

Abstract
Aims
To examine whether initiation of fibrates or statins in sulfonylurea users is associated with hypoglycaemia, and examine in vitro inhibition of cytochrome P450 (CYP) enzymes by statins, fenofibrate and glipizide.
Methods
We used healthcare data to conduct nested case-control studies of serious hypoglycaemia (i.e. resulting in hospital admission or emergency department treatment) in persons taking glipizide or glyburide, and calculated adjusted overall and time-stratified odds ratios (ORs) and 95% confidence intervals (CIs). We also characterized the in vitro inhibition of CYP enzymes by statins, fenofibrate and glipizide using fluorometric CYP450 inhibition assays, and estimated area under the concentration–time curve ratios (AUCRs) for the drug pairs.
Results
We found elevated adjusted overall ORs for glyburide-fenofibrate (OR 1.84, 95% CI 1.37, 2.47) and glyburide-gemfibrozil (OR 1.57, 95% CI 1.25, 1.96). The apparent risk did decline over time as might be expected from a pharmacokinetic mechanism. Fenofibrate was a potent in vitro inhibitor of CYP2C19 (IC50 = 0.2 μm) and CYP2B6 (IC50 = 0.7 μm) and a moderate inhibitor of CYP2C9 (IC50 = 9.7 μm). The predicted CYP-based AUCRs for fenofibrate-glyburide and gemfibrozil-glyburide interactions were only 1.09 and 1.04, suggesting that CYP inhibition is unlikely to explain such an interaction.
Conclusions
Use of fenofibrate or gemfibrozil together with glyburide was associated with elevated overall risks of serious hypoglycaemia. CYP inhibition seems unlikely to explain this observation. We speculate that a pharmacodynamic effect of fibrates (e.g. activate peroxisome proliferator-activator receptor alpha) may contribute to these apparent interactions.
Keywords: cytochrome P-450 enzyme, drug–drug interactions, fibric acids, hypoglycaemia, statins, sulfonylurea compounds
What is already known about this subject —
Drug–drug interactions involving sulfonylurea antidiabetic drugs can lead to severe hypoglycaemia, which can be life-threatening and increase the risk of dementia.
Sulfonyureas are commonly used together with fibrate and statin lipid lowering drugs.
What this study adds —
Use of fenofibrate or gemfibrozil is associated with an increased risk of serious hypoglycaemia in patients taking glyburide.
Because of the prolonged time course of the interaction and lack a compelling CYP-based mechanism, we speculate that a pharmacodynamic mechanism may be responsible for the observed increase in risk.
Introduction
Sulfonylurea antidiabetic agents including glipizide and glyburide are used by 30% of US Medicare beneficiaries with diabetes, second only to metformin 1. Users of sulfonylureas experience severe hypoglycaemia at a rate of about 2 per 100 person-years 2. Hypoglycaemia can be serious. The estimated incidence of death from hypoglycaemia in users of sulfonylureas is 43 per 100 000 person-years 3, and hypoglycaemic episodes increase the risk of dementia 4. Drug interactions are a clinically important cause of severe hypoglycaemia. For example, Juurlink et al. found that use of trimethoprim/sulfamethoxazole was associated with a 6.6-fold risk of severe hypoglycaemia in older adults using glyburide 5, and Schelleman et al. found that several anti-infectives were associated with elevated risks of hypoglycaemia in patients receiving sulfonylureas 2.
Many important drug interactions are caused by inhibition of cytochrome P450 (CYP) metabolic enzymes or drug transporters. Both glyburide and glipizide are almost completely metabolized 6. While the metabolism of glipizide has not been well characterized, the CYP enzymes responsible for the metabolism of glyburide are CYP3A (54%), CYP2C9 (30%), CYP2C19 (8%) and CYP2C8 (7%) 7.
Given the high frequency with which hypercholesterolaemia and diabetes co-occur, fibrates and statins are often taken concomitantly with sulfonylureas. For example, in the 2010 National Ambulatory Medical Care Survey 8, 56% of sulfonylurea prescriptions were accompanied by a fibrate or statin. Gemfibrozil is a potent inhibitor of CYP2C9 with a Ki of 5.8 μm 9 and a mild inhibitor of CYP2C19, CYP1A2 and CYP2C8 with Kis of 24 μm, 82 μm and 30.4 μm, respectively 10. Gemfibrozil did not show any notable inhibitory effect on CYP3A4 or CYP2D6 9, and it is not clear whether it inhibits CYP2B6. Gemfibrozil 1-O-β-glucuronide, a metabolite of gemfibrozil, is a potent, irreversible inhibitor of CYP2C8 with a KI of 20 to 52 μm and a kinact of 0.21 min−1 11. The CYP inhibition profile of fenofibrate has not been fully characterized. Although the CYP inhibitory potential of individual statins has been reported in separate studies 12–14, no study has compared their inhibition using a consistent method. Facts and Comparisons lists potential interaction between sulfonylureas (as a group) and gemfibrozil as ‘suspected’ 15. There is one published case report (with positive de-challenge and re-challenge) of hypoglycaemia following initiation of gemfibrozil in a woman receiving glyburide 16. Facts and Comparisons does not list sulfonylureas as potentially interacting with fenofibrate or statins 16.
We therefore sought to examine in a pharmacoepidemiologic study whether the initiation of commonly used fibrates or statins in patients receiving sulfonylureas is associated with severe hypoglycaemia in clinical settings, and examine the time course of the associations. Further, to investigate potential mechanisms, we sought to characterize the in vitro inhibition of major CYP enzymes by fenofibrate and statins. We did not study CYP inhibition by gemfibrozil because it has been studied extensively 9,11,17,18. Finally, because the interactions of glipizide with the cytochrome P450 system have not been fully characterized, we wished to examine glipizide’s propensity to inhibit CYP enzymes in vitro to provide clues about its metabolism.
Methods
Pharmacoepidemiologic studies
Design and setting
We performed two case-control studies nested within the Medicaid populations of California, Florida, New York, Ohio and Pennsylvania using data from 1999 to 2005. We obtained Medicare data for persons co-enrolled in Medicare to ensure complete capture of outcomes. A prior publication has reported on this study’s design and results regarding anti-infective agents 2. A schematic of the study is presented in Figure 1. The pharmacoepidemiologic studies were approved by the University of Pennsylvania’s Institutional Review Board.
Figure 1.
