

Abstract
Aims
The aim of this phase 1, single centre, open label study in four patients with solid tumours was to determine the absolute bioavailability of a 2 mg oral dose of trametinib. Trametinib is an orally bioavailable, reversible and selective allosteric inhibitor of MEK1 and MEK2 activation and kinase activity.
Methods
A microtracer study approach, in which a 5 μg radiolabelled i.v. microdose of trametinib was given concomitantly with an unlabelled 2 mg oral tablet formulation, was used to recover i.v. and oral pharmacokinetic parameters, simultaneously.
Results
The least-squares mean (90% confidence interval) absolute bioavailability of trametinib (2 mg tablet) was 72.3% (50.0%, 104.6%). Median tmax after oral administration was 1.5 h and the geometric mean terminal half-life was 11 days. The geometric mean clearance and volume of distribution after i.v. administration were 3.21 l h−1 and 976 l, respectively, resulting in a terminal elimination half-life of 11 days.
Conclusions
Trametinib absolute bioavailability was moderate to high, whereas first pass metabolism was low.
Keywords: bioavailability, intravenous, microtracer, pharmacokinetic, trametinib
What is Already Known about this Subject —
Following oral dosing of 2 mg trametinib, median tmax was 1.5 h and the mean effective half-life (t1/2) was approximately 4 days. Trametinib is greater than 95% protein bound. Trametinib accumulates about six-fold with repeat daily dosing, and the concentration–time profiles showed a flat profile at steady-state with a low peak: trough ratio.
The absolute bioavailability of trametinib has not been previously reported.
The microtracer approach has been used previously to recover i.v. and oral pharmacokinetic parameters, simultaneously, which are used to estimate absolute bioavailability.
Determination of absolute bioavailability improves our understanding of the clinical pharmacology of a compound.
What this Study Adds —
Trametinib has moderate to high bioavailability (72.3%) following oral administration of a 2 mg tablet.
The i.v. microtracer study approach revealed the contribution of high oral bioavailability with low first pass metabolism and a prolonged terminal elimination phase to the pharmacokinetics of trametinib.
Introduction
Trametinib is a reversible, highly selective allosteric inhibitor of MEK1/MEK2 activation and kinase activity 1,2 which has been approved in the United States for the treatment of BRAF V600E or V600K mutation-positive melanoma. Clinical activity of trametinib in unresectable, BRAF V600 mutation-positive melanoma was observed in phase 1, 2 and 3 studies 3–5. Significant improvement in overall survival and progression free survival over dacarbazine or paclitaxel was confirmed in the pivotal phase 3 study (METRIC) 5.
The pharmacokinetics (PK) of trametinib after oral administration were characterized in the first-time-in-human (FTIH) study 6. Following oral dosing of 2 mg trametinib, median tmax was 1.5 h and the mean effective half-life (t1/2) was approximately 4 days. Trametinib accumulates about six-fold with repeat daily dosing and the concentration–time profiles showed a flat profile at steady-state with a low peak: trough ratio.
Determination of absolute bioavailability improves our understanding of the clinical pharmacology of a compound (e.g. by characterizing the i.v. pharmacokinetics) and is required by several regulatory agencies 7. Conventional crossover studies have been fraught with the challenges and delays associated with preparing and administering a drug intravenously that has been developed for oral use. The use of i.v. radiolabelled microtracer studies has become an attractive alternative to conducting conventional crossover design absolute bioavailability studies and provides characterization of the i.v. PK of a new compound. The microtracer approach involves administration of an unlabelled oral therapeutic dose with concomitant administration of a subtherapeutic radiolabelled i.v. microdose (pharmacologically inactive). A microdose is defined as less than 1/100th of the calculated clinical dose (based on animal data) needed to yield a pharmacologic effect, up to a maximum of 100 μg 8,9. The radiolabelled microdose (<270 nCi) produces trivial exposure to ionizing radiation and does not require additional i.v. toxicity studies or extensive i.v. formulation development. The microtracer approach has the advantage of simultaneous recovery of i.v. and oral pharmacokinetic parameters to obtain within-subject, within-period comparison. As such, this approach reduces the variability of recovered pharmacokinetic parameters, reduces the number of subjects required and provides a more precise estimate of absolute bioavailability than does the use of a traditional crossover design study. I.v. radiolabelled microtracer studies have been widely implemented at GlaxoSmithKline (GSK) within clinical pharmacology programmes and across diverse therapeutic areas. Microtracer studies have been used successfully to determine the absolute bioavailability of several compounds at GSK (dabrafenib 7, SRT-2104 (GSK2245840) 10, and GSK962040 11) and elsewhere (fexofenadine, IDX899, IDX989 and saxagliptin (dapagliflozin, SCH 900518) 12–17) using either the marketed oral formulation or an oral microdose as the test treatment.
The purpose of this study was to characterize the oral and i.v. PK and to determine the absolute oral bioavailability of trametinib using simultaneous administration of an unlabelled oral dose (2 mg tablet) and a radiolabelled i.v. microdose (5 μg) in patients with solid tumours.
