

Abstract
Statin-associated muscular adverse effects cover a wide range of symptoms, including asymptomatic increase of creatine kinase serum activity and life-threatening rhabdomyolysis. Different underlying pathomechanisms have been proposed. However, a unifying concept of the pathogenesis of statin-related muscular adverse effects has not emerged so far. In this review, we attempt to categorize these mechanisms along three levels. Firstly, among pharmacokinetic factors, it has been shown for some statins that inhibition of cytochrome P450-mediated hepatic biotransformation and hepatic uptake by transporter proteins contribute to an increase of systemic statin concentrations. Secondly, at the myocyte membrane level, cell membrane uptake transporters affect intracellular statin concentrations. Thirdly, at the intracellular level, inhibition of the 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase results in decreased intracellular concentrations of downstream metabolites (e.g. selenoproteins, ubiquinone, cholesterol) and alteration of gene expression (e.g. ryanodine receptor 3, glycine amidinotransferase). We also review current recommendations for prescribers.
Keywords: adverse effects, efflux transporters, myopathy, organic anion transporters, rhabdomyolysis, statins
Introduction
Statins (3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors) are the most important class of lipid-lowering drugs. The beneficial effects of statins in coronary artery disease have been shown in large randomized trials 1,2 and statins have an acceptable safety record. However, rhabdomyolysis is a well-recognized severe, although rare, complication of statin therapy. Less severe muscular adverse effects occur more frequently. Statin use was shown to be an independent factor associated with muscular complaints in primary care patients 3.
While muscular adverse affects of lipid-lowering agents have been the subject of several review articles 4–9, our review focuses on insight into mechanisms of statin-related muscular adverse effects and recommendations to prevent complications of statin-related myopathy.
Methods
A literature search of MEDLINE was performed for articles published from 1966 through January 2014, using the following MeSH (Medical Subject Heading) terms: ‘hydroxymethylglutaryl-CoA reductase inhibitor’, ‘antilipidaemic agents’, ‘anticholesterolaemic agents’, ‘hyperlipidaemias’, ‘lipids’, ‘cholesterol’, ‘drug therapy’, ‘statin’ matched with ‘muscular diseases’, ‘muscle cramps’, ‘pain’, ‘muscles’. From the references of relevant articles, we extracted additional literature relevant to the topic.
Types of statin-associated skeletal muscular adverse effects
Commonly, muscular adverse effects of statins are collectively termed as myopathy and present a broad range of clinical symptoms and signs. The term myopathy includes muscular pain, tenderness, cramps, heaviness, stiffness or weakness 10, but no consensus on the definition exists 11 (Table 1).
Table 1.
Definitions of muscular adverse effects of statin therapy (according to ACC/AHA/NHLBI and NLA and FDA* 11 86)
| Asymptomatic CK elevation | Elevation of CK serum activity without muscular complaints |
| Myopathy† | Every kind of muscular complaints |
| Myalgia‡ | Muscle pain or weakness without CK elevation |
| Myositis‡ | Muscular complaints with CK elevation |
| Rhabdomyolysis§ | Muscular complaints with pronounced CK elevation (typically more than 10 times the ULN) with elevation of creatinine serum concentration, commonly with brown urine and myoglobinuria, histopathologic findings of myositis or myolysis and serum electrolyte abnormalities |
CK, creatine kinase; ULN, upper limit of normal.
ACC/AHA/NHLBI, American College of Cardiology/American Heart Association/National Heart, Lung and Blood Institute; FDA, U.S. Food and Drug Administration; NLA, National Lipid Association.
NLA and FDA include a 10-fold ULN creatine kinase elevation for the definition of myopathy.
not defined by the NLA and the FDA.
FDA, CK >50 times ULN and evidence of organ damage such as renal compromise; NLA, CK >10 000 IU l−1 or >10 times ULN plus an elevation in serum creatinine or medical intervention with intravenous hydration.
Frequency of myopathy and rhabdomyolysis
Different frequencies of statin myopathy have been reported. Rhabdomyolysis is a rare event. On the other hand, less severe side effects occur more frequently. For example, myalgia is reported as frequently as 2 to 10.5%. These diverging rates are probably due to variable assessment stringency 12, which includes different definitions, assessment methods and reporting biases (e.g. drug reporting systems, awareness and publicity) (see Table 2). In addition, most statin clinical trials were not designed to assess specifically muscle-related complaints 12. Only few clinical trials are of sufficient size and duration to detect rhabdomyolysis 13.
Table 2.
Examples of frequencies of muscular adverse effects associated with lipid-lowering drug therapy
| Rhabdomyolysis | 3339 cases of rhabdomyolysis, MedWatch system of the American Food and Drug Administration, observation from 1990 to 2002 4. |
| Death rate of 1.5 cases of rhabdomyolysis per 10 million statin prescriptions from 1987 to 1997. 10–50 times higher risk of fatal rhabdomyolysis on cerivastatin, positive dose-related tendency, positive association with gemfibrozil 92. | |
| Incidence of hospitalized rhabdomyolysis [per 10,000 person-years] 30: | |
| 0.44 (atorvastatin, pravastatin or simvastatin), | |
| 5.34 (cerivastatin), | |
| 2.82 (fibrate), | |
| 5.98 (fibrate + atorvastatin, pravastatin or simvastatin), | |
| 1,035 (cerivastatin + fibrate) | |
| Myalgia | 1.5% in both groups (n = 521) with hypercholesterolaemia developed myalgia during a 6 month study period (randomized double-blind simvastatin 40 vs. 80 mg day−1), no controls 93. |
| 2% of patients treated with statins (n = 3500) had myalgia (meta-analysis of patients receiving different doses and substances), no controls 94. | |
| 6%, not differing from placebo groups (randomized, simvastatin 40 mg day−1, 20,000 patients with coronary heart disease, 5 years of follow-up) 95. | |
| 7% of patients treated with statins (cross-sectional study), no controls 96. | |
| 2.7% (pravastatin, mostly 40 mg day−1) and 3.3% (atorvastatin, mostly 80 mg day−1) of patients after acute coronary syndromes (n = 4100) during 2 years of follow-up (no controls) 97. | |
| 2.2% (atorvastatin, 80 mg day−1) and 1.1% (simvastatin, 20 mg day−1) of patients with previous myocardial infarction (n = 8888), no controls 98. | |
| Myositis | No cases in the atorvastatin (80 mg day−1) or placebo group (randomized) of patients after acute coronary syndrome (n = 3000), 4 months of follow-up 99. |
| Unspecified | Muscular symptoms in 10.5% of patients on high dose statin therapy (n = 7900), cross-sectional, no controls 100. |
| 2 to 3 myopathy cases per year and 10,000 hyperlipidaemic patients taking statins, population-based follow-up, significantly differing from controls (n = 0 cases) 101. |
Risk factors
Several patient-related risk factors of statin-associated myopathy have been identified (Table 3). An increase of creatine kinase (CK) serum activity and muscle discomfort were reported with statin therapy after strong physical exertion 14. Muscle pain can also be induced by physical exertion with statin therapy without an increase of CK serum activity 15.
Table 3.
| Female gender |
| Hypothyroidism |
| Viral infections |
| Advanced age (>80 years of age in particular) |
| Small body frame and frailty |
| Multisystem disease (e.g. chronic renal insufficiency, especially due to diabetes mellitus) |
| Hepatic disorders |
| Peri-operative periods |
| Injuries |
| Alcohol abuse |
| Polypharmacotherapy |
| Specific concomitant medications (see Table 4) |
| Physical exertion |
Patients with inborn metabolic muscle diseases such as myophosphorylase deficiency (McArdle disease), carnitine palmitoyltransferase II deficiency or myoadenylate deaminase deficiency are more susceptible to the development of muscle symptoms on statin therapy. Genetic variants in these enzymes, either in a heterozygous or homozygous manner, have been found more frequently in symptomatic patients (10%) than in asymptomatic patients (3%) on statin therapy 16. An association between carrying a specific allele of dystrophia myotonica-protein kinase (DMPK) and myalgia in statin users was found 17.
The concept of a dose-dependent increased risk of statin-related muscular adverse effects is supported by the results of a meta-analysis. Overall, the observed excess of rhabdomyolysis was 4 per 10 000 patients with more intensive vs. less intensive statin therapy compared with 1 per 10 000 patients on standard statin regimens vs. control (at least 2 years follow-up). All of the excess with more intensive therapy occurred in trials of 80 mg vs. 20 mg simvastatin daily 18. Again, in the SEARCH study with more than 12 000 survivors of myocardial infarction, myopathy was observed in two (0.03%) cases among patients taking 20 mg simvastatin daily and in 53 (0.9%) cases in the 80 mg group (6 years mean follow-up) 19. As a result of these data, the Food and Drug Administration issued safety-labelling changes for simvastatin 20 (see below, Recommendation for myopathy prevention in lipid-lowering drug therapy).
