

Abstract
Aims
Two double-blind, randomized studies were conducted to assess the tolerability, pharmacokinetics and pharmacodynamics of oral TA-8995, a new cholesteryl ester transfer protein (CETP) inhibitor, in healthy subjects.
Methods
Study 1: Subjects received single doses of TA-8995 or placebo (fasted). Doses were 5, 10, 25, 50 (fed/fasted), 100 and 150 mg (Caucasian males, 18–55 years), 25 mg (Caucasian males, > 65 years and Caucasian females, 18–55 years), 25, 50, 100 and 150 mg (Japanese males, 18–55 years). Study 2: Caucasian males (18–55 years) received 1, 2.5, 10 or 25 mg once daily TA-8995 or placebo for 21–28 days. Blood and urine for pharmacokinetics and/or pharmacodynamics were collected. Tolerability was assessed by adverse events, vital signs, electrocardiograms and laboratory safety tests.
Results
Peak TA-8995 concentrations occurred approximately 4 h post-dose. Mean half-lives ranged from 81 to 166 h, without an obvious dose relationship. Exposure increased less than proportionally to dose. TA-8995 was not excreted in urine. Following 2.5 to 25 mg once daily dosing, TA-8995 demonstrated nearly complete inhibition of CETP activity (92–99%), increased high density lipoprotein-cholesterol (HDL-C) by 96 to 140% and decreased low density liporotein-cholesterol (LDL-C) by 40% to 53%. There were dose-related increases in apolipoproteins A-1 and E, HDL2-C and HDL3-C, and decreases in apolipoprotein B and lipoprotein A. There was no evidence of significant effects of age, gender, ethnicity or food on pharmacokinetics or pharmacodynamics. All doses were well tolerated.
Conclusions
TA-8995 is a potent CETP inhibitor and warrants further investigation.
Keywords: apolipoproteins, cardiovascular diseases, cholesterol, coronary disease, hypercholesterolaemia
What is already known about this subject —
Cholesteryl ester transfer protein (CETP) inhibitors have previously been shown to increase high density lipoprotein (HDL)-cholesterol and reduce low density lipoprotein (LDL)-cholesterol in humans with varying potency.
Development has been limited by adverse events (thought to be linked to off-target pharmacology) and lack of potency.
What this study adds —
Single and multiple doses of TA-8995, a novel selective CETP inhibitor, demonstrate near complete CETP inhibition and are well tolerated in healthy subjects.
There was no evidence of any off-target effects that are implicated in the toxicity of other compounds, such as changes in blood pressure, serum electrolytes or aldosterone.
Introduction
Prospective epidemiological studies have shown a strong association between low density lipoprotein-cholesterol (LDL-C) concentrations and cardiovascular disease risk 1. The subsequent application of statin therapy to decrease these atherogenic LDL-C concentrations has resulted in a marked reduction of cardiovascular-related morbidity and mortality. Every 1 mmol l−1 decrease in LDL-C results in an estimated 22% reduction of cardiovascular events and a 10% reduction of all-cause mortality 2. Notwithstanding these impressive benefits, a large residual disease burden persists that has a large impact on both individual patients as well as on global healthcare costs 3. Novel therapeutics are required to reduce further this residual cardiovascular risk in patients. Therapies that modify high density lipoprotein-cholesterol (HDL-C) concentrations would be a logical next step as a large body of evidence has shown an inverse association between HDL-C concentrations and cardiovascular disease risk ( 1.
One way to elevate HDL-C concentrations is to inhibit cholesteryl ester transfer protein (CETP). CETP is a plasma protein secreted primarily by liver and adipose tissue. CETP mediates the transfer of cholesteryl esters from HDL to apolipoprotein B-containing particles (mainly LDL and VLDL) in exchange for triglycerides, thereby decreasing the cholesterol content in HDL in favour of that in (V)LDL. Hence, CETP inhibition has been hypothesized to retain cholesteryl esters in HDL-C and decrease the cholesterol content of the atherogenic apolipoprotein B fraction. Large Mendelian randomization studies and meta-analyses have shown that CETP variants that result in lower CETP activity are associated with a decreased risk of cardiovascular disease 4–7. In addition, pharmacological CETP inhibition in rabbits resulted in reduced atherosclerotic plaque formation 8. Taken together there is ample evidence in support of CETP inhibition in humans as a therapeutic target. The rationale for CETP inhibition and pre-clinical data was recently reviewed by others 9,10.
The aims of the studies discussed in this paper were to assess the tolerability, pharmacokinetics and pharmacodynamics of a new potent CETP inhibitor, TA-8995 (a derivative of 4-{2-[benzyl-(1,2,3,4-tetrahydro-quinolin-4-yl)-amino]-pyrimidin-5-yloxy}-butyric acid), in healthy subjects.
Methods
Subjects
In both studies, subjects had to be healthy, weigh at least 50 kg (males) or 45 kg (females), and have a body mass index of 18 to 33 kg m−2 with a waist measurement of ≤91 cm. Female subjects had to be of non-childbearing potential. All subjects had to provide written informed consent and be prepared to comply with study requirements. Key exclusion criteria included HDL-C ≥ 2.59 mmol l−1 at screening or baseline and clinically relevant findings from medical history, physical examination, laboratory tests, 12-lead ECG or Holter assessment. A family history of long QT syndrome, hypokalaemia or torsades de pointes was also excluded. Use of any non-study medication was restricted as was the intake of alcohol and nicotine-containing products.
Study design
Two randomized, double-blind, placebo-controlled, parallel-group studies were conducted in healthy subjects to investigate the tolerability, pharmacokinetics and pharmacodynamics of TA-8995 (a tetrahydroquinoline). Both studies were conducted in accordance with the International Conference on Harmonization guideline for Good Clinical Practice and the principles of the Declaration of Helsinki, and were reviewed and approved by the local Ethics Committees (see Supporting Information) and Competent Authorities.
The first study (study 1) was a single dose study in 12 groups of subjects. Six groups of Caucasian male subjects aged 18 to 55 years received single oral doses of 5, 10, 25, 50, 100 and 150 mg TA-8995 or placebo under fasting conditions. The subjects who received 50 mg TA-8995/placebo also received the same dose following a high fat meal 4 weeks after the fasting dose. A further group of Caucasian males aged at least 65 years and a group of Caucasian females aged 18 to 55 years received single doses of 25 mg TA-8995 or placebo (fasted). Four groups of Japanese male subjects aged 18 to 55 years received single oral doses of 25, 50, 100 and 150 mg TA-8995 or placebo (fasted). Subjects in each group/dose level were allocated to study treatment in a ratio of six TA-8995 to two placebo. Blood samples for pharmacokinetic and pharmacodynamic assessments were collected from prior to each dose and at intervals up to 336 h post-dose. Urine was collected for pharmacokinetics from pre-dose and at intervals up to 72 h post-dose from selected dose groups. The primary pharmacodynamic endpoints included CETP activity, CETP concentration, HDL-C, LDL-C, total cholesterol and triglycerides. Secondary pharmacodynamic endpoints included apolipoproteins A1, B and E, HDL2-C and HDL3-C. Pharmacodynamic samples were not collected during the fed period, with the exception of HDL-C, LDL-C, total cholesterol and triglycerides which were collected as part of the biochemistry assessments.