Methods
Clinical study design
The study protocol (MEK115064; ClinicalTrials.gov identifier NCT01416337) and consent form were approved by a duly constituted institutional review board (Alpha Independent Review Board, San Clemente, CA, USA), and the study was conducted in accordance with good clinical practice and the guiding principles of the Declaration of Helsinki at Comprehensive Clinical Development NW, Inc (Tacoma, WA, USA). Signed and dated written informed consent was obtained from each patient before his or her participation in the study and before any study-related procedures were performed.
This was a phase 1, single centre, open label study in patients with solid tumours. After obtaining informed consent, eligible participants were admitted to the clinical research unit on day −1. After fasting overnight for 8 h, all participants received trametinib as a single 2 mg tablet (with 240 ml water) and a single i.v. dose of 5 μg (7.4 kBq, 200 nCi) [14C]-trametinib, administered 1.5 h after the oral dose (to coincide with oral tmax). The participants continued fasting for 4 h after the oral dose. The 5 μg (1 ml) i.v. microdose of [14C]-trametinib (in 5% hydroxypropyl β-cyclodextrin [Cavitron] for injection) was injected as a slow i.v. push over 1 min into the infusion line through a ‘Y’ injection port co-administered with either a 50 ml solution of normal saline or 5% dextrose administered at a rate of 10 ml min−1. All excipients used in the i.v. formulation had GRAS (generally regarded as safe) status. The i.v. infusion was given in the arm not being used for PK sampling.
Blood samples (two 6 ml samples at each time point) for analysis of trametinib, [14C]-trametinib and total radioactivity were collected at several time points through 10 h on day 1 (pre-dose and 0.5, 1, 1.5, 1.75, 2, 2.5, 3, 4, 5, 6, 8, 10 h) and at single time points on days 2, 3, 4, 5, 8 and 11 after the oral dose. The actual date and time of each blood sample collection were recorded. Blood samples were stored on wet ice for a maximum of 1 h before plasma was separated from blood by centrifugation at 4°C and stored at −70°C prior to analysis. After completion of the study, all participants entered an open label rollover study of trametinib (no washout period or follow-up visit required) and continued receiving trametinib (MEK114375, ClinicalTrials.gov identifier NCT01376310).
Dose selection
The recommended oral dose of trametinib is 2 mg once daily. The highest dosage strength (2 mg tablet) was used in this study. An i.v. dose of 5 μg was selected, which is 400-fold lower than the oral dose, thus fitting the criteria of a microdose (i.e. less than 100th of the dose needed to yield a pharmacologic effect, up to a maximum of 100 μg) 8,9. Due to the very low i.v. dose, a target detection limit of 2.5 pg ml−1 was considered acceptable to characterize the PK profile of trametinib. The lower limit achieved in developing the assay was 1.1 pg ml−1.
Study participants
Men or women were eligible if they were ≥18 years of age with solid tumours, had an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1, and had adequate organ function (total bilirubin ≤1.5 times ULN, ALT ≤2.5 times ULN, creatinine ≤1.5 times ULN, Cockroft–Gault creatinine clearance ≥50 ml min−1, 24 h urine creatinine clearance ≥50 ml min−1, LVEF ≥LLN by ECHO or MUGA). A body weight of ≥45 kg and a body mass index between 19 and 45 kg m−2 were required. Participants were excluded if they had received cancer therapy with delayed toxicity within 3 weeks, chemotherapy without delayed toxicity within 2 weeks, an investigational anticancer drug within 28 days, or any other investigational product within 30 days, five half-lives or twice the duration of the biological effect of the investigational product. In addition, patients were excluded if they had participated in a 14C human research study within 12 months preceding administration of the study medication. Consumption of food, herbal remedies or medication known to interfere with cytochrome P450 (CYP) 3A activity was prohibited, although it was later confirmed that the major pathway of elimination of trametinib is not mediated by CYP3A4. Subjects with untreated leptomeningeal or brain metastases or spinal cord compression were excluded. Subjects with a Fridericia-corrected QT interval (QTcF) or Bazett-corrected QT interval (QTcB) ≥480 ms, with history or evidence of cardiovascular risk, with a history of interstitial lung disease or pneumonitis, with a history of retinal vein occlusion or central serous retinopathy or with conditions affecting gastrointestinal absorption were excluded.
