

Abstract
Aims
HM30181 is a third generation P-glycoprotein (P-gp) inhibitor currently under development. The objectives of this study were to evaluate the effects of a single dose of HM30181 on the pharmacodynamics and pharmacokinetics of loperamide, a P-gp substrate, and to compare them with those of quinidine.
Methods
Eighteen healthy male subjects were administered loperamide alone (period 1) or with loperamide plus quinidine or HM30181 in period 2 or 3, respectively. In period 3, subjects randomly received one of three HM30181 doses: 15, 60 or 180 mg. Changes in pupil size, alertness, oxygen saturation and the oral bioavailability of loperamide were assessed in each period. In addition, the pharmacokinetics of HM30181 were determined.
Results
Pupil size, alertness and oxygen saturation did not change over time when loperamide alone or loperamide plus HM30181 was administered while HM30181 significantly increased the systemic exposure to loperamide, i.e. the geometric mean ratio (90% confidence interval) of AUC(0,tlast) for loperamide with and without HM30181 was 1.48 (1.08, 2.02). Co-administered quinidine significantly increased the systemic exposure to loperamide 2.2-fold (1.53, 3.18), which also markedly reduced pupil size, resulting in a decrease of 24.7 mm h in the area under the effect curve of pupil size change from baseline compared with loperamide alone.
Conclusions
HM30181 inhibits P-gp mainly in the intestinal endothelium, which can be beneficial because pan-inhibition of P-gp, particularly in the brain, could lead to detrimental adverse events. Further studies are warranted to investigate adequately the dose–exposure relationship of HM30181, along with its duration of effect.
Keywords: HM30181, loperamide, P-glycoprotein inhibitor, pharmacodynamics, pharmacokinetics
What is Already Known about this Subject —
The inhibition of P-glycoprotein (P-gp) has been a target of drug development programmes.
Because P-gp is found in many different tissues, non-specific P-gp inhibition may cause unexpected adverse effects due to pan-inhibition of P-gp in unintended tissues such as the brain.
HM30181, a third generation P-gp inhibitor under development, has been shown to inhibit P-gp in in vitro and animal studies.
What This Study Adds —
HM30181 increases the systemic exposure to loperamide, a P-gp substrate.
HM30181 appears to act mainly on the P-gp in the intestinal endothelium, which may lead to less adverse effects
Introduction
P-glycoprotein (P-gp) is a trans-membrane efflux protein that removes xenobiotics from cells by ATP hydrolysis, protecting the human body from exogenous substances 1. P-gp exists in various tissues including the intestine, liver, kidney and brain. P-gp interferes with the absorption of several drugs into enterocytes, whereas it facilitates drug excretion from hepatocytes and renal proximal tubular cells via the bile or urine, respectively. In addition, P-gp in the brain endothelial cell acts as a blood–brain barrier by preventing xenobiotics from entering the cerebral tissue. P-gp is overexpressed in cancer cells, which results in multidrug resistance 2,3.
Because P-gp plays an important role in drug absorption and disposition, inhibition of its function has been a target of drug development programmes. Compared with the first and second generations, the third-generation P-gp inhibitor has been known to be more selective and less interacting with other transporters, leading to fewer side effects 4–6. However, because P-gp is found in many different tissues in the body, unexpected adverse effects can still occur due to the unintended inhibition of P-gp elsewhere, of which, adverse effects in the central nervous system (CNS) can be serious.
HM30181 (N-(2-(2-(4-(2-(6,7-dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)ethyl)phenyl)-2H-tetrazol-5-yl)-4,5-dimethoxyphenyl)-4-oxo-4H-chromene-2-carboxamide) is a third generation P-gp inhibitor currently under development to enhance the oral absorption of P-gp substrates. In an in vitro study, HM30181 showed a high selectivity for P-gp and its potency was 20–50 times higher than that of tariquitar, another third generation P-gp inhibitor 7. In addition, HM30181 increased the oral bioavailability of co-administered paclitaxel by >12 times in rats 7. However, the P-gp inhibitory effect of HM30181 has not been investigated in humans.
Loperamide is an anti-diarrhoeal agent, which reduces gut motility by acting on the opiate receptor in the large intestine 8. Because loperamide is a sensitive substrate of P-gp 9, it is pumped out of the gut endothelium and the brain (i.e. the blood–brain barrier) by P-gp. This is why loperamide in the blood–brain barrier 10 is not associated with CNS effects even though it is a potent opioid 9. Therefore, if the P-gp in the blood–brain barrier is inhibited, the CNS effects of loperamide such as reduced pupil size and respiratory depression will become apparent. Loperamide is removed by hepatic metabolism mainly by CYP3A4 with minor contributions from CYP2C8 10.
The objectives of the present study were (i) to evaluate the effects of a single oral dose of HM30181 on the pharmacodynamics, particularly CNS opioid effects, and pharmacokinetics of loperamide, which is used as a probe drug for P-gp and (ii) to compare them with those of quinidine, a well-known P-gp inhibitor. The pharmacokinetic profiles of HM30181 at various doses were also evaluated.
