

Abstract
Aims
Tamoxifen is considered a pro-drug of its active metabolite endoxifen. The major metabolic enzymes involved in endoxifen formation are CYP2D6 and CYP3A. There is considerable evidence that variability in activity of these enzymes influences endoxifen exposure and thereby may influence the clinical outcome of tamoxifen treatment. We aimed to quantify the impact of metabolic phenotype on the pharmacokinetics of tamoxifen and endoxifen.
Methods
We assessed the CYP2D6 and CYP3A metabolic phenotypes in 40 breast cancer patients on tamoxifen treatment with a single dose of dextromethorphan as a dual phenotypic probe for CYP2D6 and CYP3A. The pharmacokinetics of dextromethorphan, tamoxifen and their relevant metabolites were analyzed using non-linear mixed effects modelling.
Results
Population pharmacokinetic models were developed for dextromethorphan, tamoxifen and their metabolites. In the final model for tamoxifen, the dextromethorphan derived metabolic phenotypes for CYP2D6 as well as CYP3A significantly (P < 0.0001) explained 54% of the observed variability in endoxifen formation (inter-individual variability reduced from 55% to 25%).
Conclusions
We have shown that not only CYP2D6, but also CYP3A enzyme activity influences the tamoxifen to endoxifen conversion in breast cancer patients. Our developed model may be used to assess separately the impact of CYP2D6 and CYP3A mediated drug–drug interactions with tamoxifen without the necessity of administering this anti-oestrogenic drug and to support Bayesian guided therapeutic drug monitoring of tamoxifen in routine clinical practice.
Keywords: CYP2D6, CYP3A, endoxifen, metabolic phenotype, nonmem, tamoxifen
What is already known about this subject —
CYP2D6 and CYP3A are the major metabolic pathways for bioactivation of tamoxifen to endoxifen.
Although impaired CYP2D6 activity as a result of genetic polymorphism has previously shown to result in decreased endoxifen formation, little is known about the influence of CYP3A activity on the formation of endoxifen.
What this study adds —
For the first time, a pharmacokinetic model for tamoxifen and endoxifen has been developed.
We show that both the CYP2D6 and CYP3A metabolic phenotypes significantly and relevantly impact tamoxifen bioactivation.
This implies that variability in CYP3A activity, for example due to drug–drug interactions, may be more important than previously thought.
Introduction
Anti-oestrogenic therapy with tamoxifen is an effective treatment for oestrogen receptor-positive breast cancer. Tamoxifen can be considered a pro-drug as it requires bioactivation by cytochrome P450 (CYP) enzymes. So far, 22 tamoxifen metabolites have been identified 1. The two active tamoxifen metabolites 4-hydroxy-tamoxifen and endoxifen have a 100-fold more potent anti-oestrogenic effect than the parent compound. Since endoxifen plasma concentrations well exceed 4-hydroxy-tamoxifen concentrations, endoxifen is considered to be the metabolite responsible for the clinical effect of tamoxifen and there is considerable concern that variability in endoxifen exposure affects treatment outcome 1–5.
Although multiple CYP enzymes are involved in the formation of endoxifen, the major enzymes involved in its formation are CYP2D6 and CYP3A 6,7. Endoxifen concentrations are highly variable between subjects, mainly as a result of variability in activity of these enzymes. Variability in CYP2D6 activity is partly due to genetic variation and a reduced function of the CYP2D6 alleles has been associated with lower endoxifen concentrations and a worse therapeutic outcome of tamoxifen treatment 3,7–9. CYP3A activity is also known to be highly variable, mainly as a result of environmental factors 10–12. Because endoxifen concentrations are associated with tumour response 2,4, variability in endoxifen exposure may result in variability in treatment outcome. Therefore, dose adjustments of tamoxifen based on enzyme activity or therapeutic drug monitoring of endoxifen concentrations may be warranted 3. Some have suggested to guide tamoxifen dosing on CYP2D6 genotype, to prevent subtherapeutic endoxifen concentrations 13. However, CYP2D6 genotype-based dosing does not account for all non-genetic factors, including CYP3A activity and CYP2D6 drug interactions. Hence, phenotype guided dosing and therapeutic drug monitoring are likely more successful dosing strategies. Furthermore, the role of pharmacogenetic testing to guide tamoxifen has recently been discussed extensively after the results of the BIG 1–98 and ATAC studies were presented, where it was found that CYP2D6 phenotype was a poor predictor of metabolic phenotype 14–18. Lastly, there is only limited quantitative knowledge on the impact of CYP2D6 or CYP3A phenotype on tamoxifen pharmacokinetics.
Dextromethorphan is a valuable dual phenotyping probe for both CYP2D6 and CYP3A activity 19–25. Because of the putative similar metabolic pathway of dextromethorphan and tamoxifen (see Figure 1), we have recently investigated the value of dextromethorphan as a phenotyping probe for tamoxifen metabolism. Using a non-compartmental pharmacokinetic analysis, we found an inverse correlation between the exposures of endoxifen and dextromethorphan 26. However, using this approach, we could not separately quantify the effects of CYP2D6 or CYP3A phenotypes on the formation of endoxifen. This knowledge, however, is crucial for the understanding of the relative importance of variability in CYP2D6 and CYP3A activity, e.g. as a result of pharmacokinetic interactions, for the bioactivation of tamoxifen. Furthermore, development of a pharmacokinetic model for tamoxifen and endoxifen including the dextromethorphan derived metabolic phenotypes, may be used to study pharmacokinetic interactions with tamoxifen, without the necessity to administer this anti-oestrogenic drug to study participants.
Figure 1.
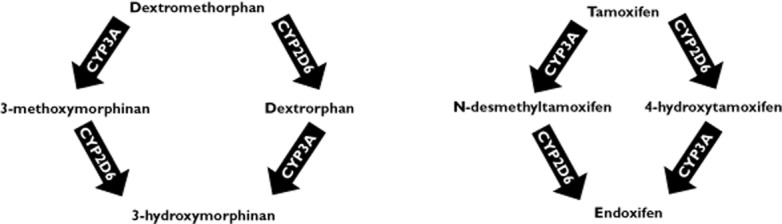
Schematic representation of dextromethorphan and tamoxifen metabolism. Scheme for both dextromethorphan and tamoxifen metabolism. Dextromethorphan is mainly converted by CYP2D6 to dextrorphan and tamoxifen is mainly converted by CYP3A into N-desmethyltamoxifen. Afterwards, dextrorphan is converted by CYP3A into 3-hydroxymorphinan and N-desmethyltamoxifen is converted by CYP2D6 into endoxifen
A model describing tamoxifen, 4-hydroxytamoxifen and N-desmethyltamoxifen pharmacokinetics has been previously published, but no population pharmacokinetic model is yet available for tamoxifen and endoxifen 27. Furthermore, previous studies have described the population pharmacokinetics of dextromethorphan and its metabolite dextrorphan, but no population pharmacokinetic model for dextromethorphan and all of its three phase I metabolites is thus far available 28,29.
Therefore, to assess the impact of CYP2D6 and CYP3A metabolic phenotypes on the pharmacokinetics of tamoxifen and endoxifen and to describe the population pharmacokinetics of dextromethorphan and its phase I metabolites, we analyzed the previously obtained data by means of a population pharmacokinetic approach.