Trametinib HPLC/MS/MS analysis
Trametinib was extracted from human plasma samples with an isotopically labelled standard ([13C6]-trametinib) by liquid-liquid extraction using ethyl acetate. Extracts were injected onto a BEH C18 column (50 × 2.1 mm, 1.7 μm; Waters Corporation, Milford, MA, USA), maintained at 65°C and eluted with a binary mobile phase gradient using 0.1% formic acid in water (A) and 0.1% formic acid in acetonitrile (B), with a constant flow rate of 0.7 ml min−1. The initial mobile phase condition of 50:50 A:B was held until 0.8 min, when the composition changed to 35:65 A:B. From 0.81 to 1.0 min, the mobile phase held at 20:80 A:B and reverted back to the initial conditions (50:50 A:B) at 1.1 min. Detection was performed by positive ion MS/MS with multiple reaction monitoring (m/z 616 to m/z 491 for trametinib). The lower limit of quantification (LLQ) for trametinib was 0.25 ng ml−1 for a 50 μl aliquot of human plasma with a higher limit of quantification (HLQ) of 250 ng ml−1. Quality control samples (QC), prepared at three different analyte concentrations (four replicates per concentration) and stored with the study samples, were analyzed with each batch of samples against separately prepared calibration standards. For the analysis to be acceptable, no more than one-third of the total QC results and no more than one-half of the results from each concentration level were to deviate from the nominal concentration by more than 15%. Data acquisition and quantitation were performed with Analyst version 1.4.2. The applicable analytical runs met all predefined run acceptance criteria.
[14C]-trametinib and total radioactivity analysis
Samples were analyzed for [14C]-trametinib and total plasma radioactivity using accelerator mass spectrometry (AMS). Prior to analysis by AMS, the carbon within the samples was harvested and converted to graphite. This involved a two stage process of sample combustion (oxidation) followed by graphitization (reduction), as previously published 18. AMS provides an isotope ratio from which total radiocarbon per mg carbon is derived. The carbon content of plasma was taken into account to allow correction of the AMS data to determine the total amount of radiocarbon in the samples. Carbon analysis was undertaken using a Costech Elemental Combustion System CHNS-O Analyzer (Costech Analytical Technologies, Inc, Valencia, CA, USA) supplied by Pelican Scientific Ltd (Cheshire, UK). The AMS was a 250 kV Single Stage Accelerator Mass Spectrometer (National Electrostatics Corp., Middleton, WI, USA), operated via NEC proprietary ‘AccelNET’ software on a Linux operating system. Post-acquisition data processing was performed using the NEC software ‘abc.’ Further details of the operating conditions have been previously published 18. The LLQ for total radioactivity was 0.95 pg equiv ml−1.
Plasma [14C]-trametinib concentrations at all time points were determined with a validated analytical LC + AMS method over the range of 1.1 to 101 pg ml−1. Trametinib (including [14C]-trametinib as a tracer) was extracted from 200 μl of human plasma by protein precipitation using three volumes of acetonitrile containing non-labelled trametinib at 2 μg ml−1 as an internal standard (IS). The samples were vortex-mixed and centrifuged at 3000 × g; the resultant supernatant was removed and dried using a vacuum centrifuge. The sample was reconstituted in 250 μl of acetonitrile/water (50:50) and 50 μl was injected onto the HPLC system. Isolation of trametinib was achieved using a Phenomenex Synergi-Polar column (4 μm packing, 4.6 × 250 mm) maintained at 30°C. The mobile phase consisted of 10 mM ammonium acetate (pH 5) (A) and acetonitrile (B). A flow rate of 1 ml min−1 was maintained for the entire run. The following 70 min gradient was used: started at 5% B, increased to 20% B over 5 min, increased to 60% B over 40 min, increased to 95% B over 5 min and maintained at 95% B for 5 min, and decreased to 5% B over 5 min. The column was then re-equilibrated at 5% B over 5 min. The area of the u.v. response of the IS was recorded, and the eluate corresponding to trametinib was collected as a discrete fraction into a quartz tube for further processing to harvest graphite and subsequent analysis by AMS. The assay had a linear dynamic range of 1.1 to 101 pg ml−1 and quantification was performed against trametinib-spiked recovery standards based on demonstrated linearity with non-weighted linear regression.
During method validation, concentrations of [14C]-trametinib in validation samples were determined on two occasions. Five of 33 validation samples failed to meet the acceptance criteria of being within 20% of the actual concentrations (25% at LLOQ). The bias was assessed to be <10% with a precision of <17.3%.
Study samples were analyzed as four separate batches with QC samples included in each batch. Individual QC results were deemed acceptable if the calculated concentration was within 20% of the actual concentration, and the analytical run was accepted if no more than one-third of the QCs exceeded the acceptable limit with at least one QC sample at each concentration within the acceptable limit. Only two of 36 samples failed to meet the established acceptance criteria (±20%) through four analytical runs.
Eight study samples (≈11%) were re-analyzed for incurred sample reproducibility (ISR) with all samples being within 20% of the mean of the original and ISR result.