The second study (study 2) was a repeated dose study in five groups of Caucasian male subjects aged 18 to 55 years. Each subject received a single oral dose of TA-8995/placebo on day 1, followed by once daily doses on days 8 to 35 (5 mg TA-8995/placebo – group 1) or days 8 to 28 (1, 2.5, 10 and 25 mg TA-8995/placebo – groups 2 to 5). All doses were administered at the study centre after a standard breakfast. Subjects in each dose group were allocated to study treatment in a ratio of 10 TA-8995 to two placebo. Blood samples for pharmacokinetic and pharmacodynamic (CETP activity, CETP concentration, HDL-C, LDL-C, total cholesterol, triglycerides) assessments were collected from prior to each dose and at intervals throughout the study until 336 h after the last dose. Secondary pharmacodynamic endpoints (including apolipoproteins A1, A2, B and E, lipoprotein a [Lp(a)], HDL2-C, HDL3-C, phospholipids, HDL-free cholesterol [HDL-FC], HDL-cholesteryl ester [HDL-CE], HDL-phospholipids [HDL-PL], HDL-triglycerides [HDL-TG] and LDL particle size) were measured at intervals until the last day of dosing. Urine was collected for pharmacokinetics from pre-dose and at intervals up to 72 h after the first and last dose.
Tolerability assessments including adverse events, blood pressure and pulse rate, ECGs, laboratory safety tests (including aldosterone) and physical examinations were conducted throughout both studies.
Details of the analytical methods for the pharmacokinetic and pharmacodynamic endpoints are presented in the Supporting Information.
Statistical analyses
The sample sizes for each study were chosen based on practical considerations rather than statistical power given that these studies represented the first studies in humans. The numbers of subjects in each group were considered to be adequate to assess the main objectives of each study. Subjects were allocated to TA-8995 or placebo in each group by means of a computer-generated randomization code. Pharmacokinetic parameters were determined by non-compartmental methods using WinNonlin software version 4.1 (Pharsight Corporation, USA). All data were listed and summarized by treatment group using descriptive statistics. The effects of food, ethnicity, age and gender on the single dose pharmacokinetics of TA-8995 were investigated in study 1 using log-transformed Cmax and AUC parameters and an analysis of variance (anova) model. The back-transformed point estimates and 90% confidence intervals for the various comparisons (ratios) are presented. In study 2, a post hoc analysis was conducted to compare the maximum percent changes from baseline at each TA-8995 dose level with pooled placebo using an anova model. All statistical analyses were conducted using SAS version 6.12 or higher (SAS Institute Inc. USA).
Results
Study 1 was conducted between March 2008 and June 2009 and study 2 was conducted between August 2009 and June 2010. A total of 96 subjects were enrolled into study 1 and 95 subjects completed the study: one Caucasian subject (50 mg TA-8995) withdrew due to personal reasons in period 1 (food effect group) and was not replaced. A total of 61 subjects were enrolled into study 2 and 59 subjects completed the study: one subject (10 mg TA-8995) was withdrawn on day 7, due to a protocol violation and one subject (placebo) withdrew on day 13 for personal reasons. No subjects were withdrawn from either study due to adverse events. Flow charts detailing subject participation are included in Supporting Information Figures S1 and S2.
Pharmacokinetics
Following single oral doses from 5 to 150 mg in study 1, TA-8995 was absorbed and peak concentrations were typically achieved 3 to 4 h post-dose (Table 1, Figure 1). Plasma concentrations increased less than proportionally to dose with eight-fold, 14-fold and 15-fold increases in AUC(0,336 h), AUC(0,24 h) and Cmax, respectively being observed for a 30-fold increase in dose. Within each dose group, variability was moderate with coefficients of variation (CV) for Cmax and AUC parameters being 8 to 39% with the exception of 100 mg TA-8995, where variability was greater over the first 24 h post-dose. Mean terminal half-life of TA-8995 varied across the dose range from 81 to 166 h, without an obvious relationship to dose. No clinically relevant effects of food, age, gender or ethnicity on TA-8995 pharmacokinetics were observed (Table 2).
Table 1.
Pharmacokinetic parameters for TA-8995 following single oral doses in Caucasian male healthy subjects under fasting conditions (study 1)
| TA-8995 dose | ||||||
|---|---|---|---|---|---|---|
| 5 mg | 10 mg | 25 mg | 50 mg | 100 mg | 150 mg | |
| Parameter (unit)* | n = 6 | n = 6 | n = 6 | n = 5 | n = 6 | n = 6 |
| Cmax (ng ml−1) | 138 (33%) | 251 (23%) | 467 (30%) | 855 (21%) | 1553 (78%) | 2090 (26%) |
| tmax (h) | 3 (2, 3) | 3 (2, 5) | 3.5 (2, 6) | 4 (3, 5) | 4 (1, 10) | 4 (3, 5) |
| AUC(0,24 h) (ng ml−1 h) | 1816 (34%) | 3580 (21%) | 5952 (26%) | 11117 (28%) | 21166 (59%) | 26403 (20%) |
| AUC(0,336 h) (ng ml−1 h) | 10351 (30%) | 19250 (32%) | 32985 (25%) | 44353 (10%) | 73359 (36%) | 78200 (23%) |
| AUC(0,∞) (ng ml−1 h) | 12078 (32%) | 22554 (39%) | 42385 (24%) | 57189 (8%) | 77323 (36%) | 84652 (29%) |
| t1/2 (h) | 125 (22%) | 124 (20%) | 162 (19%) | 166 (21%) | 80.9 (17%) | 95.9 (23%) |
Values are geometric mean (CV%) for Cmax and AUC parameters, median (minimum, maximum) for tmax and arithmetic mean (CV%) for t1/2. CV = coefficient of variation.
Figure 1.
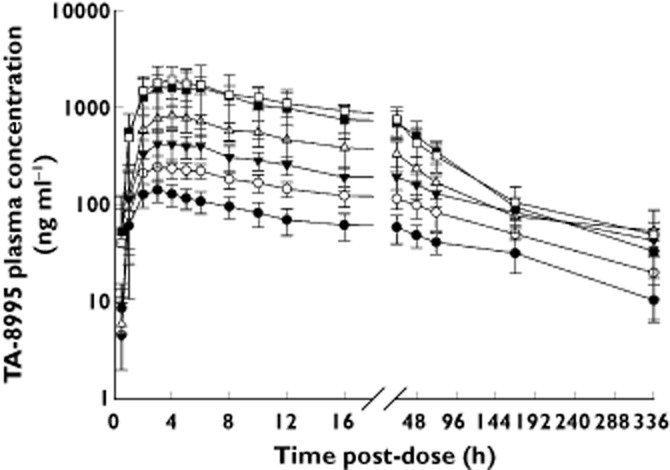
Mean (SD) TA-8995 plasma concentration vs. time profiles following single dose administration to Caucasian males under fasting conditions (study 1).  , 5 mg;
, 5 mg;  , 10 mg;
, 10 mg;  , 25 mg;
, 25 mg;  , 50 mg;
, 50 mg;  , 100 mg;
, 100 mg;  , 150 mg
, 150 mg
Table 2.