Methods
Patient selection and inclusion
Patient selection and inclusion have been described in detail elsewhere 26. In short, patients on tamoxifen treatment, older than 18 years, who were not concomitantly taking moderate or strong inhibitors or inducers of CYP3A and the drug transporters ABCB1 (P-glycoprotein) and/or ABCG2 (breast cancer resistance protein) were eligible for inclusion. All patients provided written informed consent and the study protocol was approved by the institutional review board and was registered in the Dutch Trial Registry (No. NTR1751).
Pharmacokinetic sampling and bioanalysis
Tamoxifen was dosed in concordance with the current treatment scheme of the patient. Dextromethorphan was dosed 30 mg orally as a single dose 2 h after administration of tamoxifen. For tamoxifen nine samples were obtained during a 24 h interval: at pre-dose and at 30 min and 1, 1.5, 2, 4,8, 12 and 24 h after the administration of the daily tamoxifen dose. Dextromethorphan samples were collected at pre-dose and at 30 min and 1, 1.5, 2, 4, 6, 10 and 22 h after drug administration. The plasma concentrations of these drugs and their metabolites were measured using sensitive and specific validated assays 30,31. Accuracy and precision of both these assays were respectively within 85–115% and less than 15% for all analytes. The lower limit of quantitation was 0.5 nm for dextromethorphan and its metabolites. The lower limit of quantitation for tamoxifen and endoxifen were 18 and 7 nm, respectively.
Pharmacokinetic analysis
Pharmacokinetic analysis was performed using the non-linear mixed effects modelling program nonmem version 7.2, using the first order conditional estimation method with interaction. The software program Piraña version 2.7.1 was used as an interface for nonmem and for run deployment 32. Precision of parameter estimates was estimated using the covariance step in nonmem. The minimal value of the objective function (equal to minus twice the log likelihood) provided by nonmem was used to discriminate between hierarchical models using the log likelihood ratio test. A P value of 0.05, corresponding with a change in objective function of 3.84 points with one degree of freedom was considered statistically significant when comparing hierarchical models. Model diagnostics were performed by assessing standard diagnostic plots, the normalized prediction distribution errors (NPDE) and visual predictive checks (VPC) with Perl Speaks nonmem and XPOSE4, as described earlier 33–36. Inter-individual error was described with an exponential error model and residual error was described with a proportional error model. For pharmacokinetic analysis, all parent and metabolite doses and concentrations in the dataset were converted to their molar equivalents. Concentrations below the limit of quantitation for dextromethorphan and its metabolites, (0.5 nm), that showed enough responses in the mass spectrometric detector (signal to noise ratio in the chromatogram >5) were included in the pharmacokinetic analysis.
Dextromethorphan and metabolites model development
Dextromethorphan is well known for its extensive pre-systemic metabolism. Since this pre-systemic metabolism provides crucial information on cytochrome P450 activity and correlates with systemic metabolism, it should be accounted for in the pharmacokinetic model. Therefore a semi-physiological model integrating oral availability and systemic clearance was used as previously proposed by Levi et al. for nicotine and its metabolites and for dextromethorphan and its metabolite dextrorphan by Abduljalil and coworkers 28,37,38. In short, oral absorption of dextromethorphan to the central compartment with a hypothetical metabolism compartment was described with a first order rate constant (ka) and a lag time (tlag). The hypothetical metabolism compartment was assumed to be in rapid equilibrium with the central compartment of the parent drug and the central compartment was in equilibrium with a peripheral compartment. With this model it is assumed that, after absorption, the drug goes to the metabolism compartment before reaching the central compartment. Systemic metabolism of the drug also takes place in this hypothetical compartment. After absorption, dextromethorphan is simultaneously metabolized to dextrorphan by CYP2D6 and to 3-methoxymorphinan by CYP3A. Subsequently, dextrorphan and 3-methoxymorphinan are metabolized to 3-hydroxmorphinan by CYP3A and CYP2D6, respectively. Also, dextrorphan is known to undergo phase II metabolism by glucuronidation 39, and this was accounted for by estimation of additional clearance of dextrorphan. The starting point for model development is schematically depicted in Figure 2. Differential equations specifying this model can be found in appendix 1 of this manuscript.
Figure 2.
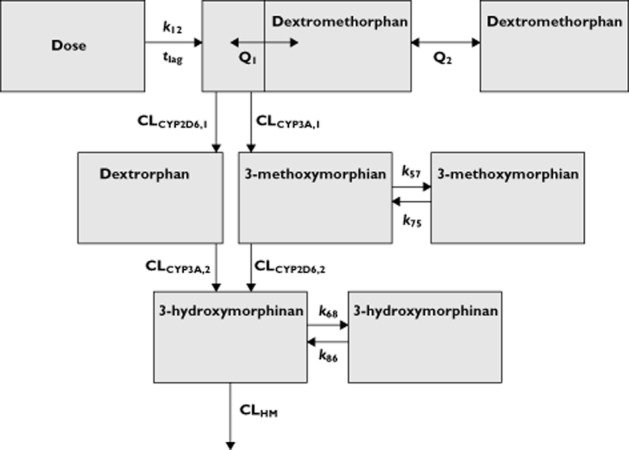
Schematic representation of the population pharmacokinetic model of dextromethorphan and its metabolites. The absorption phase is described with a rate constant (k12) and a lag time (tlag). The central dextromethorphan compartment is in rapid equilibrium with the hypothetical metabolism compartment. Drug transfer between this metabolism compartment and the central dextromethorphan compartment is described with the intercompartmental clearance parameter Q1. The equilibrium with dextromethorphan with its peripheral compartment is described with the intercompartmental clearance parameter Q2. Metabolism of dextromethorphan to dextrorphan is described with the clearance parameter CLCYP2D6,1 and its subsequent clearance to other species and simultaneous metabolism to 3-hydroxymorphinan are respectively described with CLDO and CLCYP3A,2. Metabolism of dextromethorphan to 3-methoxymorphinan is described with the clearance parameter CLCYP3A,1 and its subsequent metabolism to 3-hydroxymorphinan is described with CLCYP2D6,2. The clearance of 3-hydroxymorphinan to other species is described with clearance parameter CLHM. From this model the volume of distribution for dextromethorphan and its metabolite can be estimated. The volumes of distribution for all metabolites are fixed to a volume of 419 l to assure model identifiability
The individual post hoc estimates for the CYP2D6 and CYP3A mediated clearance terms were considered to be the respective CYP2D6 and CYP3A phenotypes for further investigation for their impact on tamoxifen pharmacokinetics (see below). Since these clearance terms are independent of concentration and reflect variability in metabolic capacity of these cytochrome P450 enzymes, they are appropriate to study as covariates for tamoxifen metabolism.
Tamoxifen and endoxifen model development
The starting point for the model describing tamoxifen and endoxifen pharmacokinetics was the population pharmacokinetic models as described earlier 27,28,37, with one central compartment for tamoxifen and a second compartment for a metabolite with first order absorption and elimination. The basic pharmacokinetic model is shown in Figure 4 and in appendix 2 of this manuscript for the differential equations specifying this model.
Figure 4.