Clinical pharmacokinetics
Analysis of plasma concentration data via non-compartmental methods was conducted using WinNonlin Professional Edition version 5.2 (Pharsight Corporation, Mountain View, CA, USA) non-compartmental Model 200 (for extravascular administration) for trametinib and non-compartmental Model 202 (for constant infusion) for [14C]-trametinib and total radioactivity by ICON Development Solutions (Marlow, UK). Standard non-compartmental PK parameters, including maximum concentration (Cmax), tmax, area under the concentration–time curve from time zero to 244 h or to infinity (AUC(0,tlast) and AUC(0,∞), respectively) and half-life (t1/2) for trametinib, [14C]-trametinib and total radioactivity, were estimated based on actual sampling times. Systemic clearance (CL) and apparent oral clearance (CL/F) were determined after i.v. and oral dosing, respectively. Vss is described as the volume of distribution at steady-state following i.v. dosing and is based on mean residence time (MRT) and CL, where Vss = MRT(0,∞) × CL. Vd,z is described as the volume of distribution during the terminal phase and is calculated as dosei.v./(λz × AUC(0,∞)i.v.), where λz is the terminal slope elimination rate constant and AUC(0,∞)i.v. following i.v. administration of trametinib is defined above.
Statistical analysis
Summary statistics are reported for each PK parameter. For calculation of absolute bioavailability, a mixed-effect model (SAS, version 9.1.3, on the Windows SAS platform) was fitted to dose-normalized, loge-transformed area under the curve (AUC(0,tlast) or AUC(0,∞)) with the subject as a random effect and treatment as a fixed effect (oral was the test, i.v. was the reference). Dose-normalized AUC(0,tlast) or AUC(0,∞) was obtained by dividing AUC(0,tlast) or AUC(0,∞) by dose for each subject. The least-squares mean difference and associated 90% confidence intervals (CIs) in AUC(0,tlast) or AUC(0,∞) between the oral and i.v. doses were obtained from the model. This least-squares mean difference was back-transformed to provide a point estimate and corresponding 90% CI for the absolute bioavailability (F). Due to the long elimination half-life and large extrapolation of area, the estimate of bioavailability is also reported based on AUC(0,tlast).
Results
Participants
Four patients (two White men and two White women;) were enrolled and underwent study procedures. All patients had metastatic (stage IV) cancer at screening, including oesophageal cancer, non-small cell lung cancer (NSCLC), ocular melanoma and bladder cancer. All patients had an ECOG performance status of 0 or 1 at screening. All received the oral and i.v. dose as planned.
Clinical pharmacokinetics
The geometric least squares mean (90% CI) absolute bioavailability (F) of the 2 mg trametinib tablet were 72.3% (50.0%, 104.6%) and 84.2% (66.1%, 107.2%) based on AUC(0,tlast) and AUC(0,∞), respectively. Individual values ranged from 45.7% to 92.8% and from 65.9% to 109% when F was determined using AUC(0,tlast) and AUC(0,∞), respectively.
Individual concentration–time profiles following oral dose and i.v. microdose are illustrated in Figure 1. Selected PK parameters for trametinib observed following oral administration and for [14C]-trametinib and total radioactivity following i.v. administration are summarized in Table 1. For a 400-fold difference in dose, trametinib geometric mean Cmax and AUC(0,tlast) were 76-fold and 289-fold lower, respectively, after i.v, microdose relative to the oral dose. Geometric mean i.v. trametinib plasma clearance, Vd,z, and Vss were 3.21 l h−1, 1060 l and 976 l, respectively, after administration of i.v. microdose. Apparent clearance following oral dose of trametinib was 3.81 l h−1.
Figure 1.
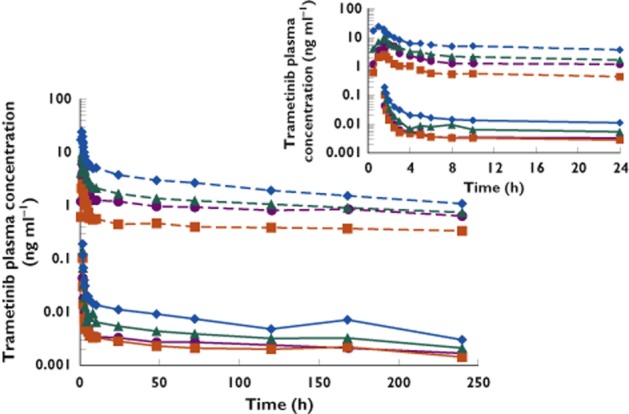
(A) Individual plasma trametinib and [14C]-trametinib concentration vs. time profiles after oral (2 mg) and i.v. (5 μg) administration. Individual patients are denoted by symbols and colours. The dashed lines represent oral trametinib; the solid lines represent i.v. [14C]-trametinib. Inset shows detail from 0 to 24 h post-dose
Table 1.
