

Abstract
Aims
The aim was to characterize the population pharmacokinetics of BYL719 in cancer patients and assess the time course of tumour response in relation to drug exposure and dosing schedule.
Methods
Plasma samples and longitudinal tumour size measurements were collected from 60 patients with advanced solid malignancies who received oral BYL719 once daily (30–450 mg) or twice daily at 120 mg or 200 mg. Non-linear mixed effect modelling was employed to develop the population pharmacokinetic and pharmacodynamic model.
Results
The pharmacokinetics were best described by a one compartment disposition model and transit compartments accounting for the lag time in absorption. The typical population oral clearance and volume of distribution estimates with their between-subject variability (BSV) were 10 l h−1 (BSV 26%) and 108 l (BSV 28%), respectively. The estimated optimal number of transit compartments was 8.1, with a mean transit time to the absorption compartment of 1.28 h (BSV 32%). The between-occasion variability in the rate and extent of absorption was 46% and 26%, respectively. Tumour growth was modelled using a turnover model characterized by a zero order growth rate of 0.581 cm week1 and a first order death rate of 0.0123 week−1. BYL719 inhibited tumour growth with an IC50 of 100 ng ml−1 (BSV 154%). Model-based predictions showed potential for additional anti-tumour activity of twice daily dosing at total daily dose below 400 mg, but a loss of efficacy if administered less frequently than once daily.
Conclusions
The proposed model provides a valuable approach for planning future clinical studies and for designing optimized dosing regimens with BYL719.
Keywords: BYL719, cancer, phosphoinositide 3-kinase, population pharmacokinetics, RECIST, transit compartment
What Is Already Known About This Subject —
BYL719 is a phosphoinositide 3-kinase inhibitor (PI3Ki) in clinical development for the treatment of cancer. There are also several other PI3Kis in clinical development.
Clinical benefit of PI3Kis according to the RECIST criteria has been reported including stable disease and partial tumour responses.
Although the RECIST classification offers a simple criterion to standardize tumour response, it limits what can be learnt about the drug’s potency and time course of effect.
What This Study Adds —
An increased understanding of BYL719’s potential for anti-tumour activity was derived from a pharmacokinetic–pharmacodynamic model that described the time course of tumour response in relation to drug exposure, which, in turn, provided an estimate of its relevant pharmacodynamic parameters.
Model-based predictions identified the potential for clinical benefit of alternative dosing regimens.
Introduction
Phosphoinositide 3-kinase (PI3Ks) are lipid kinases that are important in controlling signalling pathways involved in cell proliferation, motility, cell death and cell invasion 1,2. PI3Ks transduce signals from various growth factors and cytokines into intracellular messengers. The PI3K lipid kinase family comprises eight enzymes divided into three classes based on sequence homology comparisons. The most commonly studied are the class 1A enzymes that are activated directly by cell surface receptors such as receptor tyrosine kinases and G-protein coupled receptors 2–5. Human cells contain three genes (PIK3CA, PIK3CB and PIK3CD) that encode the catalytic subunits of class IA PI3K enzymes, termed PI3Kα, PI3Kβ and PI3Kδ, respectively 5. The major polypeptides produced by these three genes are p110α, p110β and p110δ 3–5. The α and β isoforms are ubiquitously expressed, whereas the δ isoform plays a major role in lymphocyte development, differentiation and activation 5,6. The class IB PI3K consists of only one enzyme, p110γ, which is expressed in leukocytes and in a small number of other tissues 4–6.
Mutation or amplification of PIK3CA promotes oncogenic activation of the PI3K pathway, and occurs frequently in human cancers 7–10. A high frequency of hot spot mutations has been observed in colorectal cancers, gastric cancers, brain cancers as well as breast and lung cancers 7–10. PI3K pathway activation may also be associated with resistance to chemotherapy and targeted agents in different cancers 11–13. In addition, data suggest that p110α might be the predominant isoform in vasculogenesis and p110α-specific inhibitors might block angiogenesis 14. Selective inhibition of the PI3K pathway therefore represents a promising therapeutic approach for cancer treatment 15.
BYL719, (S)-pyrrolidine-1,2-dicarboxylic acid 2-amide 1-({4-methyl-5-[2-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-pyridin-4-yl]-thiazol-2-yl}-amide), is an oral α-specific class I PI3K inhibitor 16,17. It inhibits wild-type and mutated p110α more potently than the other isoforms 16,17. Preclinical studies have shown anti-proliferative activity in cellular systems and in human tumour xenograft models with or without PIK3CA mutations, with good correlation between drug exposure and inhibition of PI3K signalling. BYL719 is currently tested in several clinical studies for the treatment of cancer, both as a single agent and in combination therapy with chemotherapeutic drugs or targeted therapies.
The ongoing clinical study CBYL719X2101 is the first in human dose escalation study of oral BYL719 in adult patients with advanced solid malignancies, whose tumours have an alteration of the PIK3CA gene 18. Its primary objective is to determine the maximum tolerated dose (MTD). At the time of this analysis, patients were dosed once daily in the dose range of 30–450 mg or twice daily at 120 mg or 200 mg. The MTD was declared at 400 mg once daily. Enrolment in the MTD expansion cohort at 400 mg once daily is ongoing. In addition, safety and tolerability of a twice daily dosing regimen is currently being explored. The most frequently observed adverse events are hyperglycaemia, gastro-intestinal related symptoms, fatigue and rash. Hyperglycaemia is considered an on-target effect of PI3K inhibition and appears to occur in a dose-related fashion 19. So far, BYL719 has been found to be safe and well tolerated, and clear evidence of target inhibition and preliminary antitumour activity was seen.
Longitudinal tumour size measurements were recorded in CBYL719X2101 for computing tumour response based on the Response Evaluation Criteria in Solid Tumours (RECIST). RECIST classifies tumour response in four categories: complete response, partial response, stable disease and progressive disease 20. Although the RECIST classification offers a simple criterion to standardize tumour response, it limits what can be learnt about the drug’s potency and time course of effect. This is because the RECIST classification ignores that repeated tumour size measurements at predefined time intervals provide a continuous scale measure of drug effect, which contains information related to the time course of drug effect. An increased understanding of drug action can be derived from a pharmacokinetic–pharmacodynamic (PK–PD) model that describes the time course of tumour response in relation to drug exposure, which, in turn, provides estimates of PD parameters that describe the specific relationships of interest. Stochastic simulations of the expected time course of drug effect can aid in identifying a drug dosage regimen and study design that will result in optimal therapeutic outcome 21,22.
The purpose of this study was to describe the population PK of BYL719 and explore the relationship of the systemic drug exposure to antitumour efficacy using available data from the first in human clinical study. The change in sum of longest tumour diameters over time was used to develop a population PK–tumour kinetic model to describe the relationship between systemic drug exposures and time course of drug effect. The PK–PD model was used to simulate the anti-tumour activity of different doses and regimens of BYL719.