Statistical comparison to assess the effect of food, age, gender and ethnicity on the pharmacokinetics of single oral doses of TA-8995 (study 1)
| Comparison (TA-8995 dose) | Parameter | Least squares means ratio | 90% confidence interval |
|---|---|---|---|
| Fed vs. fasted (50 mg) | Cmax | 108.9 | 84.5, 140.5 |
| AUC(0,336 h) | 115.8 | 106.7, 125.7 | |
| AUC(0,∞) | 98.4 | 88.4, 109.5 | |
| Elderly vs. young (25 mg) | Cmax | 111.4 | 87.9, 141.2 |
| AUC(0,336 h) | 88.1 | 69.0, 112.3 | |
| AUC(0,∞) | 92.5 | 70.4, 121.6 | |
| Female vs. male (25 mg) | Cmax | 118.0 | 77.6, 179.3 |
| AUC(0,336 h) | 101.5 | 71.3, 144.6 | |
| AUC(0,∞) | 93.8 | 67.9, 130.0 | |
| Japanese vs. Caucasian (all doses) | Cmax | 137.8 | 101.5, 187.1 |
| AUC(0,336 h) | 103.7 | 80.2, 134.3 | |
| AUC(0,∞) | 95.0 | 72.9, 123.7 |
In study 2, plasma concentrations appeared to increase approximately proportionally to dose following single doses from 1 to 25 mg, although non-proportionality was observed at steady-state: seven-fold, nine-fold and 12-fold increases in Cmin,ss, AUC(0,τ,ss) and Cmax,ss, respectively for a 25-fold increase in dose (Table 3). tmax was independent of dose with median values of 4 to 6 h post-dose. Variability was moderate following single and repeated dosing with CVs for Cmax, Cmin and AUC parameters being ≤33%. Visual inspection of trough concentrations suggests that TA-8995 approached steady-state within 1 to 2 weeks of daily dosing (Figure 2). The mean terminal half-life of TA-8995 following the last dose was 121 to 151 h and was independent of dose. A similar half-life was observed after single and repeated doses of 5 to 25 mg of TA-8995, respectively. TA-8995 accumulated with once daily dosing in a dose-dependent manner, with an approximately six-fold increase at 1 mg through to a two-fold increase at 25 mg.
Table 3.
Pharmacokinetic parameters for TA-8995 following single and repeated oral doses in Caucasian male healthy subjects (Study 2)
| TA-8995 dose (once daily) | |||||
|---|---|---|---|---|---|
| 1 mg | 2.5 mg | 5 mg | 10 mg | 25 mg | |
| Parameter (unit)* | n = 10 | n = 10 | n = 10 | n = 10 | n = 10 |
| Single dose | |||||
| Cmax (ng ml−1) | 27.0 (19%) | 65.7 (13%) | 116 (13%) | 245 (14%) | 750 (19%) |
| tmax (h) | 6 (4, 12) | 6 (4, 8) | 6 (4, 8) | 6 (4, 8) | 6 (3, 8) |
| AUC(0,24 h) (ng ml−1 h) | 434 (25%) | 1055 (12%) | 1796 (14%) | 3471 (18%) | 10437 (21%) |
| Steady-state† | |||||
| Cmax,ss (ng ml−1) | 121 (19%) | 256 (20%) | 436 (18%) | 692 (23%) | 1398 (18%) |
| tmax,ss (h) | 4 (3, 12) | 4 (2, 8) | 4 (3, 8) | 6 (4, 8) | 4 (2, 6) |
| Cmin,ss (ng ml−1) | 83.4 (19%) | 172 (20%) | 237 (20%) | 396 (33%) | 553 (26%) |
| AUC(0,τ,ss) (ng ml−1 h) | 2462 (19%) | 5013 (18%) | 7606 (20%) | 12707 (27%) | 21493 (21%) |
| t1/2,ss (h) | 138 (16%) | 151 (15%) | 148 (30%) | 131 (19%) | 121 (23%) |
| Rac | 5.8 (1.3) | 4.8 (0.9) | 4.3 (1.1) | 3.7 (0.8) | 2.1 (0.3) |
Values are geometric mean (CV%) for Cmax, Cmin and AUC parameters, median (minimum, maximum) for tmax, arithmetic mean (CV%) for t1/2, and arithmetic mean (SD) for Rac.
Assessed after the last dose: day 42 for 5 mg TA-8995 and day 35 for other doses. CV = coefficient of variation, Rac = accumulation ratio calculated as AUC(0,τ,ss)/AUC(0,24 h), where τ is the dosing interval (24 h).
Figure 2.
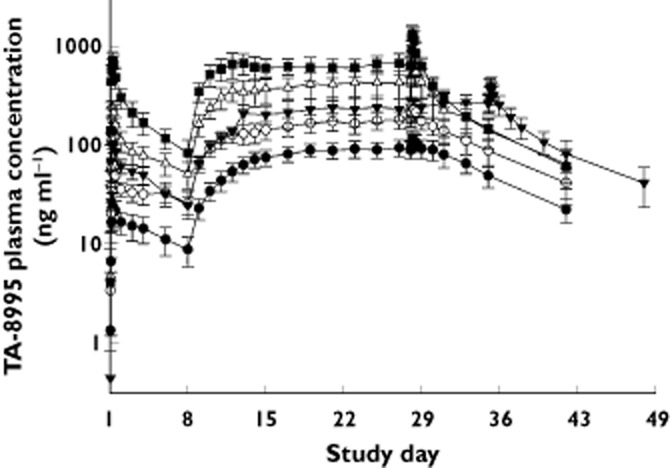
Mean (SD) TA-8995 plasma concentration vs. time profiles following single and repeated (once daily) dose administration (study 2).  , 1 mg;
, 1 mg;  , 2.5 mg;
, 2.5 mg;  , 5 mg;
, 5 mg;  , 10 mg;
, 10 mg;  , 25 mg
, 25 mg
TA-8995 was not detected in the urine in either study.
Pharmacodynamics
Baseline pharmacodynamic parameters were well balanced across treatment groups in study 1 (data not shown) and study 2 (Table 4 and Supporting Information Table S1). TA-8995 strongly inhibited CETP activity in a dose-dependent manner following both single and repeated dosing (Figure 3A). Near complete CETP inhibition was observed following a single dose of 25 mg TA-8995 (∼94%) and following repeated doses of 2.5, 5, 10 to 25 mg once daily TA-8995 (∼92 to 99%) (Table 5). This level of inhibition was maintained throughout the repeated dosing period and the maximum effect of each dose was achieved within 1 week of once daily dosing. The duration of inhibition after the last dose was dose-dependent, with activity approaching baseline levels by 2 weeks following the lowest dose (1 mg), but still being approximately 50% below baseline at 2 weeks following 10 and 25 mg dosing. Although CETP activity decreased with TA-8995 dosing, the concentration of CETP increased in a dose-dependent manner following both single and repeated dosing (Figure 3B). CETP concentration increased from baseline by 250% to 280% after 3 weeks of dosing with 10 mg and 25 mg once daily TA-8995. CETP concentrations declined in parallel for each dose, such that following the cessation of TA-8995 dosing, CETP concentrations were approaching baseline values within 2 weeks following 1 mg and 5 mg TA-8995, whereas CETP concentrations were still approximately 140% higher than baseline at 2 weeks following 10 mg and 25 mg TA-8995. The maximum percent changes in CETP activity and CETP concentrations were statistically significantly different from placebo (P < 0.0001) at all TA-8995 dose levels (1 to 25 mg). The differences in least squares means and 95% confidence intervals are presented in Supporting Information Table S2.
