

Abstract
A series of unsymmetrically substituted biphenyl compounds was designed as alpha helical proteomimetics with the aim of inhibiting the binding of coactivator proteins to the nuclear hormone receptor coactivator binding domain. These compounds were synthesized in good overall yields in seven steps starting from 2-bromoanisole. The final products were evaluated using cotransfection reporter gene assays and mammalian two-hybrid competitive inhibition assays to demonstrate their effectiveness as competitive binding inhibitors. The results from this study indicate that these proteomimetics possess the ability to inhibit coactivator-receptor interactions, but via a mixed mode of inhibition.
Keywords: Proteomimetic, Co-activator binding inhibition, Nuclear receptor
Introduction
Protein-protein interactions (PPI) play a key role in regulating many cellular pathways, particularly by providing important recognition and activation functions. Because such recognition functions are often up-regulated in disease states, disruption of the protein-protein interface is an attractive, but challenging, objective.1 Targeting these interactions with low molecular weight compounds is difficult because proteins tend to be large, have non-contiguous binding regions and often lack distinctive structural features that one can use as a basis for disruptor design. One strategy under current consideration is to use a small molecule to mimic the binding motif of one of the protein partners directly at the binding site.2 One of the most common recognition patterns involves the interaction between a short alpha helical peptidic component of one protein and a complementary groove in the second protein. Examples of such PPI include Bcl-2/Bak,3 MDM2/p53,4 and nuclear receptor/coregulator (NR-CoR) interactions.5
The alpha helix is the most common element found in proteins, comprising nearly 40% of all protein secondary structure. The structure is generated by hydrogen bonding between the carbonyls and primary amides of amino acids sited on adjacent turns of the helix.6 The concept that the central core of the helical peptide can be replaced by a non-peptidyl scaffold and that the side chains of the amino acids can be represented by appropriate substituents on that scaffold has generated a class of compounds termed “proteomimetics.” Many different helical core-replacing moieties, including indanes,7 terphenyls,8 and pyrimidines9 have been shown to mimic alpha helical structure and a number of the compounds also demonstrated the capacity to disrupt discrete PPI that require an alpha helical motif. It is unnecessary for this class of inhibitors to mimic exactly the natural, peptidyl segment, but only to provide a similar interaction with the binding subpocket.10
We previously described the synthesis of a small series of 3, 3′-symmetrically substituted bipolar-biphenyl alpha helical proteomimetics that were disruptors of estrogen receptor α (ERα) coactivator binding (see Figure 1).11 These compounds, designed to mimic the α-helical LXXLL motif of the NR Box of the ERα coactivator proteins,12 were prepared using commercially available 2-substituted phenols. The phenols were selectively brominated at the 4-position, at which point they served as intermediates for both termini of the biphenyl derivatives. The 3, 3′-dibenzyl substituted derivatives were the most effective coactivator binding inhibitors (CBIs). Furthermore, the importance of substitution pattern on the dibenzyl, as well as the dimethyl, derivative was studied.13 The objective of our current study was to incorporate larger hydrophobic sidechains into our scaffold in an attempt to make these compounds a better fit for the hydrophobic coactivator binding pocket (CBP). The synthetic approach that we envisioned relied upon the appropriate 2-substituted-4-bromophenols. Because these derivatives were not readily available, a versatile and reliable procedure for their synthesis was necessary.
Figure 1.
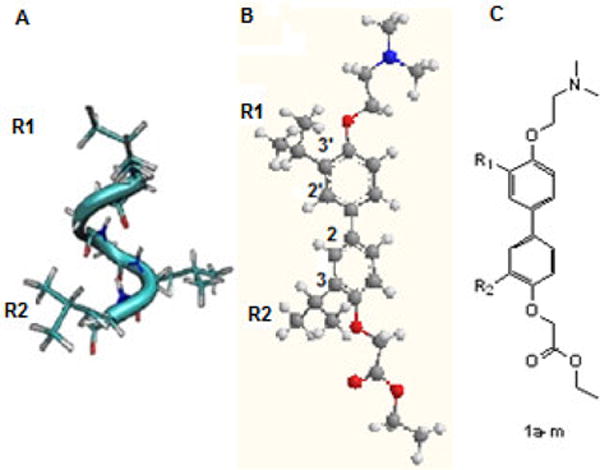
Alpha helical proteomimetic. Adapted from Williams et. al.11 A) Coactivator NR Box forms an α-helix with an LXXLL motif. B) 4,4′-Biphenyl scaffold. C) Target compounds 1a–m with varying substitution (see Table 1)
Results and Discussion
The most direct approach to the preparation of the requisite 2-substituted 4-bromophenols involves the Friedel-Crafts acylation of 4-bromophenol, followed by reduction of the ketone. This approach had a literature precedent as benzoylation of 4-chlorophenol gave the 2-benzoyl-4-chlorophenol.14 Our scheme required the bromo- rather than the chlorophenol as we would later employ Miyaura boronation, which is more difficult with chloro-substrates. Therefore, we used a Freidel-Crafts acylation with a variety of acid chlorides (Scheme 1) to attach a carbonyl at the 2-position of 4-bromophenol.15 While the reaction proceeded satisfactorily for the benzoyl derivative, other acid chlorides gave much poorer yields. However, the bromine could not be introduced at a later stage due to the possibility of acylation at para- and not the ortho-position. One possible explanation for the failure of this approach is that the free phenol can poison the AlCl3 catalyst, thereby generating a weaker aluminum phenoxide catalyst. Protection of the phenol as the methyl ether (4-bromoanisole) did not improve reaction yields. Efforts using BF3-Et2O, which would give a stable intermediate with phenolic hydroxyl, were likewise unsuccessful.16,17
Scheme 1.
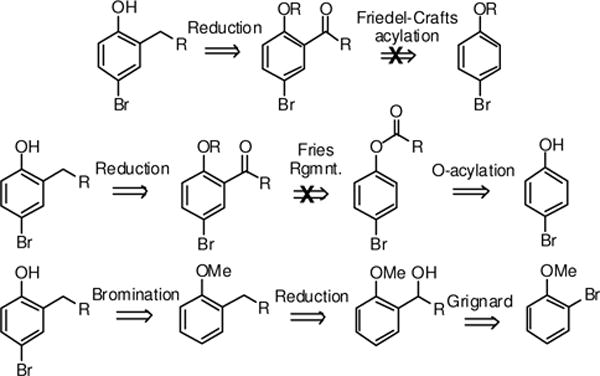
Synthetic routes to 2-substituted phenols
We next evaluated the Fries Rearrangement of the acylated phenyl esters as the method for introducing the ortho substituent. In this approach 4-bromophenol was O-acylated and the ester was subjected to conditions that promote migration of the carbonyl to the ortho-position, yielding the requisite 2-acylated-4-bromophenol.18,19 There was precedent for this strategy, however, for a very limited set of 4-halogenated phenols. Preparation of the intermediate esters proceeded in excellent yields. The Fries rearrangement reactions were again conducted using conditions typical for this reaction, however, yields were poor and inconsistent. The mechanism for this rearrangements proceeds by generation of the acylonium ion followed by its migration to the most electron-rich center. Generation of the acyl cation was observed under our reaction conditions, however, the subsequent ortho-acylation was not observed. The 4-bromo substituent had a clearly unfavorable influence on promoting the C-C bond formation at the 2-position. Whether this was due to electronic or resonance effects was unclear.
The procedure that was adopted was not as direct as the Freidel-Crafts or Fries approaches, but was applicable to a wider variety of starting materials. In this strategy we envisioned introduction of the 2-substituent by addition of the Grignard reagent derived from 2-bromoanisole to the requisite aldehyde. Subsequent reduction of the benzylic alcohol followed by para-bromination and deprotection of the methyl ether would give the desired series of 2-substituted-4-bromophenols (Scheme 2). The specific substituents were chosen to further expand the pharmacophoric model generated from the initial series in which the benzylic derivative was one of the most active coactivator binding inhibitors. The cyclohexylmethyl and heptyl derivatives would provide similar steric dimensions, and similar carbon number, but with greater conformational flexibility. Resonance may also be an influencing factor. Compared to the benzyl group, the 2-naphthylmethyl and 4-biphenyl derivatives present similar aromatic character, but with significantly increased steric demands.
Scheme 2.
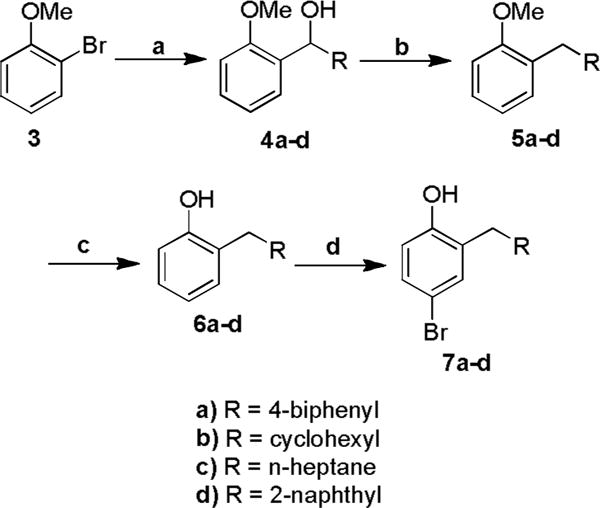
Reagents and conditions: (a) i Magnesium, iodine, tetrahydrofuran, reflux; ii R-aldehyde, rt, 91%; (b) Triethylsilane, trifluoroacetic acid, dichloromethane, rt, 87%; (c) Borontribromide, dichloromethane, rt, 95%; (d) Tetrabutylammonium tribromide, chloroform, rt, 90%.