Methods
Study population
Healthy Korean male volunteers 20–50 years of age with 80–120% of the ideal body weight were enrolled in the present study if they were without any marked past medical or medication history, based on physical examination, vital signs, 12-lead electrocardiogram and clinical laboratory tests. Subjects with dyscoria, uveitis and cataract were excluded because these conditions might obstruct or interfere with ophthalmological examination.
Study design
This study was conducted in an open label, fixed sequence, three treatment, three period, crossover design with a random assignment to a HM30181 dose in period 3. Because HM30181 has been known to have a very long half-life of 75.7–169.3 h after a single oral dose, a fixed sequence design was used in the present study such that HM30181 was administered in period 3 11. Eligible subjects were admitted to the Clinical Trials Center at Seoul National University Hospital 1 day prior to each period. In period 1, subjects received a single oral dose of loperamide at 16 mg. In period 2, a single oral dose of quinidine at 600 mg was administered, followed by a single oral dose of loperamide at 16 mg 1 h later. In period 3, subjects randomly received a single oral dose of HM30181 at 15, 60 or 180 mg in a 1:1:1 ratio, with a single oral dose of loperamide at 16 mg 1 h later. Each period was separated by a 3 day washout, enough to ensure full clearance of loperamide given in the previous period 12. P-gp inhibition by quinidine and HM30181 was investigated in periods 2 and 3, respectively, while period 1 was used as a no treatment control. Quinidine was chosen as a positive control because it is a proven P-gp inhibitor 13–16. The effects of P-gp inhibition were assessed using pharmacodynamic (changes in pupil size, alertness and oxygen saturation) and pharmacokinetic (oral bioavailability of loperamide) endpoints. Food or drinks containing caffeine, grapefruit or alcohol were not permitted throughout the study. Smoking was not allowed during the study either.
The Institutional Review Board of Seoul National University Hospital approved the study protocol (IRB No. H-0711-014-224) and informed consent was obtained from all subjects prior to study enrolment. All procedures were performed in accordance with the recommendations of the Declaration of Helsinki 17. Furthermore, the study was conducted in compliance with the current Good Clinical Practices and other applicable laws and regulatory requirements in South Korea.
Pupil size measurement
Trained study personnel measured horizontal pupil diameter using a photo slit-lamp (FS-3, Nikon, Tokyo, Japan) at 0 (i.e. pre-dose), 1, 2, 4, 6, 8, 24 and 48 h after loperamide administration while keeping the illuminance in the room constant at 0.09 lux. Before measurement, ≥2 min were allowed for subjects to adapt to the lighting conditions in the room. To stabilize accommodation, subjects were asked to see a point 100 cm away in front of them. The average of three measurements, taken 20 s apart, was regarded as the pupil size at each measurement time. All of the measurements were carried out in the same eye of each subject. Changes in pupil size from pre-dose were plotted vs. time after loperamide administration, and the area under the effect–time curve (AUEC) was calculated using the trapezoidal rule. The inter- and intra-day precisions were 4.6–7.4% and 7.0–16.9%, respectively.
Alertness measurement
Alertness was measured at 0, 1, 2, 4, 6, 8, 24 and 48 h after loperamide administration using a visual analogue scale (VAS). Subjects were asked to indicate the degree of their alertness level using a scale of 0 (full alertness) to 10 (extreme drowsiness).
Measurement of oxygen saturation
Oxygen saturation was measured in the blood at 0, 1, 2, 4, 6, 8, 24 and 48 h after loperamide administration using a bedside cardiac monitor (Solar® 8000, GE Healthcare, Little Chalfont, Buckinghamshire, UK).
Phamacokinetic blood sampling
In each period, blood samples (5 ml) were obtained at 0, 1, 2, 3, 4, 5, 6, 8, 12, 24, 36, 48, 60 and 71 h after loperamide administration. In periods 2 and 3, additional blood samples (4 ml) were taken at the same time for the analysis of quinidine or HM30181, respectively. Blood samples for loperamide and HM30181 were collected in sodium heparin tubes and those for quinidine were collected in serum separator tubes. Plasma for the analysis of loperamide and HM30181 and serum for the analysis of quinidine were obtained by centrifugation at 1600 g and stored in polypropylene tubes at −20°C until concentrations were determined.
Determination of concentrations
Plasma concentrations of HM30181 were determined using a validated liquid chromatography-tandem mass spectrometry (LC-MS/MS) method with docetaxel as an internal standard. Briefly, plasma 200 μl, methyl tert-butyl ether (MTBE) 1 ml and docetaxel 50 μl at 150 ng ml−1 were mixed thoroughly and centrifuged at 5000 g for 5 min. Supernatant (1 ml) was evaporated; the residue reconstituted with 200 μl of 30% acetonitrile and injected into the LC-MS/MS system. The lower limit of quantification (LLOQ) for HM30181 was 0.5 ng ml−1 with a linear calibration range of 0.5–50 ng ml−1. Each analytical batch had six quality control (QC) samples of known values, i.e. two for low, intermediate and high concentrations, respectively. The analytical results were accepted only if the concentrations of >four out of the six QC samples were determined within 15% of the known values. Intra- and inter-day accuracy was 90−108% and intra- and inter-day precision varied at <12.5 CV%.