Methods
Clinical study CBYL719X2101
Clinical study CBYL719X2101 was approved by the ethics committees of participating centers 18,23 and conducted in accordance with Good Clinical Practice guidelines and the Declaration of Helsinki. All patients gave written informed consent before participation. This study has been designed as a multicentre, open-label phase IA, dose escalation study with the primary objective of determining the MTD. The MTD was defined as the highest dose of BYL719 not causing dose limiting toxicities in >33% of patients in the first treatment cycle. Once the MTD has been determined an MTD-expansion arm would follow. Patients received oral, once daily BYL719 until disease progression, unacceptable toxicity, or withdrawal of consent. When adequate data had been collected to define the safety and PK of BYL719 given once daily, a twice daily dosing regimen was initiated by the addition of a new arm.
Patient population
The study population of study CBYL719X2101 consisted of adult patients with advanced solid tumours, whose tumours had a mutation or amplification of the PIK3CA gene, and whose disease had progressed despite standard therapy or for whom no standard therapy existed 18,23. Demographic characteristics of the patient population are provided in Table 1. Patients had histologically confirmed advanced tumours failing standard therapy, with a least one lesion as defined by RECIST, age ≥18 years, World Health Organization performance status ≤2, adequate bone marrow, hepatic and renal function and fasting plasma glucose concentrations ≤140 mg dl–1. Key exclusion criteria were corticosteroid treatment ≤2 weeks before starting BYL719, clinically manifest diabetes mellitus, or history of gestational diabetes, any severe or uncontrolled medical condition that could cause unacceptable risks or compromise compliance with the protocol and prior treatment with a PI3K, AKT or mTOR inhibitor with failure to benefit.
Table 1.
Summary of demographic characteristics of the patients
| Characteristic | Value |
|---|---|
| Number of patients* | 60 |
| Number of concentration data† | 1228 |
| Gender (male/female) | 12/48 |
| Age (mean ± SD) (range) (years) | 58.2 ± 10.4 (39–78) |
| Height (mean ± SD) (range) (cm) | 164 ± 8.62 (146–190) |
| Weight (mean ± SD) (range) (kg) | 74.7 ± 21.3 (36.7–178) |
| Primary tumour site (%)‡ | |
| Breast | 30 |
| Colorectal | 28 |
| Ovarian | 10 |
| Head and neck | 5 |
| Other | 27 |
| Sum of tumour diameters (mean ± SD) (range) (cm)‡ | 9.87 ± 5.37 (2.4–21.5) |
A total of 71 patients were enrolled, of whom 60 patients could be included in the model building (refer to methods).
A total of 51 patients had evaluable PK data (refer to methods). A total of 1228 plasma concentrations for which dose and sampling time were adequately recorded were included in the PKanalysis.
A total of 47 patients had evaluable PD data (refer to methods). A total of 146 tumour size measurements from 47 patients were used to establish the PD model.
PK assessments
For the once daily regimen, BYL719 was administered in the morning, approximately 1 h after a light breakfast. Patients were instructed to fast for 1 h after the administration of each BYL719 dose. Sequential blood sampling for PK was performed pre-dose and at 0.5, 1, 1.5, 2, 4, 6, 8 and 24 h post-dose on day 1 and day 8 of the first 28 day treatment cycle, and on the first day of the second treatment cycle. Additional pre-dose samples were drawn on the first day of every subsequent treatment cycle. For the twice daily regimen, BYL719 was administered in the morning and the evening (about 12 h apart), approximately 1 h after food consumption. Patients were instructed to fast for 1 h after the administration of each BYL719 dose. Sequential blood sampling was performed at pre-dose and at 0.5, 1, 1.5, 2, 3, 4, 6 and 10 h post-morning dose. Plasma concentrations of BYL719 were determined using a validated liquid chromatography-tandem mass spectrometry assay with a dynamic range of 1 to 1000 ng ml−1 (supplementary material).
Tumour assessments
Serial measurements of the largest dimension of the primary tumour were obtained by radiologic techniques at baseline, at the end of treatment cycle 2 and every 8 weeks thereafter. If multiple measurable lesions were present, the sum of the longest one dimensional measurement of each lesion was used instead. In addition, a bone scan was performed for all patients with clinical evidence of bone metastases.
Data used for model building
A total of 71 patients had been enrolled at the time of clinical data cutoff (July 2012). Individual patient PK data were considered evaluable for PK model building when there was at least one evaluable plasma sample and complete dose administration record up to and including the time of PK assessment. Individual patient PD data were considered evaluable for PD model building when there was a baseline tumour assessment and at least one post-baseline tumour assessment of the same subject, as well as a complete dose administration record up to and including the time of the last post baseline tumour assessment. A total of 11 patients had neither evaluable PK nor evaluable PD data and thus were excluded from all analyses. Of the remaining pool of 60 subjects, a subset of, respectively, 51 and 47 patients could be included in PK and PD model building. The longitudinal change in tumour measurements of all evaluable patients was described using a sequential PK–PD model fit approach (see below). First, the Bayesian post hoc estimates of the PK parameters were obtained from the final PK model, which were then fixed and used as an input function to the PD model. Patients received once daily doses of 30 mg (n = 1), 60 mg (n = 3), 90 mg (n = 6), 180 mg (n = 6), 270 mg (n = 4), 400 mg (n = 25) 450 mg (n = 9) or twice daily at 120 mg (n = 1) or 200 mg (n = 5).
Population PK analysis
A non-linear mixed effects model approach was used to estimate population parameters and their between subject (BSV) and between occasion (BOV) variability. Model development and parameter estimation were conducted using Phoenix NLME Version 1.1 (Pharsight, Mountain View, CA, USA) with first order conditional estimation – Extended Least Squares. A Bayesian approach conditioned on the population characteristics was used to estimate the individual specific parameters.
A transit compartment model (Figure 1A) in which the number of transit compartments was numerically estimated was developed as described by Savic et al. 24. In the transit model drug absorption arises as a consequence of transit through a chain of intermediate compartments before reaching the observation compartment. The analytical solution to estimate numerically the optimal number of transit compartments (N), and calculate the amount of drug in the last transit compartment AN(t) is given by equation 1.
| (1) |
where t is time post last dose, F is the absolute oral bioavailability (fixed to unity); Ktr is the first order transfer rate constant through the series of transit compartments and N! is the factorial function with argument N. To compute this solution numerically, Stirling’s approximation was employed (equation 2) 24.
| (2) |
Figure 1.
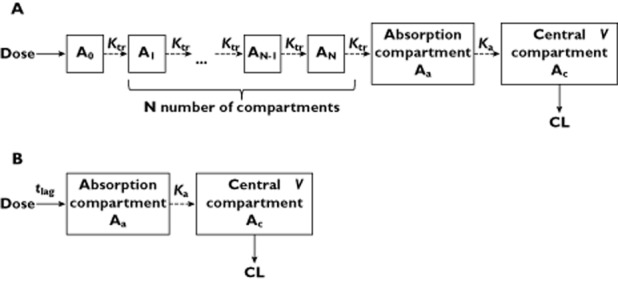
Population PK model structure of the BYL719. (A) transit model and (B) tlag model. Ktr is the first order transit transfer rate constant, Ka the first order absorption rate constant, N the total number of transit compartments, V the volume of distribution, CL the oral clearance and tlag the absorption lag time. The absorption and central compartment correspond to respectively Aa and Ac
To prevent numerical difficulties, the natural log transformation was used to describe the rate of change in drug amount in the absorption compartment Aa (equation 3).
| (3) |
where Ka is the first order absorption rate constant describing the transfer of the drug amount in the absorption compartment to the central compartment. Ktr was re-parameterized in the mean transit time (MTT), which represents the average time spent by the drug travelling from the first transit compartment to the absorption compartment (equation 4).
| (4) |
The structural disposition model was an open one compartmental model, parameterized in terms of drug clearance (CL) and the apparent volume of distribution (V). The differential equation describing the drug amount in the central compartment Ac is given by equation 5.
| (5) |
where C is the plasma concentration, calculated from V (Ac/ V).
