, Björkhem-Bergman and colleagues reported 4βHC after 10, 20 or 100 mg daily doses of RIF for 14 days resulting in 1.5-, 2.5- and 4-fold higher plasma concentrations of 4βHC, respectively [25]. Similarly, Shin and colleagues recently published metabolic markers such as 4βHC after administration of 600 mg RIF and 400 mg of KTZ after 10 days and 4 days of treatment, respectively. Results showed that after 10 days of treatment, 4βHC concentrations were 2.8-fold higher after RIF treatment with no appreciable change in the inhibition arm [26]. Results obtained from these studies are in accordance with the results described herein. In addition to the effect of RIF and KTZ for 14 days, which represents a typical multiple ascending dose study, we also showed the time course of 4βHC after stopping the treatment. Results from our placebo-controlled study therefore inform not only the effect of a 14 day treatment but also track the CYP3A activity in the post-induction/inhibition phase.
Since 4βHC is an endogenous marker formed as a result of cholesterol metabolism, the effects of concomitant concentrations of cholesterol were also assessed. The assessment of the effect of KTZ and RIF was evaluated before and after normalizing for TC at each time point. In addition, 4αHC was also used as a normalizing factor. In both cases, there was no substantial change in the overall conclusions with or without normalizing for TC or 4αHC. Some drugs, such as cholesterol lowering drugs, have a substantial effect on plasma cholesterol concentrations. The effect of these drugs on the formation of 4βHC is currently not known, but it is likely that normalizing 4βHC with TC and/or 4αHC concentrations in the same sample will alleviate this potentially confounding effect of changing cholesterol in the evaluation period.
Urinary 6βHCL : CL ratio values have also been proposed as an endogenous marker for CYP3A activity [27]. In addition, the combination of the CLf of 6βHCO and 6βHCL was proposed more recently by Peng et al. [7]. The authors reported CLf values for up to 7 days after itraconazole dosing that were slightly higher compared with our study. This finding could be due to a 14-day assessment in this study and the intrinsic differences between the effect of KTZ and itraconazole on CYP3A inhibition.
In the present study, we showed that after administration of KTZ, urinary 6βHCL : CL ratio as well as the CLf values of 6βHCO and 6βHCL have a greater dynamic range when compared with 4βHC values. While there are numerical differences in the dynamic range within each of the endogenous biomarker values after RIF administration, there was no change in the overall conclusion. Similar to the observations in this study, Mårde Arrhén et al. recently showed that after 2 weeks with different doses of RIF the results were similar between the 4βHC and urinary 6βHCL : CL ratio values. The plasma 4βHC ratio values increased in a dose dependent manner up to 4.06 at the highest RIF dose of 500 mg compared with baseline [28].
In all cases, the dynamic range after the administration of KTZ was limited and a more robust dynamic range was observed after RIF was administered indicating that none of the existing CYP3A activity endogenous biomarkers are of greater utility to evaluate reliably mild and moderate CYP3A inhibitors in relatively small numbers of subjects typically used in phase 1 or clinical pharmacology studies.
While CYP3A5 genotyping was not conducted in this study, it is well known that endogenous biomarkers such as 4βHC are affected by CYP3A5 genotype [8]. Diczfalusy et al. showed plasma concentrations of 4βHC in Tanzanian, Swedish and Korean populations with different numbers of CYP3A5*1 alleles. Within each population the concentration of 4βHC increased with the number of active CYP3A5*1 alleles suggesting that 1) there are interethnic differences in both the CYP3A4 activity and the frequency of active CYP3A5 alleles and 2) both CYP3A4 and CYP3A5 catalyze the formation of 4βHC [8].
Data from this study confirms that saliva data are a viable matrix to assess MDZ pharmacokinetics after an i.v. or potentially from a solid oral dosage form (tablet or capsule) administration. Saliva MDZ concentrations are contaminated with administered MDZ when a MDZ solution is administered orally. Given the effect of RIF on both intestinal and hepatic metabolism, the extent of MDZ exposure was substantially different between i.v. and oral MDZ. Furthermore, as evidenced by the current study for 1-OHMDZ, the hydroxyl metabolites of MDZ undergo metabolism and a sensitive bioanalytical assay is therefore needed for the assessment of MDZ pharmacokinetics after CYP3A induction to enable quantification of hydroxyl metabolites in plasma and saliva matrices.