Table 4.
Baseline primary pharmacodynamic variables (study 2)
| TA-8995 dose (once daily) | |||||||
|---|---|---|---|---|---|---|---|
| Placebo (Group 1) | Placebo (Groups 2–5) | 1 mg | 2.5 mg | 5 mg | 10 mg | 25 mg | |
| Baseline characteristics: | n = 1 | n = 8 | n = 10 | n = 10 | n = 10 | n = 10 | n = 10 |
| CETP activity (%) | 12.4 | 36.9 (9.8) | 31.1 (6.2) | 35.0 (6.4) | 30.3 (8.2) | 32.0 (9.1) | 30.7 (12.0) |
| CETP concentration (μg ml−1) | 1.7 | 3.0 (0.8) | 2.6 (0.8) | 3.0 (0.7) | 2.3 (0.5) | 2.3 (0.4) | 2.8 (0.7) |
| HDL-C (mg dl−1) | 75.0 | 47.9 (5.6) | 53.5 (12.6) | 53.2 (9.4) | 53.8 (11.9) | 52.4 (8.2) | 55.4 (15.1) |
| LDL-C (mg dl−1) | 44.0 | 115 (34.2) | 116 (20.1) | 124 (11.0) | 106 (30.9) | 119 (26.4) | 106 (36.3) |
| Total cholesterol (mg dl−1) | 139 | 181 (37.0) | 187 (23.6) | 194 (10.4) | 178 (36.5) | 186 (25.3) | 172 (40.4) |
| Triglyceride (mg dl−1) | 118 | 161 (55.9) | 164 (83.1) | 141 (48.9) | 112 (43.6) | 110 (48.7) | 122 (64.6) |
Values are mean (SD). Group 1 received 5 mg TA-8995/placebo on day 1 and days 8 to 42. Groups 2–5 received 1, 2.5, 10 and 25 mg TA-8995/placebo on day 1 and days 8 to 35.
Figure 3.

Mean (SD) percentage change from baseline in CETP activity (A), CETP concentration (B), HDL-C (C), LDL-C (D), total cholesterol (E) and triglycerides (F) following single and repeated (once daily) dosing of TA-8995 and placebo (study 2). Group 1 received TA-8995 (5 mg)/placebo on day 1 and on days 8–42. Groups 2–5 received TA-8995 (1, 2.5, 10 or 25 mg)/placebo on day 1 and on days 8–35.  , 1 mg;
, 1 mg;  , 2.5 mg;
, 2.5 mg;  , 5 mg;
, 5 mg;  , 10 mg;
, 10 mg;  , 25 mg;
, 25 mg;  , placebo (Group 1);
, placebo (Group 1);  , placebo (Group 2–5)
, placebo (Group 2–5)
Table 5.
Maximum percent change from baseline for primary pharmacodynamic variables for TA-8995 following single and repeated oral doses in Caucasian male healthy subjects (study 2)
| TA-8995 dose (once daily) | |||||||
|---|---|---|---|---|---|---|---|
| Maximum % change from baseline in: | Placebo (Group 1) | Placebo (Groups 2-5) | 1 mg | 2.5 mg | 5 mg | 10 mg | 25 mg |
| n = 1 | n = 8 | n = 10 | n = 10 | n = 10 | n = 10 | n = 10 | |
| Single dose data | |||||||
| CETP activity | 5.1 | −7.8 (7.2) | −23.4 (4.5) | −47.4 (7.8) | −63.4 (8.8) | −78.9 (8.5) | −94.4 (6.0) |
| CETP concentration | 14.5 | 17.2 (14.1) | 22.4 (14.8) | 26.6 (10.0) | 44.0 (19.6) | 74.7 (22.7) | 84.9 (25.5) |
| HDL-C concentration | 0.0 | 14.0 (14.7) | 13.3 (11.6) | 19.3 (11.4) | 28.0 (6.8) | 55.8 (10.5) | 61.4 (18.1) |
| LDL-C concentration | 0.0 | −6.3 (7.9) | −10.5 (6.4) | −14.4 (9.7) | −16.7 (9.2) | −19.3 (9.1) | −36.0 (14.7) |
| Total cholesterol | 0.0 | −4.6 (6.5) | −6.2 (3.9) | −8.4 (5.7) | −5.7 (5.8) | −2.2 (4.3) | −5.9 (6.5) |
| Triglycerides | −24.6 | −33.0 (18.0) | −32.3 (21.2) | −33.8 (14.5) | −11.5 (14.1) | −14.0 (15.2) | −27.9 (13.6) |
| Steady-state data | |||||||
| CETP activity | 21.3 | −8.9 (14.7) | −66.5 (6.4) | −91.6 (6.4) | −90.9 (7.1) | −97.6 (2.9) | −99.4 (1.1) |
| CETP concentration | 34.9 | 19.5 (18.4) | 128 (34.3) | 144 (22.3) | 215 (50.9) | 249 (49.6) | 280 (47.4) |
| HDL-C concentration | −2.7 | 10.7 (20.5) | 37.9 (20.6) | 95.6 (30.4) | 118 (33.2) | 140 (36.4) | 136 (43.9) |
| LDL-C concentration | 25.0 | −8.8 (20.8) | −29.6 (6.9) | −39.1 (14.3) | −41.7 (11.4) | −43.6 (14.6) | −53.2 (15.0) |
| Total cholesterol | 1.4 | −8.1 (18.1) | −12.7 (7.1) | −7.9 (14.2) | −1.4 (12.4) | 3.7 (18.2) | 7.5 (17.2) |
| Triglycerides | −28.8 | −46.1 (20.9) | −50.2 (16.2) | −45.8 (16.5) | −20.3 (20.2) | −22.2 (19.9) | −31.4 (25.8) |
Values are mean (SD). Group 1 received 5 mg TA-8995/placebo on day 1 and days 8 to 42. Groups 2–5 received 1, 2.5, 10 and 25 mg TA-8995/placebo on day 1 and days 8 to 35.
HDL-C concentrations increased in a dose-dependent manner following both single and repeated dosing (Figure 3C). Once daily TA-8995 at doses of 2.5 to 25 mg led to marked increases from baseline HDL-C of approximately 96% up to 140%. LDL-C concentrations decreased in a dose-dependent manner with maximum changes from baseline of approximately −40% to −53% following 2.5 to 25 mg once daily TA-8995 (Figure 3D, Table 5). The maximum percent changes from baseline were statistically significantly different from placebo (P < 0.0001) following once daily TA-8995 doses of 5 to 25 mg for HDL-C and following 10 and 25 mg for LDL-C. HDL-C and LDL-C concentrations started to return towards baseline following cessation of TA-8995 dosing consistent with the loss of CETP inhibition.