In addition to high doses of statins specific concomitant medications (Table 4) appear to increase the risk of statin-associated myopathy. Among 601 cases of statin-associated rhabdomyolysis, 99 patients concomitantly used mibefradil, 80 fibrates, 51 ciclosporin, 42 macrolide antibiotics, 33 warfarin, 26 digoxin, 12 azole antifungals 21. Some of these drugs have the potential to inhibit the metabolism of most statins leading to an increase of statin concentrations. Grapefruit juice contains inhibitors, furanocoumarin derivatives, of statin metabolism 22. There are case reports of CK increases with or without myalgia after adding ezetimibe to a well-tolerated statin therapy 23.
Table 4.
| FDA recommendations for patients on simvastatin. | |
|---|---|
| High dose of statin | See text |
| Gemfibrozil | Contraindicated with simvastatin |
| Ezetimibe (?) | – |
| Nicotinic acid | – |
| Ciclosporin | Contraindicated with simvastatin |
| Azole antifungal agents (itraconazole, ketoconazole, posaconazole) | Contraindicated with simvastatin |
| Macrolide (erythromycin, clarithromycin) and ketolide antibiotics (telithromycin) | Contraindicated with simvastatin |
| HIV protease inhibitors | Contraindicated with simvastatin |
| Danazole | Contraindicated with simvastatin |
| Verapamil, diltiazem | Do not exceed 10 mg day−1 of simvastatin |
| Amiodarone | Do not exceed 10 mg day−1 of simvastatin |
| Ranolazine | Do not exceed 20 mg day−1 of simvastatin |
| Amlodipine | Do not exceed 20 mg day−1 of simvastatin |
| Grapefruit juice | Avoid large quantities (>1 qt day−1)* of grapefruit juice |
1 qt is approximately 1 litre. FDA, U.S. Food and Drug Administration.
In a recently published population-based cohort study, coprescription of a statin with clarithromycin or erythromycin, within 30 days of the antibiotic prescription, was significantly associated with a higher risk for hospitalization with rhabdomyolysis (absolute risk increase, 0.02%; relative risk, 2.17) 24.
Suggested underlying pathomechanisms
As of now, the exact pathomechanism of statin-associated myopathy is unclear. A number of factors have been discussed. Since high dosage of statins predisposes to adverse muscular effects, pharmacokinetic factors are of interest when considering total body burden of xenobiotics. At the level of the injured organ, i.e. muscle, transport of statins across myocyte cell membranes is of additional interest. Also, mechanisms acting at the intracellular level may be distinguished
Pharmacokinetic factors
Drug metabolism
In long term statin treatment, clearance by hepatic metabolism is the principal determinant of concentration (Figure 1). Most statins undergo CYP3A4-mediated biotransformation. Atorvastatin, a substrate of CYP3A4, is converted in vivo to its lactone. In a comparative study, patients with atorvastatin-related myopathy had higher plasma concentrations of atorvastatin lactone and CYP3A4-generated metabolites, o- and p-hydroxyatorvastatin, than healthy volunteers, but not significantly different plasma concentrations of the parent compound 25. Interestingly, high atorvastatin lactone, but low hydroxylated metabolite concentrations were found in patients receiving concomitant treatment with a CYP3A4 inhibitor 26. Concomitant treatment with an inhibitor of CYP3A4 has frequently been reported in patients developing statin-associated myopathy and rhabdomyolysis 21,27. In persons taking simvastatin, lovastatin or atorvastatin, 60% of cases of rhabdomyolysis involved drugs known to inhibit CYP3A4, such as erythromycin and azole antifungals 1,28.
Figure 1.
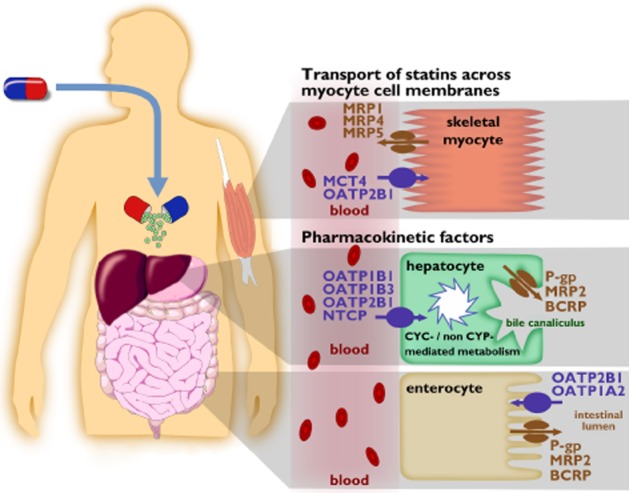
Drug targets potentially involved in statin myopathy. 1) Pharmacokinetic factors, affecting total body burden of statins (lower part). Pharmacokinetic factors include (A) uptake into hepatocytes by membrane transporters, i.e. OATP1B1, OATP1B3, OATP2B1 and NTCP, (B) hepatic biotransformation (CYP-mediated, non-CYP-mediated) to metabolites (e.g. acids, lactones) and (C) efflux transport into the bile canaliculi via P-gp, MRP2 and BCRP. Uptake of statins into enterocytes from the luminal site is mediated by OATP2B1 and OATP1A2. Efflux transporters involved in the secretion of statins from enterocytes into the intestinal lumen are P-gp, MRP2 and BCRP. 2) Factors related to the transport of statins across muscle cell membranes (upper part). In sarcolemmal membrane of human skeletal muscle MCT4 is expressed. Moreover intracellular concentrations of statins are affected by the uptake transporter proteins OATP2B1 and the efflux transporters MRP1, MRP4, and MRP5.  , uptake transporter;
, uptake transporter;  , efflux transporter
, efflux transporter
CYP3A4 inhibition, however, does not account for all cases of statin-related myopathy. Some cases were also reported with non-substrates of CYP3A4, e.g. fluvastatin (a substrate of CYP2C9) and pravastatin (no relevant CYP-dependent metabolism) 21. Gemfibrozil and its glucuronide metabolite do not inhibit CYP3A4 29. They do however inhibit CYP2C8 which is important in the metabolism of cerivastatin. Cerivastatin was withdrawn from the market in 2001. It was more frequently associated with fatal rhabdomyolysis than other statins. Interestingly, the combination of cerivastatin and gemfibrozil was associated with a particularly high risk 30. Gemfibrozil decreases considerably the area under the concentration–time curve (AUC) of the metabolite of cerivastatin formed by CYP2C8 and increases considerably the AUC of cerivastatin and its lactone, as well as the metabolite formed by CYP3A4 31. In the mechanism of interaction with gemfibrozil coadministration, glucuronidation has also been postulated as a potential factor. Gemfibrozil was shown to inhibit atorvastatin glucuronidation in vitro to a minor degree 32.
In a case series (n = 82), insufficient and low serum vitamin D concentrations have been associated with statin-treated patients with myalgia. Thirty-eight of the 82 myalgic vitamin D deficient patients were given vitamin D while continuing statins. Interestingly, in 35 of these 38 patients, myalgia disappeared 33,34. According to these findings, vitamin D deficiency was suggested to potentiate statin-induced myalgia and/or that statins cause vitamin D deficiency. Other studies found increased serum vitamin D concentrations following treatment with atorvastatin (or rosuvastatin) 35,36. 1,25-dihydroxycholecalciferol, the active metabolite of vitamin D, binds to the vitamin D receptor, activating CYP3A4 that metabolizes atorvastatin 37, so it has been suggested that low serum vitamin D might reduce CYP3A4 activity, increasing atorvastatin concentrations 38 and therefore statin toxicity. Indeed, supplementation of vitamin D in a small group of atorvastatin-treated patients (n = 16) lowered serum atorvastatin and its active metabolites 39. However, there is not enough evidence to recommend vitamin D supplementation as treatment for statin-associated muscle complaints in the absence of low vitamin D concentrations 40.
Drug transport
Transport processes play an important role in drug absorption, distribution and excretion (Figure 1). Statins have been found to be substrates of several organic anion transporting polypeptide (OATPs) transport proteins 41. Accordingly, polymorphisms of SLCO1B1 (solute carrier organic anion transporter family, member 1B1), encoding OATP1B1, an uptake transporter expressed on the sinusoidal membrane of human hepatocytes 42, were shown to affect markedly the pharmacokinetics of simvastatin but also to a lesser degree other statins 43. The SLCO1B1 c.521T>C single nucleotide polymorphism (SNP) is common (15–20% in Caucasians) and has been associated with reduced activity of OATP1B1 in vitro. In healthy volunteers carrying the SLCO1B1 c.521CC genotype, the AUC of simvastatin acid is significantly higher as compared with the c.521TC and c.521TT genotypes 44,45.
The SEARCH Collaborative Group (Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine) identified a strong association between simvastatin-related myopathy and the SLCO1B1 c.521T>C (rs4363657) genetic variant (P < 1.6 × 10−7). In this genome-wide association study, 300 000 genetic markers were screened in 85 subjects with definite and incipient myopathy and in matched controls. All of them were taking 80 mg simvastatin day–1 and were participants of a large clinical trial involving approximately 12.000 participants. The prevalence of the c.521C allele was 46% in cases of myopathy and 13% in controls. The odds ratio for myopathy was 4.5 (95% confidence interval [CI], 2.6, 7.7) in heterozygotes, and 16.9 (95% CI, 4.7, 61.1) in homozygous variant patients for c.521C allele as compared with the references sequence 46. In data from the SEARCH study, the clinical sensitivity was 70%, specificity 74%, positive predictive value 4.1% and negative predictive value 99% of a test for the C allele to predict definite or incipient myopathy during 5 years of 80 mg day−1 simvastatin use 47.