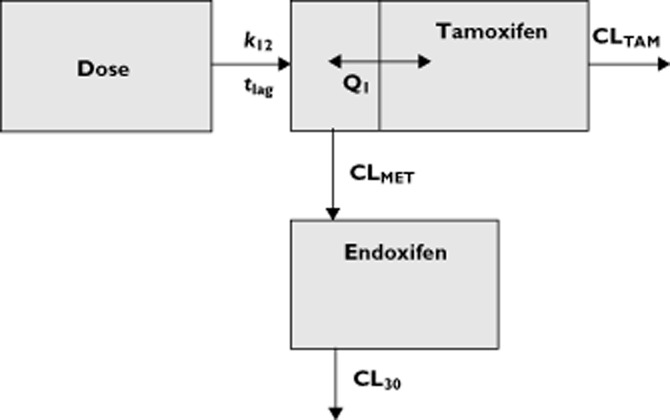
Schematic representation of the population pharmacokinetic model of tamoxifen and endoxifen. The absorption phase is described with a rate constant (k12) and a lag time (tlag). The central tamoxifen compartment is in rapid equilibrium with the hypothetical metabolism compartment. Drug transfer between this metabolism compartment and the central tamoxifen compartment is described with the intercompartmental clearance parameter Q1. Metabolism of tamoxifen to endoxifen is described with the clearance parameter CLMET. Elimination of tamoxifen to other species is described with the clearance parameter CLTAM. The volume of distribution and clearance of endoxifen are fixed to known literature values
The estimated CYP2D6 and CYP3A metabolic phenotypes from the dextromethorphan model were investigated as covariates for the clearance parameter describing formation of endoxifen (CLMET), as described with equation 1. In this equation, CLMET,POP is the population value for clearance describing the formation of endoxifen from tamoxifen and CLMET,TYPICAL is its typical value for an individual with an estimated clearance equal to its population value. Furthermore, PHENOTYPEIND is the estimated individual phenotype from the dextromethorphan population model and PHENOTYPEPOP is its corresponding population value. θ is the estimated power describing the relationship between the metabolic phenotype and tamoxifen metabolism.
| Equation 1 |
For the covariate analysis, all phenotypes were univariately tested for their influence on endoxifen formation for inclusion in an intermediate model. After inclusion of significant covariates into an intermediate model, a stepwise backward elimination procedure was carried out, retaining covariates in the model only when they were statistically significant (P < 0.05).
Results
Details of the study population have previously been published elsewhere 26. In short, 40 breast cancer patients were included. Pharmacokinetic sampling did not succeed in one subject due to obstruction of a venous cannula and thus 39 patients were available for pharmacokinetic evaluation of both tamoxifen and dextromethorphan and their metabolites. Eleven out of these 39 patients were on a once daily 40 mg tamoxifen dosing regimen and all other patients received tamoxifen 20 mg once daily. A total of 349 tamoxifen and 331 endoxifen concentrations were available for pharmacokinetic evaluation. Furthermore, a total of 293 dextromethorphan, 296 dextrorphan, 139 3-methoxymorphinan and 249 3-hydroxymorphinan concentrations were available. The demographic characteristics of all study participants are shown in Table 1.
Table 1.
Demographic characteristics of study participants
| Variable | Value |
|---|---|
| Age (years) | |
| Median | 53 |
| Range | 22–71 |
| Height (m) | |
| Median | 1.69 |
| Range | 1.56-1.79 |
| Weight (kg) | |
| Median | 72.7 |
| Range | 48.5–114 |
| Tamoxifen dose | |
| 20 mg | 70% |
| 40 mg | 30% |
Observed pharmacokinetics
The pharmacokinetics of dextromethorphan and tamoxifen showed a high inter-individual variability. Dextromethorphan metabolites appeared early and abundantly in the systemic circulation, indicating an extensive and variable pre-systemic metabolism. Furthermore, during a dosing interval, the individual endoxifen plasma concentrations were relatively constant, reflecting the long elimination half-life of both the parent drug and its active metabolite.
Dextromethorphan and metabolites pharmacokinetic model development
The starting point of our model was a model incorporating first pass metabolism and intrinsic clearance as previously described for nicotine and its metabolites as well as for dextromethorphan and its metabolite dextrorphan 28,38. Dextromethorphan absorption was variable with a slow onset. When simultaneously modelling parent and metabolite data, the volume of distribution of a metabolite or the fraction of parent drug that is converted to metabolite are unidentifiable. Therefore, to keep the model identifiable, this fraction or volume of distribution needs to be assumed 40,41. This allows proper assessment of all other relevant pharmacokinetic parameters and their variability. We therefore assumed a volume of distribution of 419 l for all dextromethorphan metabolites, as previously found for dextrorphan 28. During model development it was observed that the inter-individual variability in the clearance parameters CL2D6,1 and CL2D6,2 was highly correlated and could not be estimated independently from one another. Therefore, a single variability term was estimated for both clearance parameters. Interestingly, no covariance was observed between the inter-individual clearance parameters describing CYP3A mediated metabolism. Furthermore, the presence of peripheral compartments for 3-methoxymorphinan and 3-hydroxymorphinan was observed in the data.
The CYP2D6 clearance parameters CL2D6,1 and CL2D6,2, respectively describing the dextromethorphan to dextrorphan and the 3-methoxymorphinan to 3-hydroxymorphinan conversion, were 1560 and 1840 i h−1 with an inter-individual variability of 166% (relative SD). The CYP3A clearance parameters CLCYP3A,1 and CLCYP3A,2, respectively describing the dextromethorphan to 3-methoxymorphinan and dextrorphan to 3-hydroxymorphinan conversion, were 44.7 and 1840 l h−1 with an respective inter-individual variability of 89.8% and 53.0% (relative SD).
The parameter precision was good for all relevant parameters, as represented in the low relative standard error of the parameter estimates, indicating that all parameters could be reliably estimated. The basic pharmacokinetic model is schematically depicted in Figure 2. The corresponding pharmacokinetic parameter estimates are shown in Table 2. Lastly, diagnostic plots for dextromethorphan, dextrorphan, 3-methoxymorphinan and 3-hydroxymorphinan are shown Figure 3A, B, C and D, respectively. As observed in these plots, dextromethorphan and metabolite concentrations could be adequately estimated, allowing estimation of the CYP2D6 and CYP3A phenotypes. Although the individually predicted vs. observed concentrations plots may sometimes show a large variability, in predicted concentrations, the weighted residuals (as depicted by the NPDE) are always within acceptable limits (–3 to 3) and evenly distributed around the x-axis. The visual predictive checks also represent the large observed variability in concentrations.
Table 2.