Clinical pharmacokinetics of trametinib after concomitant oral and i.v. administration (2 mg trametinib orally, 5 μg [14C]-trametinib i.v.)
| Parameter (units) | Trametinib (oral) | [14C]-trametinib (i.v.) | Total radioactivity* (i.v.) |
|---|---|---|---|
| tmax† (h) | 1.50 | 0.08 | 0.08 |
| (1.00, 1.58) | (0.08, 0.08) | (0.08,0.08) | |
| Cmax‡ (ng ml–1) | 8.03 | 0.105 | 0.118 |
| [118] | [71] | [54] | |
| (2.56, 24.31) | (0.044, 0.190) | (0.058, 0.177) | |
| AUC(0,t)‡ (ng ml–1 h) | 248 | 0.858 | 1.68 |
| [83] | [57] | [56] | |
| (99.8, 580) | (0.546, 1.76) | (1.18, 3.60) | |
| AUC(0,∞) (ng ml–1 h) | 525 | 1.56 | 4.92§, 5.91§ |
| [36] | [31] | ||
| (333, 783)‡ | (1.26, 2.39)‡ | ||
| t1/2 (h) | 264 | 229 | 643§, 176§ |
| [62] | [39] | ||
| (130, 481)‡ | (144, 348)‡ | ||
| CL or CL/F‡ (l h–1) | 3.81 | 3.21 | ND |
| [36] | [31] | ||
| (2.56, 6.01) | (2.09, 3.97) | ||
| Vss‡ (l) | ND | 976 | ND |
| [79] | |||
| (385, 1836)‡ | |||
| Vd,z‡ (l) | ND | 1060 | ND |
| [75] | |||
| (435, 1985)‡ |
t1/2, half-life; tmax, time to reach Cmax; Vss, volume of distribution at steady state; Vd,z, volume of distribution during the terminal phase.
Total radioactivity concentration units are ng equivalents trametinib ml−1.
tmax results are presented as median (minimum, maximum).
Results are presented as geometric mean [%CV] (minimum, maximum) for n >2.
Elimination half-life and AUC(0,∞) for total radioactivity were estimated in only two of four subjects; individual subject values are listed. AUC(0,∞), area under the concentration–time curve from time zero to infinity; AUC(0,t), area under the concentration–time curve from time zero to time t; CL, systemic clearance; CL/F, apparent oral clearance; Cmax, maximum concentration; ND, not determined.
The median trametinib tmax after oral administration was 1.5 h and coincided with the tmax observed at the end of the i.v. infusion (0.08 h post-end of infusion). After tmax, a rapid distribution phase was observed as plasma concentrations decreased five-fold and 21-fold from tmax to 24 h post-oral and i.v. dose, respectively (Figure 1). The rapid distribution phase was followed by a prolonged terminal elimination phase after both oral and i.v. administration. The geometric mean terminal phase half-life for trametinib after oral dose and i.v. microdose administration was 264 h (≈11 days) and 229 h (≈10 days), respectively.
Mean profiles of [14C]-trametinib and total radioactivity plasma concentration over time are shown in Figure 2. Trametinib as parent accounted for 52% (range: 43%–63%) of the total radioactivity observed after a single i.v. dose.
Figure 2.
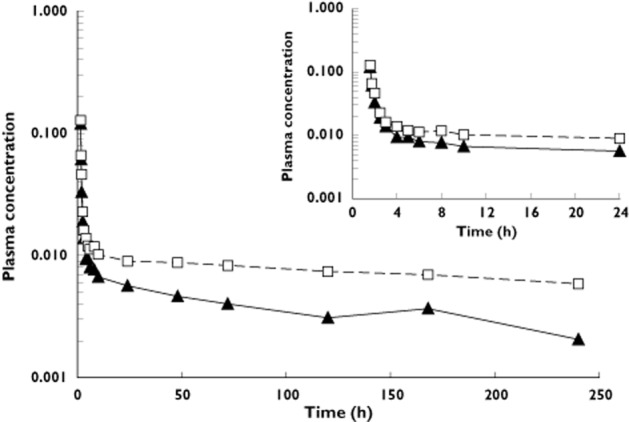
Mean plasma [14C]-trametinib (solid line, ng ml−1) and total radioactivity (dashed line, ng equivalents of trametinib ml−1) concentration vs. time profile after i.v.; 5 μg administration (n = 4). Inset shows detail from 0 to 24 h post-dose.  , [14C]-trametinib;
, [14C]-trametinib;  , total radioactivity
, total radioactivity
As shown in Figure 1, the rank order for each subject was consistent following oral and i.v. dosing, such that subjects with comparatively high exposure following oral administration also had high exposure after i.v. administration. This was further investigated by examining relationships between exposure and subject characteristics. Figure 3 shows trametinib and [14C]-trametinib AUC(0,tlast) and Cmax values by gender and by body weight. There was an apparent inverse relationship between body weight and trametinib exposure. Female subjects had higher exposure, consistent with their lower body weight.
Figure 3.

Individual AUC(0,tlast) and Cmax values for a single 2 mg oral dose of trametinib (A,C) or a single 5 μg i.v. dose of [14C]-trametinib (B,D). Symbols denote male (triangles) and female (circles) subjects
Adverse events
All reported adverse events (AEs) were grade 1 and non-recurrent and most (except headache, ecchymosis and excoriation in one subject) resolved by the time subjects transitioned to the rollover study to continue receiving trametinib. None of the AEs was considered related to treatment with the study medication. There were no withdrawals due to AEs and no reports of AEs related to changes in clinical laboratory tests, echocardiograms or vital signs. There were no reports of serious AEs.