The performance of the transit model was evaluated in comparison to the traditional lag-time model (Figure 1B). In the tlag model the rate of change in drug amount in the absorption compartment (Aa) was described using equation 6.
| (6) |
where Ka is the first order absorption rate constant describing the transfer of the drug amount in the absorption compartment (Aa) to the central compartment (Ac). The structural disposition model was identical to the transit model.
The BSV of the parameters was assumed to be log normally distributed. The relationship between a parameter and its variance was estimated using an exponential model expressed as follows:
| (7) |
where θi is the value of the parameter as predicted for the individual i; θ is the population typical value of the parameter, and ηi represents the difference in the estimated parameter for the ith subject from the population typical value. The ηi values are independently distributed random variables with a mean of zero and a variance of ω2. The hierarchical structure was constructed stepwise by first including BSV and then BOV where appropriate. Occasions were defined as scheduled visits for PK assessments: cycle 1 day 1 (OCC1), cycle 1 day 8 (OCC2) and cycle 2 day 1 (OCC3). Trough samples taken after OCC3 were pooled as OCC4.
The residual error was best described using a log additive error structure:
| (8) |
where yij is the jth observation in the ith individual; 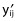 is the model’s predicted value; εij is the normally distributed additive random error with mean of zero and variance of σ2.
is the model’s predicted value; εij is the normally distributed additive random error with mean of zero and variance of σ2.
Population PK–PD analysis
The final tumour growth turnover model on the sum of longest tumour diameters is given by equation 9.
| (9) |
where tumour refers to the one-dimensional sum of individual tumour diameter measurements, Kgrowth is the zero order rate tumour growth constant, Kdeg is the first order tumour death rate constant, Emax is the maximum PD effect and IC50 is the plasma drug concentration at 50% of the maximal inhibitory effect. Both Kgrowth and Kdeg were constrained to be positive. Individual predicted drug concentrations (C) based on the tlag model and its Bayesian post hoc estimates were used as an input function to equation 9. For each patient, equation 9 was initialized by the individual baseline sum of tumour diameter measurement. The inter-individual variability of the model parameters was assumed to be log normally distributed (equation 7), while the residual error was best described using an additive error structure (equation 10).
| (10) |
where yij is the jth observation in the ith individual; 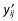 is the model’s predicted value and εij is the normally distributed additive random error with mean of zero and variance of σ2.
is the model’s predicted value and εij is the normally distributed additive random error with mean of zero and variance of σ2.
Model development and validation
The major steps of PK and PK–PD model building are listed in the supplementary material. The final model was selected based on a combination of: reduction in the objective function value (OFV), examination of goodness of fit plots, reductions in the magnitude of BSV model parameters and residual error, as well as shrinkage in random variability parameters and robust model parameter estimation. The assessment of statistical significance between nested models was based on the difference in OFV, applying the likelihood ratio test.
The stability and performance of the model were assessed by means of a non-parametric bootstrap with re-sampling (n = 500) and replacement. Estimated parameters from the bootstrap procedure were compared with those estimated from the original data set. The 2.5th and 97.5th percentiles of the sets of estimates defined the lower and upper limit of the 95% confidence intervals, respectively, for each parameter and its corresponding variability. A simulation-based diagnostic was performed by constructing visual predictive checks.
Simulation: impact of dose and regimen
Deterministic and stochastic simulations from the final PK–PD model parameters were used to assess the impact of dose and regimen on early drug response at 20 weeks, in the typical individual or in 5000 virtual patients following onset of therapy. Full adherence with the prescribed regimen and a baseline sum of longest tumour diameters of 10 cm was assumed. Individual predicted tumour size at 20 weeks following onset of therapy was calculated to classify the change in sum of tumour diameters from baseline, as per RECIST 20: progressive disease (increase of ≥20% from baseline), stable disease (increase of < 20% or decrease <30% from baseline), partial response (decrease of ≥30% from baseline) and complete response (sum of tumour diameters reaching zero).
For deterministic simulations the PK–PD parameter estimates for the typical individual were used, and a linear regression was performed of effect vs. time fixing the intercept at 10 cm and reporting the slope as the time-averaged drug effect up to 20 weeks of treatment. Within the range of dosing rates simulated, a linear effect described the change in the sum of longest tumour diameter well, at least up to 20 weeks of treatment.
Results
Population PK analysis
PK data were well described by an open one compartment model with first order elimination, as expected from the mono-exponential decline in concentrations (Figure 2). Both the tlag and transit model were found to provide an adequate fit to the data as shown by the goodness of fit plots depicted in Figure 3 (transit) and supplementary material (tlag). Overall, the individual predicted concentrations from the transit model approximated more closely the observed concentrations. Nevertheless, some bias towards underprediction of peak drug concentrations was observed for both models.
Figure 2.
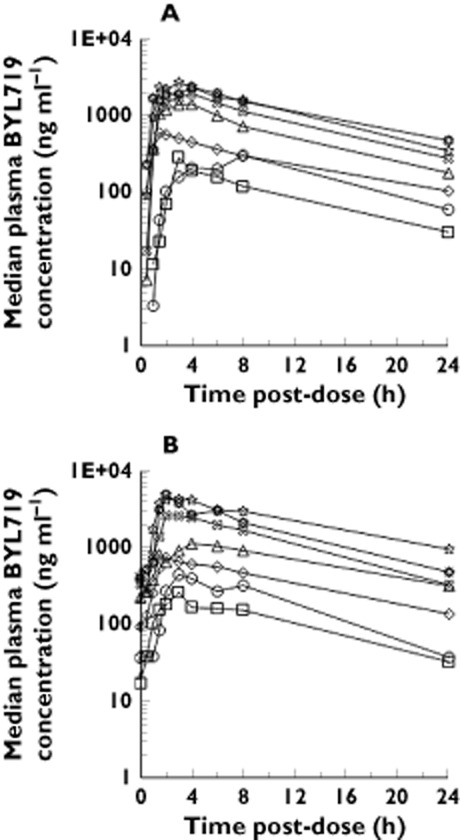
Median plasma concentration–time profile of BYL719 after (A) a single dose administration and (B) 1 month of once daily dosing. ( ) 30 mg once daily, (
) 30 mg once daily, ( ) 60 mg once daily, (
) 60 mg once daily, ( ) 90 mg once daily, (
) 90 mg once daily, ( ) 180 mg once daily, (
) 180 mg once daily, ( ) 270 mg once daily, (
) 270 mg once daily, ( ) 400 mg once daily, (
) 400 mg once daily, ( ) 450 mg once daily
) 450 mg once daily
Figure 3.