Parameter estimates for dextromethorphan and metabolites pharmacokinetic model
| Parameter | Estimate | RSE |
|---|---|---|
| Central Vd dextromethorphan (l) | 188 | 32.4% |
| Peripheral Vd dextromethorphan (l) | 1660 | 16.8% |
| k12 (h−1) | 0.213 | 7.6% |
| tlag (h) | 0.369 | 3.7% |
| CL2D6,1 (l h−1) | 1560 | 27.8% |
| CL2D6,2 (l h−1) | 362 | 46.1% |
| CL3A4,1 (l h−1) | 44.7 | 26% |
| CL3A4,2 (l h−1) | 1840 | 9.1% |
| CLHM (l h−1) | 5730 | 11.4% |
| Q1 (l h−1) | 415 | 22.1% |
| Q2 (l h−1) | 200 | 37.3% |
| k57 (h−1) | 1.11 | 19.5% |
| k75 (h−1) | 0.0815 | 28.7% |
| k68 (h−1) | 13.4 | 12.4% |
| k86 (h−1) | 0.0637 | 18.5% |
| IIV ka (shrinkage) | 31% (4%) | 39.7% |
| IIV CL2D6 (shrinkage) | 166% (-0.9%) | 29.5% |
| IIV CL3A4,1 (shrinkage) | 89.8% (19.8%) | 49.3% |
| IIV CL3A4,2 (shrinkage) | 53% (2.4%) | 27.8% |
| IIV CLHM (shrinkage) | 75.7% (6.2%) | 24.6% |
| Residual error dextromethorphan (shrinkage) | 42.9% (5.2) | 15% |
| Residual error dextrorphan (shrinkage) | 40% (7.2%) | 10.6% |
| Residual error 3-methoxymorphinan (shrinkage) | 29.7% (9.2%) | 23.2% |
| Residual error 3-hydroxymorphinan (shrinkage) | 30.7% (7%) | 12% |
| Condition number | 140 | |
IIV, inter-individual variability; RSE, relative standard error of estimate; Vd, volume of distribution.
Figure 3.
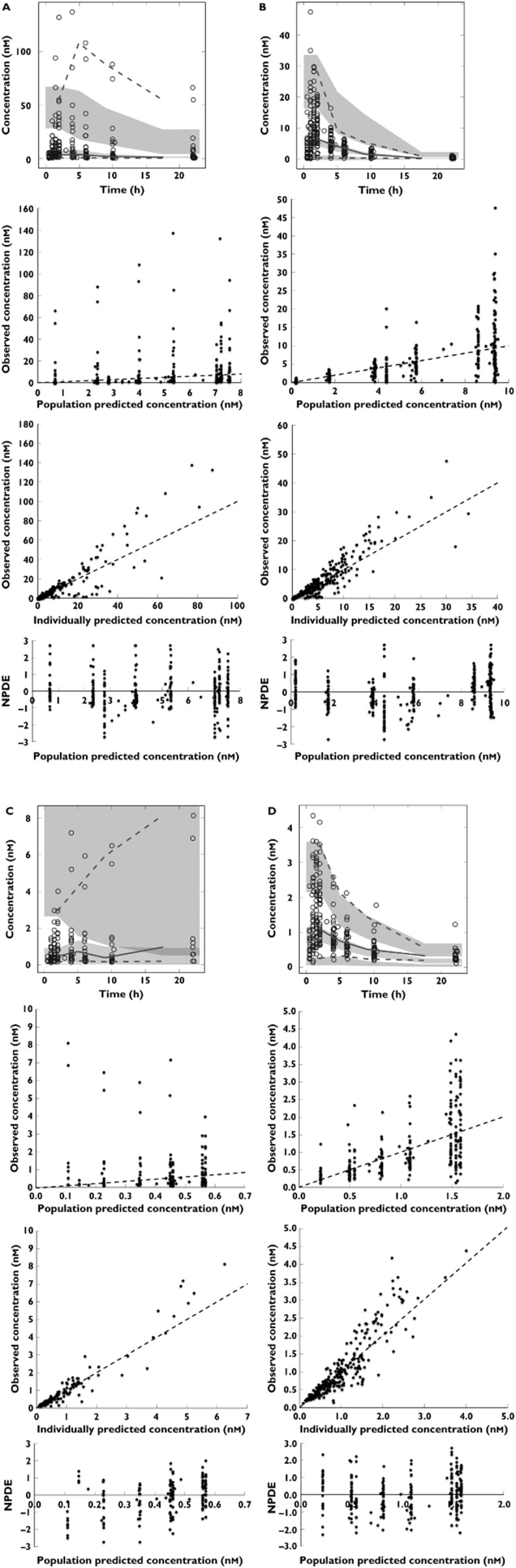
Diagnostic plots for dextromethorphan (A), dextrorphan (B), 3-methoxymorphinan (C) and 3-hydroxymorphinan (D). The upper panel of these figures shows the visual predictive checks. In these visual predictive checks the dotted lines represent the 95% intervals of the observed concentrations vs. time, the solid line represents the observed median concentration vs. time and the shaded areas represent the 95% confidence interval for the model predicted concentrations. Also, the observed concentrations are plotted as open circles. The second panel from above shows the observed concentrations vs. the population predicted concentrations, the third panel from above shows the observed concentrations vs. the individual predicted concentrations and the bottom panel shows the normalized prediction distribution error vs. the population predicted concentrations
Tamoxifen and endoxifen model development
Variable and delayed absorption was also observed for tamoxifen. After introduction of an absorption lag time, the model improved significantly and the lag time in the final model was estimated to be 0.455 h. To keep the model identifiable, the volume of distribution and clearance were fixed to known literature values of 400 l and 5.1 l h−1, respectively 42. Conversion of tamoxifen to endoxifen (CLMET) did not completely account for the clearance of tamoxifen as a result of clearance to other species. Hence, additional clearance of tamoxifen from the central compartment (CLTAM) was estimated. Post hoc estimates of volume of distribution and clearance of tamoxifen from the central compartment showed a positive correlation and estimation of their covariance significantly improved the model. The residual error of the parent drug and metabolite showed a positive correlation. The basic model describing tamoxifen and endoxifen pharmacokinetics is depicted in Figure 4. In the basic model, the clearance parameter describing conversion of tamoxifen to endoxifen was 0.300 l h−1 with an inter-individual variability of 55.2%.
Because the inter-individual variability of the CYP2D6 clearance parameters in the dextromethorphan model was highly correlated (>98%), only the post hoc estimates for CL2D6,1 were investigated for their impact as CYP2D6 phenotype. Furthermore, both post hoc estimates for CYP3A phenotype (CLCYP3A,1 and CLCYP3A,2) were tested as for their impact on tamoxifen metabolism. Univariate testing revealed that all metabolic phenotypes statistically significantly explained inter-individual variability in endoxifen formation. However, after inclusion of all metabolic phenotypes in an intermediate model and the subsequent performing of backward elimination, it was observed that the CLCYP3A,2 derived phenotype did not influence variability in endoxifen formation and removing this covariate from the model did not change other parameter estimates or the model fit. Therefore, CL2D6,1 and CLCYP3A,1 were retained as metabolic phenotypes in the final model. Table 3 shows the parameter estimates of the basic and the final model. In this table θ2D6,1 and θ3A4,1 represent the parameter estimates for the contribution of the CYP2D6 and CYP3A metabolic phenotypes, respectively, as a power function. In the final model, the residual inter-individual variability in endoxifen formation had reduced from 55.2% to 25.4%, indicating that both CYP3A and CYP2D6 metabolic phenotypes explain a major part (54%) of the variability in endoxifen formation. As observed in the dextromethorphan model, the relative standard error of the parameter estimates in the tamoxifen model was low, indicating that these could be reliably estimated.
Table 3.