This SNP also alters the pharmacokinetics of pravastatin, a hydrophilic substrate of OATP1B1. Higher pravastatin concentrations with single dosing were found in carriers of the SLCO1B1 c.521CC genotype 48,49. Also, with multiple dosing, higher plasma concentrations of pravastatin were found in carriers of a SLCO1B1 variant haplotype group 50. Haplotypes have been found to be more informative in predicting the SLCO1B1 phenotype than single SNPs 49. The frequency of the SLCO1B1*15 haplotype, in which the c.521T>C SNP exists, is significantly higher in Japanese patients who experienced myopathy after receiving pravastatin or atorvastatin than in patients without myopathy 51. In the STRENGTH (Statin Response Examined by Genetic Haplotype Markers) study, the SLCO1B1*5 haplotype was found in 37% of patients (n = 99) with a composite adverse event (discontinuation for any side effect, myalgia or CK >three times the ULN). In those without adverse events (n = 410) this haplotype was found in 25% 52. A recent analysis of data from the JUPITER (Justification for Use of Statins in Prevention) placebo-controlled trial showed no increased risk of myalgia among users of rosuvastatin who carry the rs4363657C or the rs4149056C allele in SLCO1B1 53.
In addition to metabolism, transporter proteins are also be involved in drug–drug interactions. The gemfibrozil–cerivastatin interaction may also have a basis on the transport level since gemfibrozil and its glucuronide have been shown to inhibit the OATP1B1-mediated hepatic uptake of statin acids 31.
Moreover other membrane transporters are involved in hepatic statin transport. The transporters OATP1B3, OATP2B1 and the Na+-taurocholate cotransporting polypeptide (NTCP) 42,54,55 are expressed at the sinusoidal membrane, thereby involved in the uptake of statins. The ATP dependent transporters P-glycoprotein (P-gp, ABCB1), multidrug resistance-associated protein 2 (MRP2) and breast cancer resistance protein [BCRP, ABCG2]) 54,56 are efflux transporters and are expressed at the canalicular membrane with consequences for statins. Although genetic variation of some of these uptake and efflux transporter proteins have been reported to affect pharmacokinetics of statins (e.g. pravastatin) 43, so far it is unclear on whether genetic but also epigenetic and/or non-genetic factors (e.g. drug–drug interaction) may significantly increase a patient’s susceptibility for statin-related myopathy.
Intestinal secretion is also important for the pharmacokinetics of statins. Efflux transporters are involved in the secretion from intestinal cells into the intestinal lumen. Among those, P-gp, MRP2 and BCRP are suggested to be important 43,57,58. Ciclosporin, a potent inhibitor of several membrane transporters (e.g. P-gp), increases the AUC and the peak plasma concentration of pravastatin (a statin not depending on metabolism by CYP). This interaction profile may be due to an increased bioavailability of statins by inhibition of intestinal efflux transporters by ciclosporin 31. In addition the membrane proteins OATP2B1 and OATP1A2 59,60 contribute to statin uptake from the luminal site of enterocytes. However, the clinical impact of intestinal transporter proteins on statin-related myopathy and particularly the contribution of genetic variation is unclear and needs further investigation.
Transport of statins across the myocyte cell membrane
To get into myocytes, xenobiotics may undergo passive diffusion and/or active transport. Proteins involved in the transport of drugs across cell membranes may be assumed to modulate drug concentration within myocytes by mediating influx and efflux of statins. Thus, transport protein function at the cellular level of myocytes could be involved in the development of myopathy. Statins are considered to be substrates of monocarboxylate transporter-4 (MCT4) mediating the uptake of drugs. MCT4 is expressed in skeletal muscle. Inhibition of MCT4 abolished simvastatin-induced alteration in calcium homeostasis 61 and statin-related inhibition of L-lactic acid transport is mediated by MCT4 potentially leading to alteration of muscle homeostasis 62. However, whether MCT4 and/or other transporters of statins across membranes of myocytes play a role in myopathy is unclear. More recently, the uptake transporter OATP2B1 (SLCO2B1) and the efflux transporters multidrug resistance-associated proteins 1 (MRP1, ABCC1), 4 (MRP4 ABCC4) and 5 (MRP5, ABCC5) have been shown to be expressed on the sarcolemmal membrane of human skeletal muscle fibres proposing a role for OATP2B1 in sensitizing skeletal muscle cells to statin toxicity and for the statin efflux transporters MRP1, MRP4 and MRP5 in protection for muscle from toxicity 63.
Mechanisms acting at the intracellular level
Statins inhibit HMG-CoA reductase, mediating the conversion of 3-hydroxy-3-methyl-glutaryl-CoA to mevalonate and subsequently to isopentenyl pyrophosphate (IPP). Concentrations of metabolites downstream to this HMG-CoA reductase-mediated reaction may be assumed to be decreased by statins (Figure 2). Among those are selenoproteins (a), ubiquinone (coenzyme Q10, CoQ10) (b) and prenylated proteins (c). In addition, depletion of cholesterol itself (d) may be involved as a key process. Also, impairment of mitochondrial function (e), atrogin-1 as a critical mediator (f), MHC-I (major histocompatibility complex I) expression (g) and the up-regulation of the expression of ryanodine receptor 3, a protein located in the T-tubule membrane involved in calcium release (h), have been implicated.
Impairment of the enzymatic isopentenylation of selenocystein-tRNA (Sec-tRNA) results in a decrease of available selenoproteins. This concept was generated because selenium deficiency shares common features with statin myopathy. Prominent examples of human selenoproteins are key regulators of cellular redox state. However, selenium substitution as a general co-treatment to statins is not recommended 64,65.
CoQ10 is involved in mitochondrial energy production and consequently total cell integrity 66. Polymorphisms of the CoQ2 gene, which encodes the second enzyme in the CoQ10 biosynthetic pathway, have been associated with increased odds of statin myopathy 67. An association between carrying a specific allele of CoQ2 (rs4693570) and myalgia in statin users was described 17. However, in the Clinical Practice Research Datalink study, no such association was found 68. As of now, evidence that CoQ10 has an aetiologic role in statin-associated myopathy is insufficient, and the routine use of CoQ10 in statin-treated patients is not recommended 69,70.
Farnesyl pyrophosphate and geranyl-geranyl pyrophosphate, metabolites downstream of IPP, are used for the prenylation of various proteins which are essential for cytoskeleton and cell membrane integrity 64,71.
-
The depletion of cholesterol itself has been proposed to be the key to the understanding of statin myotoxicity. Statin therapy is associated with ultrastructural damage in skeletal muscle with characteristic disruption of the T-tubular system. These findings are recapitulated by extraction of membrane cholesterol in vitro 72. It is consistent with this hypothesis that all lipid-lowering agents (i.e. statins, fibrates, nicotinic acid or others) increase the vulnerability of skeletal muscle cells by reducing the cholesterol content of cell membranes 4. However, not all findings support this hypothesis. Squalene is the direct precursor of cholesterol, and the inhibition of squalene synthase does not cause myotoxicity in in vitro systems 73,74.
Using a novel in vitro cell-based screening method for gene-by-treatment effects on transcriptional expression, glycine amidinotransferase (GATM) was recently identified as a genetic locus associated with statin-induced myopathy. It is proposed that simvastatin reduced GATM expression in carriers of the rs9806699 SNP, reducing creatine availability and creatine phosphate storage 75. Possibly, reduced creatine phosphate storage modifies skeletal muscle cellular energy pathways leading to reduced susceptibility to statin myopathy 76.
-
As an additional mechanism acting at the cellular level, simvastatin has been shown to impair mitochondrial function by interfering with the respiratory chain. Subsequently, mitochondrial membrane depolarization and Ca2+ efflux to the cytoplasm by permeability transient pore or Na+/Ca2+ exchanger may occur. This might be an important step triggering muscle fibre death 61,77. The mitochondrion plays a central role in regulating apoptosis. It has been shown that statins can induce apoptosis in a variety of cell types, including skeletal myocytes.
Simvastatin impaired ADP-stimulated mitochondrial respiration supported by complex I substrates in differentiated primary human skeletal muscle cells. Simvastatin also induced mitochondrial oxidative stress, demonstrated by increased levels of reactive oxygen species in concert with the up-regulation of the mitochondrial-mediated apoptotic mechanisms in primary human skeletal myotubes, suggesting that simvastatin induces cell death through oxidative stress 78.