Discussion
The primary objective of this study was to determine the absolute bioavailability (F) of trametinib after concomitant administration of a radiolabelled i.v. microtracer dose (5 μg) and an unlabelled oral therapeutic dose (2 mg tablet formulation). To enable determination of concentrations following administration of the 5 μg i.v. microtracer dose, an AMS assay was validated with a lower limit of quantitation of 1.1 pg ml−1. Based on AUC(0,tlast), the absolute bioavailability of trametinib was 72.3% (range, 45.7%–92.8% indicating moderate to high bioavailability of the 2 mg tablet following oral administration.
Trametinib had low i.v. plasma CL of 3.21 l h−1 (0.94 l h−1 blood clearance), which is approximately 1% of liver blood flow (where liver blood flow is 81 l h−1 19 and a blood:plasma ratio of 3.4). High absolute bioavailability and low clearance suggest low hepatic extraction and limited first pass metabolism of trametinib. A large Vss (976 l) suggests that trametinib distributes considerably to tissues. The distribution is rapid, as evidenced by the large initial decrease in concentrations after a single oral and i.v. dose (5- and 21-fold decrease within 24 h of tmax, respectively). Under steady-state conditions, a low peak:trough ratio of 1.8 was observed in subjects after once daily dosing of trametinib (2 mg) in the FTIH study. This may be attributable to the fact that the distribution processes are less pronounced with repeat dosing 6.
Subjects with comparatively high exposure following oral administration also had high exposure after i.v. administration. In addition, female subjects with lower body weight had higher exposure compared with male subjects with higher body weight, consistent with results from the population PK analysis where oral clearance (CL/F) was affected by both gender and weight, whereas the distributional clearance was affected by body weight 20.
The estimated half-lives after oral and i.v. administration were 11 and 10 days, respectively. The half-life determined in the food effect study (MEK113709 21) was approximately 5 days, likely due the shorter sampling period (up to 7 days) while the accumulation half-life was only 4 days. Using a population PK model, estimated terminal half-lives were 3.9 and 4.8 days for male and female subjects, respectively, and were consistent with the effective half-life 20. Despite a lengthy elimination phase, it is expected that steady-state exposure following repeat daily dosing of 2 mg will be reached within 15 to 20 days. This is because the AUC attributed to the long elimination phase contributes little to total AUC.
Trametinib profiles following both oral and i.v. administration showed an initial rapid distribution phase followed by a protracted elimination phase likely reflective of slow elimination from deep compartments. It has been shown that parent trametinib, but not metabolites, preferentially distributes to red blood cells and this accounts for the initial rapid distribution phase 22 and is supportive of the observation that following a single i.v. dose, parent trametinib accounts for 52% (range 43%–63%) of the total radioactivity in plasma. Following repeat dosing, as steady-state is reached, the fraction of parent trametinib in plasma accounts for >75%, while the two metabolites each accounted for approximately 10% of total radioactivity.
Given the long elimination half-life relative to the sample collection period and large extrapolation of AUC(0,∞), the primary estimate of F was based on AUC(0,tlast). In general, estimates of F using AUC(0,∞) agreed (within 17%) with estimates using AUC(0,tlast), except for one subject in whom the estimate of absolute bioavailability differed from that in the other three subjects by 44%. This subject had an elimination half-life for trametinib in plasma that was 1.5 times higher than the geometric mean half-life. For two subjects, accurate estimation of elimination half life (and AUC(0,∞)) was not possible due to a relatively flat elimination phase of the plasma concentration–time curve.
In conclusion, the i.v. microtracer study approach revealed the contribution of moderate/high oral bioavailability with low first pass metabolism and a prolonged terminal elimination phase to the PK of trametinib. Although this study had its limitations due to the study population, namely the small number of patients, the advantages of this approach was a shorter study time frame (compared with a traditional crossover design) and simultaneous collection of i.v. and oral pharmacokinetic parameters with reduced variability. In the case of trametinib, which has a long half-life, the i.v. microtracer approach is especially useful. This approach obviates the need for a lengthy washout period required in a conventional crossover study, and allows patients to receive treatment without delay by entering into the rollover study and also allows recruitment of a small number of patients while recovering the necessary PK parameters. As such, i.v. microtracer studies offer an innovative and efficient approach for assessing absolute bioavailability and may be applied across a range of therapeutic areas and compound classes.
Competing Interests Disclosure
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare CL, CP, JB, GCY, MH, FH, KO and DO are full-time paid employees and stockholders of GlaxoSmithKline. LF, an independent contractor, received payment from GlaxoSmithKline for statistical analysis related to this study and RAM’s institution received payment from GlaxoSmithKline to conduct the clinical research study.