Parameter estimates for tamoxifen and endoxifen basic and final pharmacokinetic model
| Model | Basic | Final | ||
|---|---|---|---|---|
| Parameter | Estimate | RSE | Estimate | RSE |
| k12 (h−1) | 1.96 | 17.9% | 1.90 | 20.2% |
| tlag (h) | 0.459 | 8.8% | 0.455 | 10.4% |
| Q1 (l h−1) | 32.7 | 55.7% | 61.8 | 65.4% |
| Vd tamoxifen (l) | 727 | 8.3% | 753 | 9% |
| CLTAM (l h−1) | 9.34 | 6% | 9.34 | 6.2% |
| CLMET (l h−1) | 0.300 | 17% | 0.324 | 9.8% |
| θ2D6,1 | − | − | 0.262 | 14% |
| θ3A4,1 | − | − | 0.157 | 72% |
| IIV CLTAM (shrinkage) | 37.7% (−0.5%) | 19.4% | 37.8% (−0.5%) | 19.2% |
| IIV Vd tamoxifen (shrinkage) | 26.0% (23.7%) | 53.0% | 26.7% (24.9%) | 53.9% |
| Covariance IIV CLTAM and IIV Vd tamoxifen | 63.7% | 29.9% | 61.2% | 31.2% |
| IIV CLMET (shrinkage) | 55.2% (1.7%) | 12.5% | 25.4% (3.2%) | 19.3% |
| Residual error tamoxifen (shrinkage) | 13.7% (6.3%) | 11.3% | 13.7% (6.2%) | 11.3% |
| Residual error endoxifen (shrinkage) | 18.9% (6.4%) | 10.1% | 18.9% (6.3%) | 10.1% |
| Covariance residual error | 62.2% | 22.6% | 62.2% | 22.6% |
| Objective function | −4953 | −5009 | ||
| Condition number | 51 | 69 | ||
RSE, relative standard error of estimate; Vd, volume of distribution.
Diagnostic plots of the final tamoxifen and endoxifen model, including the metabolic phenotypes, are shown in Figure 5. As observed, our model adequately captures tamoxifen and endoxifen pharmacokinetics and the variability in the clearance parameter describing endoxifen formation.
Figure 5.

Diagnostic plots for tamoxifen (A) and endoxifen (B). The upper panel of these figures shows the prediction corrected visual predictive checks. In these visual predictive checks the dotted lines represent the 95% intervals of the observed concentrations vs. time, the solid line represents the observed median concentration vs. time and the shaded areas represent the 95% confidence interval for the model predicted concentrations. Also, the observed concentrations are plotted as open circles. The second panel from above shows the observed vs. the population predicted concentrations, the third panel from above shows the observed concentrations vs. the individual predicted concentrations and the bottom panel shows the normalized prediction distribution error vs. population predicted concentrations
Discussion
We have previously shown that variability in dextromethorphan metabolism partly explained variability in tamoxifen metabolism. Using a non-compartmental analysis, we found that dextromethorphan exposure partly explained the variation in endoxifen exposure 26. These results were in concordance with the recent study by Opdam and coworkers who found that the CYP2D6 phenotype, as determined with a 13C-dextromethorphan breath test, explained a part of the variation in endoxifen concentrations 43. However, neither our recent analysis nor the study by Opdam et al. could separately quantify the effect of CYP2D6 and CYP3A activity on the metabolism of tamoxifen.
In our current analysis we could separately quantify the impact of CYP2D6 and CYP3A metabolic phenotypes on the pharmacokinetics of tamoxifen and endoxifen. We found that both phenotypes significantly and relevantly influenced endoxifen exposure. The remaining inter-individual variability in endoxifen formation reduced from 55.2% to 25.4%.
To investigate this, a population pharmacokinetic model had to be developed for dextromethorphan. Dextromethorphan is notorious for its extensive and variable pre-systemic metabolism. We integrated pre-systemic and systemic metabolism of dextromethorphan with a semi-physiological model for first pass metabolism, analogous to previous pharmacokinetic models 28,29,38. The advantage of this approach is that the individual contribution of CYP2D6 and CYP3A to dextromethorphan metabolism could be determined. Due to unavailability of data on urinary excretion of dextromethorphan and its metabolites and the unknown fraction of parent drug that is converted to metabolite, the model was kept identifiable by fixing the volumes of distribution of all dextromethorphan metabolites to a previously found volume of distribution for dextrorphan 28. This allowed us to estimate the relevant variability in all clearance parameters for metabolic conversion of dextromethorphan. All parameter estimates corresponded well with those found earlier by Abduljalil et al. 28, except for the volume of distribution of dextromethorphan, where our estimated volume of distribution was approximately two-fold lower than previously found 28. Since this represents the apparent volume of distribution, we can explain this difference by a more complete mass balance of dextromethorphan in our study due to inclusion of other metabolite data.
The dextromethorphan CYP2D6 clearance parameters were highly correlated but this was not observed for the CYP3A clearance parameters. We hypothesize that this is caused by a difference in intestinal and hepatic CYP2D6 and CYP3A expression and their role in the pre-systemic metabolism of dextromethorphan. It is known that CYP3A enzymes are highly expressed in the intestines and that this intestinal CYP3A accounts for approximately 80% of the total CYP content in the human body 44. However, CYP2D6 is hardly expressed in the intestines and the intestinal form is unlikely to play a role in intestinal first pass metabolism 44. Therefore, the variability in clearance parameters CLCYP3A,1 and CLCYP3A,2 may, respectively, reflect the variability of presystemic metabolism in the intestine and liver (suspected combined intestinal-liver CYP3A phenotype) and systemic metabolism in the liver alone (suspected hepatic CYP3A phenotype). In contrast, the clearance parameters CLCYP2D6,1 and CLCYP2D6,2 may both reflect hepatic variability in expression and may, therefore, be highly correlated. Our results confirm previous studies that found that the conversion of dextromethorphan to 3-methoxymorphinan is a suitable pathway to study for CYP3A phenotyping and that this phenotype predicts metabolic variability 20,21,25. However, it does not fully explain that variability in CLCYP3A,2 did not correlate with variability of endoxifen pharmacokinetics. We postulate that there may be two causes for this finding. The first is that concomitant use of strong CYP3A inhibitors or inducers were exclusion criteria and that therefore large differences in hepatic CYP3A activity were absent and could therefore not be identified in our population. The generalizability of our model to situations where strong metabolic inhibitors or inducers are used, therefore, needs further assessment. The second reason why the impact of the suspected hepatic CYP3A phenotype was found to be negligible, could be that the rate-limiting step in CYP3A mediated tamoxifen metabolism is described by the suspected combined intestinal-hepatic CYP3A phenotype and, therefore, additional variability caused by the suspected hepatic phenotype could not be identified.