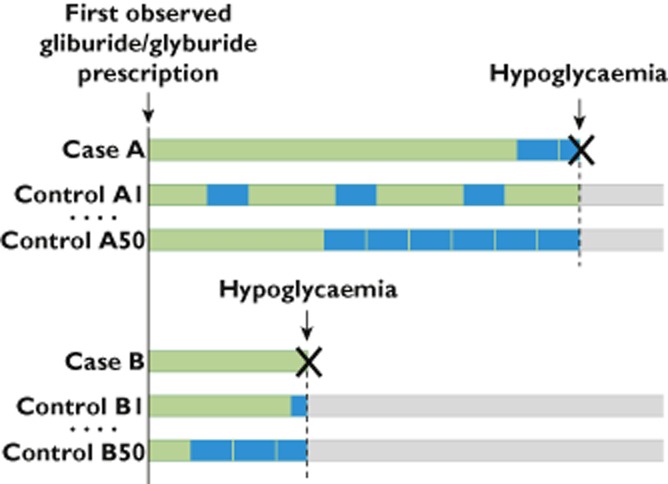
Schematic diagram of pharmacoepidemiologic studies.  , exposed to glipizide/glyburide;
, exposed to glipizide/glyburide;  , exposed to glipizide/glyburide plus fibrate/statin
, exposed to glipizide/glyburide plus fibrate/statin
Eligible person-time
All person-time exposed to glipizide or glyburide was included for all enrollees 18 years and older. We assumed that the duration of a prescription was 30 days because Medicaid prescriptions in our study states are generally dispensed in 30 day increments. Observation for one prescription was truncated when a consecutive prescription for the same study drug was dispensed. The observation period ended with the earliest of hospitalization or emergency department (ED) visit for hypoglycaemia, presumed end date of last glipizide or glyburide prescription, gap of 180 days between consecutive study prescriptions, switching between glipizide and glyburide, discontinuation of Medicaid eligibility or December 21 2005. Because we wished to study initiation of a fibrate or statin in patients already receiving a sulfonylurea, we excluded subjects in whom a fibrate or statin was dispensed on the day of or in the 90 days prior to first sulfonylurea prescription for that patient.
Identification of cases and controls
Cases were current recipients of glipizide or glyburide who were hospitalized or treated in an ED for hypoglycaemia. Cases were identified using Ginde and colleagues’ algorithm 19, which has a positive predicted value of 78% in our study population 2. The index date was the date of hospital admission or ED visit. Fifty controls without hypoglycaemia were selected at random for each case, matching on index date and state, using risk set sampling.
Exposure to fibrates or statins
Because of insufficient data to examine three drug interactions, we excluded cases and controls exposed to >1 precipitant drug on the index date. Cases and controls were considered exposed if a fibrate/statin was dispensed 1–30 days before the index date. If there were fewer than 30 exposed cases, the fibrate/statin was not examined to avoid statistically unstable estimates. In addition, to examine the time course of interactions, we calculated time since initiation of the fibrate or statin, classified as 0 to 29, 30 to 59 or 60 to 119 days. We examined pravastatin as a negative control drug since it does not inhibit CYP enzymes 20. Time windows with fewer than five exposed cases were not examined to avoid small cells.
Statistical analysis
Conditional logistic regression was used to calculate matched odds ratios (ORs) and 95% confidence intervals (CIs). In the minimally-adjusted models, age, gender, race and number of prior glipizide or glyburide prescriptions were included in the model. Potential confounding factors (demographic factors, chronic diseases, current use of drugs) were ascertained on the index date and are listed in Supplementary Table S1. Each factor that changed an OR of interest by ≥5% was included in the fully-adjusted model. To adjust for multiple comparisons, we multiplied the P values for the overall associations by the number of overall associations reported 21.
In vitro studies
Design
We used commercially available in vitro CYP enzyme inhibition assays and predicted the clinical relevance of inhibitions of CYPs by drugs used clinically to treat hyperlipidaemia or diabetes.
Chemicals and reagents
All drugs and chemicals were purchased from Toronto Research Chemicals Inc. (North York, ON, Canada). The fluorometric cytochrome P450 inhibition kits for CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6 and 3A4 were purchased from BD Biosciences (San Jose, CA, USA). Corning™ black 96-well polypropylene assay plates were purchased from Fisher Scientific (Pittsburgh, PA, USA).
Inhibition of human CYP isoforms
The half-maximal inhibitory concentrations (IC50s) of fenofibrate, statins (atorvastatin, lovastatin, pravastatin, simvastatin and simvastatin acid, the active form of simvastatin) and glipizide for recombinant human CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A4 were determined using fluorometric CYP450 inhibition assays. The assays were conducted following the manufacturer’s protocols. Briefly, the drugs were dissolved in methanol or acetonitrile. In 96-well assay plates, the drugs were diluted to a series of concentrations in a solution containing cofactors including nicotinamide adenine dinucleotide phosphate (NADP+, final concentration 1.3 mm), MgCL2 (final concentration 3.3 mm), glucose-6-phosphate (G6P, final concentration 3.3 mm) and glucose 6-phosphate dehydrogenase (final concentration 0.4 U ml−1). The mixture was pre-incubated at 37°C for 10 min. The enzymes and fluorogenic substrates were diluted to desired concentrations in sodium phosphate reaction buffer (pH 7.4, final concentration 200 mm) and mixed. Reactions were initiated with addition of the enzyme and substrate mixture to the cofactor and drug mixture. The final reaction volume of all assays was 200 μl. After incubating at 37°C for a pre-specified period of time (15 to 45 min), the reactions were stopped with addition of 75 μl quenching solution (0.5 m Tris base or 2n NaOH). Fluorescence was determined using a BioTek Synergy 2 (Winooski, VT, USA) fluorescence reader. Each of the drugs was tested at eight concentrations in duplicate. The final concentrations of fenofibrate, glipizide and pravastatin in all the assays were 0.46 μm, 1.37 μm, 4.1 μm, 12.3 μm, 37.0 μm, 111.1 μm, 333.3 μm and 1000 μm. The final concentrations of simvastatin, simvastatin acid, lovastatin and atorvastatin in the assay of CYP2B6 were 0.022 μm, 0.069 μm, 0.21 μm, 0.62 μm, 1.85 μm, 5.56 μm, 16.7 μm and 50 μm. In the assay of CYP3A4, the final concentrations of atorvastatin were 0.09 μm, 0.27 μm, 0.82 μm, 2.5 μm, 7.4 μm, 22.2 μm, 66.7 μm and 200 μm, and those of lovastatin were 0.23 μm, 0.69 μm, 2.1 μm, 6.2 μm, 18.5 μm, 55.6 μm, 166.7 μm and 500 μm. The final concentrations of simvastatin, simvastatin acid, lovastatin and atorvastatin in all the other assays were 0.05 μm, 0.14 μm, 0.41 μm, 1.2 μm, 3.7 μm, 11.1 μm, 33.3 μm and 100 μm. The final concentrations were limited by both the drug’s aqueous solubility and the enzyme’s tolerance to the organic solvent in which the drug was dissolved. To estimate IC50s, percent of inhibition was calculated using net fluorescence that was corrected for the background. The values of percent of inhibition were then fitted to a three or four parameter log-logistic model.
Estimation of area under the curve (AUC) ratios for individual inhibitor–pathway pairs
For each inhibitor–pathway pair for which an exact IC50 value was observed in our experiments, and for gemfibrozil inhibition of CYP1A2, CYP2C9 and CYP2C19, for which dissociation constants have been reported elsewhere, we estimated the area under the curve ratio (AUCR), AUCi: AUC, where AUCi is the area under a concentration–time curve of a known substrate of that pathway in the presence of an inhibitor and AUC is the area under the curve in the absence of any inhibitor. AUCR is a quantitative measure that has been widely used to assess the potential of a drug to cause undesirable clinical drug–drug interactions (DDIs) by inhibiting drug- metabolizing enzymes or drug transporters. The rationale is that if a drug inhibits a drug metabolizing enzyme, the hepatic clearance of a concomitant drug would decrease if it is metabolized by the inhibited enzyme. Consequently, the systemic exposure to the concomitant drug would increase, which may induce undesirable toxic effects. An AUCR predicts the change in the systemic exposure to the concomitant drug in the presence of an inhibitor, and thus reflects the likelihood that a DDI would occur clinically 22.