Plasma concentrations of loperamide were determined using a validated LC-MS/MS method with glipizide as internal standard. Briefly, plasma 100 μl, acetonitrile 400 μl, and glipizide 20 μl at 400 ng ml−1 were mixed thoroughly and centrifuged at 16 430 g for 5 min. The supernatant was injected into the LC-MS/MS system. LLOQ for loperamide was 0.05 ng ml−1, with a linear calibration range of 0.05–20 ng ml−1. The same QC criteria described above for HM30181 were used for the determination of loperamide concentrations. Intra- and inter-day accuracy was 94.2–103.2% and intra- and inter-day precision varied at <9.0 CV%.
Serum concentrations of quinidine were determined using TDxFLx® (Abbott laboratories, IL, USA).
Pharmacokinetic analysis
A non-compartmental pharmacokinetic analysis was performed using WinNonlin (version 5.1, Pharsight Corporation, Mountain View, CA, USA). The peak plasma concentration (Cmax) and time to Cmax (i.e. tmax) were directly obtained from the observed values. The terminal elimination rate constant (λz) was estimated by linear regression using the log-linear decline portion of the individual plasma concentration-time data. The terminal elimination half-life (t1/2) was calculated as the natural log of 2 divided by λz.
The area under the concentration–time curve (AUC) from time 0 to the last measurable time (AUC(0.tlast) was calculated using the trapezoidal rule. AUC extrapolated to infinity (AUC(0,∞)) was calculated by adding Clast/λz to AUC(0,tlast), where Clast is the last measurable concentration.
Statistical analysis
Baseline demographic characteristics were compared among HM30181 dose groups using the Kruskal–Wallis test. Repeated measures anova was used to compare the differences between periods (treatments) in the pharmacodynamic endpoints. To compare AUC(0,72 h) of loperamide between period 1 (i.e. loperamide alone as baseline) and the other periods, we performed a mixed effects analysis, in which subject was a random effect while dose, period, and the interaction between dose and period were fixed effects. SAS® 9.3 (SAS Institute Inc., NC, USA) was used for statistical data analysis and the level of statistical significance was two-sided 0.05 unless specified otherwise, which was adjusted for the multiplicity of statistical tests.
Results
Study participants
This study enrolled 18 males, all of whom completed the study (Figure 1). Their mean ± SD in age and body weight were 24.1 ± 2.3 years and 68.1 ± 8.9 kg, respectively. There was no significant difference among HM30181 dose groups in age (P = 0.196) or body weight (P = 0.144).
Figure 1.
Flowchart of study participants
Effects of HM30181 and quinidine on the pharmacodynamics and pharmacokinetics of loperamide
Pupil size
Pupil diameter was slightly decreased or remained almost the same over time when loperamide was administered alone or co-administered with HM30181, respectively, whereas it was markedly decreased when quinidine was co-administered (Figure 2A). As a result, difference in the mean change in pupil diameter was statistically significant between when loperamide was co-administered with quinidine and when it was administered alone (4, 6, 8 and 24 h post-dose) or co-administered with HM30181 (all measurement times). Likewise, the mean decrease of AUEC in pupil diameter was significantly lower when quinidine was co-administered than when loperamide was administered alone or it was co-administered with HM30181 (−44.4 vs. −19.7 or −11.4 mm h, P < 0.05, Table 1). Significant differences in the mean change in pupil diameter between quinidine co-administration and loperamide alone or HM30181 co-administration were also noted in all of the HM30181 dose groups although dose–response relationship was not apparent (Figure 2B, C and D). In addition, no significant difference in the mean change in pupil diameter was noted between loperamide alone and HM30181 co-administration throughout the entire period of measurement after loperamide administration except for up to 4 h post-dose in the HM30181 60 mg dose group (Figure 2C).
Figure 2.

Mean changes in pupil diameter over time after loperamide administration. Plots are separately drawn for all doses (A) or each dose of HM30181 (15, 60 and 180 mg in B, C and D, respectively). *P < 0.05 between loperamide plus quinidine vs. loperamide plus HM30181, +P < 0.05 between loperamide plus quinidine vs. loperamide alone and ∧P < 0.05 between loperamide plus HM30181 vs. loperamide alone. A)  , loperamide alone;
, loperamide alone;  , quinidine + loperamide;
, quinidine + loperamide;  , HM30181 + loperamide; B)
, HM30181 + loperamide; B)  , loperamide alone;
, loperamide alone;  , quinidine + loperamide;
, quinidine + loperamide;  , HM30181 15 mg + loperamide; C)
, HM30181 15 mg + loperamide; C)  , loperamide alone;
, loperamide alone;  , quinidine + loperamide;
, quinidine + loperamide;  , HM30181 60 mg + loperamide; D)
, HM30181 60 mg + loperamide; D)  , loperamide alone;
, loperamide alone;  , quinidine + loperamide;
, quinidine + loperamide;  , HM30181 180 mg + loperamide
, HM30181 180 mg + loperamide
Table 1.