The addition of the anisolyl-2-magnesium bromide to the aldehydes gave the crude products which were purified via chromatography to give secondary alcohols 4a–d (75–100 % yield).20 The intermediate alcohols 4a–d were readily reduced under mild conditions, using tetraethylsilane and trifluoroacetic acid in dichloromethane to give 5a–d (75–87 % yield).21 Deprotection with boron tribromide yielded the 2-substituted phenols 6a–d (91–95 %).22 Selective 4-bromination using tetrabutylammonium tribromide gave the 4-brominated 2-substituted phenols 7a–d (79–91 % yield).23 The 4-bromo-2-substituted bromophenols 7a–d and the unsubstituted 4-bromophenol 7e provided the key intermediates from which the two polar termini of the target proteomimetics were prepared (Scheme 3). The unsubstituted phenol 7e was included to make mono-substituted bipolar biphenyls in addition to the di-substituted bipolar biphenyls. The 4-bromophenols 7a–e underwent Miyaura boronation using PdCl2(dppf) and bis(pinacolato)diboron to give the 4-hydroxyphenylboronate esters 8a–e in 45–53 % isolated yields.24,25 Alkylation of the 4-hydroxy group of the 4-bromophenols 7a–e using sodium hydride and ethyl bromoacetate gave, after flash chromatography, ethyl 2-(4-bromo-2-substituted-phenoxy) acetates 9a–e, in 69–91 % isolated yields.
Scheme 3.
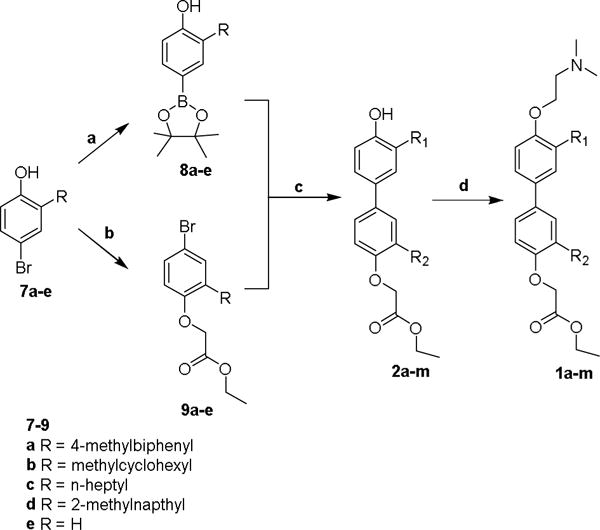
Reagents and conditions: (a) PdCl2(dppf), B2pin2, KOAc, dioxane, 80 °C; (b) 2-Ethoxycarbonylethyl bromide, NaH, THF, rt; (c) PdCl2(P(otol)3)2, P(otol)3, Na2CO3, Dichloromethane/water (3:2), reflux; (d) 2-chloro-N,N-dimethylethylamine hydrochloride, K2CO3, acetone, reflux.
The aryl bromides, 9a–e, and the aryl boronates, 8a–e, were coupled in various combinations to give multiple biphenyl phenols (2a–m) as shown in Table 1. 3-Substituted, 3′-substituted, and a 3,3′-disubstituted variants were prepared for each sidechain, as well as an unsubstituted biphenyl. Coupling under Suzuki conditions with PdCl2(P(otol)3)2, P(otol3), and sodium carbonate in dichloromethane and water gave, after purification via flash chromatography, the biphenyl phenol/esters 2a–m. P(otol3) was used because when PPh3 ligand was used, reductive elimination of a phenyl group and the aryl bromide was commonly observed, leading to a dead end side biphenyl side product. The ortho-tolyl group completely eliminated this competing side reaction.
Table 1.
Substitution patterns of phenol/ester biphenyls
| Starting Material | Biphenyl Product | Substituents | ||
|---|---|---|---|---|
| Bromide | Boranate | 3 | 3′ | |
| 9a | 8e | 2a | 4MeBP | H |
| 9e | 8a | 2b | H | 4MeBP |
| 9a | 8a | 2c | 4MeBP | 4MeBP |
| 9b | 8e | 2d | MeCy | H |
| 9e | 8b | 2e | H | MeCy |
| 9b | 8b | 2f | MeCy | MeCy |
| 9c | 8e | 2g | n-hept | H |
| 9e | 8c | 2h | H | n-hept |
| 9c | 8c | 2i | n-hept | n-hept |
| 9d | 8e | 2j | 2MeNp | H |
| 9e | 8d | 2k | H | 2MeNp |
| 9d | 8d | 2l | 2MeNp | 2MeNp |
| 9e | 8e | 2m | H | H |
4MeBP = 4-methylbiphenyl, MeCy = methylcylcohexane, 2MeNp = 2-methylnaphthalene
The biphenyl phenol/esters were then converted to the biphenyl amine/esters (1a–m) by alkylation with dimethylaminoethyl chloride.
To test the effectiveness of our proteomimetics to inhibit ERα activity, we subjected biphenyl phenol-esters 2a–m and biphenyl amine-esters 1a–m to a cotransfection reporter gene assay (Table 2). 3xERE-TATA-Luciferase and estrogen receptor (RST7–ERα) were transfected into ER-negative SKBR3 breast cancer cells along with normalization control pCMV-β-gal.26 Cells were then treated with serial twofold dilutions of our biphenyls with and without 1 nM E2 and incubated for 40 h before assaying. ERα activity was measured as a function of increasing proteomimetic concentration. We expected to see no activity in the absence of E2 since our compounds were designed to block NR-CoR interactions at the coactivator binding pocket (CBP) and not to bind at the ligand binding pocket (LBP) where E2 binds. In the presence of E2, we expected to see a decrease in ERα activity. 2a–m had little or no effect in the absence of E2 and basically no effect in the presence of E2. These results suggested that our compounds most likely were binding at the LBP, but competed weakly compared to E2. 1b, 1d, and 1j behaved as inhibitors in the presence and absence of E2 by showing a decrease in reporter gene activity of greater than 50% at concentrations below 20 μM. Complete graphs and conditions can be found in the supplemental section.
Table 2.
Co-transfection reporter gene assay
| Compound | Vehicle | 1 nM Estradiol |
|---|---|---|
| 1a | 0 | 0 |
| 1b | --- | --- |
| 1c | ++ | 0 |
| 1d | --- | --- |
| 1e | ++ | 0 |
| 1f | +++a | 0a |
| 1g | 0 | 0 |
| 1h | +++ | 0 |
| 1i | +++ | 0 |
| 1j | --- | --- |
| 1k | + | 0 |
| 1l | +++a | 0a |
| 1m | + | 0 |
| 2a | +++ | 0 |
| 2b | 0 | 0 |
| 2c | + | 0 |
| 2d | + | 0 |
| 2e | +a | 0a |
| 2f | 0 | 0 |
| 2g | 0 | 0 |
| 2h | +++ | 0 |
| 2i | 0 | 0 |
| 2j | 0 | 0 |
| 2k | ++ | 0 |
| 2l | +++ | 0 |
| 2m | + | 0 |
(+) Denotes a greater than 50% increase in reporter gene activity at compound concentration <20 μM.
(++) Denotes a greater than 50% increase in reporter gene activity at compound concentration between 20–50 μM.
(+++) Denotes a greater than 50% increase in reporter gene activity at compound concentration >50 μM.
(-) Denotes a greater than 50% decrease in reporter gene activity at compound concentration >50 μM.
(--) Denotes a greater than 50% decrease in reporter gene activity at compound concentration <between 20–50 μM.
(---) Denotes a greater than 50% decrease in reporter gene activity at compound concentration <20 μM.
(0) Denotes a less than 20% change in reporter gene activity. 4 Hydroxytamoxifen was used as an antagonist control.
Denotes cell death at concentration greater than 50 μM.
Denotes precipitation of compound from solution.
2a–m and 1a–m were then subjected to a mammalian two-hybrid competitive inhibtion assay employing the estrogen receptor (VP16-ERα) and the co-activator peptide pM-GRIP1 LxxLL2 (Table 3). Disrupting the interaction between ERα and the GRIP1 derived peptide would lead to a decrease in reporter gene activity, measured here as a reduction in luciferase bioluminescence. This would provide further evidence that these compounds are true CBIs and are inhibiting at the CBP. HepG2 cells were transfected with the activated fusion receptor VP16-ERα, the LXXLL peptide fused with yeast Gal4-DBD, and the 5xGal4Luc3 reporter gene as described.26 Cells were then treated with serial twofold dilutions of test compounds +/− 1 nM estradiol (E2) and incubated for 40 h before assaying. 1b and 1j both exhibited greater than 50% decrease in reporter gene activity at concentrations less than 20 μM. However, at concentrations above 50 μM, these compounds become toxic to the cells. Compound 1e also exhibited a decrease in reporter gene activity at concentrations less than 20 μM, but did not become toxic. Once again, 1b, 1d, and 1j showed inhibitory activity in the absence of E2. These results indicated that the compounds may be binding at both the CBP and LBP.
Table 3.
Mammalian two-hybrid competitive inhibition assay
| Compound | Vehicle | 1 nM Estradiol |
|---|---|---|
| 1a | –a | 0a |
| 1b | ---a | ---a |
| 1c | 0 | 0 |
| 1d | --- | --- |
| 1e | ++ | 0 |
| 1f | 0 | - |
| 1g | 0 | - |
| 1h | +++ | 0 |
| 1i | +++ | 0 |
| 1j | ---a | ---a |
| 1k | +b | +b |
| 1l | +++ab | 0ab |
| 1m | 0 | 0 |
| 2a | +++ab | 0ab |
| 2b | -- | -- |
| 2c | 0 | 0 |
| 2d | 0 | 0 |
| 2e | 0 | - |
| 2f | 0a | 0a |
| 2g | 0 | 0 |
| 2h | +++a | 0a |
| 2i | 0 | 0 |
| 2j | 0 | - |
| 2k | +++ab | 0ab |
| 2l | 0 | 0 |
| 2m | + | +++ |
(+) Denotes a greater than 50% increase in reporter gene activity at compound concentration <20 μM.
(++) Denotes a greater than 50% increase in reporter gene activity at compound concentration between 20–50 μM.
(+++) Denotes a greater than 50% increase in reporter gene activity at compound concentration >50 μM.
(-) Denotes a greater than 50% decrease in reporter gene activity at compound concentration >50 μM.
(--) Denotes a greater than 50% decrease in reporter gene activity at compound concentration <between 20–50 μM.
(---) Denotes a greater than 50% decrease in reporter gene activity at compound concentration <20 μM.
(0) Denotes a less than 20% change in reporter gene activity. 4 Hydroxytamoxifen was used as an antagonist control.
Denotes cell death at concentration greater than 50 μM.
Denotes precipitation of compound from solution.