A variety of mathematical models have been developed to predict AUCR. Following the FDA recommended strategy for assessing the risk of DDIs 8, we first used the most widely used model that estimates AUCR as 1 + [I]unbound /Ki,unbound, where [I]unbound is the unbound hepatic input concentration and Ki,unbound is the unbound dissociation constant of an inhibitor drug. This model requires a number of assumptions that include reversible inhibition, a constant in vivo inhibitor concentration, and that the hypothetical object drug is cleared exclusively by a single metabolic pathway that is affected by the inhibitor 23. [I]unbound was approximated by the unbound peak plasma concentration (Cmax,unbound), which was calculated as the peak total plasma concentration (Cmax) × the fraction unbound in plasma (fu), with Cmax and fu obtained from a standard 24. Ki,unbound was estimated by Ki,unbound = fu,inc × Ki, where fu,inc is the fraction of unbound inhibitor drug in in vitro incubation. The Ki values of gemfibrozil for 2C9, 2C19 and 1A2 that we used were obtained from the literature 9. The Kis for other drugs were estimated by Ki = IC50/(1 + [S]/Km), assuming reversible inhibition, where [S] and Km are the concentration and Michaelis constant of the substrates tested. The fu,inc of the drugs was predicted using the Hallifax–Houston model 25. Consistent with the recommended FDA guidelines for inhibitory drug interactions, inhibitor–pathway pairs with an AUCi: AUC of >1 and <1.25 were interpreted as ignorable, those with 1.25 ≤ AUCi: AUC <2 were considered to be clinically weak interactions, those with 2 ≤ AUCi: AUC <5 as representing moderate inhibitory interactions and those with AUCi: AUC ≥5 as representing clinically strong inhibitory interactions 8.
Estimation of overall AUCR
Because of the assumption that a hypothetical object drug is exclusively metabolized by inhibited pathway, an AUCR estimate reflects only the inhibition of a single pathway and is unspecific with interacting drugs. To account for the simultaneous inhibition of CYP3A4, CYP2C9, CYP2C19 and CYP2C8 by fenofibrate in the context of glyburide metabolism, an overall AUCR was predicted using the following equation 26:
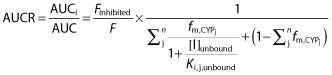 |
where [I] is the unbound plasma concentration of fenofibrate, Ki,j,unbound is the unbound dissociation constant of the jth inhibited pathway, fm,CYPj is the fraction of substrate drug metabolism by the jth inhibited pathway and was reported by Zharikova et al. (54%, 30%, 8% and 7% for CYP3A, CYP2C9, CYP2C19 and CYP2C8, respectively) 7, 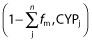 represents clearance via other CYP enzymes and/or renal clearance, F is bioavailability in the presence (denoted by Finhibited) or absence of an inhibitor. It was assumed that fenofibrate does not affect the bioavailability of glyburide due to intestinal metabolism, based on the fact that CYP3A accounts for ∼80% of the CYP450s in the gastrointestinal tract and fenofibrate is only weak inhibitor of CYP3A4. Finhibited/F was therefore conservatively assumed to be 1.
represents clearance via other CYP enzymes and/or renal clearance, F is bioavailability in the presence (denoted by Finhibited) or absence of an inhibitor. It was assumed that fenofibrate does not affect the bioavailability of glyburide due to intestinal metabolism, based on the fact that CYP3A accounts for ∼80% of the CYP450s in the gastrointestinal tract and fenofibrate is only weak inhibitor of CYP3A4. Finhibited/F was therefore conservatively assumed to be 1. 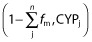 is ignorable because glyburide is almost completely metabolized. Similarly, an overall AUCR was also estimated for glyburide metabolism to account for the simultaneous inhibition of CYP2C19 and CYP2C9 by gemfibrozil.
is ignorable because glyburide is almost completely metabolized. Similarly, an overall AUCR was also estimated for glyburide metabolism to account for the simultaneous inhibition of CYP2C19 and CYP2C9 by gemfibrozil.
Results
Pharmacoepidemiologic studies
The glipizide cohort was 66% female and the median age was 64 years (interquartile range 52–74 years). The incidence rate of severe hypoglycaemia in the glipizide cohort was 2.02 per 100 person-years (95% CI: 1.97, 2.08). The glyburide cohort was 66% female and the median age was 65 years (interquartile range 53–74 years). The incidence rate of severe hypoglycaemia in the glyburide cohort was 2.47 per 100 person-years (95% CI 2.42, 2.53). Characteristics of cases and controls from the glipizide and glyburide cohorts are shown in Table 1. In the glipizide cohort, none of the adjusted overall ORs for current exposure were statistically elevated even without Bonferroni adjustment (Table 2). However, even though the overall OR for gemfibrozil-glipizide was not statistically elevated, time-stratified ORs were compatible with an elevated risk shortly after initiating gemfibrozil. In the glyburide cohort (Table 2), Bonferroni-adjusted statistically significant adjusted overall ORs were observed for fenofibrate (OR 1.84; 95% CI 1.37, 2.47; Bonferroni-adjusted P value 0.0006) and gemfibrozil (OR 1.57; 95% CI 1.25, 1.96; adjusted P value 0.0009). However, the time-stratified ORs did not start high and then decline as might be expected for a pharmacokinetic DDI.
Table 1.