Pharmacodynamic effects of loperamide by co-administered drug
| Period | Co-administered drug | Dose, mg | Number of subjects | AUEC of change in pupil diameter (mm*h) | AUEC of alertness score (VAS score*h) | AUEC of oxygen saturation (%*h) |
|---|---|---|---|---|---|---|
| 1 | None | NA | 18 | −19.7 ± 26.7 | 944.7 ± 694.7 | 4684.5 ± 27.1 |
| 2 | Quinidine | 600 | 18 | −44.4 ± 35.3† | 945.7 ± 619.2 | 4698.9 ± 31.5† |
| 3 | HM30181 | 15 | 6 | −24.2 ± 32.3 | 767.8 ± 309.5 | 4711.9 ± 20.6 |
| 60 | 6 | 1.4 ± 13.4 | 794.6 ± 832.8 | 4713.0 ± 24.3 | ||
| 180 | 6 | −11.4 ± 13.2 | 1166.0 ± 701.9 | 4717.5 ± 39.2 | ||
| All | 18 | −11.4 ± 23.0* | 909.4 ± 641.9 | 4714.1 ± 27.5*,† |
Data are mean ± SD. AUEC (the area under the effect–time curve),
P < 0.05 vs. quinidine co-administration;
P < 0.05 vs. loperamide alone.
Alertness score
No significant difference in the mean AUEC of alertness score was noted between when loperamide was administered alone and when it was co-administered with quinidine or HM30181 (Table 1).
Oxygen saturation
The mean AUEC of oxygen saturation was significantly greater when HM30181 or quinidine was co-administered with loperamide than when loperamide was administered alone (4714.1 or 4698.9 vs. 4684.5% h, Table 1).
Systemic exposure to loperamide
The systemic exposure to loperamide was significantly increased when quinidine was co-administered (Figure 3). For example, the peak concentration of loperamide was increased almost three times when co-administered with quinidine compared with when administered alone. (11.4 vs. 4.3 μg l−1, Table 2). Likewise, the geometric mean ratio (90% confidence interval) of the AUC(0,tlast) of loperamide with and without quinidine was 2.20 (1.53–3.18). Co-administered HM30181 also increased the systemic exposure to loperamide by 35–63%, but this increase was not in a dose-dependent manner.
Figure 3.
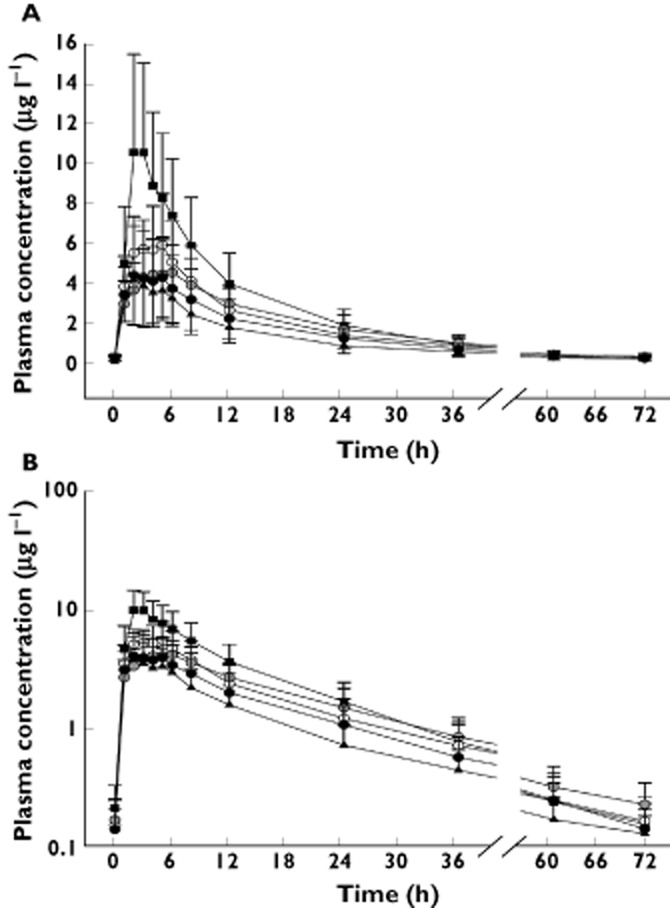
Mean plasma concentration–time profiles of loperamide. The error bars denote SDs. (A) linear scale; B) semi-logarithmic scale).  , loperamide alone;
, loperamide alone;  , quinidine + loperamide;
, quinidine + loperamide;  , HM30181 15 mg + loperamide;
, HM30181 15 mg + loperamide;  , HM30181 60 mg + loperamide;
, HM30181 60 mg + loperamide;  , HM30181 180 mg + loperamide
, HM30181 180 mg + loperamide
Table 2.