Furthermore, experiments suggest that the muscle atrophy-linked protein, atrogin-1, may be a critical mediator of muscle damage induced by statins. The background is that the ubiquitin-proteasome pathway (UPP) is the main intracellular system for protein degradation in atrophying muscle and atrogin-1 is among the UPP components. Peroxisome proliferator-activated receptor-gamma (PPAR-γ) cofactor-1 alpha (PGC-1α) is a mitochondrial biogenesis regulator. The beneficial effect of PGC-1α expression in reducing statin-associated muscle injury suggests that increased number and/or improved function of mitochondria may be central to maintain muscle integrity. Forced overexpression of PGC-1α suppresses statin-induced atrogin-1 expression and protects from statin-induced muscle damage. Statins induce marked expression of atrogin-1 in human skeletal muscle, cultured muscle cells and in animal models of statin myopathy in zebrafish. Moreover, in the absence of atrogin-1, cells and animals are less vulnerable to the toxic effects of statins 79. Statin-induced muscle damage and atrogin-1 induction is the result of a geranylgeranylation defect 80.
In patients with statin-induced necrotizing myopathy, diffuse or multifocal up-regulation of MHC-I expression (major histocompatibility complex) was also found in non-necrotic muscle fibres. This up-regulation may be due to an endoplasmatic reticulum stress response 81.
A significant muscle injury was observed among 44 myopathic patients who were receiving statins or who were discontinuing statin use (median 12 weeks ago) compared with a control group (n = 39) without muscular symptoms with or without statin use (19 and 20 patients, respectively). A typical histo-pathological appearance of statin-associated myopathy, characterized by vacuolization of the T-tubular system, was identified. Significant damage was defined as 2% or more damaged fibres per biopsy sample and was associated with a significant expression of ryanodine receptor 3 mRNA. Increased expression of ryanodine receptor 3, a protein located in the T-tubule membrane, could represent a potential defect in calcium homeostasis which could result in myofibre damage in statin users 82.
Figure 2.
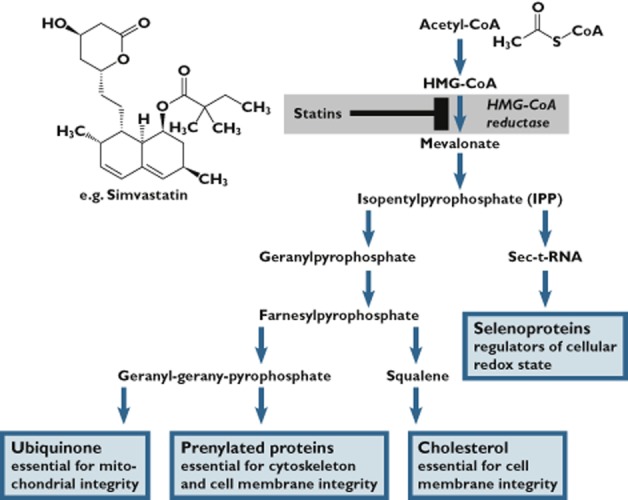
Biochemical pathways in cholesterol biosynthesis possibly related to statin-associated myopathy [adapted from 65. Reprinted from Trends in Cardiovascular Medicine, Vol. 14, Moosmann and Behl, ‘Selenoproteins, Cholesterol-Lowering Drugs, and the Consequences Revisiting of the Mevalonate Pathway’, Pages 273–281, Copyright 2004, with permission from Elsevier.]. Statins inhibit HMG-CoA reductase, mediating the conversion of 3-hydroxy-3-methyl-glutaryl-CoA to mevalonate and subsequently to isopentenyl pyrophosphate (IPP). Statins may decrease concentrations of metabolites downstream of IPP like selenoproteins, ubiquinone (coenzyme Q10, CoQ10) and prenylated proteins. The depletion of cholesterol itself may be involved as a key process
Recommendations to prevent myopathy complications in statin therapy
Drug-associated myopathy should be considered if a patient presents with unspecific muscular symptoms. While case numbers of rhabdomyolyis have been counted, only scanty information of the frequency of lower grades of muscular symptoms or asymptomatic CK elevation is available and the clinical relevance of these adverse effects is unclear. Specifically, the risk of asymptomatic CK elevation or of myopathy without CK elevation to proceed to rhabdomyolysis is unknown. A summary of the key recommendations is given in Table 5.
Table 5.
Recommendations to prevent myopathy complications (summary of recommendations according to NLA 84, ACC/AHA/NHLBI 86, Ballantyne 88 and manufacturers)
| Before treatment |
| • Inform the patient about the risks and symptoms of myopathy and of interacting drugs |
| • Evaluate muscle symptoms before starting therapy and measure CK if the patient reports muscular symptoms |
| • Obtain CK baseline in patients at high risk of muscle injury by statins and/or on medications that might affect statin metabolism (see Tables 3 and 4). If pretreatment CK is elevated >five times the ULN, repeat CK measurement within 5 to 7 days. If CK is >five-fold ULN in patients with risk factors, do not start statin therapy |
| • No recommendation is currently given (by CPIC) on whether or not SLCO1B1 genotyping should be performed in any patient prior to receiving simvastatin |
| During treatment |
| • Obtain CK in patients who develop muscular symptoms |
| • See Figure 3 |
| • If genotyping is available, restrict the use of 80 mg dose of simvastatin to patients under long term therapy (12 months or more) without muscular symptoms who are carriers of the rs4149056 TT genotype 45 |
ACC, American College of Cardiology; AHA, American Heart Association; CK, creatine kinase; CPIC, Clinical Pharmacogenomics Implementation Consortium; NHLBI, National Heart, Lung and Blood Institute; NLA, National Lipid Association; SLCO1B1, solute carrier organic anion transporter 1B1; ULN, upper limit of normal.
Recommendations before starting statin therapy
Patients should be informed about the risks and symptoms of myopathy 83 and the concomitant administration of drugs known to inhibit the metabolism of statins, e.g. protease inhibitors, ciclosporin, amiodarone, or some fibrates (Table 4). Although the National Lipid Association’s (NLA) Muscle Expert Panel does not consider obtaining a baseline CK level in all patients absolutely necessary 84, it may be reasonable in patients who are at high risk (Tables 3 and 4) of muscle injury by statins 85. Statin manufacturers recommend measuring the CK activity prior to starting lipid-lowering drug therapy if at least one of the following risk factors is present: impaired renal function, hypothyroidism, genetic myopathy in history or family history, history of statin- or fibrate-associated myopathy, history of hepatic disease and/or significant alcohol abuse and elderly patients (>70 years) with additional risk factors (Table 3). If pretreatment CK levels are elevated >five times the ULN, CK measurement should be repeated within 5 to 7 days (drug information). Manufacturers recommend not to start therapy if CK activity exceeds five-fold ULN in these patients. The NLA Muscle Safety Expert Panel strongly advocates CK values for patients on medications that might affect statin metabolism 84,85 (Table 4). A clinical advisory panel recommends the evaluation of muscle symptoms before starting therapy and 6 to 12 weeks after initiating and to obtain a CK measurement if persons have muscular soreness, tenderness or pain 86. Reference values of CK pair measurements (before and after initiating therapy), however, do not exist for patients. When interpreting those CK pairs, a high background variability, as shown in healthy blood donors without statin treatment, needs to be taken into account 87. The Clinical Pharmacogenomics Implementation Consortium (CPIC) guideline does not argue that SLCO1B1 genotyping is absolutely necessary. However, when genotyping is available, the consortium recommends restricting the use of 80 mg dose of simvastatin to patients who have been taking it for a long time (e.g. 12 months or more) without signs or symptoms of clinically significant toxic effects on muscle and are carriers of the rs4149056 TT genotype (i.e. normal myopathy risk). In carriers of the TC (intermediate myopathy risk) or CC (high myopathy risk) genotype, the FDA recommends against 80 mg of simvastatin, prescribing a lower dose, or considering an alternative statin. In CC carriers, the FDA also recommends considering routine CK surveillance 45.
Recommendations during statin therapy
In asymptomatic patients receiving statin therapy without risk factors for myopathy, it is not necessary to monitor CK serum activity 85. No specific CK monitoring recommendations are given for asymptomatic patients who have one or several risk factors for myopathy but a normal CK level before start of statin treatment.
Figure 3.