Characteristics of cases and controls exposed to glipizide or glyburide on the index date in pharmacoepidemiologic studies
| Glipizide | Glyburide | |||||
|---|---|---|---|---|---|---|
| Cases | Controls | Matched OR (95% CI) | Cases | Controls | Matched OR (95% CI) | |
| Variable | n = 5,784 | N = 289,965 | n = 7,693 | n = 384,867 | ||
| Age | ||||||
| 18–50 years | 979 (16.9%) | 54 300 (18.7%) | Reference | 952 (12.4%) | 65 540 (17.0%) | Reference |
| 50–60 years | 937 (16.2%) | 52 753 (18.2%) | 0.98 (0.90, 1.08) | 1109 (14.4%) | 67 111 (17.4%) | 1.14 (1.04, 1.24) |
| 60–70 years | 1132 (19.6%) | 65 574 (22.6%) | 0.96 (0.88, 1.05) | 1576 (20.5%) | 90 223 (23.4%) | 1.21 (1.11, 1.31) |
| 70–80 years | 1437 (24.8%) | 68 226 (23.5%) | 1.17 (1.08, 1.27) | 2104 (27.4%) | 96 520 (25.1%) | 1.52 (1.40, 1.64) |
| ≥80 years | 1299 (22.5%) | 49 112 (16.9%) | 1.49 (1.37, 1.62) | 1952 (25.4%) | 65 473 (17.0%) | 2.10 (1.94, 2.27) |
| Gender, male | 1842 (31.8%) | 97 839 (33.7%) | 0.92 (0.87, 0.97) | 2430 (31.6%) | 129 812 (33.7%) | 0.91 (0.86, 0.95) |
| Race | ||||||
| Caucasian | 2102 (36.3%) | 118 569 (40.9%) | Reference | 2918 (37.9%) | 158 652 (41.2%) | Reference |
| African American | 1608 (27.8%) | 51 468 (17.8%) | 1.78 (1.67, 1.91) | 1716 (22.3%) | 59 034 (15.3%) | 1.60 (1.50, 1.70) |
| Hispanic | 1089 (18.8%) | 63 178 (21.8%) | 0.95 (0.88, 1.03) | 1530 (19.9%) | 85 525 (22.2%) | 0.96 (0.90, 1.03) |
| Other/unknown | 985 (17.0%) | 56 750 (19.6%) | 0.96 (0.88, 1.04) | 1529 (19.9%) | 81 656 (21.2%) | 1.00 (0.94, 1.07) |
| Number of prior glipizide prescriptions | ||||||
| 0 | 629 (10.9%) | 12 601 (4.4%) | 2.83 (2.60, 3.09) | 881 (11.4%) | 16 352 (4.2%) | 3.06 (2.84, 3.29) |
| 1 | 355 (6.1%) | 12 807 (4.4%) | 1.58 (1.42, 1.77) | 420 (5.5%) | 16 214 (4.2%) | 1.48 (1.34, 1.64) |
| 2 | 304 (5.3%) | 13 469 (4.6%) | 1.30 (1.15, 1.46) | 385 (5.0%) | 17 184 (4.5%) | 1.28 (1.15, 1.43) |
| ≥3 | 4496 (77.7%) | 251 088 (86.6%) | Reference | 6007 (78.1%) | 335 117 (87.1%) | Reference |
| Dementia | 1362 (23.6%) | 38 182 (13.2%) | 2.13 (2.00, 2.27) | 1824 (23.7%) | 46 473 (12.1%) | 2.37 (2.24, 2.50) |
| Kidney disease | 1275 (22.0%) | 24 716 (8.5%) | 3.10 (2.91, 3.31) | 1289 (16.8%) | 25 916 (6.7%) | 2.82 (2.65, 2.99) |
| Use of insulin | 1066 (18.4%) | 21 790 (7.5%) | 2.82 (2.63, 3.02) | 1024 (13.3%) | 25 798 (6.7%) | 2.15 (2.01, 2.30) |
| Fibrate | ||||||
| Fenofibrate | 20 (0.4%) | 1 353 (0.5%) | 0.74 (0.48, 1.15) | 47 (0.6%) | 1 772 (0.5%) | 1.33 (0.99, 1.78) |
| Gemfibrozil | 53 (0.9%) | 2 474 (0.8%) | 1.07 (0.82, 1.41) | 81 (1.0%) | 3 417 (0.9%) | 1.19 (0.95, 1.48) |
| Statin | ||||||
| Atorvastatin | 417 (7.2%) | 25 019 (8.6%) | 0.82 (0.74, 0.91) | 566 (7.4%) | 32 099 (8.3%) | 0.87 (0.80, 0.95) |
| Fluvastatin | 21 (0.4%) | 1 606 (0.6%) | 0.66 (0.43, 1.01) | 31 (0.4%) | 2 377 (0.6%) | 0.65 (0.46, 0.93) |
| Lovastatin | 27 (0.5%) | 1 714 (0.6%) | 0.78 (0.53, 1.15) | 36 (0.5%) | 2 288 (0.6%) | 0.78 (0.56, 1.09) |
| Simvastatin | 173 (3.0%) | 8 613 (3.0%) | 1.01 (0.86, 1.18) | 252 (3.3%) | 12 163 (3.2%) | 1.04 (0.91, 1.18) |
| Pravastatin | 91 (1.6%) | 5 226 (1.8%) | 0.87 (0.71, 1.07) | 134 (1.7%) | 8 337 (2.2%) | 0.80 (0.67, 0.95) |
Table 2.
Fully adjusted association between initiation of a fibrate or statin (exposed vs. unexposed) and the odds of hospitalization for hypoglycaemia in patients receiving glipizide or glyburide
| Glipizide users | Glyburide users | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Fibrate/statin | Overall adjusted odds ratio* (95% CI) | Bonferroni-corrected P value for overall association | Adjusted odds ratio* (95% CI) 0–29 days after fibrate/statin initiation | Adjusted odds ratio* (95% CI) 30–59 days after fibrate/statin initiation | Adjusted odds ratio* (95% CI) 60–119 days after fibrate/statin initiation | Fully adjusted odds ratio (95% CI)* ≥120 days after fibrate/statin initiation | Overall adjusted odds ratio* (95% CI) | Bonferroni-corrected P value for overall association | Adjusted odds ratio* (95% CI) 0–29 days after fibrate/statin initiation | Adjusted odds ratio* (95% CI) 30–59 days after fibrate/statin initiation | Adjusted odds ratio* (95% CI) 60–119 days after fibrate/statin initiation | Fully adjusted odds ratio* (95% CI) ≥ 120 days after fibrate/statin initiation |
| Fenofibrate | Insufficient data | – | Insufficient data | Insufficient data | Insufficient data | Insufficient data | 1.84 (1.37, 2.47) | 0.0006† | Insufficient data | 2.08 (0.91, 4.74) | 2.44 (1.29, 4.63) | 1.88 (1.29, 2.76) |
| Gemfibrozil | 1.32 (0.99–1.74) | 0.56 | 1.79 (1.02, 3.13) | 1.69 (0.79–3.60) | 0.71 (0.29, 1.71) | 1.29 (0.88, 1.89) | 1.57 (1.25, 1.96) | 0.0009† | 1.36 (0.80, 2.32) | 1.11 (0.49, 2.49) | 1.65 (0.97, 2.81) | 1.72 (1.28, 2.30) |
| Atorvastatin | 0.92 (0.83–1.02) | >0.99 | 0.97 (0.73, 1.29) | 0.87 (0.61, 1.24) | 1.02 (0.79, 1.32) | 0.89 (0.79, 1.01) | 1.01 (0.92, 1.10) | >0.99 | 1.25 (0.99, 1.57) | 1.09 (0.83, 1.45) | 1.01 (0.80, 1.28) | 0.96 (0.86, 1.07) |
| Fluvastatin | Insufficient data | – | Insufficient data | Insufficient data | Insufficient data | Insufficient data | 0.81 (0.57, 1.16) | >0.99 | 0.95 (0.39, 2.31) | Insufficient data | 1.17 (0.52, 2.63) | 0.76 (0.48, 1.21) |
| Lovastatin | Insufficient data | – | Insufficient data | Insufficient data | Insufficient data | Insufficient data | 0.95 (0.68, 1.33) | >0.99 | 0.97 (0.48, 1.96) | 0.98 (0.40, 2.39) | Insufficient data | 1.14 (0.73, 1.78) |
| Simvastatin | 1.10 (0.94–1.29) | >0.99 | 1.28 (0.87, 1.89) | 0.67 (0.36, 1.25) | 1.06 (0.70, 1.61) | 1.14 (0.94, 1.38) | 1.17 (1.03, 1.33) | 0.20 | 1.52 (1.13, 2.06) | 0.96 (0.61, 1.52) | 0.95 (0.65, 1.39) | 1.17 (1.00, 1.38) |
| Pravastatin | 0.97 (0.79–1.20) | >0.99 | 0.83 (0.44, 1.55) | 1.17 (0.62, 2.19) | 1.19 (0.70, 2.02) | 0.93 (0.71, 1.22) | 0.91 (0.77, 1.09) | >0.99 | 1.12 (0.72, 1.76) | 0.57 (0.27, 1.20) | 0.97 (0.61, 1.55) | 0.91 (0.73, 1.12) |
Matched on index date and state and adjusted for age, gender, race, number of prior glipizide or glyburide prescriptions, dementia, kidney disease, and use of insulin.