Pharmacokinetic parameters of loperamide by period
| Period | Co-administered drug | Dose, mg | n | tmax (h) | Cmax (μg l−1) | t1/2 (h) | AUC(0,tlast) (μg l−1 h) | Ratio* (90% confidence interval) |
|---|---|---|---|---|---|---|---|---|
| 1 | None | 18 | 2.0 [1.0, 6.0] | 4.3 ± 2.1 | 18.4 ± 4.4 | 63.8 ± 27.4 | 1 (reference) | |
| 2 | Quinidine | 600 | 18 | 3.0 [2.0, 3.0] | 11.4 ± 5.1 | 14.6 ± 1.9 | 139.2 ± 56.0 | 2.20 (1.53, 3.18) |
| 3 | HM30181 | 15 | 6 | 2.5 [2.0, 5.0] | 6.7 ± 2.6 | 15.5 ± 1.7 | 98.7 ± 37.9 | 1.46 (0.96, 2.22) |
| 60 | 6 | 4.5 [3.0, 6.0] | 4.7 ± 1.5 | 18.5 ± 1.7 | 100.7 ± 37.6 | 1.63 (0.92, 2.91) | ||
| 180 | 6 | 2.5 [2.0, 5.0] | 4.8 ± 2.2 | 15.8 ± 1.6 | 79.7 ± 38.4 | 1.35 (0.68, 2.71) | ||
| All | 18 | 3.0 [2.0, 6.0] | 5.4 ± 2.3 | 16.6 ± 2.1 | 93.0 ± 37.0 | 1.48 (1.08, 2.02) |
Data are mean ± SD except for tmax, for which median [min, max] is shown.
Geometric mean ratios of the area under the concentration–time curve from time 0 to the last measurable time (AUC(0,tlast)) of loperamide for HM30181 or quinidine co-administration vs. loperamide alone.
Pharmacokinetics of HM30181
The plasma concentration of HM30181 peaked at 13–37 h post-dose and then declined slowly with half-lives of 85.0 ± 92.4 (mean ± SD), 55.9 ± 19.5 and 88.0 ± 57.4 h in the 15, 60 and 180 mg dose groups, respectively (Figure 4, Table 3). Although the Cmax and AUC values of HM30181 were increased as the dose increased, they were not dose-proportional. The 90% confidence intervals for the coefficients of the slopes in the power model were 0.1446, 0.5012 and 0.2991, 0.5590 for Cmax and AUC(0,tlast), respectively, similar to those seen in a previous study 11.
Figure 4.
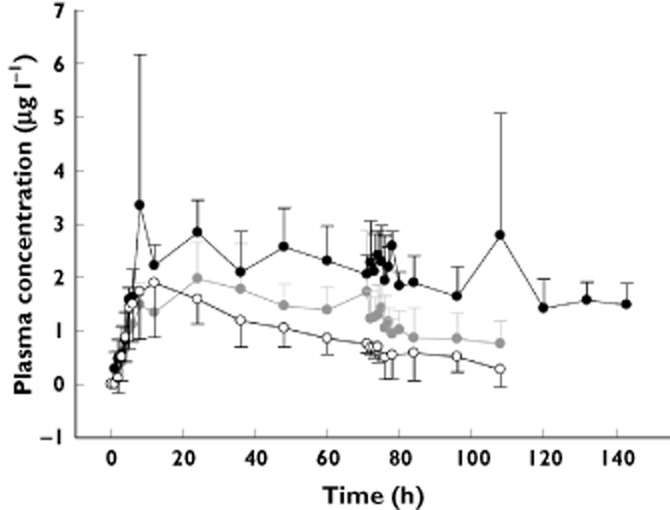
Mean plasma concentration–time profiles of HM30181.The error bars denote SDs.  , HM30181 15 mg;
, HM30181 15 mg;  , HM30181 60 mg;
, HM30181 60 mg;  , HM30181 180 mg
, HM30181 180 mg
Table 3.
Pharmacokinetic parameters of HM30181 by dose
| Dose (mg) | n | tmax (h) | Cmax (μg l−1) | AUC(0,tlast) (μg l−1 h) |
|---|---|---|---|---|
| 15 | 6 | 12.0 [8, 24] | 2.06 ± 0.94 | 105.26 ± 46.59 |
| 60 | 6 | 30.0 [8, 74.8] | 2.37 ± 0.88 | 157.73 ± 56.99 |
| 180 | 6 | 24.0 [8, 107.8] | 4.85 ± 2.6 | 292.83 ± 41.7 |
| All | 18 | 24.0 [8, 107.8] | 3.10 ± 2.03 | 185.27 ± 93.36 |
Data are mean ± SD except for tmax, for which median [min, max] is shown.