To provide insight into the mechanism of inhibition, the ERα cotransfection reporter gene assay was repeated with 1b, 1d, and 1j and increasing concentrations of E2 (Figure 2). If these compounds are true CBIs, one should expect an inhibition of the maximum response without a shift in the E2 EC50. Compounds 1b and 1j showed an increased E2 EC50 at increasing concentrations as well as a slight decrease in activity at maximum concentrations, with overall IC50 of 3.1 μM and 3.2 μM respectively. These findings suggest that these compounds are directly competing with E2 for binding at the LBP. Compound 1d showed an increased E2 EC50 at increasing concentrations with an IC50 of 12.7 μM, but a decreased activity at maximum concentration. This result indicates that 1e may have a mixed mode of inhibition, although the apparent affinity for the CBP is significantly lower than for LBP.
Figure 2.
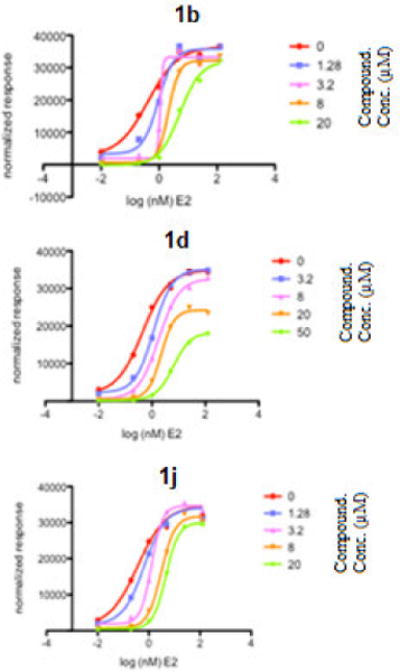
ERα competitive binding assay with estradiol
None of the n-heptyl compounds had any inhibitory effect on ERα-coactivator interaction, which may be due to the extreme flexibility of the group which does not allow the biphenyl to penetrate the binding pocket and adopt the proper conformation. Furthermore, none of di-substituted compounds showed any inhibitory activity. This could be because these compounds are too large to fit in the ERα CBP. These compounds will also be tested in the future as androgen receptor (AR)-coactivator inhibitors, as the AR has a larger hydrophobic groove at its CBP.
The results from this study were compared with those previously reported11,13 and the comparison provides a clearer picture of the potential of the biphenyl scaffold as the basis for co-regulatory protein inhibition. The earlier study examined a limited set of 2,2′-,2,3′-, and 3,2′-disubstituted biphenyl derivatives from which 6 compounds demonstrated significant (low μM) activity against the CBP of ER- or AR-LBP. Those compounds also showed little if any competitive activity against the hormone binding site, as compared 1d derivative, the best compound in the current series. These observations highlight the influence of the 2- or 2′-substituents on selectivity of binding as well as upon CBP binding affinity. The absence of 2- or 2′-substitution relieves conformational constraints at the 1,1-biphenyl junction and thereby allows the compounds to assume a more planar structure. More planar conformations demonstrate estrogen binding site affinity as demonstrated by simple biphenyls as well as phenyl carboranes. The presence of a substituent at either the 2- or 2′-position imparts enough conformational restraint (torsional strain) to substantially reduce or eliminate binding at the estrogen binding site. Affinity to the CBP as well as a preference for ERα vs AR results from the identity of the groups at the 3- and 3′-positions as well as at the 2- and 2′-positions. Based upon the results this and previous studies, it would appear that the optimal derivatives would have a staggered conformation with either a 2, 3′ or 3, 2′ substitution pattern. The remaining 3- or 3′-position would then be substituted with a variety of functional groups selected to provide enhanced affinity and CBP selectivity. Introduction of the terminal groups would then provide the proper orientation within the binding site as well as enhanced affinity. It is clear from these, as well as previous results, that the amine terminus is necessary for CBP inhibition as the phenolic biphenyls did not show any inhibitory activity. The previous study was limited by the commercial availability of 2- and 3-substituted 4-bromophenol precursors. With the development of the methods described in this report, it is now possible to explore in greater detail the effects of individual substituents on binding affinity and selectivity.
Conclusions
In summary we have described the facile preparation of a novel series of bipolar biphenyl proteomimetics derived from 2-bromoanisoles. The Grignard reaction can be utilized to with a wide variety of aldehydes to give 2-substuted phenols which can subsequently be converted to aryl bromides and aryl boronates. These intermediates readily undergo Suzuki coupling to generate the desired intermediate phenolic biphenyls and ultimately the biphenyl amino-esters. Evaluation of the products using cotransfection reporter gene assays and mammalian two-hybrid competitive inhibition assays, demonstrated that only 1b, 1d, and 1j possessed significant inhibitory activity. Of these, only 1d appeared to bind at the CBP, however it retained a competitive inhibition at the LBP. Combining these results with those from previous studies provides a rational basis for designing subsequent generations of CBIs. These studies suggest that future agents should combine the ligand domain binding reducing effects of 2-2′-substitution with the affinity and selectivity-enhancing effects of 3-3′-substituents. Studies to demonstrate the effectiveness of this strategy are in progress and will be described in subsequent publications.
Experimental section
General procedures
All commercially available reagents were purchased from Sigma-Aldrich and used without further purification. Solvents were distilled and reactions requiring inert conditions were performed under N2 or argon. Column chromatography was performed using silica gel unless otherwise indicated. Flash chromatography was performed using the Argonaut Flash Master Solo with an FC204 fraction collector. Thin layer chromatography was used to monitor reactions using Selecto Scientific 200 micron silica gel flexible TLC plates. 1H was recorded on a Varian Unity-INOVA 500MHz spectrometer. 13C NMR were recorded on a Varian 400MHz spectrometer. High resolution mass spectral data were obtained by direct flow injection (injection volume = 1, 5 or 50 ìL) ElectroSpray Ionization (ESI) on a Waters Qtof API US instrument in the positive mode at the Boston University Chemical Instrumentation Center.
Synthesis of 1-(2-methoxyphenyl)heptan-1-ol (4c)
Magnesium shavings (0.547 g, 22.5 mmol) were added to a 3-neck round bottom flask fitted with a condenser. The entire apparatus was flame-dried, a chip of iodine was added and the system was sealed and flushed with Ar. THF (20 mL) and 2-bromoanisole (3) (2.805 g, 15 mmol) were added via syringe. The mixture turned red initially and then gray after 10 minutes. The reaction was heated at reflux for 6 hours, cooled to room temperature and heptanal (0.571 g, 5 mmol) was added via syringe. The reaction was stirred at room temperature overnight, turning white-gray, and quenched with saturated ammonium chloride. The mixture was then eluted with ethyl acetate and washed with water (x2), brine (x2) and dried over magnesium sulfate. The solvent was evaporated and the crude product (4.4 g yellow oil) was purified via by chromatography on silica gel (Hexane/EtOAc, 90:10). The desired product (0.938 g, 85%) was isolated as a clear yellow oil. 1H NMR (500 MHz, CDCl3): δ = 7.33 (dd, J = 7.5, 2 Hz, 1H), 7.27 (td, J = 7, 1.5 Hz, 1H), 6.98 (td, J = 7, 1 Hz, 1H), 6.91 (d, J = 8 Hz, 1H), 4.89 (q, J = 7 Hz, 1H), 3.87 (s, 3H), 2.65 (d, J = 5 Hz, 1H), 1.76–1.84 (m, 2H), 1.47–1.51 (m, 1H), 1.28–1.38 (m, 6H), 0.91 (t, J = 6.5 Hz, 3H) ppm.
4-Biphenyl (2-methoxyphenyl)methanol (4a)
Same synthesis 4c. Product isolated in 91% yield as a white solid. 1H NMR (500 MHz, CDCl3): δ = 7.61 (dd, J = 8.5, 1.5 Hz, 2H), 7.58 (dd, J = 9.0, 2.0 Hz, 2H), 7.49 (d, J = 8.0 Hz, 2H), 7.45 (t, J = 7.5 Hz, 2H), 7.36 (td, J = 8.0, 2.0 Hz, 1H), 7.34 (dd, J = 7.5, 1.5 Hz, 1H), 7.30 (td, J = 8.0, 1.0 Hz, 1H), 7.00 (td, J = 7.5, 1.0 Hz, 1H), 6.92 (d, J = 8.5 Hz, 1H), 6.14 (d, J = 5.5, 1H), 3.84 (s, 3H), 3.27 (d, J = 5.5 Hz, 1H) ppm.
Cyclohexyl (2-methoxyphenyl)methanol (4b)
Same synthesis as 4c. Product isolated in quantitative yield as a yellow oil. 1H NMR (500 MHz, CDCl3): δ = 7.33 (dd, J = 7.5, 2 Hz, 1H), 7.27 (td, J = 7.0, 1.5 Hz, 1H), 6.98 (td, J = 7.0, 1.0 Hz, 1H), 6.91 (d, J = 8.0 Hz, 1H)Hz, 1H), 3.84 (s, 3H), 3.27 (d, J = 5.5 Hz, 1H) ppm.
(2-methoxyphenyl)(naphthalen-2-yl)methanol (4d)
Same synthesis as 4c. Product isolated in 75% yield as a white solid. 1H NMR (500 MHz, CDCl3): δ = 7.87 (s, 1H), 7.79–7.83 (m, 2H), 7.78 (s, 1H), 7.48 (dd, J = 8.5, 1.5 Hz, 1H), 7.42–7.46 (m, 2H), 7.27 (td, J = 8.5, 1.5 Hz, 1H), 7.24 (d, J = 7.5 Hz, 1H), 6.94 (t, J = 7.5 Hz, 1H), 6.90 (d, J = 8 Hz, 1H), 6.23 (s, 1H), 3.82 (s, 3H) ppm.
Synthesis of 1-heptyl-2-methoxybenzene (5c)
Triethylsilane (2.290 g, 19.7 mmol) was added to 4c (0.868 g, 3.94 mmol) in a solution of dichloromethane. Trifluoroacetic acid (2.246 g, 19.7 mmol) was added via syringe and the reaction mixture was stirred under nitrogen at room temperature for one hour. The mixture was quenched with saturated sodium bicarbonate and washed with dichloromethane (3x). The organic layers were combined and washed with brine. The organic layer was separated and dried over magnesium sulfate which was then filtered off. The solvent was evaporated and the crude product (1.4 g brown oil) was purified by flash chromatography on silica gel (100% Hexane). The desired product (0.680 g, 84%) was isolated as a yellow oil. 1H NMR (500 MHz, CDCl3): δ = 7.20 (t, J = 8.0 Hz, 1H), 7.17 (d, J = 9.0 Hz, 1H), 6.91 (td, J = 7.0, 1.5 Hz, 1H), 6.87 (d, J = 8.5 Hz, 1H), 3.84 (s, 3H), 2.63–2.66 (m, 2H), 1.57–1.65 (m, 2H), 1.32–1.37 (m, 8H), 0.91–0.94 (m, 3H) ppm.