Indicates overall adjusted Bonferroni-corrected P value is less than 0.05. CI, confidence interval.
Inhibition of human cytochrome P450 isoforms
IC50s are shown in Table 3 and individual inhibition curves are presented in Figure 2. Surprisingly, we found that fenofibrate was a relatively potent inhibitor of CYP2B6 (IC50 ± SEM = 0.7 ± 0.2 μm) and CYP2C19 (IC50 = 0.2 ± 0.1 μm), which has not been reported previously. Fenofibrate was also a moderate inhibitor of CYP2C8 (IC50 = 4.8 ± 1.7 μm) and CYP2C9 (IC50 = 9.7 ± 40.7 μm) and a weak inhibitor of CYP3A4 (IC50 = 142.1 ± 114.3 μm). Glipizide was a mild inhibitor of CYP2C9 (IC50 = 24 ± 7.2 μm) and a weak inhibitor of CYP2C8 (IC50 = 338.2 ± 26.8 μm) and CYP3A4 (IC50 = 102 ± 33.7 μm). In general, the IC50s that we found for statins were consistent with those reported previously 12–14.
Table 3.
Half-maximal inhibitory concentrations (IC50; μm, ± SEM) of fenofibrate, selected statins, and glipizide for selected recombinant human cytochrome P450s
| 1A2 | 2B6 | 2C8 | 2C9 | 2C19 | 2D6 | 3A4 | |
|---|---|---|---|---|---|---|---|
| Fenofibrate | >1000 | 0.7 ± 0.2 | 4.8 ± 1.7 | 9.7 ± 40.7 | 0.2 ± 0.1 | >1000 | 142.1 ± 114.3 |
| Atorvastatin | >100 | >50 | 55.7 ± 19.6 | 7.3 ± 0.7 | 81.5 ± 14.9 | >100 | 5.2 ± 1.9 |
| Lovastatin | >100 | >50 | 27.5 ± 1.5 | 6.6 ± 0.6 | 32.7 ± 5.5 | 40 ± 22 | 23.4 ± 5.3 |
| Pravastatin | >1000 | >1000 | >1000 | >1000 | >1000 | >1000 | >1000 |
| Simvastatin | >100 | 46 | 28 ± 13.4 | 8.1 ± 1.3 | 55.8 ± 23.1 | 47 ± 5.7 | 4.8 ± 2.5 |
| Simvastatin acid | >100 | >50 | 76.5 ± 6.2 | 16.5 ± 6 | >100 | >100 | 42.6 ± 4.3 |
| Glipizide | >1000 | >1000 | 338.2 ± 26.8 | 24 ± 7.2 | >1000 | >1000 | 102 ± 33.7 |
Figure 2.
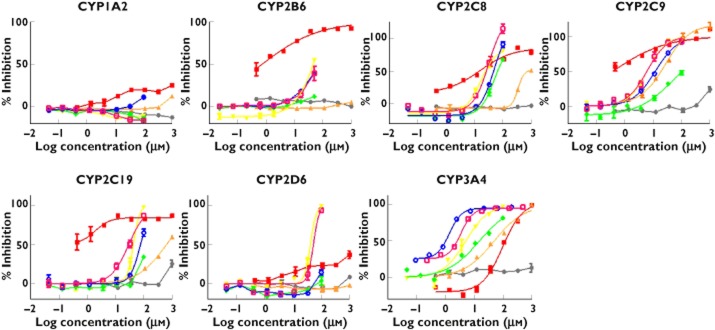
Degree of enzyme inhibition by log concentration of inhibitor found in in vitro experiments.  , pravastatin;
, pravastatin;  , fenofibrate;
, fenofibrate;  , glipizide;
, glipizide;  , simvastatin;
, simvastatin;  , simvastatin acid;
, simvastatin acid;  , atorvastatin;
, atorvastatin;  , lovastatin
, lovastatin
Predicted likelihood of clinically relevant interactions
The estimated AUCR based on the sensitive 1 + [I]unbound/Ki unbound method for fenofibrate was 2.98 for drugs metabolized by CYP2C19 and 1.54 for drugs metabolized by CYP2B6 (Table 4). These AUCRs suggest that drugs metabolized primarily by CYP2C19 or CYP2B6 may have clinically relevant interactions with fenofibrate, while the predicted risks of interactions for the remaining drug–pathway pairs are ignorable. These AUCRs apply only to inhibition of a single pathway and are non-specific with regard to substrate drugs.
Table 4.
Inhibitory constants, maximum serum concentrations and predicted area under the curve ratios of cytochrome P450 metabolic pathways by selected fibrates, statins and sulfonylureas
| Inhibitor | CYP pathway | Inhibitory constant (Ki, unbound) (μm) | Maximum serum concentration (Cmax, unbound) (μm) 18 | Area under the curve ratio (AUCR = AUCi:AUC) |
|---|---|---|---|---|
| Atorvastatin | 2C19 | 4.17 | 0.0005 | 1.00 |
| 2C8 | 2.65 | 1.00 | ||
| 2C9 | 0.35 | 1.00 | ||
| 3A4 | 0.63 | 1.00 | ||
| Fenofibrate | 2B6 | 0.44 | 0.2384 | 1.54 |
| 2C19 | 0.12 | 2.98 | ||
| 2C8 | 2.24 | 1.11 | ||
| 2C9 | 4.61 | 1.05 | ||
| 3A4 | 106.03 | 1.00 | ||
| Gemfibrozil | 1A2 | 98.64 | 0.6 | 1.01 |
| 2C19 | 17.41 | 1.03 | ||
| 2C9 | 4.21 | 1.14 | ||
| Glipizide | 2C8 | 130.47 | 0.0167 | 1.00 |
| 2C9 | 9.45 | 1.00 | ||
| 3A4 | 62.99 | 1.00 | ||
| Lovastatin | 2C19 | 16.30 | 0.0001 | 1.00 |
| 2C8 | 12.76 | 1.00 | ||
| 2C9 | 3.12 | 1.00 | ||
| 2D6 | 14.85 | 1.00 | ||
| 3A4 | 1.34 | 1.00 | ||
| Simvastatin | 2B6 | 28.15 | 0.0014 | 1.00 |
| 2C19 | 27.89 | 1.00 | ||
| 2C8 | 13.03 | 1.00 | ||
| 2C9 | 3.84 | 1.00 | ||
| 2D6 | 17.50 | 1.00 | ||
| 3A4 | 3.57 | 1.00 |
Note that AUCRs greater than 1.1 are in bold.