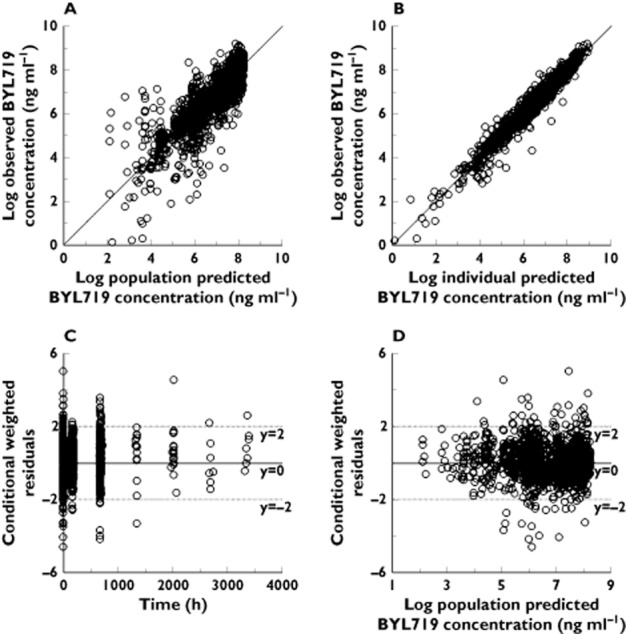
Goodness of fit plots for the population PK transit model. (A) Plot of the population-predicted vs. observed BYL719 plasma concentration. (B) Plot of individual-predicted vs. observed BYL719 plasma concentration. (C) Plot of the conditional weighted residual error (CWRES) vs. time. (D) Plot of the conditional weighted residual error (CWRES) vs. population predicted BYL719 plasma concentration. The solid line in panel A and B is the line of identity (observed=predicted)
The population PK parameters along with their variability and relative standard errors are summarized in Table 2. Oral drug clearance was estimated to be 10.1 l h−1 and 11.5 l h−1, and oral volume of distribution was 108 l and 118 l in the transit and tlag model, respectively. The terminal elimination half-life in the typical individual was estimated at 7.1 h in the tlag model, and 7.4 h in the transit model. The population disposition parameters were similar to the non-compartmental analysis (supplementary material). The close agreement of drug disposition parameters between both models indicates the lack of relevant bias on drug disposition from drug input mis-specification in the tlag model 25, and confirms the validity of the transit model assumption that the whole of the preceding dose has been absorbed when the next oral dose is given 26. As expected, the differences in the population estimates were more pronounced for the absorption parameters (Table 2). Ka was about four-fold larger in the transit model and MTT was about two-fold larger than the absorption lag time (Table 2).
Table 2.
Population PK parameters of BYL719 and bootstrap validation
| Lag time model | Transit model | |||||
|---|---|---|---|---|---|---|
| Parameter*† | Estimate | RSE (%) | 95% confidence interval limits† | Estimate | RSE (%) | 95% confidence interval limits‡ |
| OFV | 1969 | – | – | 1508 | – | – |
| Disposition | ||||||
| CL (l h−1) | 11.5 | 9.1 | (10.4, 12.6) | 10.1 | 4.6 | (9.28, 11.0) |
| V (l) | 118 | 4.9 | (101, 135) | 108 | 6.4 | (97.3, 120) |
| BSV CL (%) | 26.7 | 3.6 | (20.1, 34.7) | – | – | |
| BSV V (%) | 41 | 3.1 | (26.0, 50.0) | 27.6 | 24.3 | (20.8, 33.1) |
| Absorption | ||||||
| F | – | – | – | 1 (fixed) | – | – |
| Ka (1 h−1) | 0.784 | 4.3 | (0.607, 0.996) | 3.36 (fixed) | – | – |
| tlag (h) | 0.489 (fixed) | – | – | – | – | – |
| MTT (h) | – | – | – | 1.28 | 6.4 | (1.12, 1.49) |
| n | – | – | – | 8.09 | 4.6 | (6.22, 10.0) |
| BSV F (%) | – | – | – | 26.0 | 29.1 | (17.9, 32.8) |
| BOV F (%) | – | – | – | 25.3 | 20.2 | (18.1, 31.6) |
| BSV Ka (%) | 52.4 | 3.4 | (26.9, 87.4) | – | – | – |
| BOV Ka (%) | 62.8 | 3.4 | (42.9, 81.0) | – | – | – |
| BOV tlag (%) | 55.9 | 3.3 | (44.8, 67.0) | – | – | – |
| BSV MTT (%) | – | – | – | 31.6 | 41.2 | (16.3, 41.0) |
| BOV MTT (%) | – | – | – | 46.2 | 17.1 | (39.5, 52.6) |
| Residual error (%) | 36.6 | 1.5 | (31.4, 40.9) | 31.6 | 2.4 | (26.4, 36.5) |
PK parameters are as defined in equation 1 to 6 (Methods). BSV, between-subject variability; BOV, between-occasion variability; OFV, objective function value; RSE, relative standard error.
Major model building steps are provided in the Supplementary material.
Calculated from 500 bootstrapped resamples.
BSV was tested on all model parameters, followed by stepwise elimination when their estimation was not adequately supported by the data (supplementary material). Overall, the BSV was highest for the absorption parameters, being low to moderate for the disposition parameters (Table 2). Comparison of model diagnostics of the transit model, parameterized in different ways, favoured the variability in oral drug clearance to be modelled using a relative bioavailability parameter F (equation 1), rather than by CL (equation 5), suggesting that the underlying BSV in oral drug clearance might reside more within the drug’s bio-availability than in its systemic clearance. Interestingly, the BSV on F in the transit model (26.0 %) was similar to the BSV on oral clearance in the tlag model (26.7%), indicating that similar variance estimates in oral drug clearance were captured in both models.
Intra-subject comparison of systemic drug exposure after single and repeated oral dose administration (AUC(0,∞) vs. AUC(0,τ) at steady-state) revealed relevant BOV in oral drug exposure, but not in t1/2 (data not shown), indicating BOV in oral drug clearance is predominantly affected through F. The stepwise addition of a BOV on MTT and F in the transit model resulted in significant improvement of the model fit (supplementary material). Similarly, in the tlag model, the stepwise addition of a BOV on the absorption parameters (tlag and Ka) and oral drug clearance (CL) resulted in a significant drop in OFV.
The median parameter estimates from the bootstrap procedure were in close agreement with their respective values from the final population model (Table 2) and those derived from model independent analysis (supplementary material). The models were stable to perturbation of the initial estimates. The results of the VPC show that the observed concentration range and variance at the median and the 5th and 95th percentiles was well predicted for both final models (supplementary material).
Population PK–PD analysis
The longitudinal change in sum of tumour diameters over time was used to develop a population PK–PD model to describe the relationship between systemic drug exposures and time course of drug effect. Overall, the tumour kinetic model fitted the observed longitudinal change in sum of tumour diameters well as shown by the goodness of fit plots depicted in Figure 4. The VPC of the final model is provided in the supplementary material. Table 3 lists the population PD and tumour growth parameters along with their 95% confidence intervals. The IC50 for tumour growth inhibition was estimated to be 101 ng ml−1, corresponding to a free drug estimate of about 10 ng ml−1 (average free fraction in plasma was 10.8 ± 1.64 % 27). The calculated IC80 by the Hill equation was 404 ng ml−1. The IC50 was found to be associated with a large BSV of 154 %. This is not surprising as it is the only parameter where a random effect was supported and is therefore carrying all the PD BSV. Bayesian post hoc estimates of IC50 were not found to correlate with dose, suggesting that drug effect was reasonably well described and separated from the underlying system parameters (data not shown). The final model parameter estimates for the bootstrap validation were in good agreement with the estimates of the final model (Table 3).