1-(4-biphenylmethyl)-2-methoxybenzene (5a)
Same synthesis as 5c, but from 4a. Product isolated in 87% yield as a white solid. 1H NMR (500 MHz, CDCl3): δ = 7.56 (dd, J = 8.5, 1 H, 2H), 7.49 (d, J = 8.5 Hz, 2H), 7.40 (t, J = 7.5 Hz, 2H), 7.30 (t, J = 7.5 Hz, 1H), 7.27 (d, J = 7.0 Hz, 2H), 7.20 (td, J = 7.0, 2.0 Hz, 1H), 7.11 (dd, J = 7.0, 1.5 Hz, 1H), 6.89 (td, J = 7.5, 1.0 Hz, 1H), 6,87 (d, J = 8.5 Hz, 1H), 4.01 (s, 2H), 3.81 (s, 3H) ppm.
1-(cyclohexylmethyl)-2-methoxybenzene (5b)
Same synthesis as 5c, but from 4b. Product isolated in 75% yield as a clear oil. 1H NMR (500 MHz, CDCl3): δ = 7.16 (td, J = 7.5, 1.5 Hz, 1H), 7.07 (dd, J = 7.0, 2.0 Hz, 1H), 6.86 (t, J = 7.0 Hz, 1H), 6.83 (d, J = 7.5 Hz, 1H), 3.80 (s, 3H), 2.48 (d, J = 2.0 Hz, 2H).
2-(2-methoxybenzyl)naphthalene (5d)
Same synthesis as 5c, but from 4d. Product isolated in 76% yield as a white solid. 1H NMR (500 MHz, CDCl3): δ = 7.95 (d, J = 7.5 Hz, 1H), 7.91 (d, J = 8 Hz, 1H), 7.90 (d, J = 7.5 Hz, 1H), 7.81 (s, 1H), 7.59 (td, J = 8.0, 1.5 Hz, 1H), 7.56 (d, J = 1.5 Hz, 1H), 7.55 (td, J = 8.0, 1.5 Hz, 1H), 7.37 (td, J = 8.0, 1.5 Hz, 1H), 7.28 (d, J = 7.5 Hz, 1H), 7.05 (td, J = 7.5, 1.0 Hz, 1H), 7.02 (d, J = 8.5 Hz, 1H), 4.32 (s, 2H), 3.95 (s, 3H) ppm.
Synthesis of 2-heptylphenol (6c)
Boron tribromide (1.771 g, 7.07 mmol) was added slowly via syringe to 5c (0.486 g, 2.36 mmol) in a solution of dichloromethane. The reaction was stirred at room temperature overnight and was quenched with water. The mixture was eluted with dichloromethane, washed with brine (x2) and dried over magnesium sulfate. The solvent was evaporated and the crude mixture (0.527 g brown oil) was purified by flash chromatography on silica gel (Hexane/EtOAc, 80:20). The desired product (0.433 g, 95%) was isolated as a yellow oil. 1H NMR (500 MHz, CDCl3): δ = 7.17 (d, J = 7.5 Hz, 1H), 7.11 (t, J = 7.5 Hz, 1H), 6.92 (t, J = 7.5 Hz, 1H), 6.79 (d, J = 8.0 Hz, 1H), 5.21 (s, 1H), 2.65 (t, J = 8.0 Hz, 2H), 1.67 (5let, J = 7.5 Hz, 2H), 1.33–1.41 (m, 8H), 0.94 (t, J = 7.0 Hz, 3H) ppm.
2-(4-biphenylmethyl)phenol (6a)
Same synthesis as 6c, but from 5a. Product isolated in 94% as a white solid. 1H NMR (500 MHz, CDCl3): δ = 7.65 (d, J = 7.5 Hz, 1H), 7.60 (d, J = 6.5 Hz, 1H), 7.50 (t, J = 7.5 Hz, 2H), 7.41 (t, J = 7.0 Hz, 1H), 7.39 (d, J = 7.5 Hz, 2H), 7.24 (d, J = 7.0 Hz, 1H), 7.21 (td, J = 7.5, 1.5 Hz, 1H), 6.99 (td, J = 7.5, 1 Hz, 1H), 6.87 (d, J = 7.5 Hz, 1H), 5.18 (s, 1H), 4.12 (s, 3H) ppm.
2-(cyclohexylmethyl)phenol (6b)
Same synthesis as 6c, but from 5b. Product isolated in 95% yield. 1H NMR (500 MHz, CDCl3): δ = 7.07 (t, J = 7.5 Hz, 1H), 7.06 (d, J = 7.5 Hz, 1H), 6.85 (t, J = 8.0 Hz, 1H), 6.76 (d, J = 7.0 Hz, 1H), 4.70 (s, 3H), 2.48 (d, J = 7.0 Hz, 2H), 1.60–1.75 (m, 5H), 1.50–1.60 (m, 1H), 1.11–1.25 (m, 3H), 0.92–1.02 (m, 2H) ppm.
2-((naphthalen-2-yl)methyl) phenol (6d)
Same synthesis as 6c, but from 5d. Product isolated in 91% yield as a yellow oily solid. 1H NMR (500 MHz, CDCl3): δ = 7.76 (dd, J = 8.5, 2.5 Hz, 1H), 7.72 (d, J = 8.0 Hz, 1H), 7.71 (d, J = 9.0 Hz, 1H), 7.63 (s, 1H), 7.37–7.42 (m, 2H), 7.35 (dd, J = 8.5, 2.0 Hz, 1H), 7.10 (d, J = 7.5 Hz, 1H), 7.09 (t, J = 7.5 Hz, 1H), 6.84 (td, J = 8.0, 1.0 Hz, 1H), 6.79 (d, J = 8.0 Hz, 1H), 6.05 (br s, 1H), 4.14 (s, 2H) ppm.
Synthesis of 4-bromo-2-heptylphenol (7c)
Tetrabutylammonium tribromide (1.147 g, 2.38 mmol) was added to a solution of 6c (0.381 g, 1.98 mmol) in chloroform. The solution was stirred at room temperature overnight. The solution was then evaporated and the crude product was eluted with ether and washed with water (x2), 1 N HCl (x2), and brine (x2). The organic layer was separated and dried over magnesium sulfate. The solvent was evaporated and the crude mixture (0.474 g brown oil) was purified by flash chromatography on silica gel (100% Hexane). The desired product (0.426 g, 79%) was isolated as a yellow oil. 1H NMR (500 MHz, CDCl3): δ = 7.24 (d, J = 2.5 Hz, 1H), 7.15 (dd, J = 8.5, 3.0 Hz, 1H), 6.23 (d, J = 9.0 Hz, 1H), 5.14 (s, 1H), 2.56 (t, J = 7.5 Hz, 2H), 1.56–1.64 (m, 2H), 1.24–1.40 (m, 8H), 0.90 (t, J = 7.0 Hz, 3H) ppm.
4-bromo-2-(4-biphenylmethyl)phenol (7a)
Same synthesis as 7c, but from 6a. Product isolated in 79% yield as a white solid. 1H NMR (500 MHz, CDCl3): δ = 7.61 (d, J = 7.5 Hz, 2H), 7.56 (d, J = 6. 7c, but 5 Hz, 2H), 7.46 (t, J = 7.5 Hz, 2H), 7.37 (t, J = 7.5 Hz, 1H), 7.32 (d, J = 5.0 Hz, 2H), 7.31 (s, 1H), 7.27 (dd, J = 8.5, 1.5 Hz, 1H), 6.72 (d, J = 8.0 Hz, 1H), 4.86 (br s, 1H), 4.02 (s, 2H) ppm.
4-bromo-2-(cyclohexylmethyl)phenol (7b)
Same synthesis as 7c, but from 6b. Product isolated in 77% yield as a light yellow oil. 1H NMR (500 MHz, CDCl3): δ = 7.17 (dd, J = 7, 2.5 Hz, 1H), 7.15 (d, J = 2.5 Hz, 1H), 6.64 (d, J = 8.5 Hz, 1H), 4.20 (br s, 1H), 2.43 (d, J = 7.5 Hz, 2H), 1.61–1.72 (m, 5H), 1.5–1.59 (m, 1H), 1.12–1.23 (m, 3H), 0.91–1.02 (m, 2H) ppm.
4-bromo-2-((naphthalen-2-yl)methyl)phenol (7d)
Same synthesis as 7c, but from 6d. Product isolated in 90% yield as a yellow oil. 1H NMR (500 MHz, CDCl3): δ = 7.80 (d, J = 9.0 Hz, 1H), 7.78 (d, J = 8.0 Hz, 1H), 7.76 (d, J = 9.0 Hz, 1H), 7.64 (s, 1H), 7.46 (td, J = 8.0, 1.5 Hz, 1H), 7.44 (td, J = 7.0, 2.0 Hz, 1H), 7.34 (dd, J = 8.0, 2.0 Hz, 1H), 7.26 (d, J = 2.5 Hz, 1H), 7.22 (dd, J = 8.5, 2.5 Hz, 1H), 6.67 (d, J = 8.5 Hz, 1H), 5.19 (br s, 1H), 4.10 (s, 3H) ppm.