To assess the effect of the simultaneous inhibition of the pathways involved in glyburide metabolism by fenofibrate, we considered the contribution of various of CYP enzymes to glyburide metabolism. The CYP enzymes responsible for the metabolism of glyburide include CYP3A, CYP2C9, CYP2C19 and CYP2C8. The percentages contributed by these pathways are 54%, 30%, 8% and 7%, respectively 7. Given this, the AUCR for glyburide with vs. without fenofibrate was 1.09, suggesting that the CYP-based interaction between fenofibrate and glyburide is negligible. Similarly, the AUCR for glyburide with vs. without gemfibrozil was 1.04, also suggesting a negligible CYP-based interaction between gemfibrozil and glyburide.
Discussion
Among users of glyburide, we found an overall elevated risk of severe hypoglycaemia associated following initiation of fenofibrate (OR 1.94; 95% CI 1.37, 2.47; Bonferroni-corrected P value 0.0006) and gemfibrozil (OR 1.57; 95% CI 1.25, 1.96; Bonferoni-corrected P value 0.0009). Although prior studies have found that this risk is highest in the first 30 days of sulfonylurea therapy 27,28, the large majority of patients experiencing severe hypoglycaemia in our study and previous studies 27,28 had been receiving a sulfonylurea for more than 1 month. Glyburide is metabolized primarily by CYP3A and CYP2C9 29,30. We found that fenofibrate is a weak in vitro inhibitor of CYP3A4 and a moderate in vitro inhibitor of CYP2C9, while prior studies have found that gemfibrozil is a potent inhibitor of CYP2C9 9. The predicted AUC for glyburide was only 1.09-fold and 1.04-fold as high in the presence vs. absence of fenofibrate and gemfibrozil, respectively, which suggests an ignorable risk of a CYP-based interaction. This suggests that inhibition of CYPs is unlikely to be the mechanism underlying the observed increase in hypoglycaemia risk. However, our AUC predictions were based on in vitro data obtained using individual recombinant CYP enzymes and fluorescent probe substrates that might compromise the accuracy of prediction, and considered CYP inhibition in the liver only. Future studies using a more physiologically relevant system, including both human liver and intestinal microsomes, may provide a better understanding on the effects of fenofibrate and gemfibrozil on the metabolism of glyburide.
Interactions of glyburide with fenofibrate, gemfibrozil and simvastatin could also occur at the level of organic anion transporter polypeptides (OATPs) which have been suggested to play an important role in the pharmacokinetics of glyburide 31,32. Glyburide is a substrate of OATP2B1 33 and was able to inhibit OATP1B1-mediated uptake of pitavastatin in vitro 34, suggesting that it may also be a substrate of OATP1B1. The co-administration of rifampicin, a potent inhibitor of OATP1B1, 1A2 and 1B3 35, increased the AUC of glyburide by 2.2-fold in humans 31, suggesting an important role of OATPs in the disposition of glyburide. In baboons, OATPs were found to play a critical role in the hepatic uptake and metabolism of glyburide 32. Fenofibrate and gemfibrozil inhibited OATP1B1-mediated uptake of estradiol-17β-D-glucuronide with an IC50 of about 20 μm 36 and a Ki of 25 μm, respectively 36. Gemfibrozil also inhibits OATP2B1 and OATP1B3 37,38. Simvastatin acid, the active metabolite of simvastatin, is a known substrate of OATP1B1 38. It is possible that fenofibrate, gemfibrozil and simvastatin acid inhibit OATP-mediated hepatic uptake of glyburide, resulting in reduced hepatic clearance and thus higher systemic exposure to glyburide. This hypothesis, however, seems at odds with the absence of an observable interaction of glyburide with pravastatin, given that pravastatin is also known as a substrate of OATP1B1 39. Future in vitro studies using human hepatocytes are needed to evaluate further the effects of fenofibrate, gemfibrozil and simvastatin acid on the hepatic uptake of glyburide.
Although the metabolism of glipizide has not been fully characterized, CYP2C9 is believed to play a predominant role 40,41. OATP1B1 may also play a role in the hepatic uptake of glipizide, as glipizide inhibited OATP1B1-mediated uptake of rosuvastatin and estradiol-17β-D-glucuronide in vitro 42. Drug interactions with glyburide and glipizide might occur when the actions of CYP2C9 or OATP1B1 are altered by concomitant drugs.
The observation that the associations did not appear to decline over time may argue against a pharmacokinetic mechanism. Both fenofibrate and gemfibrozil activate peroxisome proliferator activator receptor α (PPARα), which can affect glucose metabolism 43. Fenofibrate has also been found in some, but not all, studies to reduce blood glucose, an effect that has been speculatively attributed to mechanisms including PPARα activation, reduction of triglycerides and stimulation of β-oxidation in skeletal muscles 44. These mechanisms might reduce glucose over months rather than days, and thus may be more consistent with the observed pharmacoepidemiologic results. Thus, pharmacodynamic mechanisms may provide a more plausible explanation than CYP inhibition for the associations that we observed.
We found that fenofibrate is a relatively potent in vitro inhibitor of CYP2B6 and CYP2C19, which has not been reported previously. The AUCR estimates for individual pathways suggest that fenofibrate may have clinically important interactions with drugs that are metabolized extensively by CYP2C19 (AUCR = 2.98) or CYP2B6 (AUCR = 1.54). However, we are unaware of reports of clinical drug interactions between fenofibrate and substrates of CYP2C19 and CYP2B6. This may be because the AUCR approach is likely to over-predict the risk of clinical DDIs, as pointed out by others 23. The lack of documented interactions may also be due to the lack of studies of such interactions. Future studies are needed to examine further the inhibitory effect of fenofibrate on the metabolism of CYP2C19 and CYP2B6.
A principal strength of these studies was coordination of pharmacoepidemiologic and in vitro research. This coordination permitted the elucidation of mechanisms of interactions suggested by the pharmacoepidemiologic data. Such a translational science model of using multiple research systems to study a given phenomenon from different perspectives will improve the study of DDIs 45 and other phenomena. Other strengths include the large sample size and use of validated outcomes in the pharmacoepidemiologic studies, and use of high throughput assays to evaluate inhibition of CYPs in a consistent way that allows the evaluation of a large number of possible mechanisms in an efficient manner.
Weaknesses include an insufficient number of events to evaluate some potential interactions of interest, the potential for residual confounding (e.g. patients receiving pravastatin may differ from patients receiving fibrates with respect to hypoglycaemia risk and therefore it may not be perfect as a control drug), the use of recombinant CYP enzymes that are artificial constructs rather than human liver microsomes, the use fluorogenic substrates rather than traditional probe substrates, and that AUCR was estimated using a static model. Further, Medicaid enrollees are more likely to be female and non-White than the general population. That said, the Medicaid population is a large and important population in its own right, and biological relationships identified in Medicaid enrollees have consistently been replicated in other populations. Future pharmacoepidemiologic studies should seek to confirm our findings.
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare that this study was supported by the National Institutes of Health (grants R01DK102694, R01AG025152, R01GM74217, UL1TR000003, U10HD063094, and T32GM008425) and the Agency for Healthcare Research and Quality (Grant R01HS019818-01). DAF has received research support for investigator-initiated studies from Pfizer Inc and Novartis and SH has consulted for Abbott Laboratories, Novartis Pharmaceuticals, Bayer, Astra-Zeneca and Bristol-Myers Squibb, received research support from Astra-Zeneca and Bristol-Myers Squibb, and received institutional support from Pfizer Inc. to support pharmacoepidemiology training. There are no other relationships or activities that could appear to have influenced the submitted work.