In conclusion, this study shows that plasma 4βHC can be used as a minimally invasive probe to assess CYP3A induction, which may be useful especially in early drug development. While the utility of 4βHC is more limited when it comes to the assessment of CYP3A inhibitors, results from this study show that the biomarker has utility when studying strong CYP3A inducers and has a sufficient dynamic range to also detect mild and moderate inducers. The major advantage of plasma 4βHC is that it is the only known CYP3A biomarker available that does not require urine collection. However, it must be acknowledged that information regarding the contribution of intestinal CYP3A to 4βHC formation, as well as the role of transporters in governing 4βHC disposition, is lacking.
As confirmed in this report, the best utility of 4βHC assessment is during multiple dose studies in early clinical development which typically last for 7 days or longer. Further studies incorporating this biomarker in evaluating the effect of multiple dosing of new molecular entities on CYP3A activity and comparing those results to that of RIF on 4βHC concentrations observed in this study will be important to understand the full utility of this biomarker in drug development.
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare support from Bristol-Myers Squibb Co for the submitted work. All authors were stockholders and/or employees of Bristol-Myers Squibb Co. at the time the study was conducted and analyzed. There are no other relationships or activities that could appear to have influenced the submitted work.
The authors thank Dr Michael Furlong (Bioanalytical Sciences), Mr Fizal Nabie (Clinical Biomarkers) and Ms Melanie Pe Benito (Clinical Science Operations) for their assistance during the study. The authors would also like to acknowledge gratefully Dr Sabiha Mondal and the staff at PPD, LLC and the study participants.
Supporting Information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Figure S1 Plot of 6β-hydroxycortisol : cortisol ratios vs. MDZ AUC (0,∞) ratio [MDZ+perpetrator/MDZ alone] after i.v. (A) and oral (B) MDZ administration
Figure S2 Plot of formation clearance of 6β-hydroxycortisone and 6β-hydroxycortisol vs. MDZ AUC(0,∞) ratio [MDZ+perpetrator/MDZ alone] after i.v. (A) and oral (B) MDZ administration
Figure S3 Plot of 4β-hydroxycholesterol percent change from baseline vs. ratio of total formation clearance (day 14/day 1) in (A) linear scale and (B) logarithmic scale
Figure S4 Plot of 4β-hydroxycholesterol percent change from baseline vs. ratio of urinary metabolite ratio of 6β-hydroxycortisol (day 14/day 1) in (A) linear scale and (B) logarithmic scale
Table S1 Schematic of planned study design
Table S2 Summary statistics for plasma MDZ pharmacokinetics parameters
Table S3 Statistical summary of saliva MDZ pharmacokinetic parameters
Table S4 Statistical summary of plasma 1-OHMDZ pharmacokinetic parameters
Table S5 Statistical summary of urinary 6β-hydroxycortisol : cortisol ratio
Table S6 Statistical summary of formation clearance of 6β-hydroxycortisone, 6β-hydroxycortisol and combination of 6β-hydroxycortisone and 6β-hydroxycortisol
References
- 1.United States Food and Drug Administration. 2012. Guidance for Industry Drug Interaction Studies – Study Design, Data Analysis, Implications for Dosing, and Labeling Recommendations, February.
- 2.European Medicines Agency. 2012. Guideline on the Investigation of Drug Interactions CPMP/EWP/560/95/Rev. 1.
- 3.Lund E, Diczfalusy U, Björkhem I. On the mechanism of oxidation of cholesterol at C-7 in a lipoxygenase system. J Biol Chem. 1992;267:12462–12467. [PubMed] [Google Scholar]
- 4.Breuer O. Identification and quantitation of cholest-5-ene-3β,4β diol in rat liver and human plasma. J Lipid Res. 1995;36:2275–2281. [PubMed] [Google Scholar]
- 5.Dresser GK, Spence JD, Bailey DG. Pharmacokinetic-pharmacodynamic consequences and clinical relevance of cytochrome P450 3A4 inhibition. Clin Pharmacokinet. 2000;38:41–57. doi: 10.2165/00003088-200038010-00003. [DOI] [PubMed] [Google Scholar]