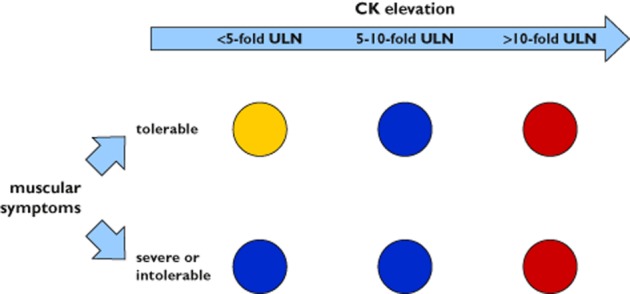
Recommendations to prevent myopathy complications during statin therapy (summary of recommendations according to NLA 84, ACC/AHA/NHLBI 86, Ballantyne 88 and manufacturers). Yellow  – statin therapy can be continued at the same or reduced doses with symptoms used as the clinical guide to stop or continue therapy. Repeat CK within 1 week. Blue
– statin therapy can be continued at the same or reduced doses with symptoms used as the clinical guide to stop or continue therapy. Repeat CK within 1 week. Blue  – discontinue the statin until the patient is asymptomatic. Once the patient is asymptomatic, the same statin can then be restarted at the same dose to test the reproducibility of symptoms, at a lower dose with or without other lipid-lowering medications, or a different statin can be used instead. Red
– discontinue the statin until the patient is asymptomatic. Once the patient is asymptomatic, the same statin can then be restarted at the same dose to test the reproducibility of symptoms, at a lower dose with or without other lipid-lowering medications, or a different statin can be used instead. Red  – stop statin therapy. ACC, American College of Cardiology; AHA, American Heart Association; CK, creatine kinase; NHLBI, National Heart, Lung and Blood Institute; NLA, National Lipid Association; ULN, upper limit of normal
– stop statin therapy. ACC, American College of Cardiology; AHA, American Heart Association; CK, creatine kinase; NHLBI, National Heart, Lung and Blood Institute; NLA, National Lipid Association; ULN, upper limit of normal
Therapy should be discontinued if muscular symptoms with CK elevation exceeding five-fold ULN (e.g. drug information Sortis®, Zocor®), or severe muscular symptoms occur (with or without CK elevation five-fold ULN), or CK elevation is 10-fold ULN (with or without muscular symptoms), or signs of rhabdomyolysis are present. CK measurement should be obtained in symptomatic patients 85.
Manufacturers recommend to discontinue statin therapy if CK activity is five times ULN or if CK elevation is less than five times ULN and significant muscular symptoms exist (e.g. simvastatin or atorvastatin). If CK activity is found to be elevated less than five-fold ULN, repeating the test within 1 week is recommended 88. The NLA Muscle Expert Panel recommends, if the patient has intolerable muscle symptoms, to discontinue the statin, regardless of CK level, until the patient is asymptomatic. Once the patient is asymptomatic, the same statin can then be restarted at the same dose to test the reproducibility of symptoms, at a lower dose with or without other lipid-lowering medications or a different statin can be used instead 11,84. In a randomized, double-blind, trial, 199 patients with previous symptomatic myopathy after receiving a statin (other than fluvastatin) received either fluvastatin 80 mg day−1 or ezetimibe 10 mg day−1 or both and were followed for 12 weeks. In the fluvastatin group (n = 69), 4% discontinued therapy due to recurrent myopathic symptoms. 8% and 3% of the patients discontinued therapy due to recurrent symptoms in the ezetimibe group (n = 66) and in the group receiving both fluvastatin and ezetimibe (n = 64), respectively 89. Alternative therapeutic strategies, however lacking prospective evaluation, may include altered dosing regimens (e.g. non-daily dosing) or once a week rosuvastatin 90 or the addition of a non-statin such as ezetimibe or bile acid-binding resin 11,91.
In patients with tolerable muscle symptoms and no or mild CK elevation (CK<10 times the ULN), the Muscle Expert Panel recommends that statin therapy can be continued at the same or reduced doses with symptoms used as the clinical guide to stop or continue therapy. If the patient has tolerable muscle complaints but moderate (CK levels ≥10-fold ULN, but <50-fold ULN) or severe (CK levels ≥50-fold ULN) CK elevations or clinically important rhabdomyolysis, statin therapy should be stopped 84.
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare no financial relationships with any organizations that might have an interest in the submitted work in the previous 3 years and no other relationships or activities that could appear to have influenced the submitted work.
The skilful technical assistance of Mr Bernd Borstel is gratefully acknowledged. The work has been supported by the Robert Bosch Stiftung, Stuttgart, Germany, the Federal Ministry for Education and Research (BMBF, Berlin, Germany) grant 03 IS 2061C and 0315755 and the Deutsche Forschungsgemeinschaft KFO 274 (grant SCHW858/1-1).
References
- Law MR, Wald NJ, Rudnicka AR. Quantifying effect of statins on low density lipoprotein cholesterol, ischaemic heart disease, and stroke: systematic review and meta-analysis. BMJ. 2003;326:1423. doi: 10.1136/bmj.326.7404.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaRosa JC, He J, Vupputuri S. Effect of statins on risk of coronary disease: a meta-analysis of randomized controlled trials. JAMA. 1999;282:2340–2346. doi: 10.1001/jama.282.24.2340. [DOI] [PubMed] [Google Scholar]
- Mosshammer D, Lorenz G, Meznaric S, Schwarz J, Muche R, Morike K. Statin use and its association with musculoskeletal symptoms-025EFa cross-sectional study in primary care settings. Fam Pract. 2009;26:88–95. doi: 10.1093/fampra/cmp006. [DOI] [PubMed] [Google Scholar]
- Thompson PD, Clarkson P, Karas RH. Statin-associated myopathy. JAMA. 2003;289:1681–1690. doi: 10.1001/jama.289.13.1681. [DOI] [PubMed] [Google Scholar]
- Sinzinger H, Wolfram R, Peskar BA. Muscular side effects of statins. J Cardiovasc Pharmacol. 2002;40:163–171. doi: 10.1097/00005344-200208000-00001. [DOI] [PubMed] [Google Scholar]
- Sathasivam S, Lecky B. Statin induced myopathy. BMJ. 2008;337:a2286. doi: 10.1136/bmj.a2286. [DOI] [PubMed] [Google Scholar]
- Kashani A, Phillips CO, Foody JM, Wang Y, Mangalmurti S, Ko DT, Krumholz HM. Risks associated with statin therapy: a systematic overview of randomized clinical trials. Circulation. 2006;114:2788–2797. doi: 10.1161/CIRCULATIONAHA.106.624890. [DOI] [PubMed] [Google Scholar]
- Evans M, Rees A. Effects of HMG-CoA reductase inhibitors on skeletal muscle: are all statins the same? Drug Saf. 2002;25:649–663. doi: 10.2165/00002018-200225090-00004. [DOI] [PubMed] [Google Scholar]
- Armitage J. The safety of statins in clinical practice. Lancet. 2007;370:1781–1790. doi: 10.1016/S0140-6736(07)60716-8. [DOI] [PubMed] [Google Scholar]
- Finsterer J. [Medically induced myopathia] Nervenarzt. 2006;77:682–686. doi: 10.1007/s00115-006-2080-4. [DOI] [PubMed] [Google Scholar]
- Joy TR, Hegele RA. Narrative review: statin-related myopathy. Ann Intern Med. 2009;150:858–868. doi: 10.7326/0003-4819-150-12-200906160-00009. [DOI] [PubMed] [Google Scholar]
- Bays H. Statin safety: an overview and assessment of the data–2005. Am J Cardiol. 2006;97:6C–26C. doi: 10.1016/j.amjcard.2005.12.006. [DOI] [PubMed] [Google Scholar]
- Davidson MH, Clark JA, Glass LM, Kanumalla A. Statin safety: an appraisal from the adverse event reporting system. Am J Cardiol. 2006;97:32C–43C. doi: 10.1016/j.amjcard.2005.12.008. [DOI] [PubMed] [Google Scholar]
- Thompson PD, Zmuda JM, Domalik LJ, Zimet RJ, Staggers J, Guyton JR. Lovastatin increases exercise-induced skeletal muscle injury. Metabolism. 1997;46:1206–1210. doi: 10.1016/s0026-0495(97)90218-3. [DOI] [PubMed] [Google Scholar]
- Sinzinger H, Schmid P, O’Grady J. Two different types of exercise-induced muscle pain without myopathy and CK-elevation during HMG-Co-enzyme-A-reductase inhibitor treatment. Atherosclerosis. 1999;143:459–460. doi: 10.1016/s0021-9150(98)00310-4. [DOI] [PubMed] [Google Scholar]