One might postulate that because tamoxifen and dextromethorphan share a metabolic pathway and were simultaneously present in the body, they may have influenced each other’s pharmacokinetics. In our study neither of these drugs showed saturation pharmacokinetics. Also, no pharmacokinetic interactions are known between these frequently studied drugs. Although we think metabolic competition is therefore an unlikely phenomenon, it cannot be ruled out completely. To our knowledge, we are the first to develop a population pharmacokinetic model for tamoxifen and its metabolite endoxifen. The estimates for the apparent oral volume of distribution and clearance of tamoxifen were slightly higher than values reported earlier 27,45,46. An explanation may be decreased a bioavailability, resulting in a higher apparent oral clearance and volume of distribution. The cause for this difference, however, remains to be elucidated. In the human body, tamoxifen is first metabolized to N-desmethyltamoxifen and 4-hydroxytamoxifen before being metabolized to endoxifen. Our model directly described the formation of endoxifen from tamoxifen and due to the long elimination half-life of endoxifen, its clearance was not identifiable and was fixed to a known literature value. Therefore, variability in the clearance parameter describing the conversion of tamoxifen to endoxifen should be considered the sum of variability in all clearance parameters describing the metabolism of tamoxifen, N-desmethyl-tamoxifen, 4-hydroxy-tamoxifen and variability in clearance of endoxifen. A limitation of our analysis may thus be that we cannot predict concentrations of the intermediate metabolites of tamoxifen. However, endoxifen remains the metabolite of interest because it is responsible for the clinical effects of tamoxifen. With our current approach we could adequately study the impact of metabolic phenotype on the formation of endoxifen.
An interesting finding from our analysis is that both CYP2D6 and CYP3A phenotypes predict endoxifen exposure. The finding that CYP2D6 activity correlates with endoxifen concentrations is in concordance with previous findings 3,8,43 and variability in cytochrome activity explained a large part, but not all, inter-individual variability in endoxifen formation. To the best of our knowledge we are the first to show in vivo that not only variability in CYP2D6 activity influences endoxifen exposure, but also variability in CYP3A activity influences endoxifen exposure. The clinical relevance of this finding certainly needs further assessment, as it suggests that CYP3A inhibition may result in decreased tamoxifen efficacy.
Although CYP2D6 and CYP3A are the major metabolic pathways in tamoxifen bioactiviation, other phase I metabolic enzymes, including CYP2B6, CYP2C9, CYP2C19 and CYP1A2, are thought to be involved as well 7. Since dextromethorphan is not necessarily a substrate for all these enzymes, this may explain the remaining inter-individual variability in endoxifen formation. Nonetheless, our findings illustrate that dextromethorphan may be used as a harmless probe substrate for investigation of CYP2D6 and CYP3A mediated interactions with tamoxifen and that CYP3A activity also dictates endoxifen exposure. Also, because CYP3A is major determinant of endoxifen formation, we think the role of CYP2D6 genotyping alone for dose guiding of tamoxifen is not sufficient. We think that with all the current knowledge, tamoxifen dosing may be best guided by monitoring tamoxifen and endoxifen concentrations. For example, after the start of tamoxifen treatment ‘poor tamoxifen bioactivators’ may be identified using the population pharmacokinetic model for tamoxifen and endoxifen.
In short, we developed a population pharmacokinetic model for dextromethorphan and all of its phase I metabolites, as well as for tamoxifen and endoxifen incorporating CYP2D6 and CYP3A metabolic phenotypes. These metabolic phenotypes explained a part of the variability in endoxifen formation. Interestingly, besides the CYP2D6 activity, CYP3A activity had a major impact on the conversion of tamoxifen to endoxifen. We think that further research for the clinical relevancy of pharmacokinetic interactions influencing the CYP3A enzyme during tamoxifen treatment is, therefore, necessary. Also, dextromethorphan may be used as a harmless probe to predict pharmacokinetic interactions with CYP2D6 or CYP3A substrates like tamoxifen without the necessity to administer a potentially harmful drug. Prospective validation of our model for such a use, however, is warranted.
Appendix 1
The differential equations specifying the dextromethorphan and metabolites model as schematically depicted in Figure 2 are as described in the next column.
depot compartment
tamoxifen central compartment
dextrorphan central compartment
dextromethorphan peripheral compartment
3-methoxymorphinan central compartment
3-hydroxymorphinan central compartment
3-methoxymorphinan peripheral compartment
3-hydroxymorphinan peripheral compartment
where Ak and Ck = Ak/Vk denote the amount and concentration of the species associated with the kth compartment. The drug concentration in the hypothetical liver compartment is denoted by CL and is described with the following equation, as previously derived by Levi et al. and Abduljalil et al. 28,37,38:
Appendix 2
The differential equations specifying the tamoxifen and endoxifen model as schematically depicted in Figure 4 are as described on the next page.
depot compartment
tamoxifen central compartment
endoxifen central compartment
where Ak and Ck = Ak/Vk denote the amount and concentration of the species associated with the kth compartment. The drug concentration in the hypothetical liver compartment is denoted by CL and is described with the following equation, as previously derived by Levi et al. and Abduljalil et al. 28,37,38:
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare no support from any organization for the submitted work, no financial relationships with any organizations that might have an interest in the submitted work in the previous 3 years and no other relationships or activities that could appear to have influenced the submitted work.
References
- Murdter TE, Schroth W, Bacchus-Gerybadze L, Winter S, Heinkele G, Simon W, Fasching PA, Fehm T, Eichelbaum M, Schwab M, Brauch H. Activity levels of tamoxifen metabolites at the estrogen receptor and the impact of genetic polymorphisms of phase I and II enzymes on their concentration levels in plasma. Clin Pharmacol Ther. 2011;89:708–717. doi: 10.1038/clpt.2011.27. [DOI] [PubMed] [Google Scholar]