Synthesis of 2-heptyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenol (8c)
7c (0.235 g, 0.867 mmol), potassium acetate (0.256 g, 2.6 mmol), and PdCl2dppf (0.043 g, 6 mol %) were added to a round bottom flask and flushed with nitrogen. Dry dioxane (20 mL) was added via syringe and the reaction was stirred at 80 °C for 2 hours. Bis(pinacolato)diboron (0.242 g, 0.953 mmol) in dry dioxane (5 mL) was added via syringe and the reaction was stirred at 80 °C overnight. The solvent was evaporated, filtered though Celite with ethylacetate and evaporated. The crude mixture was eluted with ethylacetate, washed with saturated ammonium chloride, water (x2), and brine (x2), and dried over magnesium sulfate. The solvent was evaporated and the crude mixture (0.600 g brown oil) was purified by flash chromatography on silica gel (Hexane/EtOAc 95:5). The desired product (0.125 g, 45%) was isolated as a yellow oil. 1H NMR (500 MHz, CDCl3): δ = 7.63 (s, 1H), 7.56 (dd, J = 7.0, 1.5 Hz, 1H), 6.77 (d, J = 8.0 Hz, 1H), 5.82 (s, 1H), 2.63 (t, J = 7.5 Hz, 2H), 1.59–1.62 (m, 2H), 1.38 (s, 12H), 1.26–1.42 (m, 8H), 0.92 (t, J = 7.0 Hz, 3H) ppm.
2-(4-biphenylmethyl)-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenol (8a)
Synthesis same as 8c, but from 7a. Product isolated in 50% yield as a white solid. 1H NMR (400 MHz, Acetone-d6): δ = 8.80 (s, 1H), 7.59–7.64 (m, 3H), 7.51–7.57 (m, 3H), 7.42 (t, J = 6.8 Hz, 2H), 7.37 (d, J = 8 Hz, 1H), 7.32 (d, J = 7.2 Hz, 1H), 6.93 (d, J = 7.6 Hz, 1H), 4.06 (s, 2H), 1.29 (s, 12H) ppm.
2-(cyclohexylmethyl)-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenol (8b)
Synthesis same as 8c, but from 7b. Product isolated in 50% yield as a yellow oil. 1H NMR (400 MHz, CDCl3): δ =
4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-2-((naphthalen-2-yl)methyl)phenol (8d)
Synthesis same as 8c, but from 7d. Product isolated in 53% yield as a yellow oil. 1H NMR (500 MHz, CDCl3): δ = 7.70–7.80 (m, 4H), 7.63 (s, 1H), 7.62 (dd, J = 8.5, 2.0 Hz, 1H), 7.39–7.45 (m, 2H), 7.37 (dd, J = 9.0, 2.0 Hz, 1H), 6.75 (d, J = 8.0 Hz, 1H), 5.52 (br s, 1H), 4.14 (s, 2H), 1.32 (s, 12H) ppm.
Synthesis of ethyl 2-(4-bromo-2-heptylphenoxy)acetate (9c)
Sodium hydride (60% in oil, 0.04 g, 0.986 mmol) was added slowly to a solution of 7c (0.191 g, 0.704 mmol) in dry THF (20 mL). The solution was stirred at room temperature for 1.5 hours. Ethyl bromoacetate (0.165 g, 0.986 mmol) was added and the reaction was stirred overnight at room temperature. The solvent was evaporated and the white crude material was eluted with ethyl acetate, washed with saturated sodium bicarbonate, water (x2), brine (x2), and dried over magnesium sulfate. The solvent was evaporated and the crude mixture was (0.324 g clear, yellow oil) was purified by flash chromatography on silica gel (100% Hexane). The desired product (0.219 g, 87%) was isolated as a clear oil. 1H NMR (500 MHz, CDCl3): δ = 7.29 (d, J = 2.0 Hz, 1H), 7.24 (dd, J = 8.5, 2.5 Hz, 1H), 6.60 (d, J = 8.0 Hz, 1H), 4.62 (s, 2H), 4.28 (q, J = 8.0 Hz, 2H), 2.66(t, J = 7.5 Hz, 2H), 1.58–1.66 (m, 2H), 1.26–1.38 (m, 8H), 0.91 (t, J = 7.0 Hz, 3H) ppm.
Ethyl 2-(4-bromo-2-(4-biphenylmethyl)phenoxy)acetate (9a)
Same synthesis as 9c, but from 7a. Product isolated in 69% yield as a white solid. 1H NMR (400 MHz, CDCl3): δ = 7.57 (d, J = 7.2 Hz, 2H), 7.42 (t, J = 7.6 Hz, 2H), 7.29–7.35 (m, 3H), 7.23–7.35 (m, 2H), 6.62 (d, J = 5.6 Hz, 2H), 4.60 (s, 2H), 4.26 (q, J = 7.2 Hz, 2H), 4.04 (s, 2H), 1.27 (t, J = 7.2 Hz, 3H) ppm.
Ethyl 2-(4-bromo-2-(cyclohexylmethyl)phenoxy)acetate (9b)
Same synthesis as 9c, but from 7b. Product isolated in 91% yield as a yellow oil. 1H NMR (500 MHz, CDCl3): δ = 7.19–7.23 (m, 2H), 6.57 (d, J = 9.0 Hz, 1H), 4.59 (s, 2H), 4.25 (q, J = 7.0 Hz, 2H), 2.51 (d, J = 8.0 Hz, 2H), 1.54–1.71 (m, 6H), 1.28 (t, J = 7.5 Hz, 3H), 1.12–1.22 (m, 3H), 0.92–1.02 (m, 2H) ppm.
Ethyl 2-(4-bromo-2-((naphthalen-2-yl)methyl) phenoxy)acetate (9d)
Same synthesis as 9c, but from 7d. Product isolated in 83% yield as a yellow oily solid. 1H NMR (500 MHz, CDCl3): δ = 7.78 (d, J = 8.5 Hz, 1H), 7.76 (d, J = 8.5 Hz, 1H), 7.74 (d, J = 8.5 Hz, 1H), 7.68 (s, 1H), 7.43 (t, J = 7.5 Hz, 1H), 7.40 (t, J = 8.0 Hz, 1H), 7.37 (d, J = 8.0 Hz, 1H), 7.22–7.27 (m, 2H), 6.60 (d, J = 8.0 Hz, 1H), 4.57 (s, 2H), 4.24 (q, J = 7.0 Hz, 2H), 4.15 (s, 2H), 1.26 (t, J = 7.0 Hz, 3H) ppm.
Synthesis of Ethyl 2-(1,1′-biphenyl-4′-ol-4-oxy) acetate (2m)
9e (0.167 g, 0.45 mmol), sodium carbonate (0.095 g, 0.9 mmol), PdCl2(P(otol)3)2 (0.018 g, 5 mol %), and P(otol)3 (0.007 g, 5 mol %) added to test tube and flushed with argon. Solvent was added via syringe (Dichloromethane/water 3 mL: 2 mL) and the test tube was sealed and heated at 80 °C for 30 minutes. Reaction was cooled to room temperature, 8e (0.198 g, 0.9 mmol) was added; test tube was resealed, heated at 80 °C, and stirred overnight. The solvent was evaporated and the crude material was eluted with ethyl acetate, washed with saturated ammonium chloride, brine (x2) water (x2), and dried over magnesium sulfate. Solvent was evaporated and the crude material was purified by flash chromatography on silica gel (Hexane/EtOAc, 80:20). The desired product was isolated (0.105 g, 86%) as a white solid (m.p. 131–132 °C). 1H NMR (400 MHz, CDCl3): δ = 7.45 (d, J = 8.8 Hz, 2H), 7.49 (d, J = 8.8 Hz, 2H), 6.95 (d, J = 8.0 Hz, 2H), 6.87 (d, J = 8.0 Hz, 2H), 5.41(br s, 1H), 4.67 (s, 2H), 4.30 (q, J = 7.2 Hz, 2H), 1.32 (t, J = 7.2 Hz, 3H) ppm; 13C NMR (100 MHz, CDCl3): δ = 169.5, 157.1, 155.2, 134.8, 133.4, 128.2, 128.0, 155.9, 115.1, 65.7, 61.8, 14.4 ppm.
Ethyl 2-(1,1′-biphenyl-3-(4-biphenylmethyl)-4′-ol-4-oxy) acetate (2a)
Same synthesis as 2m with 8e and 9a. Product isolated in 78 % yield as a white (m.p. 182–183 °C). 1H NMR (400 MHz, CDCl3/CD3OD): δ = 7.35 (d, J = 7.2 Hz, 2H), 7.28 (d, J = 8.0 Hz, 2H), 7.05–7.21 (m, 9H), 6.64 (d, J = 8.8 Hz, 2H), 6.59 (d, J = 8.0 Hz, 1H), 4.44 (s, 2H), 4.27 (s, 1H), 4.05 (q, J = 8.8 Hz, 2H), 3.91 (s, 2H), 1.09 (t, J = 7.8 Hz, 3H) ppm; 13C NMR (100 MHz, CDCl3/CD3OD): δ = 169.6, 156.1, 154.7, 141.1, 140.1, 138.7, 134.8, 132.3, 130.4, 129.4, 129.0, 128.6, 127.8, 126.9, 126.9, 126.8, 125.4, 115.5, 112.0, 65.8, 61.4, 35.7, 13.9 ppm.
Ethyl 2-(1,1′-biphenyl-3′-(4-biphenylmethyl)-4′-ol-4-oxy) acetate (2b)
Same synthesis as 2m with 8a and 9e. Product isolated in 73 % yield as a white solid solid (m.p. 155–156 °C). 1H NMR (400 MHz, CDCl3/CD3OD): δ = 7.49 (d, J = 6.8 Hz, 2H), 7.43 (d, J = 8.0 Hz, 2H), 7.30–7.38 (m, 5H), 7.27 (d, J = 8.4 Hz, 2H), 7.20 (s, 1H), 7.19 (dd, J = 8.0, 2.0 Hz, 1H), 6.85 (d, J = 8.8 Hz, 2H), 6.80 (d, J = 8.0 Hz, 1H), 4.56 (s, 2H), 4.20 (q, J = 7.6 Hz, 2H), 3.40 (s, 2H), 1.23 (t, J = 8.4 Hz, 3H) ppm; 13C NMR (100 MHz, CDCl3/CD3OD): δ = 169.6, 156.8, 154.2, 141.3, 140.3, 138.9, 135.1, 132.5, 129.4, 129.2, 128.8, 128.0, 127.9, 127.2, 127.1, 127.1, 125.9, 115.6, 115.0, 65.6, 61.7, 35.7, 14.2 ppm.