- 6.Ged C, Rouillon JM, Pichard L, Combalbert J, Bressot N, Bories P, Michel H, Beaune P, Maurel P. The increase in urinary excretion of 6 β-hydroxycortisol as a marker of human hepatic cytochrome P450IIIA induction. Br J ClinPharmacol. 1989;28:373–387. doi: 10.1111/j.1365-2125.1989.tb03516.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peng CC, Templeton I, Thummel KE, Davis C, Kunze KL, Isoherranen N. Evaluation of 6β-hydroxycortisol, 6β-hydroxycortisone, and a combination of the two as endogenous probes for inhibition of CYP3A4 in vivo. Clin Pharmacol Ther. 2011;89:888–895. doi: 10.1038/clpt.2011.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Diczfalusy U, Miura J, Roh H-K, Mirghani RA, Sayi J, Larsson H, Bodin KG, Allqvist A, Jande M, Kim J-W, Aklillu E, Gustafsson LL, Bertilsson L. 4b-hydroxycholesterol is a new endogenous CYP3A marker: relationship to CYP3A5 genotype, quinine 3-hydroxylation and sex in Koreans, Swedes and Tanzanians. Pharmacogenet Genomics. 2008;18:201–208. doi: 10.1097/FPC.0b013e3282f50ee9. [DOI] [PubMed] [Google Scholar]
- 9.Bodin K, Bretillon L, Aden Y, Bertilsson L, Broomé U, Einarsson C, Diczfalusy U. Antiepileptic drugs increase plasma levels of 4β -hydroxycholesterol in humans. J Biol Chem. 2001;276:38685–38689. doi: 10.1074/jbc.M105127200. [DOI] [PubMed] [Google Scholar]
- 10.Goodenough AK, Onorato JM, Ouyang Z, Chang S, Rodrigues AD, Kasichayanula S, Huang SP, Turley W, Burrell R, Bifano M, Jemal M, LaCreta F, Tymiak A, Wang-Iverson D. Quantification of 4-beta-hydroxycholesterol in human plasma using automated sample preparation and LC-ESI-MS/MS analysis. Chem Res Toxicol. 2011;19:1575–1585. doi: 10.1021/tx2001898. [DOI] [PubMed] [Google Scholar]
- 11.Yang Z, Rodrigues AD. Does the long plasma half-life of 4beta-hydroxycholesterol impact its utility as a cytochrome P450 3A (CYP3A) metric? J Clin Pharmacol. 2010;50:1330–1338. doi: 10.1177/0091270009360041. [DOI] [PubMed] [Google Scholar]
- 12.Diczfalusy U, Nylén H, Elander P, Bertilsson L. 4β-hydroxycholesterol, an endogenous marker of CYP3A4/5 activity in humans. Br J Clin Pharmacol. 2011;71:183–189. doi: 10.1111/j.1365-2125.2010.03773.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Diczfalusy U, Kanebratt KP, Bredberg E, Andersson TB, Böttiger Y, Bertilsson L. 4β-hydroxycholesterol as an endogenous marker for CYP3A4/5 activity: stability and half-life of elimination after induction with rifampicin. Br J Clin Pharmacol. 2009;67:38–43. doi: 10.1111/j.1365-2125.2008.03309.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bodin K, Andersson U, Rystedt E, Ellis E, Norlin M, Pikuleva I, Eggertsen G, Björkhem I, Diczfalusy U. Metabolism of 4β-hydroxycholesterol in humans. J Biol Chem. 2002;277:31534–31540. doi: 10.1074/jbc.M201712200. [DOI] [PubMed] [Google Scholar]
- 15.Olkkola KT, Ahonen J. Midazolam and other benzodiazepines. Hand Exp Pharmacol. 2008;182:335–360. doi: 10.1007/978-3-540-74806-9_16. [DOI] [PubMed] [Google Scholar]
- 16.Nordt SP, Clark RF. Midazolam: a review of therapeutic uses and toxicity. J Emerg Med. 1997;15:357–365. doi: 10.1016/s0736-4679(97)00022-x. [DOI] [PubMed] [Google Scholar]
- 17.Link B, Haschke M, Grignaschi N, Bodmer M, Aschmann YZ, Wenk M, Krähenbühl S. Pharmacokinetics of intravenous and oral midazolam in plasma and saliva in humans: usefulness of saliva as matrix for CYP3A phenotyping. Br J Clin Pharmacol. 2008;66:473–484. doi: 10.1111/j.1365-2125.2008.03201.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Soars MG, Petullo DM, Eckstein JA, Kasper SC, Wrighton SA. An assessment of UDP-glucuronosyltransferase induction using primary human hepatocytes. Drug Metab Dispos. 2004;32:140–148. doi: 10.1124/dmd.32.1.140. [DOI] [PubMed] [Google Scholar]
- 19.Tomalik-Scharte D, Lütjohann D, Doroshyenko O, Frank D, Jetter A, Fuhr U. Plasma 4beta-hydroxycholesterol: an endogenous CYP3A metric? Clin Pharmacol Ther. 2009;86:147–153. doi: 10.1038/clpt.2009.72. [DOI] [PubMed] [Google Scholar]
- 20.FDA. 2012. FDA Draft Guidance for Industry Drug Interaction Studies – Study Design, Data Analysis, Implications for Dosing, and Labeling Recommendations, February.