Figure 4.
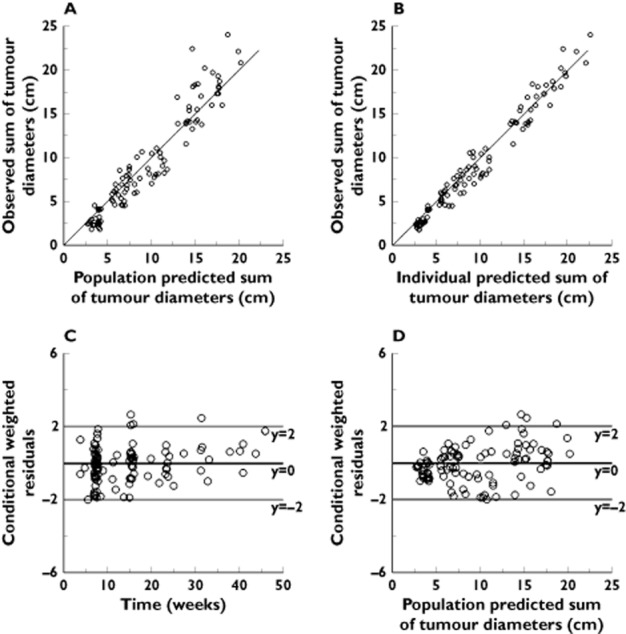
Goodness of fit plots for the PK–PD model describing the longitudinal data of change in sum of tumour diameter. (A) Plot of the population-predicted vs. observed sum of tumour diameters. (B) Plot of individual-predicted vs. observed sum of tumour diameters. (C) Plot of the conditional weighted residual error (CWRES) vs. time. (D) Plot of the conditional weighted residual error (CWRES) vs. population predicted sum of tumour diameters. The solid line in panel A and B is the line of identity (observed=predicted)
Table 3.
Population PD parameters of BYL719 and bootstrap validation
| Parameter* | Estimate | RSE (%) | 95% confidence interval limits‡ |
|---|---|---|---|
| Kgrowth (cm week−1) | 0.581 (fixed)† | – | – |
| Kdeg (week−1) | 0.0123 | 5.2 | (0.00784, 0.0236) |
| IC50 (ng ml−1) | 101 | 7.6 | (40.6, 179) |
| BSV IC50 (%) | 154 | 20.6 | (80.6, 261) |
| Emax | 1 (fixed)† | – | – |
| Residual error (cm) | 1.27 | 7.1 | (0.942, 1.48) |
PK parameters are as defined in equation 9 (Methods). BSV, between-subject variability; OFV, objective function value; RSE, relative standard error.
Major model building steps and rationale for fixing Kgrowth and Emax is provided in the supplementary material.
Calculated from 500 bootstrapped resamples.
Simulation: impact of dose and regimen
Deterministic and stochastic simulations from the final PK–PD model were used to assess the impact of dose and regimen on drug response at 20 weeks following onset of therapy. At a total daily dose of 400 mg, the proportion of patients with a trough concentration exceeding 101 ng ml−1 (IC50) was comparable in the once and twice daily regimens (90% vs. 99%), while the proportion of patients with a trough concentration exceeding 404 ng ml−1 (IC80) was considerably larger in the twice daily regimen (55% vs. 90%) (Figure 5). Accordingly, in the typical individual, no sizable difference in anti-tumour activity is expected up to 20 weeks post-onset of therapy when the drug is given once daily or twice daily at a total daily dose of 400 mg (Table 4).
Figure 5.
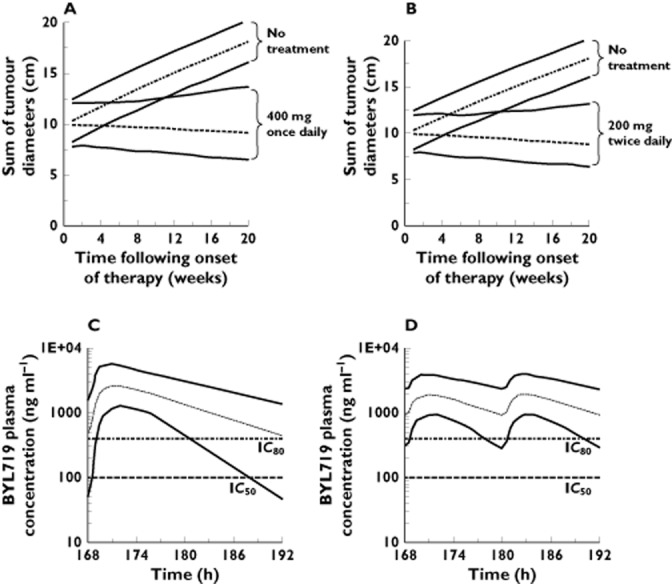
Simulation of the time course of drug effect in 5000 patients taking BYL719 (A) 400 mg once daily vs. no treatment, or (B) 200 mg twice daily vs. no treatment. All patients were assumed to have a baseline sum of tumour diameters of 10 cm. Simulation of the BYL719 plasma concentration time profile in 5000 patients taking BYL719 (C) 400 mg once daily or (D) 200 mg twice daily. The dashed line is the 50th percentile (median). The lower and upper solid lines represent the 5th and 95th percentile of simulated data. Horizontal reference lines indicate the estimated IC50 (101 ng ml−1) and IC80 (404 ng ml−1) of BYL719 on tumour growth
Table 4.
Simulation of absolute and relative drug effect at different dosing rates
| Total daily dose 400 mg | Total daily dose 300 mg | Total daily dose 200 mg | Total daily dose 100 mg | ||||||
|---|---|---|---|---|---|---|---|---|---|
| QD | QOD | BID | QD | BID | QD | BID | QD | BID | |
| Drug effect (mm week−1) | −0.663 | −0.145 | −0.758 | −0.531 | −0.647 | −0.293 | −0.441 | 0.267* | 0.0790 |
| % relative to 400 mg QD | 100 | 22 | 114 | 80 | 98 | 44 | 67 | −40 | −12 |
| % relative to 300 mg QD | 100 | 122 | 55 | 83 | −50 | −15 | |||
| % relative to 200 mg QD | 100 | 151 | −91 | −27 | |||||
Values presented are predicted response at 20 weeks after onset of therapy (refer to Methods). BID, twice daily (total daily dose given in two instalments with a dosing interval of 12 h); QD, once daily (total daily dose given at interval of 24 h); QOD, 800 mg once every other day (dosing interval of 48 h).
Flip from growth to regression occurs between a total daily dose of 135 and 136 mg in QD and total daily dose of 108 to 110 mg in BID.