Discussion
Our results show that co-administered HM30181 does not enhance the CNS opioid effects of loperamide, such as the decrease in pupil size and respiratory depression while it moderately increases the systemic exposure to loperamide 1.48-fold (90% confidence interval 1.08, 2.02, Table 2) although it was dose-independent. In contrast, co-administered quinidine significantly increased the systemic exposure to loperamide by >two times, which was also associated with a markedly decreased pupil size. Several studies have reported that the CNS opioid effects of loperamide could be enhanced by the co-administration of a P-gp inhibitor. For example, loperamide showed respiratory depression which was measured by the respiratory response to carbon dioxide rebreathing when the quinidine was concomitantly received 13. In another study, co-administered quinidine and loperamide resulted in a significant decrease in pupil size 18, which is consistent with our results. In this study, the genetic polymorphism of P-gp also affected the pharmacokinetics of loperamide and its CNS opioid effects 18.
The metabolites of loperamide such as desmethylloperamide and didesmethylloperamide were not measured in the present study 19. Therefore, the possibility that the increased systemic exposure to loperamide might have been caused by the inhibitory effect of HM3018 on the CYP3A4, which is known to be the primary enzyme system in the metabolism of loperamide 10, cannot be ruled out. However, the inhibitory effect of HM30181 on the gut P-gp could have been the primary driver for the observed increase in the systemic exposure to loperamide based on the following reasons. First, HM30181 did not inhibit the CYP3A4 in human liver microsomes at such a high concentration of 50 μm or 34 437 μg l−1, which is >7100 times the mean Cmax in the highest HM30181 dose group in the present study 20. Second, HM30181 showed a potent and specific inhibitory effect on P-gp in the ATPase assay using membrane vesicles enriched in MDR1 or other ABC transporters 7. Finally, the elimination half-life of loperamide was comparable between loperamide alone and HM30181 co-administration (Figure 3, Table 2).
The results of present study suggest that P-gp in the intestine and brain is inhibited much more by quinidine than HM30181 while the latter is still able to increase significantly the systemic exposure to a P-gp substrate. It has been shown that P-gp inhibition in the brain, compared with other tissues, requires higher doses or concentrations of the inhibitor 21,22. Interestingly, HM30181 did not inhibit the blood–brain barrier in rats after a single intravenous injection at 21 mg kg−1 23, which is equivalent to 237 mg for a 70 kg adult. Because the bioavailability of HM30181 is very low at only 0.3% 10, the highest oral dose in the present study, i.e. 180 mg, will be certainly much lower than the dose that might inhibit the murine blood–brain barrier.Therefore, the absence of the CNS opioid effects by loperamide in this study when HM30181 was co-administered may be because the HM30181 concentration was not sufficiently high enough to inhibit P-gp in the brain. Furthermore, because HM30181 has a low water solubility (i.e. 1 g in 4556 l, at pH 3.8) 7, the P-gp inhibitory action of HM30181 could be local, i.e. in the gut lumen, which was also suggested in a previous study 24. Collectively, the moderate inhibition of P-gp by HM30181, thought to be primarily in the intestine, may be construed as beneficial because it enables more specific P-gp inhibition, leading to fewer adverse events.
The present study also showed that HM30181 had a rather flat dose–response profile over the range of 15 to 180 mg (Tables 1 and 2; Figures 2 and 3). This finding may suggest that the P-gp inhibition effect of HM30181 was saturated at doses lower than or equal to 15 mg. Further studies are warranted to investigate adequately the dose–response relationship profile of HM30181, particularly at doses <15 mg.
The oxygen saturation results, i.e. oxygen saturation was increased when quinidine or HM30181 was co-administered compared with when loperamide alone was administered (Table 1), were contrary to our expectation. Given the limited accuracy of the pulse oximetry method 25 and the fact that the mean oxygen saturation was within normal range in all the groups (i.e. 97.6–98.2%), we speculated the clinical meaning of the differences was minimal.
The present study had several limitations. First, as described above, the metabolites of loperamide were not measured. Considering CYP2C8 and CYP3A4 metabolizing enzymes are involved in the elimination of loperamide 10, the increased systemic exposure to loperamide in the present could have been partly because those enzymes were inhibited by co-administered HM30181 or quinidine. However, HM30181 did not inhibit the CYP enzymes in a previous hepatic microsomal study 20, whereas quinidine is a weak inhibitor of CYP2C8 26,27. Therefore, it was less likely that co-administered HM30181 affected the elimination of loperamide by inhibiting the CYP enzymes while quinidine might have. In the latter, however, the degree of inhibition would be still small, given that quinidine is a weak CYP2C8 inhibitor and another enzyme is also involved (i.e. CYP3A4). This notion is further supported by the fact that the half-life of loperamide was comparable in the present study between when it was administered alone and when it was co-administered with HM30181 or quinidine (i.e. 14.6–18.4 h, Table 2). Another major limitation is the small number of subjects in the present study. It should be also noted that HM30181 was cleared very slowly from the body with a very long half-life of 56–88 h. Therefore, more studies need to be conducted to determine how long the P-gp inhibition effect of HM30181 will be maintained.