There were trends indicating dose-related increases in apolipoproteins A1 and E, HDL2-C and HDL3-C and decreases in apolipoprotein B and Lp(a) concentrations (Supporting Information Figure S3). Variability was high for all of those variables. Nevertheless the data suggest that maximum effects may have been achieved with doses of 5 to 10 mg once daily TA-8995. There was no dose-related trend in apolipoprotein A2 or phospholipids (Supporting Information Figure S3), but there were dose-related increases in HDL-FC, HDL-CE and HDL-PL and decreases in HDL-TG across the dose range 1 to 10 mg with no further changes noted at 25 mg TA-8995 (data not shown). There were no noteworthy changes in LDL particle size (data not shown).
No consistent dose-related changes were observed for total cholesterol and triglycerides following TA-8995 dosing (Figure 3E, F).
Similar trends were observed for the pharmacodynamic variables in study 1 following single doses of TA-8995 from 1 to 150 mg, where dose-related decreases in CETP activity and LDL-C concentration and increases in CETP and HDL-C concentrations were observed (data not shown). No consistent effects on total cholesterol or triglycerides were observed in study 1. In addition, there was no evidence of any clinically relevant effect of food, age, gender or ethnicity on the pharmacodynamic variables.
Tolerability
Single doses of TA-8995 up to 150 mg and repeated doses up 25 mg once daily were well tolerated in all subjects. There were no serious adverse events and no subjects withdrew because of adverse events. The only adverse events reported by more than two subjects in either study were headache, epistaxis, pharyngolaryngeal pain, rhinorrhoea, nasopharyngitis, upper abdominal pain and myalgia. The incidence of those events was not dose-related (see Supporting Information Tables S3 and S4).
There were no clinically significant effects on blood pressure or heart rate, ECG variables, physical examination or laboratory safety tests (other than those discussed under pharmacodynamics). In particular, TA-8995 had no effect on serum electrolyte or aldosterone concentrations.
Discussion
Despite the evidence supporting the potential of CETP inhibition in reducing cardiovascular morbidity, clinical development of CETP inhibitors has not been straightforward. The first compound to progress to phase 3 clinical trials was torcetrapib. Torcetrapib was shown to increase HDL-C by 72% and decrease LDL-C by 25%, but it was subsequently withdrawn from development owing to safety concerns including an unexpected increase in cardiovascular events and death when in combination with atorvastatin, compared with atorvastatin alone 11. Although the mechanism of those events is not fully understood, there is increasing evidence that they might have been due to off-target effects of torcetrapib such as increased blood pressure, changes in electrolytes (increases in sodium and bicarbonate and decreases in potassium) and increases in aldosterone, consistent with mineralocorticoid activity 11–15. There is also some evidence from animal studies that torcetrapib increases expression of endothelin-1, which has been postulated to have contributed to the apparent increase in vascular deaths and possibly the (non-significant) increase in cancer deaths in the ILLUMINATE trial 16,17. Subsequently another CETP inhibitor, dalcetrapib, entered phase 2B clinical trials. Dalcetrapib was shown to be a weak inhibitor that increased HDL-C by 30–40% with minimal effects on LDL-C concentrations but did not appear to exhibit the off-target effects of torcetrapib 18–20 Recently, dalcetrapib development has also been terminated on the grounds of futility i.e. it did not reduce the risk of recurrent cardiovascular events 18.
Two more CETP inhibitors, anacetrapib and evacetrapib, are currently in phase 3 clinical trials. Data from phase 2 studies suggest that both are potent CETP inhibitors without mineralocorticoid activity. Anacetrapib 200 mg once daily has been shown to increase HDL-C by 97% and decrease LDL-C by 36% in healthy subjects 21 and 100 mg once daily anacetrapib in combination with statins has been shown to increase HDL-C by 149% and decrease LDL-C by 45% in patients 22. Evacetrapib (500 mg once daily monotherapy in patients) has been shown to increase HDL-C by 129% and decrease LDL-C by 36% 23. No outcome data are available currently to confirm whether this apparent improved potency over torcetrapib and dalcetrapib translates into a reduction in cardiovascular risk.
Data from these initial clinical trials of TA-8995 in healthy subjects have shown that it is also a potent CETP inhibitor with near maximal inhibition of CETP activity being observed with once daily doses of 2.5 mg TA-8995 and above. Inhibition of CETP activity was associated with increases in HDL-C of up to 140% and decreases in LDL-C of up to 44% (53% observed with 25 mg) with once daily doses of 10 mg TA-8995. Similar changes were observed for 200 mg anacetrapib (which increased HDL-C by 97% when given fasted and 123% when fed and decreased LDL-C by 36% [fasted] to 56% [fed]) 21 and 500 mg evacetrapib (which increased HDL-C by 129%, and decreased LDL-C by 36%) 23. The effects of TA-8995 were maintained throughout the dosing period and then returned steadily towards baseline after cessation of TA-8995 dosing. Despite the reduction in CETP activity observed with TA-8995, CETP concentration increased during the dosing period but declined during the follow-up period. That apparent induction of CETP mass has also been noted with other CETP inhibitors 33.
There was no dose-related effect of TA-8995 on total cholesterol concentration, presumably because of the conflicting effects of TA-8995 on the various sub-components including HDL-C, LDL-C and their related lipoproteins. There was also no dose-related effect on triglyceride concentrations. Similar effects have been reported for anacetrapib 21 although evacetrapib has been shown to reduce triglycerides at high doses (500 mg). Whilst there is strong evidence that decreased HDL-C and increased LDL-C are indicators of increased cardiovascular risk, the relevance of changes in triglycerides is still unclear 1.
TA-8995 also appeared to increase the HDL-C apolipoproteins A1 and E as well as the HDL subfractions HDL2-C and HDL3-C and phospholipids, and decrease the LDL-C apolipoprotein B as well as Lp(a). Decreased apolipoprotein A1 and elevated apolipoprotein B are recognized markers of cardiovascular disease 24–26.
In total, such changes in atherogenic lipoproteins, accompanied by an increase of the HDL-2 fraction and apolipoproteins A1 and E, support the hypothesis that TA-8995 treatment would decrease cardiovascular disease risk.
The potential of TA-8995 to lower Lp(a) is also of particular interest, as increased Lp(a) concentrations have been found to be a causal factor for cardiovascular disease, myocardial infarction and aortic stenosis and there is an urgent need for drugs to treat high Lp(a) 27–31. A longitudinal cohort study demonstrated that lipid apheresis combined with lipid-lowering medication in 120 patients with coronary artery disease significantly decreased Lp(a) concentrations (by approximately 75%) and also significantly reduced the rates of major adverse coronary events by 86% (>95% for myocardial infarction) 32. Although no randomized studies have yet shown that lowering Lp(a) decreases cardiovascular risk, the data are encouraging and, as a result, a reimbursement structure is now in place in Germany to treat high risk patients with lipoprotein apheresis.
A recent study of anacetrapib suggests that its pharmacodynamic effects may persist for many weeks after cessation of therapy 33. In particular, increases in HDL-C and decreases in LDL-C were still apparent 8 weeks after the end of an 8 week dosing period. Those effects appear to be related to the persistence of anacetrapib in plasma, and the authors conclude that anacetrapib may have a terminal half-life of 3 to 4 weeks. That is much longer than the 60–80 h previously quoted in healthy subjects 21. Whilst a difference in the pharmacokinetics of anacetrapib between healthy subjects and patients cannot be excluded, it is also possible that the sampling period in the phase 1 studies (up to 120 h after the last dose) was too short to characterize adequately the true terminal half-life. In the studies described here, TA-8995 concentrations were measured for 336 h after the final dose, so the calculated half-life of 120–150 h after repeated dosing probably represents the true terminal phase. Likewise, the steady return to baseline of the affected pharmacodynamic variables after cessation of dosing, also suggests that long term residual activity of TA-8995 is unlikely.