Thanks are due to Steven Corless and Mike Hobbs (DMPK, GlaxoSmithKline) for the analysis of samples by AMS and LC + AMS, Sherry Wang and Ciara Rodgers (DMPK, GlaxoSmithKline) for analysis by LC/MS/MS, Alexandra Piepszak (GlaxoSmithKline, data management), Veronique Fauvel and Christopher Jefferds (ICON Clinical Research) who assisted with the conduct of the trial, Ken Martin (Comprehensive Clinical Development) and the patients who participated in this study. Funding for this study was provided by GlaxoSmithKline (NCT01416337). All listed authors meet the criteria for authorship set forth by the International Committee for Medical Journal Editors. Editorial support in the form of collating author comments, copyediting, referencing, and graphic services was provided by Clinical Thinking and was funded by GlaxoSmithKline.
References
- Abe H, Kikuchi S, Hayakawa K, Iida T, Nagahashi N, Maeda K, Sakamoto J, Matsumoto N, Miura T, Matsumura K, Seki N, Inaba T, Kawasaki H, Yamaguchi T, Kakefuda R, Nanayama T, Kurachi H, Hori Y, Yoshida T, Kakegawa J, Watanabe Y, Gilmartin AG, Richter MC, Moss KG, Laquerre SG. Discovery of a highly potent and selective MEK inhibitor: GSK1120212 (JTP-74057 DMSO solvate) ACS Med Chem Lett. 2011;2:320–324. doi: 10.1021/ml200004g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmartin AG, Bleam MR, Groy A, Moss KG, Minthorn EA, Kulkarni SG, Rominger CM, Erskine S, Fisher KE, Yang J, Zappacosta F, Annan R, Sutton D, Laquerre SG. GSK1120212 (JTP-74057) is an inhibitor of MEK activity and activation with favorable pharmacokinetic properties for sustained in vivo pathway inhibition. Clin Cancer Res. 2011;17:989–1000. doi: 10.1158/1078-0432.CCR-10-2200. [DOI] [PubMed] [Google Scholar]
- Falchook GS, Lewis KD, Infante JR, Gordon MS, Vogelzang NJ, DeMarini DJ, Sun P, Moy C, Szabo SA, Roadcap LT, Peddareddigari VG, Lebowitz PF, Le NT, Burris HAI, Messersmith WA, O’Dwyer PJ, Kim KB, Flaherty K, Bendell JC, Gonzalez R, Kurzrock R, Fecher LA. Activity of the oral MEK inhibitor trametinib in patients with advanced melanoma: a phase 1 dose-escalation trial. Lancet Oncol. 2012;13:782–789. doi: 10.1016/S1470-2045(12)70269-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KB, Kefford R, Pavlick AC, Infante JR, Ribas A, Sosman JA, Fecher LA, Millward M, McArthur GA, Hwu P, Gonzalez R, Ott PA, Long GV, Gardner OS, Ouellet D, Xu Y, DeMarini DJ, Le NT, Patel K, Lewis KD. Phase II study of the MEK1/MEK2 inhibitor trametinib in patients with metastatic BRAF-mutant cutaneous melanoma previously treated with or without a BRAF inhibitor. J Clin Oncol. 2013;31:482–489. doi: 10.1200/JCO.2012.43.5966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaherty KT, Robert C, Hersey P, Nathan P, Garbe C, Milhem M, Demidov LV, Hassel JC, Rutkowski P, Mohr P, Dummer R, Trefzer U, Larkin JM, Utikal J, Dreno B, Nyakas M, Middleton MR, Becker JC, Casey M, Sherman LJ, Wu FS, Ouellet D, Martin AM, Patel K, Schadendorf D. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012;367:107–114. doi: 10.1056/NEJMoa1203421. [DOI] [PubMed] [Google Scholar]
- Infante JR, Fecher LA, Falchook GS, Nallapareddy S, Gordon MS, Becerra C, DeMarini DJ, Cox DS, Xu Y, Morris SR, Peddareddigari VG, Le NT, Hart L, Bendell JC, Eckhardt G, Kurzrock R, Flaherty K, Burris HAI, Messersmith WA. Safety, pharmacokinetic, pharmacodynamic, and efficacy data for the oral MEK inhibitor trametinib: a phase 1 dose-escalation trial. Lancet Oncol. 2012;13:773–781. doi: 10.1016/S1470-2045(12)70270-X. [DOI] [PubMed] [Google Scholar]
- Denton CL, Minthorn E, Carson SW, Young GC, Richards-Peterson LE, Botbyl J, Han C, Morrison RA, Blackman SC, Ouellet D. Concomitant oral and intravenous pharmacokinetics of dabrafenib, a BRAF inhibitor, in patients with BRAF V600 mutation-positive solid tumors. J Clin Pharmacol. 2013;53:955–961. doi: 10.1002/jcph.127. [DOI] [PubMed] [Google Scholar]
- Department of Health and Human Services; Center for Drug Evaluation and Research (CDER) 2003. FDA guidance for industry: bioavailability and bioequivalence studies for orally administered drug products – general considerations. Available at http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm070124.pdf (last accessed 26 February 2013)