The authors thank Qufei Wu and Qing Liu for their programming and statistical analysis, and Maria Kalai for assistance with manuscript preparation.
Supporting Information
Additional Supporting Information may be found in the online version of this article at the publisher’s website:
Potential confounding factors that were considered in the pharmacoepidemiologic study
References
- Margolis DJ, Leonard CE, Razzaghi H, Hoffstad OJ, Freeman CP, de Nava KL, Molina T, Tan Y, Bartman BA. Utilization of Antidiabetic Drugs among Medicare Beneficiaries with Diabetes, 2006–2009: Data Points #9. Rockville, MD: Data Points Publication Series; 2011. [PubMed] [Google Scholar]
- Schelleman H, Bilker WB, Brensinger CM, Wan F, Hennessy S. Anti-infectives and the risk of severe hypoglycemia in users of glipizide or glyburide. Clin Pharmacol Ther. 2010;88:214–222. doi: 10.1038/clpt.2010.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrington WG, Levy JB. Metformin: effective and safe in renal disease? Int Urol Nephrol. 2008;40:411–417. doi: 10.1007/s11255-008-9371-6. [DOI] [PubMed] [Google Scholar]
- Whitmer RA, Karter AJ, Yaffe K, Quesenberry CP, Jr, Selby JV. Hypoglycemic episodes and risk of dementia in older patients with type 2 diabetes mellitus. JAMA. 2009;301:1565–1572. doi: 10.1001/jama.2009.460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juurlink DN, Mamdani M, Kopp A, Laupacis A, Redelmeier DA. Drug-drug interactions among elderly patients hospitalized for drug toxicity. JAMA. 2003;289:1652–1658. doi: 10.1001/jama.289.13.1652. [DOI] [PubMed] [Google Scholar]
- Balant L, Fabre J, Zahnd GR. Comparison of the pharmacokinetics of glipizide and glibenclamide in man. Eur J Clin Pharmacol. 1975;8:63–69. doi: 10.1007/BF00616416. [DOI] [PubMed] [Google Scholar]
- Zharikova OL, Fokina VM, Nanovskaya TN, Hill RA, Mattison DR, Hankins GD, Ahmed MS. Identification of the major human hepatic and placental enzymes responsible for the biotransformation of glyburide. Biochem Pharmacol. 2009;78:1483–1490. doi: 10.1016/j.bcp.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Food and Drug Administration. 2012. pp. 1–75. Drug interaction studies – study design, data analysis, implications for dosing, and labeling recommendations.
- Wen X, Wang JS, Backman JT, Kivisto KT, Neuvonen PJ. Gemfibrozil is a potent inhibitor of human cytochrome P450 2C9. Drug Metab Dispos. 2001;29:1359–1361. [PubMed] [Google Scholar]
- Kajosaari LI, Laitila J, Neuvonen PJ, Backman JT. Metabolism of repaglinide by CYP2C8 and CYP3A4 in vitro: effect of fibrates and rifampicin. Basic Clin Pharmacol Toxicol. 2005;97:249–256. doi: 10.1111/j.1742-7843.2005.pto_157.x. [DOI] [PubMed] [Google Scholar]
- Ogilvie BW, Zhang D, Li W, Rodrigues AD, Gipson AE, Holsapple J, Toren P, Parkinson A. Glucuronidation converts gemfibrozil to a potent, metabolism-dependent inhibitor of CYP2C8: implications for drug-drug interactions. Drug Metab Dispos. 2006;34:191–197. doi: 10.1124/dmd.105.007633. [DOI] [PubMed] [Google Scholar]
- Cohen LH, van Leeuwen RE, van Thiel GC, van Pelt JF, Yap SH. Equally potent inhibitors of cholesterol synthesis in human hepatocytes have distinguishable effects on different cytochrome P450 enzymes. Biopharm Drug Dispos. 2000;21:353–364. doi: 10.1002/bdd.249. [DOI] [PubMed] [Google Scholar]
- Sakaeda T, Fujino H, Komoto C, Kakumoto M, Jin JS, Iwaki K, Nishiguchi K, Nakamura T, Okamura N, Okumura K. Effects of acid and lactone forms of eight HMG-CoA reductase inhibitors on CYP-mediated metabolism and MDR1-mediated transport. Pharm Res. 2006;23:506–512. doi: 10.1007/s11095-005-9371-5. [DOI] [PubMed] [Google Scholar]
- Tornio A, Pasanen MK, Laitila J, Neuvonen PJ, Backman JT. Comparison of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors (statins) as inhibitors of cytochrome P450 2C8. Basic Clin Pharmacol Toxicol. 2005;97:104–108. doi: 10.1111/j.1742-7843.2005.pto_134.x. [DOI] [PubMed] [Google Scholar]
- 2013. Facts & Comparisons eAnswers.
- Ahmad S. Gemfibrozil: interaction with glyburide. South Med J. 1991;84:102. [PubMed] [Google Scholar]
- Honkalammi J, Niemi M, Neuvonen PJ, Backman JT. Gemfibrozil is a strong inactivator of CYP2C8 in very small multiple doses. Clin Pharmacol Ther. 2012;91:846–855. doi: 10.1038/clpt.2011.313. [DOI] [PubMed] [Google Scholar]
- Jenkins SM, Zvyaga T, Johnson SR, Hurley J, Wagner A, Burrell R, Turley W, Leet JE, Philip T, Rodrigues AD. Studies to further investigate the inhibition of human liver microsomal CYP2C8 by the acyl-beta-glucuronide of gemfibrozil. Drug Metab Dispos. 2011;39:2421–2430. doi: 10.1124/dmd.111.041947. [DOI] [PubMed] [Google Scholar]
- Ginde AA, Blanc PG, Lieberman RM, Camargo CA., Jr Validation of ICD-9-CM coding algorithm for improved identification of hypoglycemia visits. BMC Endocr Disord. 2008;8:4. doi: 10.1186/1472-6823-8-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett DW, Chando TJ, Didonato GC, Singhvi SM, Pan HY, Weinstein SH. Biotransformation of pravastatin sodium in humans. Drug Metab Dispos. 1991;19:740–748. [PubMed] [Google Scholar]
- Miller RG. Simultaneous Statistical Inference. 2. New York: Springer-Verlag; 1981. [Google Scholar]
- Wienkers LC, Heath TG. Predicting in vivo drug interactions from in vitro drug discovery data. Nat Rev Drug Discov. 2005;4:825–833. doi: 10.1038/nrd1851. [DOI] [PubMed] [Google Scholar]
- Einolf HJ. Comparison of different approaches to predict metabolic drug-drug interactions. Xenobiotica. 2007;37:1257–1294. doi: 10.1080/00498250701620700. [DOI] [PubMed] [Google Scholar]
- Brunton LL, Chabner B, Knollmann A, Bjorn C. Goodman & Gilman’s The Pharmacological Basis of Therapeutics, 12e. 12: The McGraw-Hill Companies; 2012. [Google Scholar]
- Hallifax D, Houston JB. Binding of drugs to hepatic microsomes: comment and assessment of current prediction methodology with recommendation for improvement. Drug Metab Dispos. 2006;34:724–726. doi: 10.1124/dmd.105.007658. [DOI] [PubMed] [Google Scholar]