Pharmacokinetic data suggest that TA-8995 is well absorbed after oral administration with peak plasma concentrations typically observed within 4 to 6 h post-dose. Importantly there was no evidence of a clinically relevant effect of food on either the pharmacokinetics or pharmacodynamics of TA-8995. This was in contrast with the significant food effect observed with anacetrapib where a high fat meal increases AUC(0,∞) and Cmax by six- and eight-fold, respectively, compared with fasting conditions 34. Non-proportional pharmacokinetics of TA-8995 were observed with lower than expected increases in Cmax and AUC relative to increasing dose. Despite its long half-life, TA-8995 trough concentrations appeared to reach steady-state within 1 to 2 weeks of repeated dosing. TA-8995 is not renally cleared, as no drug could be detected in the urine.
Overall the data from these two studies suggest that TA-8995 is well tolerated at doses higher than those needed for adequate CETP inhibition. Importantly there was no evidence of any of the off-target effects that are implicated in the toxicity of torcetrapib, such as changes in blood pressure, serum electrolytes (including sodium, potassium and bicarbonate) or aldosterone.
These studies confirm that TA-8995 is a potent inhibitor of CETP activity in humans. This inhibition leads to significant elevations in HDL-C and apolipoprotein A1 and reductions in LDL-C and apolipoprotein B. Promising trends in other markers of cardiovascular risk, such as Lp(a), were also observed. In addition, although TA-8995 appears to have similar potency to other CETP inhibitors in clinical development, it may offer benefits in terms of dose size, lack of food effects and lack of prolonged residual effects on cessation of dosing. Further studies are warranted in patients with cardiovascular risk factors to determine whether these findings translate into clinical benefit. A phase 2 multicentre, randomized, double-blind, placebo-controlled study of TA-8995 in patients with mild dyslipidaemia, alone and in combination with statin therapy, is now in progress.
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare NM, SM, KT, SK, AK are employed by Mitsubishi Tanabe Pharma Corporation who sponsored the submitted work. ML, DF, CS are employed by Mitsubishi Pharma Europe Ltd who was the EU representative of the sponsor of the submitted work. SW was the Principal Investigator and MB was the co-investigator for study 1 and both were employed by Hammersmith Medicines Research who received funding from Mitsubishi Tanabe Pharma Corporation to conduct the study. WW was the Principal Investigator for study 2 and was employed by Momentum Pharma Services GmbH who received funding from Mitsubishi Tanabe Pharma Corporation to conduct the study. JF, PR are employed by Xention Ltd who are contracted to Dezima BV, which now owns the licence for TA-8995. JJPK is employed by, and holds shares in, Dezima BV. SvD is employed by Dezima BV. There are no financial relationships with any other organizations that might have an interest in the submitted work in the previous 3 years. There are no other relationships or activities that could appear to have influenced the submitted work.
The authors would like to thank Samantha Abel (Valley Writing Solutions Ltd, UK) for her help with the preparation of this manuscript.
Funding Sources
These studies were sponsored by Mitsubishi Tanabe Pharma Corporation, Tokyo, Japan.
Supporting Information
Additional Supporting Information may be found in the online version of this article at the publisher’s web-site:
Figure S1 Participant flow (Study 1)
Figure S2 Participant flow (Study 2)
Figure S3 Mean (SD) percent change from baseline in secondary pharmacodynamic variables (Study 2). Timepoints have been staggered for presentation purposes
Table S1 Mean (SD) baseline secondary pharmacodynamic variables (Study 2)
Table S2 Summary of statistical analyses (Study 2)
Table S3 Treatment-emergent adverse events occurring in more than two (2%) subjects (Study 1)
Table S4 Treatment-emergent adverse events occurring in more than two (3%) subjects (Study 2)
Supporting Information S1 Ethics committee details and analytical methods
References
- The Emerging Risk Factors Collaboration. Major lipids, apolipoproteins, and risk of vascular disease. JAMA. 2009;302:1993–2000. doi: 10.1001/jama.2009.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cholesterol Treatment Trialists (CTT) Collaboration. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170000 participants in 26 randomised trials. Lancet. 2010;13:1670–1681. doi: 10.1016/S0140-6736(10)61350-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roger VL, Go AS, Lloyd-Jones DM, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Makuc DM, Marcus GM, Marelli A, Matchar DB, Moy CS, Mozaffarian D, Mussolino ME, Nichol G, Paynter NP, Soliman EZ, Sorlie PD, Sotoodehnia N, Turan TN, Virani SS, Wong ND, Woo D, Turner MB American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics – 2012 Update: a report from the American Heart Association. Circulation. 2012;125:e2–e220. doi: 10.1161/CIR.0b013e31823ac046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johannsen TH, Frikke-Schmidt R, Schou J, Nordestgaard BG, Tybjærg-Hansen A. Genetic inhibition of CETP, ischemic vascular disease and mortality, and possible adverse effects. J Am Coll Cardiol. 2012;60:2041–2048. doi: 10.1016/j.jacc.2012.07.045. [DOI] [PubMed] [Google Scholar]