Ethyl 2-(1,1′-biphenyl-3,3′-(di(4-biphenylmethyl))-4′-ol-4-oxy) acetate (2c)
Same synthesis as 2m with 8a and 9a. Product isolated in a 70 % yield as a yellow solid (m.p. 185–186 °C). 1H NMR (400 MHz, Acetone-d6): δ = 7.61 (d, J = 7.2 Hz, 4H), 7.53 (d, J = 8.8 Hz, 2H), 7.52 (d, J = 8.0 Hz, 2H), 7.34–7.47 (m, 11H), 7.27–7.34 (m, 3H), 6.94 (d, J = 8.8 Hz, 1H), 6.92 (d, J = 8.0 Hz, 1H), 4.78 (s, 2H), 4.24 (q, J = 7.2 Hz, 2H), 4.11 (s, 2H) 4.06 (s, 2H), 1.27 (t, J = 7.2 Hz, 3H) ppm; 13C NMR (100 MHz, Acetone-d6): δ =
Ethyl 2-(1,1′-biphenyl-3-cyclohexylmethyl -4′-ol-4-oxy) acetate (2d)
Same synthesis as 2m with 8e and 9b. Product isolated in quantitative yield as a yellow solid (m.p. 152–153 °C). 1H NMR (400 MHz, CDCl3/CD3OD): δ = 7.40 (d, J = 8.0 Hz, 2H), 7.26–7.32 (m, 2H), 6.88 (d, J = 8.8 Hz, 2H), 6.75 (d, J = 9.6 Hz, 1H), 4.66 (s, 2H), 4.27 (q, J = 7.2 Hz, 2H), 4.22 (br s, 1H), 2.60 (d, J = 6.8 Hz, 2H), 1.58–1.76 (m, 6H), 1.31 (t, J = 7.0 Hz, 3H), 1.12–1.24 (m, 3H), 0.94–1.08 (m, 2H) ppm; 13C NMR (100 MHz, CDCl3/CD3OD): δ = 169.8, 156.0, 155.1, 134.2, 132.6, 130.7, 129.6, 127.9, 124.8, 115.6, 111.6, 65.8, 61.5, 38.5, 38.2, 33.4, 26.7, 26.4, 14.1 ppm.
Ethyl 2-(1,1′-biphenyl-3′-cyclohexylmethyl -4′-ol-4-oxy) acetate (2e)
Same synthesis as 2m with 8b and 9e. Product isolated in 73 % yield as a white solid (m.p. 140–141 °C). 1H NMR (400 MHz, CDCl3): δ = 7.46 (d, J = 8.8 Hz, 2H), 7.24 (br s, 2H), 6.96 (d, J = 8.0 Hz, 2H), 6.82 (d, J = 8.0 Hz, 1H), 4.78 (s, 1H), 4.65 (s, 2H), 4.29 (q, J = 7.2 Hz, 2H), 2.53 (d, J = 7.6 Hz, 2H), 1.62–1.78 (m, 6H), 1.32 (t, J = 7.0 Hz, 3H), 1.13–1.24 (m, 3H), 0.96–1.06 (m, 2H) ppm; 13C NMR (100 MHz, CDCl3): δ =
Ethyl 2-(1,1′-biphenyl-3,3′-di(cyclohexylmethyl) -4′-ol-4-oxy) acetate (2f)
Same synthesis as 2m with 8b and 9b. Product isolated in 50% yield as a white solid (m.p. 118–119 °C). 1H NMR (400 MHz, Acetone-d6): δ = 8.19 (br s, 1H), 7.31–7.37 (m, 2H), 7.29 (d, J = 2.0 Hz, 1H), 7.25 (dd, J = 8.0, 2.0 Hz, 1H), 6.88 (d, J = 8.4 Hz, 2H), 4.74 (s, 2H), 4.22 (q, J = 7.2 Hz, 2H), 2.61 (d, J = 6.8 Hz, 2H), 2.56 (d, J = 6.8 Hz, 2H), 1.58–1.76 (m, 12H), 1.26 (t, J = 7.6 Hz, 3H), 1.12–1.23 (m, 6H), 0.96–1.18 (m, 4H) ppm; 13C NMR (100 MHz, Acetone-d6): δ = 168.9, 155.4, 154.7, 134.2, 132.1, 130.2, 129.5, 129.3, 127.9, 125.1, 124.9, 115.5, 111.8, 65.4, 60.8, 38.5, 38.4, 38.3, 38.2, 33.4, 33.4, 26.7, 26.4, 13.9 ppm.
Ethyl 2-(1,1′-biphenyl-3-n-heptyl -4′-ol-4-oxy) acetate (2g)
Same synthesis as 2m with 8e and 9c. Product isolated in 95% yield as an oily white solid (m.p. 69–70 °C). 1H NMR (400 MHz, CDCl3): δ = 7.49 (d, J = 8.8 Hz, 2H), 7.32 (d, J = 1.6 Hz, 1H), 7.27 (dd, J = 8.8, 2.8 Hz, 1H), 6.85 (d, J = 8.8 Hz, 2H), 6.75 (d, J = 8.8 Hz, 1H), 5.37 (br s, 1H), 4.68 (s, 2H), 4.29 (q, J = 7.1 Hz, 2H), 2.72 (t, J = 7.6 Hz, 2H), 1.65 (m, 2H), 1.24–1.42 (m, 8H), 1.32 (t, J = 6.8 Hz, 3H), 0.89 (t, J = 7.0 Hz, 3H) ppm; 13C NMR (100 MHz, CDCl3): δ = 169.7, 155.1, 155.0, 134.4, 133.8, 132.5, 128.9, 128.2, 125.0, 115.8, 111.6, 65.9, 61.6, 32.1, 30.6, 30.2, 29.9, 29.5, 22.9, 14.4, 14.4 ppm.
Ethyl 2-(1,1′-biphenyl-3′-n-heptyl -4′-ol-4-oxy) acetate (2h)
Same synthesis as 2m with 8c and 9e. Product isolated in 84% yield as a white solid (m.p. 128–129 °C). 1H NMR (400 MHz, CDCl3): δ = 7.46 (d, J = 8.0 Hz, 2H), 7.29 (d, J = 2.0 Hz, 1H), 7.23 (dd, J = 8.0, 2.0 Hz, 1H), 6.95 (d, J = 8.8 Hz, 2H), 6.80 (d, J = 8.0 Hz, 1H), 4.88 (br s, 1H), 4.65 (s, 2H), 4.29 (q, J = 8.8 Hz, 2H), 2.64 (t, J = 8.0 Hz, 2H), 1.60–1.69 (m, 2H), 1.23–1.42 (m, 8 H), 1.31 (t, J = 8.4 Hz, 3H), 0.88 (t, J = 6.6 Hz, 2H) ppm; 13C NMR (100 MHz, CDCl3): δ = 169.4, 157.0, 153.0, 135.1, 133.5, 129.2, 128.8, 128.1, 125.5, 115.7, 115.1, 65.8, 61.7, 32.1, 30.4, 30.1, 29.8, 29.5, 22.9, 14.4, 14.4 ppm.
Ethyl 2-(1,1′-biphenyl-3,3′-di-n-heptyl-4′-ol-4-oxy) acetate (2i)
Same synthesis as 2m with 8c and 9c. Product isolated in 86% yield as an oily yellow solid (m.p. 73–74 °C). 1H NMR (400 MHz, CDCl3): δ = 7.32 (d, J = 2.4 Hz, 1H), 7.28 (d, J = 2.0 Hz, 1H), 7.27 (dd, J = 10.0, 2.8 Hz, 1H), 7.23 (dd, J = 8.6, 1.4 Hz, 1H), 6.79 (d, J = 8.0 Hz, 1H), 6.74 (d, J = 8.8 Hz, 1H), 5.04 (br s, 1H), 4.67 (s, 2H), 4.28 (q, J = 6.8 Hz, 2H), 2.72 (t, J = 7.6 Hz, 2H), 2.64 (t, J = 7.6 Hz, 2H), 1.58–1.70 (m, 4H), 1.20–1.43 (m, 16H), 1.31 (t, J = 7.2 Hz, 3H), 0.88 (t, J = 7.0 Hz, 6H) ppm; 13C NMR (100 MHz, CDCl3): δ = 169.6, 155.1, 153.0, 134.7, 133.8, 132.4, 129.1, 128.9, 128.9, 125.6, 125.1, 115.7, 111.7, 66.0, 61.6, 32.1, 32.1, 30.7, 30.3, 30.2, 29.9, 29.8, 29.5, 29.5, 22.9, 22.9, 14.4, 14.4, 14.4 ppm.
Ethyl 2-(1,1′-biphenyl-3-(naphthalen-2-yl)methyl-4′-ol-4-oxy) acetate (2j)
Same synthesis as 2m with 8e and 9d. Product isolated in 80% yield as a white solid (m.p. 154–155 °C). 1H NMR (400 MHz, CDCl3/CD3OD): δ = 7.60–7.68 (m, 3H), 7.57 (s, 1H), 7.26–7.33 (m, 5H), 7.15 (d, J = 6.8 Hz, 1H), 7.14 (s, 1H), 6.78 (d, J = 8.0 Hz, 2H), 6.76 (d, J = 8.8 Hz, 1H), 4.50 (s, 2H), 4.15 (q, J = 7.2 Hz, 2H), 4.07 (s, 2H), 1.18 (t, J = 6.2 Hz, 3H) ppm; 13C NMR (100 MHz, CDCl3/CD3OD): δ = 169.6, 156.7, 154.3, 138.7, 135.1, 133.8, 132.3, 132.2, 129.2, 127.9, 127.9, 127.8, 127.8, 127.6, 127.6, 127.0, 125.8, 125.8, 125.2, 115.5, 114.9, 65.6, 61.7, 36.1, 14.1 ppm.
Ethyl 2-(1,1′-biphenyl-3′-(naphthalen-2-yl)methyl-4′-ol-4-oxy) acetate (2k)
Same synthesis as 2m with 8d and 9e. Product isolated in 95% yield as a white solid. (m.p. 162–163 °C). 1H NMR (400 MHz, CDCl3/CD3OD): δ = 7.72–7.81 (m, 4H), 7.39–7.48 (m, 3H), 7.32–7.38 (m, 4H), 6.84 (d, J = 7.2 Hz, 2H), 6.82 (d, J = 9.6 Hz, 1H), 4.65 (s, 2H), 4.27 (q, J = 7.2 Hz, 2H), 4.26 (s, 2H), 1.23 (t, J = 7.2 Hz, 3H) ppm; 13C NMR (100 MHz, CDCl3/CD3OD): δ = 169.6, 156.1, 154.8, 138.5, 134.9, 133.7, 132.4, 132.1, 130.5, 129.2, 127.9, 127.9, 127.8, 127.6, 127.6, 127.2, 125.8, 125.6, 125.2, 115.6, 112.1, 66.0, 61.5, 36.4, 14.1 ppm.