Relative anti-tumour effect decreased when total daily dose was decreased, indicating dose-dependency (Table 4). Increasing the dosing interval to every 2 days is predicted to result in a considerable loss of drug effect. Interestingly, simulation predicts a larger difference between the once and twice daily regimens in favour of the twice daily regimen at lower total daily doses (Table 4). For the average patient there is a 51% advantage comparing 100 mg twice daily with 200 mg once daily whilst the advantage is 22% comparing 150 mg twice daily with 300 mg once daily. Thus, if dose reduction from a starting dose of 400 mg once daily is clinically indicated moving to a twice daily regimen may be considered.
RECIST classification of simulated tumour response at 20 weeks was found to be a less sensitive metric to detect the impact of dosing rate on the early clinical outcome at 20 weeks (supplementary material). This may be caused by a loss of information when creating categories from a random variable that is continuously changing with time. RECIST criteria classified most patients as having stable disease despite an underlying difference in treatment response. RECIST classification of simulated response confirms the anticipated gradual loss of efficacy with dose increments from the MTD, which can, at least partly, be offset by switching to a twice daily regimen.
Discussion
Several approaches to modelling tumour dynamics using different tumour load metrics can be found in the literature 28–31. A concise mathematical treatment of alternative models has also been published 32. None of these uses the simple turnover model that we have applied here. Whilst our model may appear to have some basis in mechanism, we regard our implementation as empirical. We have initialized the individual subject tumour size to the observed baseline value and this implementation implies, given the final parameters, that the drug effect is a function of the baseline size with larger effect possible at higher baseline values compared with lower ones. Also, the model cannot be generalized beyond the range of the baseline tumour sizes represented in this population (2–20 cm). One underlying reason for this is that whilst both growth and regression of the tumour is possible at different drug effects in this range, there is a natural boundary at a baseline tumour size of Kgrowth/Kdeg (about 47 cm) where even in the absence of any drug, growth of the tumour cannot occur. Despite these limitations we feel that the model is useful and that the uses we have made of the model add insight and will benefit the further development of BYL719.
Simulations from the PK–PD model confirmed the presence of a concentration–effect relationship in the dose range of 100–400 mg once daily, with a respective increase and decrease in number of partial tumour responses and progressive disease with increasing dose. At 400 mg once daily, the fraction of patients experiencing stable disease or a partial response was simulated to be respectively 78% and 9% at 20 weeks following onset of therapy. At the time of writing this manuscript, several patients had achieved partial tumour responses and many patients had stable disease. We acknowledge the potential for an upward bias in the simulations may exist, as complete adherence to the prescribed regimen was assumed.
The investigation of schedule dependence in the drug effect through simulation revealed little difference in tumour shrinkage between 400 mg once daily and 200 mg twice daily, while a considerable loss of effect is predicted with less frequent dosing. While more frequent regimens might provide more sustained target inhibition, preliminary safety data suggest a possible higher frequency or intensity of hyperglycaemia may exist when a total dose of 400 mg is given twice as opposed to once daily (manuscript in preparation). The inhibition of PI3K is known to enhance the sensitivity of the pancreatic β-cell to glucose concentrations in the post-prandial range 19,33. Insulin resistance may develop in peripheral tissues as a consequence of PI3K inhibition, and hyperglycaemia may develop when adequate secretion of the hormone, in proportion to insulin resistance, fails to occur 19,33. We might speculate that if the concentration–effect relationship for both effects are one and the same, then a relatively faster dynamic onset and offset of the effect on the glucose-insulin axis relative to that on tumour size may allow for maximizing the risk–benefit by using an adequate dosing scheme which alters time above IC50 and IC80. For example, at a total daily dose of 400 mg, the proportion of patients experiencing sustained pathway inhibition (trough >IC80) was simulated to be considerably larger with 200 mg given twice daily compared with 400 mg given once a day (Figure 5), whilst the latter regimen appears better tolerated based on preliminary safety data (manuscript in preparation). Thus, assuming the incidence and severity of drug related hyperglycaemia might correlate with the underlying state of glucose homeostasis in the individual, step-wise dose de-escalation from the MTD and/or a different treatment schedule might be a useful strategy to improve efficacy as well as individual tolerability to the treatment. Simulations suggest a larger difference in anti-tumour activity exists between the once and twice daily regimens, in favour of the twice daily regimen, at total daily doses below 400 mg. In such cases moving from a once daily to a twice daily regimen may offer a larger benefit in terms of change in tumour size. This has supported the continued interest in investigating the tolerability and efficacy of the twice daily regimen in our clinical study.
As there was no serial tumour measurement made before starting treatment, tumour turnover parameters could be confounded by drug effect, and should be interpreted with caution. Nevertheless, the free BYL719 plasma concentration causing 50% of maximal reduction in tumour growth rate of 10 ng ml−1 was is in close agreement with the in vitro-based inhibitory potential against the kinase activity of recombinant PI3Kα (4.41 ng ml−1) 27 and in Rat1-myr-p110α cells (free drug IC50 of 18 ng ml−1). Despite the enrolment of patients with PI3KCA mutated tumours, individual response to treatment was found to vary greatly, as indicated by the large BSV (154%) on the IC50. It is acknowledged that a different IC50 or BSV may exist in a more homogenous population with respect to cancer type, prior treatment or when BYL719 is used in combination therapy with other anti-cancer agents.
The oral PK of BYL719 were best described by a one compartment disposition model and transit compartments accounting for the lag time in absorption. The typical population oral clearance and volume of distribution estimates with their BSV were 10 l h−1 (BSV 26%) and 108 l (BSV 28%), respectively. Oral clearance of BYL719 was found to be well below human hepatic blood flow (about 90 l h−1) 34. It seems therefore reasonable to conclude that BYL719 is a low clearance drug and hepatic first pass extraction is not expected to restrict oral bioavailability to a significant extent. The projected human total body systemic clearance from interspecies scaling was 8–14 l h−1, in close agreement to the oral clearance observed in this study, suggesting that BYL719 might demonstrate high absolute oral bioavailability, although this remains to be demonstrated in a dedicated study.
Time-varying conditions with respect to food content and fasting time may explain, at least partly, the BOV in both the rate and extent of absorption. Of particular significance are the preclinical observations in bile duct-cannulated rats that a role for biliary and direct gastrointestinal secretion may exist for BYL719, with 20% of parent drug recovered in bile and faeces after intravenous bolus 27. An entero-hepatic shunt, potentially triggered by food, may underlie the moderate to high BOV in oral drug exposure and is likely to do so through changes in F over time. Moreover, food could also possibly increase oral absorption, for BYL719 has a remarkable six-fold increase in solubility in intestinal fluids of the fed vs. fasted state. Not surprisingly, model diagnostics of the transit model favoured parameterization with random variability on relative F rather than on disposition clearance.
In conclusion, an increased understanding of BYL719’s potential for anti-tumour activity was derived from a PK–PD model that described the time course of tumour response in relation to drug exposure in the first 20 weeks post-onset of therapy. The investigation of schedule dependence in the drug effect through simulation revealed little difference in tumour shrinkage between 400 mg once daily and 200 mg twice daily, while a considerable loss of effect was predicted with less frequent dosing. Model-based predictions showed potential for additional anti-tumour activity of twice daily dosing at a total daily dose below 400 mg. The PK–PD model provided a valuable approach for planning future clinical studies and for designing optimized dosing regimens with BYL719. The good PK properties and evidence for a concentration–effect relationship support the further clinical development of BYL719.