- Vladutiu GD, Simmons Z, Isackson PJ, Tarnopolsky M, Peltier WL, Barboi AC, Sripathi N, Wortmann RL, Phillips PS. Genetic risk factors associated with lipid-lowering drug-induced myopathies. Muscle Nerve. 2006;34:153–162. doi: 10.1002/mus.20567. [DOI] [PubMed] [Google Scholar]
- Ruano G, Windemuth A, Wu AH, Kane JP, Malloy MJ, Pullinger CR, Kocherla M, Bogaard K, Gordon BR, Holford TR, Gupta A, Seip RL, Thompson PD. Mechanisms of statin-induced myalgia assessed by physiogenomic associations. Atherosclerosis. 2011;218:451–456. doi: 10.1016/j.atherosclerosis.2011.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baigent C, Blackwell L, Emberson J, Holland LE, Reith C, Bhala N, Peto R, Barnes EH, Keech A, Simes J, Collins R. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170 000 participants in 26 randomised trials. Lancet. 2010;376:1670–1681. doi: 10.1016/S0140-6736(10)61350-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armitage J, Bowman L, Wallendszus K, Bulbulia R, Rahimi K, Haynes R, Parish S, Peto R, Collins R. Intensive lowering of LDL cholesterol with 80 mg versus 20 mg simvastatin daily in 12,064 survivors of myocardial infarction: a double-blind randomised trial. Lancet. 2010;376:1658–1669. doi: 10.1016/S0140-6736(10)60310-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan A, Colman E. Weighing the benefits of high-dose simvastatin against the risk of myopathy. N Engl J Med. 2011;365:285–287. doi: 10.1056/NEJMp1106689. [DOI] [PubMed] [Google Scholar]
- Omar MA, Wilson JP. FDA adverse event reports on statin-associated rhabdomyolysis. Ann Pharmacother. 2002;36:288–295. doi: 10.1345/aph.1A289. [DOI] [PubMed] [Google Scholar]
- Huynh T, Cordato D, Yang F, Choy T, Johnstone K, Bagnall F, Hitchens N, Dunn R. HMG CoA reductase-inhibitor-related myopathy and the influence of drug interactions. Intern Med J. 2002;32:486–490. doi: 10.1046/j.1445-5994.2002.00264.x. [DOI] [PubMed] [Google Scholar]
- Fux R, Morike K, Gundel UF, Hartmann R, Gleiter CH. Ezetimibe and statin-associated myopathy. Ann Intern Med. 2004;140:671–672. doi: 10.7326/0003-4819-140-8-200404200-00034. [DOI] [PubMed] [Google Scholar]
- Patel AM, Shariff S, Bailey DG, Juurlink DN, Gandhi S, Mamdani M, Gomes T, Fleet J, Hwang YJ, Garg AX. Statin toxicity from macrolide antibiotic coprescription: a population-based cohort study. Ann Intern Med. 2013;158:869–876. doi: 10.7326/0003-4819-158-12-201306180-00004. [DOI] [PubMed] [Google Scholar]
- Hermann M, Bogsrud MP, Molden E, Asberg A, Mohebi BU, Ose L, Retterstol K. Exposure of atorvastatin is unchanged but lactone and acid metabolites are increased several-fold in patients with atorvastatin-induced myopathy. Clin Pharmacol Ther. 2006;79:532–539. doi: 10.1016/j.clpt.2006.02.014. [DOI] [PubMed] [Google Scholar]
- Kantola T, Kivisto KT, Neuvonen PJ. Effect of itraconazole on the pharmacokinetics of atorvastatin. Clin Pharmacol Ther. 1998;64:58–65. doi: 10.1016/S0009-9236(98)90023-6. [DOI] [PubMed] [Google Scholar]
- Pasanen MK, Backman JT, Neuvonen PJ, Niemi M. Frequencies of single nucleotide polymorphisms and haplotypes of organic anion transporting polypeptide 1B1 SLCO1B1 gene in a Finnish population. Eur J Clin Pharmacol. 2006;62:409–415. doi: 10.1007/s00228-006-0123-1. [DOI] [PubMed] [Google Scholar]
- Law M, Rudnicka AR. Statin safety: a systematic review. Am J Cardiol. 2006;97:52C–60C. doi: 10.1016/j.amjcard.2005.12.010. [DOI] [PubMed] [Google Scholar]
- Prueksaritanont T, Tang C, Qiu Y, Mu L, Subramanian R, Lin JH. Effects of fibrates on metabolism of statins in human hepatocytes. Drug Metab Dispos. 2002;30:1280–1287. doi: 10.1124/dmd.30.11.1280. [DOI] [PubMed] [Google Scholar]
- Graham DJ, Staffa JA, Shatin D, Andrade SE, Schech SD, La Grenade L, Gurwitz JH, Chan KA, Goodman MJ, Platt R. Incidence of hospitalized rhabdomyolysis in patients treated with lipid-lowering drugs. JAMA. 2004;292:2585–2590. doi: 10.1001/jama.292.21.2585. [DOI] [PubMed] [Google Scholar]
- Neuvonen PJ, Niemi M, Backman JT. Drug interactions with lipid-lowering drugs: mechanisms and clinical relevance. Clin Pharmacol Ther. 2006;80:565–581. doi: 10.1016/j.clpt.2006.09.003. [DOI] [PubMed] [Google Scholar]
- Goosen TC, Bauman JN, Davis JA, Yu C, Hurst SI, Williams JA, Loi CM. Atorvastatin glucuronidation is minimally and nonselectively inhibited by the fibrates gemfibrozil, fenofibrate, and fenofibric acid. Drug Metab Dispos. 2007;35:1315–1324. doi: 10.1124/dmd.107.015230. [DOI] [PubMed] [Google Scholar]
- Ahmed W, Khan N, Glueck CJ, Pandey S, Wang P, Goldenberg N, Uppal M, Khanal S. Low serum 25 (OH) vitamin D levels (<32 ng mL−1) are associated with reversible myositis-myalgia in statin-treated patients. Transl Res. 2009;153:11–16. doi: 10.1016/j.trsl.2008.11.002. [DOI] [PubMed] [Google Scholar]
- Lee P, Greenfield JR, Campbell LV. Vitamin D insufficiency–a novel mechanism of statin-induced myalgia? Clin Endocrinol (Oxf) 2009;71:154–155. doi: 10.1111/j.1365-2265.2008.03448.x. [DOI] [PubMed] [Google Scholar]
- Yavuz B, Ertugrul DT, Cil H, Ata N, Akin KO, Yalcin AA, Kucukazman M, Dal K, Hokkaomeroglu MS, Yavuz BB, Tutal E. Increased levels of 25 hydroxyvitamin D and 1,25-dihydroxyvitamin D after rosuvastatin treatment: a novel pleiotropic effect of statins? Cardiovasc Drugs Ther. 2009;23:295–299. doi: 10.1007/s10557-009-6181-8. [DOI] [PubMed] [Google Scholar]
- Perez-Castrillon JL, Vega G, Abad L, Sanz A, Chaves J, Hernandez G, Duenas A. Effects of atorvastatin on vitamin D levels in patients with acute ischemic heart disease. Am J Cardiol. 2007;99:903–905. doi: 10.1016/j.amjcard.2006.11.036. [DOI] [PubMed] [Google Scholar]
- Thummel KE, Brimer C, Yasuda K, Thottassery J, Senn T, Lin Y, Ishizuka H, Kharasch E, Schuetz J, Schuetz E. Transcriptional control of intestinal cytochrome P-4503A by 1alpha,25-dihydroxy vitamin D3. Mol Pharmacol. 2001;60:1399–1406. doi: 10.1124/mol.60.6.1399. [DOI] [PubMed] [Google Scholar]
- Perez-Castrillon JL, Abad Manteca L, Vega G, Del Pino Montes J, de Luis D, Duenas Laita A. Vitamin D levels and lipid response to atorvastatin. Int J Endocrinol. 2010;2010:320721. doi: 10.1155/2010/320721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz JB. Effects of vitamin D supplementation in atorvastatin-treated patients: a new drug interaction with an unexpected consequence. Clin Pharmacol Ther. 2009;85:198–203. doi: 10.1038/clpt.2008.165. [DOI] [PubMed] [Google Scholar]
- Gupta A, Thompson PD. The relationship of vitamin D deficiency to statin myopathy. Atherosclerosis. 2011;215:23–29. doi: 10.1016/j.atherosclerosis.2010.11.039. [DOI] [PubMed] [Google Scholar]
- Shitara Y, Horie T, Sugiyama Y. Transporters as a determinant of drug clearance and tissue distribution. Eur J Pharm Sci. 2006;27:425–446. doi: 10.1016/j.ejps.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Nies AT, Niemi M, Burk O, Winter S, Zanger UM, Stieger B, Schwab M, Schaeffeler E. Genetics is a major determinant of expression of the human hepatic uptake transporter OATP1B1, but not of OATP1B3 and OATP2B1. Genome Med. 2013;5:1. doi: 10.1186/gm405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemi M. Transporter pharmacogenetics and statin toxicity. Clinical. Pharmacol Ther. 2010;87:130–133. doi: 10.1038/clpt.2009.197. [DOI] [PubMed] [Google Scholar]
- Pasanen MK, Neuvonen M, Neuvonen PJ, Niemi M. SLCO1B1 polymorphism markedly affects the pharmacokinetics of simvastatin acid. Pharmacogenet Genomics. 2006;16:873–879. doi: 10.1097/01.fpc.0000230416.82349.90. [DOI] [PubMed] [Google Scholar]