Ethyl 2-(1,1′-biphenyl-3,3′-(di(naphthalen-2-yl))methyl-4′-ol-4-oxy) acetate (2l)
Same synthesis as 2m with 8d and 9d. Product isolated in 40% yield as a yellow solid (m.p. 142–144 °C). 1H NMR (400 MHz, CDCl3/CD3OD): δ = 7.73–7.90 (m, 8H), 7.51 (dd, J = 8.8, 1.6 Hz, 1H), 7.39–7.48 (m, 7H), 7.34 (dd, J = *8.0, 2.0 Hz, 1H), 7.26 (dd, J = 8.0, 2.0 Hz, 1H), 6.93 (d, J = 8.8 Hz, 1H), 6.91 (d, J = 8.8 Hz, 1H), 4.76 (s, 2H), 4.23 (q, J = 7.6 Hz, 2H), 4.22 (s, 2H), 4.17 (s, 2H), 1.25 (t, J = 7.2 Hz, 3H) ppm; 13C NMR (100 MHz, CDCl3/CD3OD): δ = 169.4, 155.1, 153.5, 138.6, 137.7, 134.7, 133.9, 133.8, 132.4, 132.3, 130.7, 129.7, 129.5, 128.5, 128.1, 128.0, 127.9, 127.8, 127.8, 127.8, 127.8, 127.6, 127.4, 127.4, 127.0, 126.5, 126.3, 126.0, 125.9, 125.7, 125.4, 116.3, 112.1, 66.1, 61.6, 36.9, 36.6, 14.4 ppm.
Synthesis of Ethyl 2-(1,1′-biphenyl-4′-[2-(dimethylamino) ethoxy]-4-oxy) acetate (1m)
2m (0.027 mg, 0.1 mmol), potassium carbonate (0.014 mg, 0.5 mmol), and acetone (2 mL) were added to a test tube which was flushed with argon, sealed and heated at reflux for 1 hour. The reaction was then cooled to room temperature, 2-dimethylaminoethylchloride hydrochloride (0.015 mg, 0.3 mmol) was added, and the reaction was heated to reflux overnight. Solvent was evaporated, and the crude mixture was eluted with ethyl acetate, washed with water (x2), brine (x2), and dried with magnesium sulfate. Solvent was evaporated and the crude mixture (0.032 g white solid) was purified by flash chromatography on silica gel (Hexane/EtOAc, 50:50). The desired product was isolated as a white solid (0.026 g, 76% yield). 1H NMR (400 MHz, Acetone-d6): δ = 7.53 (d, J = 8.8 Hz, 2H), 7.52 (d, J = 8.8 Hz, 2H), 6.99 (d, J = 8.8 Hz, 4H), 4.75 (s, 2H), 4.22 (q, J = 7.2 Hz, 2H), 4.11(t, J = 6.4 Hz, 2H), 2.69 (t, J = 5.8 Hz, 2H), 2.28 (s, 6H), 1.26 (t, J = 7.4 Hz, 3H) ppm; 13C NMR (100 MHz, Acetone-d6): δ = 168.7, 158.5, 157.6, 134.1, 133.1, 127.7, 127.6, 115.1, 115.0, 66.6, 65.2, 60.8, 58.3, 45.5, 13.8 ppm.
Ethyl 2-(1,1′-biphenyl-3-(4-biphenylmethyl)-4′-[2-(dimethylamino) ethoxy]-4-oxy) acetate (1a)
Same synthesis as 1m from 2a. Product isolated in 31% yield as a white solid (m.p. 76–77 °C). 1H NMR (400 MHz, Acetone-d6): δ = 7.62 (d, J = 7.2 Hz, 2H), 7.50–7.60 (m, 5H), 7.40–7.48 (m, 5H), 7.32 (t, J = 7.6 Hz, 1H), 7.04 (d, J = 8.8 Hz, 1H), 6.99 (d, J = 8.8 Hz, 2H), 4.73 (s, 2H), 4.21 (q, J = 7.2 Hz, 2H), 4.13 (t, J = 5.8 Hz, 2H), 4.07 (s, 2H), 2.72 (t, J = 5.8 Hz, 2H), 2.28 (s, 6H), 1.25 (t, J = 7.0 Hz, 3H) ppm; 13C NMR (100 MHz, Acetone-d6): δ = 168.8, 157.5, 156.2, 141.2, 140.9, 138.7, 134.3, 133.1, 130.4, 129.7, 129.0, 128.8, 127.7, 127.2, 126.9, 126.9, 125.7, 115.1, 112.2, 66.9, 65.2, 60.8, 58.4, 45.6, 35.9, 13.8 ppm.
Ethyl 2-(1,1′-biphenyl-3′-(4-biphenylmethyl)-4′-[2-(dimethylamino) ethoxy]-4-oxy) acetate (1b)
Same synthesis as 1m from 2b.. Product isolated in 70% yield as a white solid (m.p. 100–101 °C). 1H NMR (400 MHz, Acetone-d6): δ = 7.62 (d, J = 7.6 Hz, 2H), 7.55 (d, J = 8.8 Hz, 2H), 7.45–7.55 (m, 5H), 7.38–7.45 (m, 3H), 7.32 (t, J = 7.6 Hz, 1H), 6.98 (d, J = 8.8 Hz, 3H), 4.79 (s, 2H), 4.25 (q, J = 7.2 Hz, 2H), 4.14 (s, 2H), 4.09 (t, J = 6.0 Hz, 2H), 2.67 (t, J = 6.2 Hz, 2H), 2.26 (s, 6H), 1.28 (t, J = 7.4 Hz, 3H) ppm; 13C NMR (100 MHz, Acetone-d6): δ = 168.8, 158.5, 155.2, 141.2, 140.8, 138.7, 134.2, 133.2, 130.7, 129.8, 129.0, 128.9, 127.7, 127.2, 126.9, 126.9, 125.5, 115.0, 112.4, 66.6, 65.6, 60.9, 58.3, 45.5, 35.6, 13.9 ppm.
Ethyl 2-(1,1′-biphenyl-3,3′-di(4-biphenylmethyl)-4′-[2-(dimethylamino) ethoxy]-4-oxy) acetate (1c)
Same synthesis as 1m from 2c. Product isolated in 35% yield as a clear oil. 1H NMR (400 MHz, Acetone-d6): δ = 7.60 (d, J = 8.0 Hz, 4H), 7.48–7.56 (m, 16H), 7.31 (t, J = 7.6 Hz, 2H), 7.02 (d, J = 8.0 Hz, 1H), 6.96 (d, J = 8.8 Hz, 1H), 4.78 (s, 2H), 4.24 (q, J = 7.6 Hz, 2H), 4.12 (t, J = 6.2 Hz, 2H), 4.12 (s, 2H), 4.05 (s, 2H), 2.71 (t, J = 5.8 Hz, 2H), 2.72 (s, 6H), 1.27 (t, J = 7.0 Hz, 3H) ppm; 13C NMR (100 MHz, Acetone-d6): δ = 168.8, 156.2, 155.2, 141.2, 140.8, 140.7, 138.7, 138.7, 134.3, 130.7, 130.3, 129.8, 129.7, 129.0, 129.0, 128.8, 128.8, 127.2, 127.2, 126.9, 126.9, 126.9, 126.9, 125.7, 125.6, 112.4, 112.1, 66.9, 65.6, 60.9, 58.4, 45.6, 35.8, 35.6, 13.9 ppm.
Ethyl 2-(1,1′-biphenyl-3-cyclohexylmethyl -4′-[2-(dimethylamino) ethoxy]-4-oxy) acetate (1d)
Same synthesis as 1m from 2d. Product isolated in 36% yield as a yellow oil.. 1H NMR (400 MHz, Acetone-d6): δ = 13C NMR (100 MHz, Acetone-d6): δ = 168.
Ethyl 2-(1,1′-biphenyl-3′-cyclohexylmethyl -4′-[2-(dimethylamino) ethoxy]-4-oxy) acetate (1e)
Same synthesis as 1m from 2e. Product isolated in 67% yield as a clear oil. 1H NMR (400 MHz, Acetone-d6): δ = 7.53 (d, J = 8.8 Hz, 2H), 7.39 (dd, J = 7.2, 2.0 Hz, 1H), 7.34 (d, J = 2.0 Hz, 1H), 6.99 (d, 8.8 Hz, 2H), 4.74 (s, 2H), 4.22 (q, J = 6.8 Hz, 2H), 4.12 (t, J = 6.0 Hz, 2H), 2.72 (t, J = 6.2 Hz, 2H), 2.55 (d, J = 6.8 Hz, 2H), 2.30 (s, 6H), 1.58–1.72 (m, 6H), 1.26 (t, J = 7.0 Hz, 3H), 1.10–1.22 (m, 3H), 0.94–1.06 (m, 2H) ppm; 13C NMR (100 MHz, Acetone-d6): δ = 168.8, 157.5, 156.6, 134.5, 132.5, 130.1, 129.2, 127.6, 125.1, 115.1, 111.8, 67.0, 65.2, 60.8, 58.5, 45.7, 38.5, 38.4, 33.4, 26.7, 26.4, 13.8 ppm.
Ethyl 2-(1,1′-biphenyl-3,3′-(cyclohexylmethyl) -4′-[2-(dimethylamino) ethoxy]-4-oxy) acetate (1f)
Same synthesis as 1m from 2f. Product isolated in 65% yield as a yellow oil. 1H NMR (400 MHz, Acetone-d6): δ = 7.32–7.40 (m, 4H), 6.98 (d, J = 8.8 Hz, 1H), 6.90 (d, J = 8.8 Hz, 1H), 4.75 (s, 2H), 4.22 (q, J = 7.2 Hz, 2H), 4.11 (t, J = 6.6 Hz, 2H), 2.72 (t, J = 6.0 Hz, 2H), 2.61 (d, J = 6.4 Hz, 2H), 2.55 (d, J = 6.8 Hz, 2H), 2.30 (s, 6H), 1.58–1.76 (m, 12H), 1.27 (t, J = 6.8 Hz, 3H), 1.12–1.24 (m, 6H), 0.94–1.09 (m, 4H) ppm; 13C NMR (100 MHz, Acetone-d6): δ = 168.9, 156.8, 155.5, 133.9, 132.8, 130.3, 130.0, 129.4, 129.2, 125.2, 125.0, 111.9, 111.8, 67.0, 65.4, 60.8, 58.5, 45.7, 38.5, 38.5, 38.4, 38.3, 33.4, 33.4, 26.7, 26.4, 26.4, 26.4, 13.8 ppm.