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf. S. De Buck, A. Jakab, M. Boehm, B. Douglas and C. Quadt are employees of Novartis Pharma AG. The authors declare no conflict of interest directly relevant to the content of this publication.
This study was supported by Novartis Pharma A.G. The authors would like to thank all laboratory staff of the Drug Metabolism and Bioanalytics group for providing the BYL719 plasma concentration data and metabolism data used in this study. The authors are also grateful to Antonin Schmitt for his advice and valuable comments. We also acknowledge the clinical investigators and study coordinators at Massachusetts General Hospital Cancer Center (M. Frisbie, C. Caldwell), University of Texas MD Anderson Cancer Center (A. Gonzalez, J. Bendell, F. Janku, T. Helgason, D. Westerhold), Sarah Cannon Research Institute (S. Burris, J. Bendell, J. Infante, T. Jew), Vanderbilt-Ingram Cancer Center (J. Berlin, W. Van Meulen), Vall d’Hebron Institute of Oncology (J. Tabernero, J. Rodon, G. Argiles, L. Martinez), University of Oxford Churchill Hospital (M. Middleton, A. Gupta, W. Saka, N. Hayward), Nederlands Kanker Instituut-Antoni van Leeuwenhoek Ziekenhuis (J. Schellens, M. Roelvink, S. Marchetti, E. Harms) and Universitätsklinikum Essen (M. Schuler) for conducting the study and technicians for technical support.
Supporting Information
Additional Supporting Information may be found in the online version of this article at the publisher’s web-site:
Goodness of fit plots for the population PK tlag model of BYL719. (A) Plot of the population-predicted vs. observed BYL719 plasma concentration. (B) Plot of individual-predicted vs. observed BYL719 plasma concentration. (C) Plot of the conditional weighted residual error (CWRES) vs. time. (D) Plot of the conditional weighted residual error (CWRES) vs. population predicted BYL719 plasma concentration. The solid line in panels A and B is the line of identity (observed=predicted)
Visual predictive check of the final tlag model stratified after (A) a single and (B) multiple doses of BYL719. Results of the visual predictive check of the final transit model stratified after (C) a single and (D) multiple doses of BYL719. A total of 500 simulations were performed based on each subject’s fixed effect parameters and at the time points specified in the data set. The open circles represent the observed concentrations. The solid lines represent the 5th, 50th and 95th percentiles of the simulated data. The dotted lines represent the 90% confidence interval about the 5th, 50th and 95th percentiles
Visual predictive check of the final PK–PD model. A total of 500 simulations were performed based on each subject’s fixed effect parameters and at the time points specified in the data set over 32 weeks. The open circles represent the observed concentrations. The solid lines represent the 5th, 50th and 95th percentiles of the simulated data. The top and bottom dotted line represent respectively the upper 90% confidence interval about 95th percentile and the lower 90% confidence interval about the 5th percentile of the simulated data
Non-compartmental PK analysis
RECIST response simulation
Determination of BYL719 in plasma
Model building steps
References
- Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606–619. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- Vivanco I, Sawyers CL. The phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- Courtney KD, Corcoran RB, Engelman JA. The PI3K pathway as drug target in human cancer. J Clin Oncol. 2010;28:1075–1083. doi: 10.1200/JCO.2009.25.3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fruman DA, Meyers RE, Cantley LC. Phosphoinositide kinases. Annu Rev Biochem. 1998;67:481–507. doi: 10.1146/annurev.biochem.67.1.481. [DOI] [PubMed] [Google Scholar]
- Katso R, Okkenhaug K, Ahmadi K, White S, Timms J, Waterfield MD. Cellular function of phosphoinositide 3-kinases: implications for development, homeostasis, and cancer. Annu Rev Cell Dev Biol. 2001;17:615–675. doi: 10.1146/annurev.cellbio.17.1.615. [DOI] [PubMed] [Google Scholar]
- Okkenhaug K, Vanhaesebroeck B. PI3K in lymphocyte development, differentiation and activation. Nat Rev Immunol. 2003;3:317–330. doi: 10.1038/nri1056. [DOI] [PubMed] [Google Scholar]
- Ikenoue T, Kanai F, Hikiba Y, Obata T, Tanaka Y, Imamura J, Ohta M, Jazag A, Guleng B, Tateishi K, Asaoka Y, Matsumura M, Kawabe T, Omata M. Functional analysis of PIK3CA gene mutations in human colorectal cancer. Cancer Res. 2005;65:4562–4567. doi: 10.1158/0008-5472.CAN-04-4114. [DOI] [PubMed] [Google Scholar]
- Isakoff SJ, Engelman JA, Irie HY, Luo J, Brachmann SM, Pearline RV, Cantley LC, Brugge JS. Breast cancer-associated PIK3CA mutations are oncogenic in mammary epithelial cells. Cancer Res. 2005;65:10992–11000. doi: 10.1158/0008-5472.CAN-05-2612. [DOI] [PubMed] [Google Scholar]
- Liu Z, Roberts TM. Human tumor mutants in the p110alpha subunit of PI3K. Cell Cycle. 2006;5:675–677. doi: 10.4161/cc.5.7.2605. [DOI] [PubMed] [Google Scholar]
- Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, Yan H, Gazdar A, Powell SM, Riggins GJ, Willson JK, Markowitz S, Kinzler KW, Vogelstein B, Velculescu VE. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554. doi: 10.1126/science.1096502. [DOI] [PubMed] [Google Scholar]
- Brognard J, Clark AS, Ni Y, Dennis PA. Akt/protein kinase B is constitutively active in non-small cell lung cancer cells and promotes cellular survival and resistance to chemotherapy and radiation. Cancer Res. 2001;61:3986–3997. [PubMed] [Google Scholar]
- Hu L, Hofmann J, Lu Y, Mills GB, Jaffe RB. Inhibition of phosphatidylinositol 3′-kinase increases efficacy of paclitaxel in in vitro and in vivo ovarian cancer models. Cancer Res. 2002;62:1087–1092. [PubMed] [Google Scholar]
- Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009;8:627–644. doi: 10.1038/nrd2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graupera M, Guillermet-Guibert J, Foukas LC, Phng LK, Cain RJ, Salpekar A, Pearce W, Meek S, Millan J, Cutillas PR, Smith AJ, Ridley AJ, Ruhrberg C, Gerhardt H, Vanhaesebroeck B. Angiogenesis selectively requires the p110alpha isoform of PI3K to control endothelial cell migration. Nature. 2008;453:662–666. doi: 10.1038/nature06892. [DOI] [PubMed] [Google Scholar]
- Willems L, Tamburini J, Chapuis N, Lacombe C, Mayeux P, Bouscary D. PI3K and mTOR signaling pathways in cancer: new data on targeted therapies. Curr Oncol Rep. 2012;14:129–138. doi: 10.1007/s11912-012-0227-y. [DOI] [PubMed] [Google Scholar]
- Brana I, Siu LL. Clinical development of phosphatidylinositol 3-kinase inhibitors for cancer treatment. BMC Med. 2012;10:161. doi: 10.1186/1741-7015-10-161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furet P, Guagnano V, Fairhurst RA, Imbach-Weese P, Bruce I, Knapp M, Fritsch C, Blasco F, Blanz J, Aichholz R, Hamon J, Fabbro D, Caravatti G. Discovery of NVP-BYL719 a potent and selective phosphatidylinositol-3 kinase alpha inhibitor selected for clinical evaluation. Bioorg Med Chem Lett. 2013;23:3741–3748. doi: 10.1016/j.bmcl.2013.05.007. [DOI] [PubMed] [Google Scholar]
- Juric D, Rodon J, Gonzalez-Angulo AM, Burris HA, Bendell JC, Berlin JD, Middleton MR, Bootle D, Boehm M, Schmitt A, Rouyrre N, Quadt C, Baselga J. BYL719, a next generation PI3K alpha specific inhibitor: preliminary safety, PK, and efficacy results from the first-in-human study. In Proceedings of the 103rd Annual Meeting of the American Association for Cancer Research: March 31-April 4 2012.