In conclusion, P-gp inhibition by HM30181 appears to occur mainly at the intestinal level. This specificity in the location of P-gp inhibition is rather beneficial because pan-inhibition of P-gp can be harmful 13,28, as shown not only previously, but also in the present study. For the optimal use of HM30181 in various clinical applications, however, further studies are needed to investigate thoroughly the dose–response relationship of HM30181, particularly at doses <15 mg. In addition, based on the long half-life of HM30181, those studies should address how long the P-gp inhibition effect of HM30181 will be continued.
Conflict of Interest/Disclosure
All authors have completed the Unified Competing Interest form and declare TK, HL, KL, SL, SY, SS, IJ, KY and JC had support from Hanmi Pharm. Co. Ltd. for the submitted work, TE had support from Training program grant of the Korea Healthcare Technology R&D Project, Ministry for Health and Welfare Affairs of Republic of Korea (A070001) and KP and HH, who were participated in study design and manuscript review, are in the employment of Hanmi Pharm. Co. Ltd.
References
- Marzolini C, Kim RB. Placental transfer of antiretroviral drugs. Clin Pharmacol Ther. 2005;78:118–122. doi: 10.1016/j.clpt.2005.05.002. [DOI] [PubMed] [Google Scholar]
- Varma MV, Ashokraj Y, Dey CS, Panchagnula R. P-glycoprotein inhibitors and their screening: a perspective from bioavailability enhancement. Pharmacol Res. 2003;48:347–359. doi: 10.1016/s1043-6618(03)00158-0. [DOI] [PubMed] [Google Scholar]
- Thiebaut F, Tsuruo T, Hamada H, Gottesman MM, Pastan I, Willingham MC. Cellular localization of the multidrug-resistance gene product P-glycoprotein in normal human tissues. Proc Natl Acad Sci U S A. 1987;84:7735–7738. doi: 10.1073/pnas.84.21.7735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K, Wu J, Li X. Recent advances in the research of P-glycoprotein inhibitors. Biosci Trends. 2008;2:137–146. [PubMed] [Google Scholar]
- Thomas H, Coley HM. Overcoming multidrug resistance in cancer: an update on the clinical strategy of inhibiting P-glycoprotein. Cancer Control. 2003;10:159–165. doi: 10.1177/107327480301000207. [DOI] [PubMed] [Google Scholar]
- Krishna R, Mayer LD. Multidrug resistance (MDR) in cancer. Mechanisms, reversal using modulators of MDR and the role of MDR modulators in influencing the pharmacokinetics of anticancer drugs. Eur J Pharm Sci. 2000;11:265–283. doi: 10.1016/s0928-0987(00)00114-7. [DOI] [PubMed] [Google Scholar]
- Kwak JO, Lee SH, Lee GS, Kim MS, Ahn YG, Lee JH, Kim SW, Kim KH, Lee MG. Selective inhibition of MDR1 (ABCB1) by HM30181 increases oral bioavailability and therapeutic efficacy of paclitaxel. Eur J Pharmacol. 2010;627:92–98. doi: 10.1016/j.ejphar.2009.11.008. [DOI] [PubMed] [Google Scholar]
- Heykants J, Michiels M, Knaeps A, Brugmans J. Loperamide (R 18 553), a novel type of antidiarrheal agent. Part 5: the pharmacokinetics of loperamide in rats and man. Arzneimittelforschung. 1974;24:1649–1653. [PubMed] [Google Scholar]
- Schinkel AH, Wagenaar E, Mol CA, van Deemter L. P-glycoprotein in the blood-brain barrier of mice influences the brain penetration and pharmacological activity of many drugs. J Clin Invest. 1996;97:2517–2524. doi: 10.1172/JCI118699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DE. Loperamide: a pharmacological review. Rev Gastroenterol Disord. 2007;7(Suppl. 3):S11–18. [PubMed] [Google Scholar]
- Kim TE, Gu N, Yoon SH, Cho JY, Park KM, Shin SG, Jang IJ, Yu KS. Tolerability and pharmacokinetics of a new P-glycoprotein inhibitor, HM30181, in healthy Korean male volunteers: single- and multiple-dose randomized, placebo-controlled studies. Clin Ther. 2012;34:482–494. doi: 10.1016/j.clinthera.2012.01.003. [DOI] [PubMed] [Google Scholar]
- Killinger JM, Weintraub HS, Fuller BL. Human pharmacokinetics and comparative bioavailability of loperamide hydrochloride. J Clin Pharmacol. 1979;19:211–218. doi: 10.1002/j.1552-4604.1979.tb01654.x. [DOI] [PubMed] [Google Scholar]
- Sadeque AJ, Wandel C, He H, Shah S, Wood AJ. Increased drug delivery to the brain by P-glycoprotein inhibition. Clin Pharmacol Ther. 2000;68:231–237. doi: 10.1067/mcp.2000.109156. [DOI] [PubMed] [Google Scholar]