- Voight BF, Peloso GM, Orho-Melander M, Frikke-Schmidt R, Barbalic M, Jensen MK, Hindy G, Hólm H, Ding EL, Johnson T, Schunkert H, Samani NJ, Clarke R, Hopewell JC, Thompson JF, Li M, Thorleifsson G, Newton-Cheh C, Musunuru K, Pirruccello JP, Saleheen D, Chen L, Stewart A, Schillert A, Thorsteinsdottir U, Thorgeirsson G, Anand S, Engert JC, Morgan T, Spertus J, Stoll M, Berger K, Martinelli N, Girelli D, McKeown PP, Patterson CC, Epstein SE, Devaney J, Burnett MS, Mooser V, Ripatti S, Surakka I, Nieminen MS, Sinisalo J, Lokki ML, Perola M, Havulinna A, de Faire U, Gigante B, Ingelsson E, Zeller T, Wild P, de Bakker PI, Klungel OH, Maitland-van der Zee AH, Peters BJ, de Boer A, Grobbee DE, Kamphuisen PW, Deneer VH, Elbers CC, Onland-Moret NC, Hofker MH, Wijmenga C, Verschuren WM, Boer JM, van der Schouw YT, Rasheed A, Frossard P, Demissie S, Willer C, Do R, Ordovas JM, Abecasis GR, Boehnke M, Mohlke KL, Daly MJ, Guiducci C, Burtt NP, Surti A, Gonzalez E, Purcell S, Gabriel S, Marrugat J, Peden J, Erdmann J, Diemert P, Willenborg C, König IR, Fischer M, Hengstenberg C, Ziegler A, Buysschaert I, Lambrechts D, Van de Werf F, Fox KA, El Mokhtari NE, Rubin D, Schrezenmeir J, Schreiber S, Schäfer A, Danesh J, Blankenberg S, Roberts R, McPherson R, Watkins H, Hall AS, Overvad K, Rimm E, Boerwinkle E, Tybjaerg-Hansen A, Cupples LA, Reilly MP, Melander O, Mannucci PM, Ardissino D, Siscovick D, Elosua R, Stefansson K, O’Donnell CJ, Salomaa V, Rader DJ, Peltonen L, Schwartz SM, Altshuler D, Kathiresan S. Plasma HDL cholesterol and risk of myocardial infarction: a Mendelian randomisation study. Lancet. 2012;380:572–580. doi: 10.1016/S0140-6736(12)60312-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson A, Di Angelantonio E, Sarwar N, Erqou S, Saleheen D, Dullaart RPF, Keavney B, Ye Z, Danesh J. Association of cholesteryl ester transfer protein genotypes with CETP mass and activity, lipid levels, and coronary risk. JAMA. 2008;299:2777–2788. doi: 10.1001/jama.299.23.2777. [DOI] [PubMed] [Google Scholar]
- Ridker PM, Pare G, Parker AN, Zee RYL, Miletich JP, Chasman DI. Polymorphism in the CETP gene region, HDL cholesterol, and risk of future myocardial infarction: genomewide analysis among 18 245 initially healthy women from the Women’s Genome Health Study. Circ Cardiovasc Genet. 2009;2:26–33. doi: 10.1161/CIRCGENETICS.108.817304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto H, Yonemori F, Wakitani K, Minowa T, Maeda K, Shinkai H. A cholesteryl ester transfer protein inhibitor attenuates atherosclerosis in rabbits. Nature. 2000;406:203–207. doi: 10.1038/35018119. [DOI] [PubMed] [Google Scholar]
- Barter PJ, Rye KA. Cholesteryl ester transfer protein inhibition as a strategy to reduce cardiovascular risk. J Lipid Res. 2012;53:1755–1766. doi: 10.1194/jlr.R024075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochem AE, Kuivenhoven JA, Stroes ESG. The promise of cholesteryl ester transfer protein (CETP) inhibition in the treatment of cardiovascular disease. Curr Pharm Des. 2013;19:3143–3149. doi: 10.2174/1381612811319170022. [DOI] [PubMed] [Google Scholar]
- Barter PJ, Caulfield M, Eriksson M, Grundy SM, Kastelein JJP, Komajda M, Lopez-Sendon J, Mosca L, Tardi J-C, Waters DD, Shear CL, Revkin JH, Buhr KA, Fisher MR, Tall AR, Brewer B the ILLUMINATE investigatores. Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med. 2007;357:21009–22122. doi: 10.1056/NEJMoa0706628. [DOI] [PubMed] [Google Scholar]
- Kastelein JJP, van Leuven SI, Burgess L Evans GW, Kuivenhoven JA, Barter PJ, Revkin JH, Grobbee DE, Riley WA, Shear CL, Duggan WT, Bots ML RADIANCE 1 Investigators. Effect of torcetrapib on carotid atherosclerosis in familial hypercholesterolemia. N Engl J Med. 2007;356:1620–1630. doi: 10.1056/NEJMoa071359. [DOI] [PubMed] [Google Scholar]
- Nicholls SJ, Tuzcu EM, Brennan DM, Tardif J-C, Nissen SE. Cholesteryl ester transfer protein inhibition, high-density lipoprotein raising, and progression of coronary atherosclerosis. Insights from ILLUSTRATE (Investigation of Lipid Level Management Using Coronary Ultrasound to Assess Reduction of Atherosclerosis by CETP Inhibition and HDL Elevation) Circulation. 2008;118:2506–2514. doi: 10.1161/CIRCULATIONAHA.108.790733. [DOI] [PubMed] [Google Scholar]
- Vergeer M, Bots ML, van Leuven SI, Basart DC, Sijbrands EJ, Evans GW, Grobbee DE, Visseren FL, Stalenhoef AF, Stroes ES, Kastelein JJP. Cholesteryl ester transfer protein inhibitor torcetrapib and off-target toxicity: pooled analysis of the rating atherosclerotic disease change by imaging with a new CETP inhibitor (RADIANCE) trials. Circulation. 2008;118:2515–2522. doi: 10.1161/CIRCULATIONAHA.108.772665. [DOI] [PubMed] [Google Scholar]
- Forrest MJ, Bloomfield D, Briscoe RJ, Brown PN, Cumiskey A-M, Ehrhart J, Hershey JC, Keller WJ, Ma X, McPherson HE, Messina E, Peterson LB, Sharif-Rodriguez W, Siegl PKS, Sinclair PJ, Sparrow CP, Stevenson AS, Sun S-Y, Tsai C, Vargas H, Walker M, West SH, White V, Woltmann RF. Torcetrapib-induced blood pressure elevation is independent of CETP inhibition and is accompanied by increasing circulating levels of aldosterone. Br J Pharmacol. 2008;154:1465–1473. doi: 10.1038/bjp.2008.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simic B, Hermann M, Shaw SG, Bigler L, Stalder U, Dörries C, Besler C, Lüscher TF, Ruschitzka F. Torcetrapib impairs endothelial function in hypertension. Eur Heart J. 2012;33:1615–1624. doi: 10.1093/eurheartj/ehr348. [DOI] [PubMed] [Google Scholar]
- Barter PJ, Rye K-A, Beltangady MS, Ports WC, Duggan WT, Boekholdt SM, DeMicco DA, Kastelein JJ, Shear CL. Relationship between atorvastatin dose and the harm caused by torcetrapib. J Lipid Res. 2012;53:2436–2442. doi: 10.1194/jlr.P026328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz GG, Olsson AG, Abt M, Ballantyne CM, Barter PJ, Brumm J, Chaitman BR, Holme IM, Kallend D, Leiter LA, Leitersdorf E, McMurray JJ, Mundl H, Nicholls SJ, Shah PK, Tardif JC, Wright RS dal-OUTCOMES Investigators. Effects of dalcetrapib in patients with recent acute coronary syndrome. N Engl J Med. 2012;367:2089–2099. doi: 10.1056/NEJMoa1206797. [DOI] [PubMed] [Google Scholar]
- Stein EA, Stroes ES, Steiner G, Buckley BM, Capponi AM, Burgess T, Niesor EJ, Kallend D, Kastelein JJ. Safety and tolerability of dalcetrapib. Am J Cardiol. 2009;104:82–91. doi: 10.1016/j.amjcard.2009.02.061. [DOI] [PubMed] [Google Scholar]