- Zawalich WS, Zawalich KC. A link between insulin resistance and hyperinsulinemia: inhibitors of phosphatidylinositol 3-kinase augment glucose-induced insulin secretion from islets of lean, but not obese, rats. Endocrinology. 2000;141:3287–3295. doi: 10.1210/endo.141.9.7636. [DOI] [PubMed] [Google Scholar]
- Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, Kaplan R, Lacombe D, Verweij J. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1) Eur J Cancer. 2009;45:228–247. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- Derendorf H, Lesko LJ, Chaikin P, Colburn WA, Lee P, Miller R, Powell R, Rhodes G, Stanski D, Venitz J. Pharmacokinetic/pharmacodynamic modeling in drug research and development. J Clin Pharmacol. 2000;40:1399–1418. [PubMed] [Google Scholar]
- Lesko LJ, Rowland M, Peck CC, Blaschke TF. Optimizing the science of drug development: opportunities for better candidate selection and accelerated evaluation in humans. Pharm Res. 2000;17:1335–1344. doi: 10.1023/a:1007574217260. [DOI] [PubMed] [Google Scholar]
- Available at http://clinicaltrials.gov/show/NCT01219699 (last accessed 14 April 2014)
- Savic RM, Jonker DM, Kerbusch T, Karlsson MO. Implementation of a transit compartment model for describing drug absorption in pharmacokinetic studies. J Pharmacokinet Pharmacodyn. 2007;34:711–726. doi: 10.1007/s10928-007-9066-0. [DOI] [PubMed] [Google Scholar]
- Nerella NG, Block LH, Noonan PK. The impact of lag time on the estimation of pharmacokinetic parameters. I. One-compartment open model. Pharm Res. 1993;10:1031–1036. doi: 10.1023/a:1018970924508. [DOI] [PubMed] [Google Scholar]
- Shen J, Boeckmann A, Vick A. Implementation of dose superimposition to introduce multiple doses for a mathematical absorption model (transit compartment model) J Pharmacokinet Pharmacodyn. 2012;39:251–262. doi: 10.1007/s10928-012-9247-3. [DOI] [PubMed] [Google Scholar]
- Data on file at Novartis Institutes for Biomedical Research. Drug metabolism and Pharmacokinetics (DMPK) Europe, Basel, Switzerland.
- Bruno R, Claret L. On the use of change in tumor size to predict survival in clinical oncology studies: toward a new paradigm to design and evaluate phase II studies. Clin Pharmacol Ther. 2009;86:136–138. doi: 10.1038/clpt.2009.97. [DOI] [PubMed] [Google Scholar]
- Claret L, Girard P, Hoff PM, Van CE, Zuideveld KP, Jorga K, Fagerberg J, Bruno R. Model-based prediction of phase III overall survival in colorectal cancer on the basis of phase II tumor dynamics. J Clin Oncol. 2009;27:4103–4108. doi: 10.1200/JCO.2008.21.0807. [DOI] [PubMed] [Google Scholar]
- Tham LS, Wang L, Soo RA, Lee SC, Lee HS, Yong WP, Goh BC, Holford NH. A pharmacodynamic model for the time course of tumor shrinkage by gemcitabine + carboplatin in non-small cell lung cancer patients. Clin Cancer Res. 2008;14:4213–4218. doi: 10.1158/1078-0432.CCR-07-4754. [DOI] [PubMed] [Google Scholar]
- Wang Y, Sung C, Dartois C, Ramchandani R, Booth BP, Rock E, Gobburu J. Elucidation of relationship between tumor size and survival in non-small-cell lung cancer patients can aid early decision making in clinical drug development. Clin Pharmacol Ther. 2009;86:167–174. doi: 10.1038/clpt.2009.64. [DOI] [PubMed] [Google Scholar]
- Stein A. 2011. Dynamic models of metastatic tumor growth. 27th Annual Workshop on Mathematical Problems in Industry, New Jersey Institute of Technology, June 13–17, Available at http://eaton.math.rpi.edu/Faculty/Schwendeman/Workshop/MPIReports/2011/Novartis11.pdf (last accessed 14 April 2014)
- Eto K, Yamashita T, Tsubamoto Y, Terauchi Y, Hirose K, Kubota N, Yamashita S, Taka J, Satoh S, Sekihara H, Tobe K, Iino M, Noda M, Kimura S, Kadowaki T. Phosphatidylinositol 3-kinase suppresses glucose-stimulated insulin secretion by affecting post-cytosolic [Ca(2+)] elevation signals. Diabetes. 2002;51:87–97. doi: 10.2337/diabetes.51.1.87. [DOI] [PubMed] [Google Scholar]
- Wilkinson GR. Clearance approaches in pharmacology. Pharmacol Rev. 1987;39:1–47. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Goodness of fit plots for the population PK tlag model of BYL719. (A) Plot of the population-predicted vs. observed BYL719 plasma concentration. (B) Plot of individual-predicted vs. observed BYL719 plasma concentration. (C) Plot of the conditional weighted residual error (CWRES) vs. time. (D) Plot of the conditional weighted residual error (CWRES) vs. population predicted BYL719 plasma concentration. The solid line in panels A and B is the line of identity (observed=predicted)
Visual predictive check of the final tlag model stratified after (A) a single and (B) multiple doses of BYL719. Results of the visual predictive check of the final transit model stratified after (C) a single and (D) multiple doses of BYL719. A total of 500 simulations were performed based on each subject’s fixed effect parameters and at the time points specified in the data set. The open circles represent the observed concentrations. The solid lines represent the 5th, 50th and 95th percentiles of the simulated data. The dotted lines represent the 90% confidence interval about the 5th, 50th and 95th percentiles
Visual predictive check of the final PK–PD model. A total of 500 simulations were performed based on each subject’s fixed effect parameters and at the time points specified in the data set over 32 weeks. The open circles represent the observed concentrations. The solid lines represent the 5th, 50th and 95th percentiles of the simulated data. The top and bottom dotted line represent respectively the upper 90% confidence interval about 95th percentile and the lower 90% confidence interval about the 5th percentile of the simulated data
Non-compartmental PK analysis
RECIST response simulation
Determination of BYL719 in plasma
Model building steps