- 21.Lütjohann D, Marinova M, Schneider B, Oldenburg J, von Bergmann K, Bieber T, Björkhem I, Diczfalusy U. 4beta-hydroxycholesterol as a marker of CYP3A4 inhibition in vivo – effects of itraconazole in man. Int J Clin Pharmacol Ther. 2009;47:7097–7115. doi: 10.5414/cpp47709. [DOI] [PubMed] [Google Scholar]
- 22.Tsunoda SM, Velez RL, von Moltke LL, Greenblatt DJ. Differentiation of intestinal and hepatic cytochrome P450 3A activity with use of midazolam as an in vivo probe: effect of ketoconazole. Clin Pharmacol Ther. 1999;66:461–471. doi: 10.1016/S0009-9236(99)70009-3. [DOI] [PubMed] [Google Scholar]
- 23.Gylling H, Vanhanen H, Miettinen TA. Effects of ketoconazole on cholesterol precursors and low density lipoprotein kinetics in hypercholesterolemia. J Lipid Res. 1993;34:59–67. [PubMed] [Google Scholar]
- 24.Zhao P, Ragueneau-Majlessi I, Zhang L, Strong JM, Reynolds KS, Levy RH, Thummel KE, Huang SM. Quantitative evaluation of pharmacokinetic inhibition of CYP3A substrates by ketoconazole: a simulation study. J Clin Pharmacol. 2009;49:351–359. doi: 10.1177/0091270008331196. [DOI] [PubMed] [Google Scholar]
- 25.Björkhem-Bergman L, Bäckström T, Nylén H, Rönquist-Nii Y, Bredberg E, Andersson TB, Bertilsson L, Diczfalusy U. Comparison of endogenous 4β-hydroxycholesterol with midazolam as markers for CYP3A4 induction by rifampicin. Drug Metab Dispos. 2013;41:1488–1493. doi: 10.1124/dmd.113.052316. [DOI] [PubMed] [Google Scholar]
- 26.Shin KH, Choi MH, Lim KS. Evaluation of endogenous metabolic markers of hepatic CYP3A activity using metabolic profiling and midazolam clearance. Clin Pharmacol Ther. 2013;94:601–609. doi: 10.1038/clpt.2013.128. [DOI] [PubMed] [Google Scholar]
- 27.Boulton DW, Arnaud P, DeVane CL. A single dose of methadone inhibits cytochrome P-4503A activity in healthy volunteers as assessed by the urinary cortisol ratio. Br J Clin Pharmacol. 2001;51:350–354. doi: 10.1046/j.1365-2125.2001.01360.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mårde Arrhén Y, Nylén H, Lövgren-Sandblom A, Kanebratt KP, Wide K, Diczfalusy U. A comparison of 4β-hydroxycholesterol/cholesterol and 6β-hydroxycortisol/cortisol as markers of CYP3A4 induction. Br J Clin Pharmacol. 2013;75:1536–1540. doi: 10.1111/bcp.12016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Plot of 6β-hydroxycortisol : cortisol ratios vs. MDZ AUC (0,∞) ratio [MDZ+perpetrator/MDZ alone] after i.v. (A) and oral (B) MDZ administration
Figure S2 Plot of formation clearance of 6β-hydroxycortisone and 6β-hydroxycortisol vs. MDZ AUC(0,∞) ratio [MDZ+perpetrator/MDZ alone] after i.v. (A) and oral (B) MDZ administration
Figure S3 Plot of 4β-hydroxycholesterol percent change from baseline vs. ratio of total formation clearance (day 14/day 1) in (A) linear scale and (B) logarithmic scale
Figure S4 Plot of 4β-hydroxycholesterol percent change from baseline vs. ratio of urinary metabolite ratio of 6β-hydroxycortisol (day 14/day 1) in (A) linear scale and (B) logarithmic scale
Table S1 Schematic of planned study design
Table S2 Summary statistics for plasma MDZ pharmacokinetics parameters
Table S3 Statistical summary of saliva MDZ pharmacokinetic parameters
Table S4 Statistical summary of plasma 1-OHMDZ pharmacokinetic parameters
Table S5 Statistical summary of urinary 6β-hydroxycortisol : cortisol ratio
Table S6 Statistical summary of formation clearance of 6β-hydroxycortisone, 6β-hydroxycortisol and combination of 6β-hydroxycortisone and 6β-hydroxycortisol