- Coezy E, Borgna JL, Rochefort H. Tamoxifen and metabolites in MCF7 cells: correlation between binding to estrogen receptor and inhibition of cell growth. Cancer Res. 1982;42:317–323. [PubMed] [Google Scholar]
- Madlensky L, Natarajan L, Tchu S, Pu M, Mortimer J, Flatt SW, Nikoloff DM, Hillman G, Fontecha MR, Lawrence HJ, Parker BA, Wu AH, Pierce JP. Tamoxifen metabolite concentrations, CYP2D6 genotype, and breast cancer outcomes. Clin Pharmacol Ther. 2011;89:718–725. doi: 10.1038/clpt.2011.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong IY, Teft WA, Ly J, Chen YH, Alicke B, Kim RB, Choo EF. Determination of clinically therapeutic endoxifen concentrations based on efficacy from human MCF7 breast cancer xenografts. Breast Cancer Res Treat. 2013;19:61–69. doi: 10.1007/s10549-013-2530-1. [DOI] [PubMed] [Google Scholar]
- Newman WG, Hadfield KD, Latif A, Roberts SA, Shenton A, McHague C, Lalloo F, Howell S, Evans DG. Impaired tamoxifen metabolism reduces survival in familial breast cancer patients. Clin Cancer Res. 2008;14:5913–5918. doi: 10.1158/1078-0432.CCR-07-5235. [DOI] [PubMed] [Google Scholar]
- Desta Z, Ward BA, Soukhova NV, Flockhart DA. Comprehensive evaluation of tamoxifen sequential biotransformation by the human cytochrome P450 system in vitro: prominent roles for CYP3A and CYP2D6. J Pharmacol Exp Ther. 2004;310:1062–1075. doi: 10.1124/jpet.104.065607. [DOI] [PubMed] [Google Scholar]
- Hoskins JM, Carey LA, McLeod HL. CYP2D6 and tamoxifen: DNA matters in breast cancer. Nat Rev Cancer. 2009;9:576–586. doi: 10.1038/nrc2683. [DOI] [PubMed] [Google Scholar]
- Borges S, Desta Z, Li L, Skaar TC, Ward BA, Nguyen A, Jin Y, Storniolo AM, Nikoloff DM, Wu L, Hillman G, Hayes DF, Stearns V, Flockhart DA. Quantitative effect of CYP2D6 genotype and inhibitors on tamoxifen metabolism: implication for optimization of breast cancer treatment. Clin Pharmacol Ther. 2006;80:61–74. doi: 10.1016/j.clpt.2006.03.013. [DOI] [PubMed] [Google Scholar]
- Goetz MP, Knox SK, Suman VJ, Rae JM, Safgren SL, Ames MM, Visscher DW, Reynolds C, Couch FJ, Lingle WL, Weinshilboum RM, Fritcher EG, Nibbe AM, Desta Z, Nguyen A, Flockhart DA, Perez EA, Ingle JN. The impact of cytochrome P450 2D6 metabolism in women receiving adjuvant tamoxifen. Breast Cancer Res Treat. 2007;101:113–121. doi: 10.1007/s10549-006-9428-0. [DOI] [PubMed] [Google Scholar]
- Lee HS, Goh BC, Fan L, Khoo YM, Wang L, Lim R, Ong AB, Chua C. Phenotyping CYP3A using midazolam in cancer and non-cancer Asian patients. Br J Clin Pharmacol. 2003;55:270–277. doi: 10.1046/j.1365-2125.2003.01767.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharasch ED, Jubert C, Senn T, Bowdle TA, Thummel KE. Intraindividual variability in male hepatic CYP3A4 activity assessed by alfentanil and midazolam clearance. J Clin Pharmacol. 1999;39:664–669. doi: 10.1177/00912709922008290. [DOI] [PubMed] [Google Scholar]
- Binkhorst L, van Gelder T, Mathijssen RH. Individualization of tamoxifen treatment for breast carcinoma. Clin Pharmacol Ther. 2012;92:431–433. doi: 10.1038/clpt.2012.94. [DOI] [PubMed] [Google Scholar]
- Irvin WJ, Jr, Walko CM, Weck KE, Ibrahim JG, Chiu WK, Dees EC, Moore SG, Olajide OA, Graham ML, Canale ST, Raab RE, Corso SW, Peppercorn JM, Anderson SM, Friedman KJ, Ogburn ET, Desta Z, Flockhart DA, McLeod HL, Evans JP, Carey LA. Genotype-guided tamoxifen dosing increases active metabolite exposure in women with reduced CYP2D6 metabolism: a multicenter study. J Clin Oncol. 2011;29:3232–3239. doi: 10.1200/JCO.2010.31.4427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brauch H, Schroth W, Goetz MP, Murdter TE, Winter S, Ingle JN, Schwab M, Eichelbaum M. Tamoxifen use in postmenopausal breast cancer: CYP2D6 matters. J Clin Oncol. 2013;31:176–180. doi: 10.1200/JCO.2012.44.6625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rae JM, Drury S, Hayes DF, Stearns V, Thibert JN, Haynes BP, Salter J, Sestak I, Cuzick J, Dowsett M. CYP2D6 and UGT2B7 genotype and risk of recurrence in tamoxifen-treated breast cancer patients. J Natl Cancer Inst. 2012;104:452–460. doi: 10.1093/jnci/djs126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regan MM, Leyland-Jones B, Bouzyk M, Pagani O, Tang W, Kammler R, Dell’orto P, Biasi MO, Thurlimann B, Lyng MB, Ditzel HJ, Neven P, Debled M, Maibach R, Price KN, Gelber RD, Coates AS, Goldhirsch A, Rae JM, Viale G. CYP2D6 genotype and tamoxifen response in postmenopausal women with endocrine-responsive breast cancer: the breast international group 1-98 trial. J Natl Cancer Inst. 2012;104:441–451. doi: 10.1093/jnci/djs125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rae JM, Regan M, Leyland-Jones B, Hayes DF, Dowsett M. CYP2D6 genotype should not be used for deciding about tamoxifen therapy in postmenopausal breast cancer. J Clin Oncol. 2013;31:2753–2755. doi: 10.1200/JCO.2013.49.4278. [DOI] [PubMed] [Google Scholar]
- Rae JM, Regan MM, Thibert JN, Gersch C, Thomas D, Leyland-Jones B, Viale G, Pusztai L, Hayes DF, Skaar T, Van PC. Concordance between CYP2D6 genotypes obtained from tumor-derived and germline DNA. J Natl Cancer Inst. 2013;19:1332–1334. doi: 10.1093/jnci/djt204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacqz-Aigrain E, Funck-Brentano C, Cresteil T. CYP2D6- and CYP3A-dependent metabolism of dextromethorphan in humans. Pharmacogenetics. 1993;3:197–204. doi: 10.1097/00008571-199308000-00004. [DOI] [PubMed] [Google Scholar]
- Yu A, Haining RL. Comparative contribution to dextromethorphan metabolism by cytochrome P450 isoforms in vitro: can dextromethorphan be used as a dual probe for both CTP2D6 and CYP3A activities? Drug Metab Dispos. 2001;29:1514–1520. [PubMed] [Google Scholar]
- Jones DR, Gorski JC, Hamman MA, Hall SD. Quantification of dextromethorphan and metabolites: a dual phenotypic marker for cytochrome P450 3A4/5 and 2D6 activity. J Chromatogr B Biomed Appl. 1996;678:105–111. doi: 10.1016/0378-4347(95)00431-9. [DOI] [PubMed] [Google Scholar]
- Wieling J, Tamminga WJ, Sakiman EP, Oosterhuis B, Wemer J, Jonkman JH. Evaluation of analytical and clinical performance of a dual-probe phenotyping method for CYP2D6 polymorphism and CYP3A4 activity screening. Ther Drug Monit. 2000;22:486–496. doi: 10.1097/00007691-200008000-00020. [DOI] [PubMed] [Google Scholar]
- Min DI, Ku YM, Vichiendilokkul A, Fleckenstein LL. A urine metabolic ratio of dextromethorphan and 3-methoxymorphinan as a probe for CYP3A activity and prediction of cyclosporine clearance in healthy volunteers. Pharmacotherapy. 1999;19:753–759. doi: 10.1592/phco.19.9.753.31536. [DOI] [PubMed] [Google Scholar]
- Ducharme J, Abdullah S, Wainer IW. Dextromethorphan as an in vivo probe for the simultaneous determination of CYP2D6 and CYP3A activity. J Chromatogr B Biomed Appl. 1996;678:113–128. doi: 10.1016/0378-4347(95)00574-9. [DOI] [PubMed] [Google Scholar]