- Kharasch ED, Hoffer C, Whittington D, Sheffels P. Role of P-glycoprotein in the intestinal absorption and clinical effects of morphine. Clin Pharmacol Ther. 2003;74:543–554. doi: 10.1016/j.clpt.2003.08.011. [DOI] [PubMed] [Google Scholar]
- Kharasch ED, Hoffer C, Whittington D. The effect of quinidine, used as a probe for the involvement of P-glycoprotein, on the intestinal absorption and pharmacodynamics of methadone. Br J Clin Pharmacol. 2004;57:600–610. doi: 10.1111/j.1365-2125.2003.02053.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skarke C, Jarrar M, Erb K, Schmidt H, Geisslinger G, Lotsch J. Respiratory and miotic effects of morphine in healthy volunteers when P-glycoprotein is blocked by quinidine. Clin Pharmacol Ther. 2003;74:303–311. doi: 10.1016/S0009-9236(03)00220-0. [DOI] [PubMed] [Google Scholar]
- Yamamoto Y, Inoue Y, Matsuda K, Takahashi Y, Kagawa Y. Influence of concomitant antiepileptic drugs on plasma lamotrigine concentration in adult Japanese epilepsy patients. Biol Pharm Bull. 2012;35:487–493. doi: 10.1248/bpb.35.487. [DOI] [PubMed] [Google Scholar]
- Skarke C, Jarrar M, Schmidt H, Kauert G, Langer M, Geisslinger G, Lotsch J. Effects of ABCB1 (multidrug resistance transporter) gene mutations on disposition and central nervous effects of loperamide in healthy volunteers. Pharmacogenetics. 2003;13:651–660. doi: 10.1097/00008571-200311000-00001. [DOI] [PubMed] [Google Scholar]
- Tayrouz Y, Ganssmann B, Ding R, Klingmann A, Aderjan R, Burhenne J, Haefeli WE, Mikus G. Ritonavir increases loperamide plasma concentrations without evidence for P-glycoprotein involvement. Clin Pharmacol Ther. 2001;70:405–414. doi: 10.1067/mcp.2001.119212. [DOI] [PubMed] [Google Scholar]
- Paek IB, Kim SY, Kim MS, Kim J, Lee G, Lee HS. Characterization of human liver cytochrome P-450 enzymes involved in the O-demethylation of a new P-glycoprotein inhibitor HM-30181. J Toxicol Environ Health A. 2007;70:1356–1364. doi: 10.1080/15287390701434307. [DOI] [PubMed] [Google Scholar]
- Kurnik D, Sofowora GG, Donahue JP, Nair UB, Wilkinson GR, Wood AJ, Muszkat M. Tariquidar, a selective P-glycoprotein inhibitor, does not potentiate loperamide’s opioid brain effects in humans despite full inhibition of lymphocyte P-glycoprotein. Anesthesiology. 2008;109:1092–1099. doi: 10.1097/ALN.0b013e31818d8f28. [DOI] [PubMed] [Google Scholar]
- Choo EF, Kurnik D, Muszkat M, Ohkubo T, Shay SD, Higginbotham JN, Glaeser H, Kim RB, Wood AJ, Wilkinson GR. Differential in vivo sensitivity to inhibition of P-glycoprotein located in lymphocytes, testes, and the blood-brain barrier. J Pharmacol Exp Ther. 2006;317:1012–1018. doi: 10.1124/jpet.105.099648. [DOI] [PubMed] [Google Scholar]
- Bauer F, Wanek T, Mairinger S, Stanek J, Sauberer M, Kuntner C, Parveen Z, Chiba P, Müller M, Langer O, Erker T. Interaction of HM30181 with P-glycoprotein at the murine blood-brain barrier assessed with positron emission tomography. Eur J Pharmacol. 2012;696:18–27. doi: 10.1016/j.ejphar.2012.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha YJ, Lee H, Gu N, Kim TE, Lim KS, Yoon SH, Chung JY, Jang IJ, Shin SG, Yu KS, Cho JY. Sustained increase in the oral bioavailability of loperamide after a single oral dose of HM30181, a P-glycoprotein inhibitor, in healthy male participants. Basic Clin Pharmacol Toxicol. 2013;113:419–424. doi: 10.1111/bcpt.12108. [DOI] [PubMed] [Google Scholar]
- Sinex JE. Pulse oximetry: principles and limitations. Am J Emerg Med. 1999;17:59–67. doi: 10.1016/s0735-6757(99)90019-0. [DOI] [PubMed] [Google Scholar]
- Ong CE, Coulter S, Birkett DJ, Bhasker CR, Miners JO. The xenobiotic inhibitor profile of cytochrome P4502C8. Br J Clin Pharmacol. 2000;50:573–580. doi: 10.1046/j.1365-2125.2000.00316.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsky RL, Gaman EA, Obach RS. Examination of 209 drugs for inhibition of cytochrome P450 2C8. J Clin Pharmacol. 2005;45:68–78. doi: 10.1177/0091270004270642. [DOI] [PubMed] [Google Scholar]
- Darby RA, Callaghan R, McMahon RM. P-glycoprotein inhibition: the past, the present and the future. Curr Drug Metab. 2011;12:722–731. doi: 10.2174/138920011798357006. [DOI] [PubMed] [Google Scholar]