- Guest EJ, Rowland-Yeo K, Rostami-Hodjegan A, Tucker GT, Houston JB, Galetin A. Assessment of algorithms for predicting drug-drug interactions via inhibition mechanisms: comparison of dynamic and static models. Br J Clin Pharmacol. 2011;71:72–87. doi: 10.1111/j.1365-2125.2010.03799.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shorr RI, Ray WA, Daugherty JR, Griffin MR. Incidence and risk factors for serious hypoglycemia in older persons using insulin or sulfonylureas. Arch Intern Med. 1997;157:1681–1686. [PubMed] [Google Scholar]
- van Staa T, Abenhaim L, Monette J. Rates of hypoglycemia in users of sulfonylureas. J Clin Epidemiol. 1997;50:735–741. doi: 10.1016/s0895-4356(97)00024-3. [DOI] [PubMed] [Google Scholar]
- Zhou L, Naraharisetti SB, Liu L, Wang H, Lin YS, Isoherranen N, Unadkat JD, Hebert MF, Mao Q. Contributions of human cytochrome P450 enzymes to glyburide metabolism. Biopharm Drug Dispos. 2010;31:228–242. doi: 10.1002/bdd.706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin OQ, Tomlinson B, Chow MS. CYP2C9, but not CYP2C19, polymorphisms affect the pharmacokinetics and pharmacodynamics of glyburide in Chinese subjects. Clin Pharmacol Ther. 2005;78:370–377. doi: 10.1016/j.clpt.2005.06.006. [DOI] [PubMed] [Google Scholar]
- Zheng HX, Huang Y, Frassetto LA, Benet LZ. Elucidating rifampin’s inducing and inhibiting effects on glyburide pharmacokinetics and blood glucose in healthy volunteers: unmasking the differential effects of enzyme induction and transporter inhibition for a drug and its primary metabolite. Clin Pharmacol Ther. 2009;85:78–85. doi: 10.1038/clpt.2008.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tournier N, Saba W, Cisternino S, Peyronneau MA, Damont A, Goutal S, Dubois A, Dolle F, Scherrmann JM, Valette H, Kuhnast B, Bottlaender M. Effects of selected OATP and/or ABC transporter inhibitors on the brain and whole-body distribution of glyburide. AAPS J. 2013;15:1082–1090. doi: 10.1208/s12248-013-9514-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh H, Yamashita F, Tsujimoto M, Murakami H, Koyabu N, Ohtani H, Sawada Y. Citrus juices inhibit the function of human organic anion-transporting polypeptide OATP-B. Drug Metab Dispos. 2005;33:518–523. doi: 10.1124/dmd.104.002337. [DOI] [PubMed] [Google Scholar]
- Hirano M, Maeda K, Shitara Y, Sugiyama Y. Drug-drug interaction between pitavastatin and various drugs via OATP1B1. Drug Metab Dispos. 2006;34:1229–1236. doi: 10.1124/dmd.106.009290. [DOI] [PubMed] [Google Scholar]
- Kalliokoski A, Niemi M. Impact of OATP transporters on pharmacokinetics. Br J Pharmacol. 2009;158:693–705. doi: 10.1111/j.1476-5381.2009.00430.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki M, Li B, Louie SW, Pudvah NT, Stocco R, Wong W, Abramovitz M, Demartis A, Laufer R, Hochman JH, Prueksaritanont T, Lin JH. Effects of fibrates on human organic anion-transporting polypeptide 1B1-, multidrug resistance protein 2- and P-glycoprotein-mediated transport. Xenobiotica. 2005;35:737–753. doi: 10.1080/00498250500136676. [DOI] [PubMed] [Google Scholar]
- Noe J, Portmann R, Brun ME, Funk C. Substrate-dependent drug-drug interactions between gemfibrozil, fluvastatin and other organic anion-transporting peptide (OATP) substrates on OATP1B1, OATP2B1, and OATP1B3. Drug Metab Dispos. 2007;35:1308–1314. doi: 10.1124/dmd.106.012930. [DOI] [PubMed] [Google Scholar]
- Chen C, Mireles RJ, Campbell SD, Lin J, Mills JB, Xu JJ, Smolarek TA. Differential interaction of 3-hydroxy-3-methylglutaryl-coa reductase inhibitors with ABCB1, ABCC2, and OATP1B1. Drug Metab Dispos. 2005;33:537–546. doi: 10.1124/dmd.104.002477. [DOI] [PubMed] [Google Scholar]
- Hsiang B, Zhu Y, Wang Z, Wu Y, Sasseville V, Yang WP, Kirchgessner TG. A novel human hepatic organic anion transporting polypeptide (OATP2). Identification of a liver-specific human organic anion transporting polypeptide and identification of rat and human hydroxymethylglutaryl-CoA reductase inhibitor transporters. J Biol Chem. 1999;274:37161–37168. doi: 10.1074/jbc.274.52.37161. [DOI] [PubMed] [Google Scholar]
- Kidd RS, Straughn AB, Meyer MC, Blaisdell J, Goldstein JA, Dalton JT. Pharmacokinetics of chlorpheniramine, phenytoin, glipizide and nifedipine in an individual homozygous for the CYP2C9*3 allele. Pharmacogenetics. 1999;9:71–80. doi: 10.1097/00008571-199902000-00010. [DOI] [PubMed] [Google Scholar]
- Tan B, Zhang YF, Chen XY, Zhao XH, Li GX, Zhong DF. The effects of CYP2C9 and CYP2C19 genetic polymorphisms on the pharmacokinetics and pharmacodynamics of glipizide in Chinese subjects. Eur J Clin Pharmacol. 2010;66:145–151. doi: 10.1007/s00228-009-0736-2. [DOI] [PubMed] [Google Scholar]
- van de Steeg E, Greupink R, Schreurs M, Nooijen IH, Verhoeckx KC, Hanemaaijer R, Ripken D, Monshouwer M, Vlaming ML, Degroot J, Verwei M, Russel FG, Huisman MT, Wortelboer HM. Drug-drug interactions between rosuvastatin and oral antidiabetic drugs occurring at the level of OATP1B1. Drug Metab Dispos. 2013;41:592–601. doi: 10.1124/dmd.112.049023. [DOI] [PubMed] [Google Scholar]
- Gross B, Staels B. PPAR agonists: multimodal drugs for the treatment of type-2 diabetes. Best Pract Res Clin Endocrinol Metab. 2007;21:687–710. doi: 10.1016/j.beem.2007.09.004. [DOI] [PubMed] [Google Scholar]
- Tsimihodimos V, Miltiadous G, Daskalopoulou SS, Mikhailidis DP, Elisaf MS. Fenofibrate: metabolic and pleiotropic effects. Curr Vasc Pharmacol. 2005;3:87–98. doi: 10.2174/1570161052773942. [DOI] [PubMed] [Google Scholar]
- Hennessy S, Flockhart DA. The need for translational research on drug-drug interactions. Clin Pharmacol Ther. 2012;91:771–773. doi: 10.1038/clpt.2012.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Potential confounding factors that were considered in the pharmacoepidemiologic study