- Wilke RA, Ramsey LB, Johnson SG, Maxwell WD, McLeod HL, Voora D, Krauss RM, Roden DM, Feng Q, Cooper-Dehoff RM, Gong L, Klein TE, Wadelius M, Niemi M. The clinical pharmacogenomics implementation consortium: CPIC guideline for SLCO1B1 and simvastatin-induced myopathy. Clin Pharmacol Ther. 2012;92:112–117. doi: 10.1038/clpt.2012.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link E, Parish S, Armitage J, Bowman L, Heath S, Matsuda F, Gut I, Lathrop M, Collins R. SLCO1B1 variants and statin-induced myopathy – a genomewide study. N Engl J Med. 2008;359:789–799. doi: 10.1056/NEJMoa0801936. [DOI] [PubMed] [Google Scholar]
- Stewart A. SLCO1B1 Polymorphisms and statin-induced myopathy. PLoS Curr. 2013;4:5. doi: 10.1371/currents.eogt.d21e7f0c58463571bb0d9d3a19b82203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemi M, Pasanen MK, Neuvonen PJ. SLCO1B1 polymorphism and sex affect the pharmacokinetics of pravastatin but not fluvastatin. Clin Pharmacol Ther. 2006;80:356–366. doi: 10.1016/j.clpt.2006.06.010. [DOI] [PubMed] [Google Scholar]
- Niemi M, Schaeffeler E, Lang T, Fromm MF, Neuvonen M, Kyrklund C, Backman JT, Kerb R, Schwab M, Neuvonen PJ, Eichelbaum M, Kivisto KT. High plasma pravastatin concentrations are associated with single nucleotide polymorphisms and haplotypes of organic anion transporting polypeptide-C (OATP-C, SLCO1B1) Pharmacogenetics. 2004;14:429–440. doi: 10.1097/01.fpc.0000114750.08559.32. [DOI] [PubMed] [Google Scholar]
- Igel M, Arnold KA, Niemi M, Hofmann U, Schwab M, Lutjohann D, von Bergmann K, Eichelbaum M, Kivisto KT. Impact of the SLCO1B1 polymorphism on the pharmacokinetics and lipid-lowering efficacy of multiple-dose pravastatin. Clin Pharmacol Ther. 2006;79:419–426. doi: 10.1016/j.clpt.2006.01.010. [DOI] [PubMed] [Google Scholar]
- Morimoto K, Oishi T, Ueda S, Ueda M, Hosokawa M, Chiba K. A novel variant allele of OATP-C (SLCO1B1) found in a Japanese patient with pravastatin-induced myopathy. Drug Metab Pharmacokinet. 2004;19:453–455. doi: 10.2133/dmpk.19.453. [DOI] [PubMed] [Google Scholar]
- Voora D, Shah SH, Spasojevic I, Ali S, Reed CR, Salisbury BA, Ginsburg GS. The SLCO1B1*5 genetic variant is associated with statin-induced side effects. J Am Coll Cardiol. 2009;54:1609–1616. doi: 10.1016/j.jacc.2009.04.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danik JS, Chasman DI, MacFadyen JG, Nyberg F, Barratt BJ, Ridker PM. Lack of association between SLCO1B1 polymorphisms and clinical myalgia following rosuvastatin therapy. Am Heart J. 2013;165:1008–1014. doi: 10.1016/j.ahj.2013.01.025. [DOI] [PubMed] [Google Scholar]
- Niemi M. Transporter pharmacogenetics and statin toxicity. Clin Pharmacol Ther. 2010;87:130–133. doi: 10.1038/clpt.2009.197. [DOI] [PubMed] [Google Scholar]
- Ho RH, Tirona RG, Leake BF, Glaeser G, Lee W, Lemke CJ, Wang Y, Kim RB. Drug and bile acid transporters in rosuvastatin hepatic uptake: function, expression, and pharmacogenetics. Gastroenterology. 2006;130:1793–1806. doi: 10.1053/j.gastro.2006.02.034. [DOI] [PubMed] [Google Scholar]
- Nies AT, Schwab M, Keppler D. Interplay of conjugating enzymes with OATP uptake transporters and ABCC/MRP efflux pumps in the elimination of drugs. Expert Opin Drug Metab Toxicol. 2008;4:545–568. doi: 10.1517/17425255.4.5.545. [DOI] [PubMed] [Google Scholar]
- Itagaki S, Chiba M, Kobayashi M, Hirano T, Iseki K. Contribution of multidrug resistance-associated protein 2 to secretory intestinal transport of organic anions. Biol Pharm Bull. 2008;31:146–148. doi: 10.1248/bpb.31.146. [DOI] [PubMed] [Google Scholar]
- Generaux GT, Bonomo FM, Johnson M, Doan KM. Impact of SLCO1B1 (OATP1B1) and ABCG2 (BCRP) genetic polymorphisms and inhibition on LDL-C lowering and myopathy of statins. Xenobiotica. 2011;41:639–651. doi: 10.3109/00498254.2011.562566. [DOI] [PubMed] [Google Scholar]
- Kobayashi D, Nozawa T, Imai K, Nezu J, Tsuji A, Tamai I. Involvement of human organic anion transporting polypeptide OATP-B (SLC21A9) in pH-dependent transport across intestinal apical membrane. J Pharmacol Exp Ther. 2003;306:703–708. doi: 10.1124/jpet.103.051300. [DOI] [PubMed] [Google Scholar]
- Glaeser H, Bailey DG, Dresser GK, Gregor JC, Schwarz UI, McGrath JS, Jolicoeur E, Lee W, Leake BF, Tirona RG, Kim RB. Intestinal drug transporter expression and the impact of grapefruit juice in humans. Clin Pharmacol Ther. 2007;81:362–370. doi: 10.1038/sj.clpt.6100056. [DOI] [PubMed] [Google Scholar]
- Sirvent P, Bordenave S, Vermaelen M, Roels B, Vassort G, Mercier J, Raynaud E, Lacampagne A. Simvastatin induces impairment in skeletal muscle while heart is protected. Biochem Biophys Res Commun. 2005;338:1426–1434. doi: 10.1016/j.bbrc.2005.10.108. [DOI] [PubMed] [Google Scholar]
- Kobayashi M, Otsuka Y, Itagaki S, Hirano T, Iseki K. Inhibitory effects of statins on human monocarboxylate transporter 4. Int J Pharm. 2006;317:19–25. doi: 10.1016/j.ijpharm.2006.02.043. [DOI] [PubMed] [Google Scholar]
- Knauer MJ, Urquhart BL, Meyer Zu Schwabedissen HE, Schwarz UI, Lemke CJ, Leake BF, Kim RB, Tirona RG. Human skeletal muscle drug transporters determine local exposure and toxicity of statins. Circ Res. 2010;106:297–306. doi: 10.1161/CIRCRESAHA.109.203596. [DOI] [PubMed] [Google Scholar]
- Moosmann B, Behl C. Selenoprotein synthesis and side-effects of statins. Lancet. 2004;363:892–894. doi: 10.1016/S0140-6736(04)15739-5. [DOI] [PubMed] [Google Scholar]
- Moosmann B, Behl C. Selenoproteins, cholesterol-lowering drugs, and the consequences: revisiting of the mevalonate pathway. Trends Cardiovasc Med. 2004;14:273–281. doi: 10.1016/j.tcm.2004.08.003. [DOI] [PubMed] [Google Scholar]
- Schmitz G, Drobnik W. Pharmacogenomics and pharmacogenetics of cholesterol-lowering therapy. Clin Chem Lab Med. 2003;41:581–589. doi: 10.1515/CCLM.2003.088. [DOI] [PubMed] [Google Scholar]
- Oh J, Ban MR, Miskie BA, Pollex RL, Hegele RA. Genetic determinants of statin intolerance. Lipids Health Dis. 2007;6:7. doi: 10.1186/1476-511X-6-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr DF, O’Meara H, Jorgensen AL, Campbell J, Hobbs M, McCann G, van Staa T, Pirmohamed M. SLCO1B1 genetic variant associated with statin-induced myopathy: a proof-of-concept study using the clinical practice research datalink. Clin Pharmacol Ther. 2013;94:695–701. doi: 10.1038/clpt.2013.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcoff L, Thompson PD. The role of coenzyme Q10 in statin-associated myopathy: a systematic review. J Am Coll Cardiol. 2007;49:2231–2237. doi: 10.1016/j.jacc.2007.02.049. [DOI] [PubMed] [Google Scholar]
- Mas E, Mori TA. Coenzyme Q(10) and statin myalgia: what is the evidence? Curr Atheroscler Rep. 2010;12:407–413. doi: 10.1007/s11883-010-0134-3. [DOI] [PubMed] [Google Scholar]
- Baker SK. Molecular clues into the pathogenesis of statin-mediated muscle toxicity. Muscle Nerve. 2005;31:572–580. doi: 10.1002/mus.20291. [DOI] [PubMed] [Google Scholar]
- Draeger A, Monastyrskaya K, Mohaupt M, Hoppeler H, Savolainen H, Allemann C, Babiychuk EB. Statin therapy induces ultrastructural damage in skeletal muscle in patients without myalgia. J Pathol. 2006;210:94–102. doi: 10.1002/path.2018. [DOI] [PubMed] [Google Scholar]
- Dirks AJ, Jones KM. Statin-induced apoptosis and skeletal myopathy. Am J Physiol Cell Physiol. 2006;291:C1208–1212. doi: 10.1152/ajpcell.00226.2006. [DOI] [PubMed] [Google Scholar]
- Johnson TE, Zhang X, Bleicher KB, Dysart G, Loughlin AF, Schaefer WH, Umbenhauer DR. Statins induce apoptosis in rat and human myotube cultures by inhibiting protein geranylgeranylation but not ubiquinone. Toxicol Appl Pharmacol. 2004;200:237–250. doi: 10.1016/j.taap.2004.04.010. [DOI] [PubMed] [Google Scholar]