- European Medicines Agency (EMEA); Committee for Medicinal Products for Human Use (CHMP) 2013. Position paper on non-clinical safety studies to support clinical trials with a single microdose, 2004. Available at http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_000400.jsp&mid=WC0b01ac0580029570 (last accessed 26 February 2013)
- Hoffmann E, Wald J, Lavu S, Roberts J, Beaumont C, Haddad J, Elliott P, Westphal C, Jacobson E. Pharmacokinetics and tolerability of SRT2104, a first-in-class small molecule activator of SIRT1, after single and repeated oral administration in man. Br J Clin Pharmacol. 2013;75:186–196. doi: 10.1111/j.1365-2125.2012.04340.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasist Johnson LS, Young MA, Stevens LA, Cozens SJ, Collier J, Robertson DC, Dukes GE. A microtracer study of GSK962040, a motilin receptor agonist, to support dosing regimens in the critical care setting. Clin Pharmacol Ther. 2013;93:S80. [Google Scholar]
- Gao L, Li J, Kasserra C, Song Q, Arjomand A, Hesk D, Chowdhury SK. Precision and accuracy in the quantitative analysis of biological samples by accelerator mass spectrometry: application in microdose absolute bioavailability studies. Anal Chem. 2011;83:5607–5616. doi: 10.1021/ac2006284. [DOI] [PubMed] [Google Scholar]
- Xu XS, Dueker SR, Christopher LJ, Lohstroh PN, Keung CF, Cao KK, Bonacorsi SJ, Cojocaru L, Shen JX, Humphreys WG, Stouffer B, Arnold ME. Overcoming bioanalytical challenges in an onglyza intravenous [(14)C]microdose absolute bioavailability study with accelerator MS. Bioanalysis. 2012;4:1855–1870. doi: 10.4155/bio.12.171. [DOI] [PubMed] [Google Scholar]
- Boulton DW, Kasichayanula S, Keung CF, Arnold ME, Christopher LJ, Xu XS, Lacreta F. Simultaneous oral therapeutic and intravenous 14C-microdoses to determine the absolute oral bioavailability of saxagliptin and dapagliflozin. Br J Clin Pharmacol. 2013;75:763–768. doi: 10.1111/j.1365-2125.2012.04391.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappin G, Shishikura Y, Jochemsen R, Weaver RJ, Gesson C, Houston B, Oosterhuis B, Bjerrum OJ, Rowland M, Garner C. Pharmacokinetics of fexofenadine: evaluation of a microdose and assessment of absolute oral bioavailability. Eur J Pharm Sci. 2010;40:125–131. doi: 10.1016/j.ejps.2010.03.009. [DOI] [PubMed] [Google Scholar]
- Sarapa N, Hsyu PH, Lappin G, Garner RC. The application of accelerator mass spectrometry to absolute bioavailability studies in humans: simultaneous administration of an intravenous microdose of 14C-nelfinavir mesylate solution and oral nelfinavir to healthy volunteers. J Clin Pharmacol. 2005;45:1198–1205. doi: 10.1177/0091270005280051. [DOI] [PubMed] [Google Scholar]
- Zhou XJ, Garner RC, Nicholson S, Kissling CJ, Mayers D. Microdose pharmacokinetics of IDX899 and IDX989, candidate HIV-1 non-nucleoside reverse transcriptase inhibitors, following oral and intravenous administration in healthy male subjects. J Clin Pharmacol. 2009;49:1408–1416. doi: 10.1177/0091270009343698. [DOI] [PubMed] [Google Scholar]
- Young GC, Corless S, Felgate CC, Colthup PV. Comparison of a 250 kV single-stage accelerator mass spectrometer with a 5 MV tandem accelerator mass spectrometer – fitness for purpose in bioanalysis. Rapid Commun Mass Spectrom. 2008;22:4035–4042. doi: 10.1002/rcm.3829. [DOI] [PubMed] [Google Scholar]
- Rowland M, Tozer TN. Clinical Pharmacokinetics: Concepts and Applications. 2. Philadelphia, PA: Lea & Febiger; 1989. [Google Scholar]
- Kassir N, Mouksassi M, Cox DS, DeMarini DJ, Gardner OS, Sherman L, Crist WA, Ouellet D. Population pharmacokinetics (PK) of trametinib (GSK1120212), a MEK inhibitor, in subjects with cancer. Clin Pharmacol Ther. 2013;93:S69. [Google Scholar]
- Cox DS, Papdopoulos KP, Fang L, Bauman J, LoRusso P, Tolcher AW, Patnaik A, Pendry C, Orford K, Ouellet D. Evaluation of the effects of food on the single-dose pharmacokinetics of trametinib, a first-in-class MEK inhibitor, in patients with cancer. J Clin Pharmacol. 2013;53:946–954. doi: 10.1002/jcph.115. [DOI] [PubMed] [Google Scholar]
- Ho MY, Morris MJ, Pirhalla JL, Bauman JW, Pendry CB, Orford KW, Morrison RA, Cox DS. Trametinib, a first-in-class oral MEK inhibitor mass balance study with limited enrollment of two male subjects with advanced cancers. Xenobiotica. 2013;44:352–368. doi: 10.3109/00498254.2013.831143. [DOI] [PubMed] [Google Scholar]