- Lüscher TF, Taddei S, Kaski JC, Jukema JW, Kallend D, Münzel T, Kastelein JJ, Deanfield JE dal-VESSEL Investigators. Vascular effects and safety of dalcetrapib in patients with or at risk of coronary heart disease: the dal-VESSEL randomized clinical trial. Eur Heart J. 2012;33:857–865. doi: 10.1093/eurheartj/ehs019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishna R, Bergman AJ, Fallon M, Cote J, Van Hoydonck P, Laethem T, Gendrano IN, 3rd, Van Dyck K, Hilliard D, Laterza O, Snyder K, Chavez-Eng C, Lutz R, Chen J, Bloomfield DM, De Smet M, Van Bortel LM, Gutierrez M, Al-Huniti N, Dykstra K, Gottesdiener KM, Wagner JA. Multiple-dose pharmacodynamics and pharmacokinetics of anacetrapib, a potent cholesteryl ester transfer protein (CETP) inhibitor, in healthy subjects. Clin Pharmacol Ther. 2008;84:679–683. doi: 10.1038/clpt.2008.109. [DOI] [PubMed] [Google Scholar]
- Cannon CP, Shah S, Dansky HM, Davidson M, Brinton EA, Gotto AM, Stepanavage M, Liu SX, Gibbons P, Ashraf TB, Zafarino J, Mitchel Y, Barter P Determining the Efficacy and Tolerability Investigators. Safety of anacetrapib in patients with or at high risk for coronary heart disease. N Engl J Med. 2010;363:2406–2415. doi: 10.1056/NEJMoa1009744. [DOI] [PubMed] [Google Scholar]
- Nicholls SJ, Brewer HB, Kastelein JJ, Krueger KA, Wang MD, Shao M, Hu B, McErlean E, Nissen SE. Effects of the CETP inhibitor evacetrapib administered as monotherapy or in combination with statins on HDL and LDL cholesterol. JAMA. 2011;306:2099–2109. doi: 10.1001/jama.2011.1649. [DOI] [PubMed] [Google Scholar]
- Dansky HM, Bloomfield D, Gibbons P, Liu S, Sisk CM, Tribble D, McKenney JM, Littlejohn TW, 3rd, Mitchel Y. Efficacy and safety after cessation of treatment with the cholesteryl ester transfer protein inhibitor anacetrapib (MK-0859) in patients with primary hypercholesterolemia or mixed hyperlipidemia. Am Heart J. 2011;162:708–716. doi: 10.1016/j.ahj.2011.07.010. [DOI] [PubMed] [Google Scholar]
- Florvall G, Basu S, Larsson A. Apolipoprotein A1 is a stronger prognostic marker than HDL and LDL cholesterol for cardiovascular disease and mortality in elderly men. J Gerontol A Biol Sci Med Sci. 2006;61:1262–1266. doi: 10.1093/gerona/61.12.1262. [DOI] [PubMed] [Google Scholar]
- Walldiius G, Jungner I. Rationale for using apolipoprotein B and apolipoproteins A-1 as indicators of cardiac risk and as targets for lipid-lowering therapy. Eur Heart J. 2005;26:210–212. doi: 10.1093/eurheartj/ehi077. [DOI] [PubMed] [Google Scholar]
- Barter PJ, Ballantyne CM, Carmena R, Castro Cabezas M, Chapman MJ, Couture P, de Graaf J, Durrington PN, Faergeman O, Frohlich J, Furberg CD, Gagne C, Haffner SM, Humphries SE, Jungner I, Krauss RM, Kwiterovich P, Marcovina S, Packard CJ, Pearson TA, Reddy KS, Rosenson R, Sarrafzadegan N, Sniderman AD, Stalenhoef AF, Stein E, Talmud PJ, Tonkin AM, Walldius G, Williams KM. Apo B versus cholesterol in estimating cardiovascular risk and in guiding therapy: report of the thirty-person/ten-country panel. J Intern Med. 2006;259:247–258. doi: 10.1111/j.1365-2796.2006.01616.x. [DOI] [PubMed] [Google Scholar]
- Nordestgaard BG, Chapman MJ, Ray K, Borén J, Andreotti F, Watts GF, Ginsberg H, Amarenco P, Catapano A, Descamps OS, Fisher E, Kovanen PT, Kuivenhoven JA, Lesnik P, Masana L, Reiner Z, Taskinen MR, Tokgözoglu L, Tybjærg-Hansen A European Atherosclerosis Society Consensus Panel. Lipoprotein(a) as a cardiovascular risk factor: current status. Eur Heart J. 2010;31:2844–2853. doi: 10.1093/eurheartj/ehq386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamstrup PR, Tybjaerg-Hansen A, Nordestgaard BG. Lipoprotein(a) and risk of myocardial infarction – genetic epidemiologic evidence of causality. Scand J Clin Lab Invest. 2011;71:87–93. doi: 10.3109/00365513.2010.550311. [DOI] [PubMed] [Google Scholar]
- The Emerging Risk Factors Collaboration. Lipoprotein(a) concentration and the risk of coronary heart disease, stroke and nonvascular mortality. JAMA. 2009;302:412–423. doi: 10.1001/jama.2009.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamstrup PR, Benn M, Tybjaerg-Hansen A, Nordestgaard BG. Extreme lipoprotein(a) levels and risk of myocardial infarction in the general population: the Copenhagen city heart study. Circulation. 2008;117:176–184. doi: 10.1161/CIRCULATIONAHA.107.715698. [DOI] [PubMed] [Google Scholar]
- Thanassoulis G, Campbell CY, Owens DS, Smith JG, Smith AV, Pelos GM, Kerr KF, Pechlivanis S, Budoff MJ, Harris TB, Malhotra R, O’Brien KD, Kamstrup PR, Nordestgaard BG, Tybjaerg-Hansen A, Allison MA, Aspelund T, Criqui MH, Heckbert SR, Hwang S-J, Liu Y, Sjogren M, van der Pals J, Kälsch H, Mühleisen TW, Nöthen MM, Cupples LA, Caslake M, Di Angelantonio E, Danesh J, Rotter JI, Sigurdsson S, Wong Q, Erbel R, Kathiresan S, Melander O, Gudnason V, O’Donnell CJ, Post WS CHARGE Extracoronary Calcium Working Group. Genetic associations with valvular calcification and aortic stenosis. N Engl J Med. 2013;368:503–512. doi: 10.1056/NEJMoa1109034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeger BR, Richter Y, Nagel E, Heigl F, Vogt A, Roeseler E, Parhofer K, Ramlow W, Koch M, Utermann G, Labarrere CA, Seidel D Group of Clinical Investigators. Longitudinal cohort study on the effectiveness of lipid apheresis treatment to reduce high lipoprotein(a) levels and prevent major adverse coronary events. Nat Clin Pract Cardiovasc Med. 2009;6:229–239. doi: 10.1038/ncpcardio1456. [DOI] [PubMed] [Google Scholar]
- Krishna GA, Panebianco D, Cote J, Bergman AJ, Van Hoydonck P, Laethem T, Van Dyck K, Chen J, Chavez-Eng C, Archer L, Lutz R, Hilliard D, Snyder K, Jin B, Van Bortel L, Lasseter KC, Al-Huniti N, Dykstra K, Gottesdiener K, Wagner JA. Single dose pharmacokinetics and pharmacodynamics of anacetrapib, a potent cholesteryl ester transfer protein (CETP) inhibitor, in healthy subjects. Br J Clin Pharmacol. 2009;68:535–545. doi: 10.1111/j.1365-2125.2009.03465.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Participant flow (Study 1)
Figure S2 Participant flow (Study 2)
Figure S3 Mean (SD) percent change from baseline in secondary pharmacodynamic variables (Study 2). Timepoints have been staggered for presentation purposes
Table S1 Mean (SD) baseline secondary pharmacodynamic variables (Study 2)
Table S2 Summary of statistical analyses (Study 2)
Table S3 Treatment-emergent adverse events occurring in more than two (2%) subjects (Study 1)
Table S4 Treatment-emergent adverse events occurring in more than two (3%) subjects (Study 2)
Supporting Information S1 Ethics committee details and analytical methods