- Funck-Brentano C, Boelle PY, Verstuyft C, Bornert C, Becquemont L, Poirier JM. Measurement of CYP2D6 and CYP3A4 activity in vivo with dextromethorphan: sources of variability and predictors of adverse effects in 419 healthy subjects. Eur J Clin Pharmacol. 2005;61:821–829. doi: 10.1007/s00228-005-0051-5. [DOI] [PubMed] [Google Scholar]
- de Graan AJ, Teunissen SF, de Vos FY, Loos WJ, van Schaik RH, de Jongh FE, de Vos AI, van Alphen RJ, van der Holt B, Verweij J, Seynaeve C, Beijnen JH, Mathijssen RH. Dextromethorphan as a phenotyping test to predict endoxifen exposure in patients on tamoxifen treatment. J Clin Oncol. 2011;29:3240–3246. doi: 10.1200/JCO.2010.32.9839. [DOI] [PubMed] [Google Scholar]
- Hutson PR, Love RR, Havighurst TC, Rogers E, Cleary JF. Effect of exemestane on tamoxifen pharmacokinetics in postmenopausal women treated for breast cancer. Clin Cancer Res. 2005;11:8722–8727. doi: 10.1158/1078-0432.CCR-05-0915. [DOI] [PubMed] [Google Scholar]
- Abduljalil K, Frank D, Gaedigk A, Klaassen T, Tomalik-Scharte D, Jetter A, Jaehde U, Kirchheiner J, Fuhr U. Assessment of activity levels for CYP2D6*1, CYP2D6*2, and CYP2D6*41 genes by population pharmacokinetics of dextromethorphan. Clin Pharmacol Ther. 2010;88:643–651. doi: 10.1038/clpt.2010.137. [DOI] [PubMed] [Google Scholar]
- Moghadamnia AA, Rostami-Hodjegan A, Bdul-Manap R, Wright CE, Morice AH, Tucker GT. Physiologically based modelling of inhibition of metabolism and assessment of the relative potency of drug and metabolite: dextromethorphan vs. dextrorphan using quinidine inhibition. Br J Clin Pharmacol. 2003;56:57–67. doi: 10.1046/j.1365-2125.2003.01853.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loos WJ, de Graan AJ, de Bruijn P, van Schaik RH, van Fessem MA, Lam MH, Mathijssen RH, Wiemer EA. Simultaneous quantification of dextromethorphan and its metabolites dextrorphan, 3-methoxymorphinan and 3-hydroxymorphinan in human plasma by ultra performance liquid chromatography/tandem triple-quadrupole mass spectrometry. J Pharm Biomed Anal. 2011;54:387–394. doi: 10.1016/j.jpba.2010.08.033. [DOI] [PubMed] [Google Scholar]
- Teunissen SF, Jager NG, Rosing H, Schinkel AH, Schellens JH, Beijnen JH. Development and validation of a quantitative assay for the determination of tamoxifen and its five main phase I metabolites in human serum using liquid chromatography coupled with tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2011;879:1677–1685. doi: 10.1016/j.jchromb.2011.04.011. [DOI] [PubMed] [Google Scholar]
- Keizer RJ, van Benten M, Beijnen JH, Schellens JH, Huitema AD. Pirana and PCluster: a modeling environment and cluster infrastructure for NONMEM. Comput Methods Programs Biomed. 2011;101:72–79. doi: 10.1016/j.cmpb.2010.04.018. [DOI] [PubMed] [Google Scholar]
- Bergstrand M, Hooker AC, Wallin JE, Karlsson MO. Prediction-corrected visual predictive checks for diagnosing nonlinear mixed-effects models. AAPS J. 2011;13:143–151. doi: 10.1208/s12248-011-9255-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comets E, Brendel K, Mentre F. Computing normalised prediction distribution errors to evaluate nonlinear mixed-effect models: the npde add-on package for R. Comput Methods Programs Biomed. 2008;90:154–166. doi: 10.1016/j.cmpb.2007.12.002. [DOI] [PubMed] [Google Scholar]
- Jonsson EN, Karlsson MO. Xpose – an S-PLUS based population pharmacokinetic/pharmacodynamic model building aid for NONMEM. Comput Methods Programs Biomed. 1999;58:51–64. doi: 10.1016/s0169-2607(98)00067-4. [DOI] [PubMed] [Google Scholar]
- Lindbom L, Ribbing J, Jonsson EN. Perl-speaks-NONMEM (PsN) – a Perl module for NONMEM related programming. Comput Methods Programs Biomed. 2004;75:85–94. doi: 10.1016/j.cmpb.2003.11.003. [DOI] [PubMed] [Google Scholar]
- Levi M, Dempsey DA, Benowitz NL, Sheiner LB. Population pharmacokinetics of nicotine and its metabolites I. Model development. J Pharmacokinet Pharmacodyn. 2007;34:5–21. doi: 10.1007/s10928-006-9027-z. [DOI] [PubMed] [Google Scholar]
- Levi M, Dempsey DA, Benowitz NL, Sheiner LB. Prediction methods for nicotine clearance using cotinine and 3-hydroxy-cotinine spot saliva samples II. Model application. J Pharmacokinet Pharmacodyn. 2007;34:23–34. doi: 10.1007/s10928-006-9026-0. [DOI] [PubMed] [Google Scholar]
- Hendrickson HP, Gurley BJ, Wessinger WD. Determination of dextromethorphan and its metabolites in rat serum by liquid-liquid extraction and liquid chromatography with fluorescence detection. J Chromatogr B Analyt Technol Biomed Life Sci. 2003;788:261–268. doi: 10.1016/s1570-0232(02)01042-5. [DOI] [PubMed] [Google Scholar]
- Shivva V, Korell J, Tucker IG, Duffull SB. An approach for identifiability of population pharmacokinetic–pharmacodynamic models. CPT Pharmacometr Syst Pharmacol. 2013;19:e49. doi: 10.1038/psp.2013.25. doi: 10.1038/psp.2013.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans ND, Godfrey KR, Chapman MJ, Chappell MJ, Aarons L, Duffull SB. An identifiability analysis of a parent-metabolite pharmacokinetic model for ivabradine. J Pharmacokinet Pharmacodyn. 2001;28:93–105. doi: 10.1023/a:1011521819898. [DOI] [PubMed] [Google Scholar]
- Ahmad A, Shahabuddin S, Sheikh S, Kale P, Krishnappa M, Rane RC, Ahmad I. Endoxifen, a new cornerstone of breast cancer therapy: demonstration of safety, tolerability, and systemic bioavailability in healthy human subjects. Clin Pharmacol Ther. 2010;88:814–817. doi: 10.1038/clpt.2010.196. [DOI] [PubMed] [Google Scholar]
- Opdam FL, Dezentje VO, den Hartigh J, Modak AS, Vree R, Batman E, Smorenburg CH, Nortier JW, Gelderblom H, Guchelaar HJ. The use of the (13)C-dextromethorphan breath test for phenotyping CYP2D6 in breast cancer patients using tamoxifen: association with CYP2D6 genotype and serum endoxifen levels. Cancer Chemother Pharmacol. 2013;71:593–601. doi: 10.1007/s00280-012-2034-4. [DOI] [PubMed] [Google Scholar]
- Paine MF, Hart HL, Ludington SS, Haining RL, Rettie AE, Zeldin DC. The human intestinal cytochrome P450 ‘pie’. Drug Metab Dispos. 2006;34:880–886. doi: 10.1124/dmd.105.008672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickschen K, Willmann S, Thelen K, Lippert J, Hempel G, Eissing T. Physiologically based pharmacokinetic modeling of tamoxifen and its metabolites in women of different CYP2D6 phenotypes provides new insight into the tamoxifen mass balance. Front Pharmacol. 2012;3:1–15. doi: 10.3389/fphar.2012.00092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lien EA, Anker G, Ueland PM. Pharmacokinetics of tamoxifen in premenopausal and postmenopausal women with breast cancer. J Steroid Biochem Mol Biol. 1995;55:229–231. doi: 10.1016/0960-0760(95)00169-z. [DOI] [PubMed] [Google Scholar]