- Mangravite LM, Engelhardt BE, Medina MW, Smith JD, Brown CD, Chasman DI, Mecham BH, Howie B, Shim H, Naidoo D, Feng Q, Rieder MJ, Chen YD, Rotter JI, Ridker PM, Hopewell JC, Parish S, Armitage J, Collins R, Wilke RA, Nickerson DA, Stephens M, Krauss RM. A statin-dependent QTL for GATM expression is associated with statin-induced myopathy. Nature. 2013;502:377–380. doi: 10.1038/nature12508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballard KD, Thompson PD. Does reduced creatine synthesis protect against statin myopathy? Cell Metab. 2013;18:773–774. doi: 10.1016/j.cmet.2013.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirvent P, Mercier J, Vassort G, Lacampagne A. Simvastatin triggers mitochondria-induced Ca2+ signaling alteration in skeletal muscle. Biochem Biophys Res Commun. 2005;329:1067–1075. doi: 10.1016/j.bbrc.2005.02.070. [DOI] [PubMed] [Google Scholar]
- Kwak HB, Thalacker-Mercer A, Anderson EJ, Lin CT, Kane DA, Lee NS, Cortright RN, Bamman MM, Neufer PD. Simvastatin impairs ADP-stimulated respiration and increases mitochondrial oxidative stress in primary human skeletal myotubes. Free Radic Biol Med. 2012;52:198–207. doi: 10.1016/j.freeradbiomed.2011.10.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanai J, Cao P, Tanksale P, Imamura S, Koshimizu E, Zhao J, Kishi S, Yamashita M, Phillips PS, Sukhatme VP, Lecker SH. The muscle-specific ubiquitin ligase atrogin-1/MAFbx mediates statin-induced muscle toxicity. J Clin Invest. 2007;117:3940–3951. doi: 10.1172/JCI32741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao P, Hanai J, Tanksale P, Imamura S, Sukhatme VP, Lecker SH. Statin-induced muscle damage and atrogin-1 induction is the result of a geranylgeranylation defect. FASEB J. 2009;23:2844–2854. doi: 10.1096/fj.08-128843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Needham M, Fabian V, Knezevic W, Panegyres P, Zilko P, Mastaglia FL. Progressive myopathy with up-regulation of MHC-I associated with statin therapy.[see comment] Neuromuscul Disord. 2007;17:194–200. doi: 10.1016/j.nmd.2006.10.007. [DOI] [PubMed] [Google Scholar]
- Mohaupt MG, Karas RH, Babiychuk EB, Sanchez-Freire V, Monastyrskaya K, Iyer L, Hoppeler H, Breil F, Draeger A. Association between statin-associated myopathy and skeletal muscle damage. CMAJ. 2009;181:E11–18. doi: 10.1503/cmaj.081785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotto AM., Jr Statins, cardiovascular disease, and drug safety. Am J Cardiol. 2006;97:3C–5C. doi: 10.1016/j.amjcard.2005.12.005. [DOI] [PubMed] [Google Scholar]
- Thompson PD, Clarkson PM, Rosenson RS. An assessment of statin safety by muscle experts. Am J Cardiol. 2006;97:69C–76C. doi: 10.1016/j.amjcard.2005.12.013. [DOI] [PubMed] [Google Scholar]
- McKenney JM, Davidson MH, Jacobson TA, Guyton JR. Final conclusions and recommendations of the National Lipid Association Statin Safety Assessment Task Force. Am J Cardiol. 2006;97:89C–94C. doi: 10.1016/j.amjcard.2006.02.030. [DOI] [PubMed] [Google Scholar]
- Pasternak RC, Smith SC, Jr, Bairey-Merz CN, Grundy SM, Cleeman JI, Lenfant C. ACC/AHA/NHLBI clinical advisory on the use and safety of statins. Stroke. 2002;33:2337–2341. doi: 10.1161/01.str.0000034125.94759.41. [DOI] [PubMed] [Google Scholar]
- Moßhammer D, Muche R, Menzel D, Ring C, Wernet D, Meisner C, Gleiter C, Lorenz G, Mörike K. Pairs of creatine kinase serum activity. Open Clin Chem J. 2009;2:12–15. [Google Scholar]
- Ballantyne CM, Corsini A, Davidson MH, Holdaas H, Jacobson TA, Leitersdorf E, Marz W, Reckless JP, Stein EA. Risk for myopathy with statin therapy in high-risk patients. Arch Intern Med. 2003;163:553–564. doi: 10.1001/archinte.163.5.553. [DOI] [PubMed] [Google Scholar]
- Stein EA, Ballantyne CM, Windler E, Sirnes PA, Sussekov A, Yigit Z, Seper C, Gimpelewicz CR. Efficacy and tolerability of fluvastatin XL 80 mg alone, ezetimibe alone, and the combination of fluvastatin XL 80 mg with ezetimibe in patients with a history of muscle-related side effects with other statins. Am J Cardiol. 2008;101:490–496. doi: 10.1016/j.amjcard.2007.09.099. [DOI] [PubMed] [Google Scholar]
- Ruisinger JF, Backes JM, Gibson CA, Moriarty PM. Once-a-week rosuvastatin (2.5 to 20 mg) in patients with a previous statin intolerance. Am J Cardiol. 2009;103:393–394. doi: 10.1016/j.amjcard.2008.09.095. [DOI] [PubMed] [Google Scholar]
- Harper CR, Jacobson TA. Evidence-based management of statin myopathy. Curr Atheroscler Rep. 2010;12:322–330. doi: 10.1007/s11883-010-0120-9. [DOI] [PubMed] [Google Scholar]
- Staffa JA, Chang J, Green L. Cerivastatin and reports of fatal rhabdomyolysis. N Engl J Med. 2002;346:539–540. doi: 10.1056/NEJM200202143460721. [DOI] [PubMed] [Google Scholar]
- Stein EA, Davidson MH, Dobs AS, Schrott H, Dujovne CA, Bays H, Weiss SR, Melino MR, Stepanavage ME, Mitchel YB Grp EDSUS. Efficacy and safety of simvastatin 80 mg/day in hypercholesterolemic patients. Am J Cardiol. 1998;82:311–316. doi: 10.1016/s0002-9149(98)00421-4. [DOI] [PubMed] [Google Scholar]
- Bakker-Arkema RG, Nawrocki JW, Black DM. Safety profile of atorvastatin-treated patients with low LDL-cholesterol levels. Atherosclerosis. 2000;149:123–129. doi: 10.1016/s0021-9150(99)00294-4. [DOI] [PubMed] [Google Scholar]
- Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20 536 high-risk individuals: a randomised placebocontrolled trial. Lancet. 2002;360:7–22. [Google Scholar]
- Franc S, Dejager S, Bruckert E, Chauvenet M, Giral P, Turpin G. A comprehensive description of muscle symptoms associated with lipid-lowering drugs. Cardiovasc Drugs Ther. 2003;17:459–465. doi: 10.1023/b:card.0000015861.26111.ab. [DOI] [PubMed] [Google Scholar]
- Cannon CP, Braunwald E, McCabe CH, Rader DJ, Rouleau JL, Belder R, Joyal SV, Hill KA, Pfeffer MA, Skene AM. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med. 2004;350:1495–1504. doi: 10.1056/NEJMoa040583. [DOI] [PubMed] [Google Scholar]
- Pedersen TR, Faergeman O, Kastelein JJ, Olsson AG, Tikkanen MJ, Holme I, Larsen ML, Bendiksen FS, Lindahl C, Szarek M, Tsai J. High-dose atorvastatin vs usual-dose simvastatin for secondary prevention after myocardial infarction: the IDEAL study: a randomized controlled trial. JAMA. 2005;294:2437–2445. doi: 10.1001/jama.294.19.2437. [DOI] [PubMed] [Google Scholar]
- Schwartz GG, Olsson AG, Ezekowitz MD, Ganz P, Oliver MF, Waters D, Zeiher A, Chaitman BR, Leslie S, Stern T. Effects of atorvastatin on early recurrent ischemic events in acute coronary syndromes: the MIRACL study: a randomized controlled trial. JAMA. 2001;285:1711–1718. doi: 10.1001/jama.285.13.1711. [DOI] [PubMed] [Google Scholar]
- Bruckert E, Hayem G, Dejager S, Yau C, Begaud B. Mild to moderate muscular symptoms with high-dosage statin therapy in hyperlipidemic patients–the PRIMO study. Cardiovasc Drugs Ther. 2005;19:403–414. doi: 10.1007/s10557-005-5686-z. [DOI] [PubMed] [Google Scholar]
- Gaist D, Rodriguez LA, Huerta C, Hallas J, Sindrup SH. Lipid-lowering drugs and risk of myopathy: a population-based follow-up study. Epidemiology. 2001;12:565–569. doi: 10.1097/00001648-200109000-00017. [DOI] [PubMed] [Google Scholar]
- Wortmann RL. Lipid-lowering agents and myopathy. Curr Opin Rheumatol. 2002;14:643–647. doi: 10.1097/00002281-200211000-00002. [DOI] [PubMed] [Google Scholar]
- Rosenson RS. Current overview of statin-induced myopathy. Am J Med. 2004;116:408–416. doi: 10.1016/j.amjmed.2003.10.033. [DOI] [PubMed] [Google Scholar]