Ethyl 2-(1,1′-biphenyl-3-n-heptyl -4′-[2-(dimethylamino) ethoxy]-4-oxy) acetate (1g)
Same synthesis as 1m from 2g. Product isolated in 93% yield as a clear oil. 1H NMR (400 MHz, Acetone-d6): δ = 7.53 (d, J = 8.8 Hz, 2H), 7.39 (s, 1H), 7.38 (dd, J = 8.8, 2.4 Hz, 1H), 6.99 (d, J = 8.8 Hz, 3H), 4.74 (s, 2H), 4.22 (q, J = 7.0 Hz, 2H), 4.12 (t, J = 6.2 Hz, 2H), 2.73 (t, J = 6.0 Hz, 2H), 2.67 (t, J = 7.8 Hz, 2H), 2.30 (s, 6H), 1.59–1.68 (m, 2H), 1.28–1.40 (m, 8H), 1.26 (t, J = 7.2 Hz, 3H), 0.88 (t, J = 7.2 Hz, 3H) ppm; 13C NMR (100 MHz, Acetone-d6): δ = 168.9, 158.4, 155.4, 133.8, 131.9, 128.4, 127.7, 124.9, 115.0, 111.9, 66.5, 65.4, 60.8, 58.2, 45.5, 32.0, 30.5, 30.2, 29.6, 22.7, 13.8, 13.7 ppm.
Ethyl 2-(1,1′-biphenyl-3′-n-heptyl -4′-[2-(dimethylamino) ethoxy]-4-oxy) acetate (1h)
Same synthesis as 1m from 2h. Product isolated in 53% yield as a clear oil. 1H NMR (400 MHz, Acetone-d6): δ = 7.53 (d, J = 8.8 Hz, 2H), 7.39 (s, 1H), 7.38 (dd, J = 8.0, 2.4 Hz, 1H), 6.99 (d, J = 8.8 Hz, 3H), 4.74 (s, 2H), 4.22 (q, J = 7.0 Hz, 2H), 4.12 (t, J = 6.2 Hz, 2H), 2.73 (t, J = 5.6 Hz, 2H), 2.67 (t, J = 7.8 Hz, 2H), 2.30 (s, 6H), 1.58–1.68 (m, 2H), 1.28–1.40 (m, 8H), 1.26 (t, J = 7.2 Hz, 3H), 0.88 (t, J = 6.6 Hz, 3H) ppm; 13C NMR (100 MHz, Acetone-d6): δ = 168.8, 157.5, 156.3, 134.5, 132.8, 131.6, 128.3, 127.6, 125.1, 115.1, 111.8, 67.0, 65.2, 60.8, 58.5, 45.7, 32.0, 30.6, 30.3, 29.7, 29.3, 22.7, 13.8, 13.7 ppm.
Ethyl 2-(1,1′-biphenyl-3,3′-di-n-heptyl -4′-[2-(dimethylamino) ethoxy]-4-oxy) acetate (1i)
Same synthesis as 1m from 2i. Product isolated in 72% yield as a clear oil. 1H NMR (400 MHz, Acetone-d6): δ = 7.33–7.44 (m, 4H), 6.97 (d, J = 8.0 Hz, 1H), 6.89 (d, J = 8.0 Hz, 1H), 4.76 (s, 2H), 4.22 (q, J = 6.8 Hz, 2H), 4.12 (t, J = 6.2 Hz, 2H), 2.70–2.77 (m, 4H), 2.67 (t, J = 8.0 Hz, 2H), 1.59–1.72 (m, 4H), 1.30–1.43 (m, 16H), 1.27 (t, J = 7.2 Hz, 3H), 0.89 (t, J = 7.2 Hz, 3H) ppm; 13C NMR (100 MHz, Acetone-d6): δ = 168.8, 156.2, 155.3, 134.2, 133.2, 131.8, 131.6, 128.4, 128.3, 125.1, 124.9, 111.9, 111.8, 66.9, 65.4, 60.8, 58.5, 45.6, 32.0, 32.0, 30.6, 30.5, 30.3, 30.2, 29.7, 29.7, 29.3, 29.3, 22.7, 22.7, 13.8, 13.7, 13.7 ppm.
Ethyl 2-(1,1′-biphenyl-3-(naphthalen-2-yl)methyl -4′-[2-(dimethylamino) ethoxy]-4-oxy) acetate (1j)
Same synthesis as 1m from 2j. Product isolated in 63% yield as a white solid (m.p. 90–91 °C). 1H NMR (400 MHz, Acetone-d6): δ = 7.89 (s, 1H), 7.78–7.84 (m, 3H), 7.57 (dd, J = 8.0, 2.0 Hz, 1H), 7.52 (d, J = 2.4 Hz, 1H), 7.48 (d, J = 8.8 Hz, 2H), 7.38–7.45 (m, 3H), 6.99 (d, J = 8.0 Hz, 1H), 6.96 (d, J = 8.8 Hz, 2H), 4.80 (s, 2H), 4.26 (s, 2H), 4.24 (q, J = 7.2 Hz, 2H), 4.09 (t, J = 5.8 Hz, 2H), 2.66 (t, J = 5.8 Hz, 2H), 2.26 (s, 6H), 1.27 (t, J = 6.4 Hz, 3H) ppm; 13C NMR (100 MHz, Acetone-d6): δ = 168.8, 158.5, 155.3, 139.1, 134.1, 134.2, 133.2, 132.4, 130.6, 128.9, 128.1, 127.9, 127.7, 127.7, 127.7, 127.3, 126.0, 125.6, 125.3, 115.0, 112.4, 66.6, 65.6, 60.9, 58.3, 45.5, 36.1, 13.8 ppm.
Ethyl 2-(1,1′-biphenyl-3′-(naphthalen-2-yl)methyl -4′-[2-(dimethylamino) ethoxy]-4-oxy) acetate (1k)
Same synthesis as 1m from 2k. Product isolated in 44% yield as a clear oil. 1H NMR (400 MHz, Acetone-d6): δ = 7.76–7.84 (m, 4H), 7.46–7.54 (m, 4H), 7.38–7.44 (m, 3H), 7.04 (d, J = 8.8 Hz, 1H), 6.97 (d, J = 8.8 Hz, 2H), 4.73 (s, 2H), 4.21 (q, J = 7.0 Hz, 2H), 4.20 (s, 2H), 4.12 (t, J = 6.0 Hz, 2H), 2.71 (t, J = 6.0 Hz, 2H), 2.26 (s, 6H), 1.25 (t, J = 7.0 Hz, 3H) ppm; 13C NMR (100 MHz, Acetone-d6): δ = 168.9, 157.5, 156.3, 139.2, 134.2, 134.0, 133.0, 132.4, 130.3, 128.8, 128.1, 127.8, 127.7, 127.7, 127.7, 127.3, 126.0, 125.7, 125.3, 115.1, 112.2, 66.9, 65.2, 60.8, 58.5, 45.6, 36.4, 13.8 ppm.
Ethyl 2-(1,1′-biphenyl-3,3′-di(naphthalen-2-yl)methyl-4′-[2-(dimethylamino) ethoxy]-4-oxy) acetate (1l)
Same synthesis as 1m from 2l. Product isolated in 58% yield as a clear oil. 1H NMR (400 MHz, Acetone-d6): δ = 7.85 (s, 1H), 7.83 (s, 1H), 7.73–7.82 (m, 6H), 7.35–7.54 (m, 10H), 6.99 (d, J = 8.8 Hz, 1H), 6.94 (d, J = 8.0 Hz, 1H), 4.77 (s, 2H), 4.23 (s, 2H), 4.22 (q, J = 7.2 Hz, 2H), 4.16 (s, 2H), 4.10 (t, J = 6.0 Hz, 2H), 2.69 (t, J = 6.0 Hz, 2H), 2.25 (s, 6H), 1.25 (t, J = 6.4 Hz, 3H) ppm; 13C NMR (100 MHz, Acetone-d6): δ = 168.9, 156.3, 155.2, 139.1, 139.0, 134.3, 134.0, 133.1, 132.4, 132.4, 130.6, 130.2, 129.0, 128.9, 128.1, 128.1, 127.9, 127.8, 127.7, 127.7, 127.7, 127.7, 127.3, 127.2, 126.0, 126.0, 125.8, 125.6, 125.3, 125.3, 112.4, 112.1, 66.9, 65.6, 60.8, 58.4, 45.5, 36.4, 36.1, 13.8 ppm.
Supplementary Material
Acknowledgments
This work was supported in part by the Department of Defense PCRP Concept grant W81XWH-04-1-0647 (to R.N.H.), PCRP Training Award W81XWH-09-1-0208 (to P.T.W.), and Public Health Service award CA139818 (to D.P.M).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cummings CG, Hamilton AD. Disrupting protein–protein interactions with non-peptidic, small molecule α-helix mimetics. Current Opinion in Chemical Biology. 2010;14:341. doi: 10.1016/j.cbpa.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 2.Fry DC. Drug-Like Inhibitors of PPIs: A Structural Examination of Effective Protein Mimicry. Current Protein and Peptide Science. 2008;9:240. doi: 10.2174/138920308784533989. [DOI] [PubMed] [Google Scholar]
- 3.Sattler M, L H, Nattesheim D, Meadoes RP, Harlan JE, Eberstadt M, Yoon HS, Shuker SB, Change BS, Minn AJ, Thompson CB, Fesik SW. Science. 1997;275:983. doi: 10.1126/science.275.5302.983. [DOI] [PubMed] [Google Scholar]
- 4.Kussie PH, G S, Marechal V, Elenbaas B, Moreau J, Levine AJ, Pavletich N. Science. 1996;274:948. doi: 10.1126/science.274.5289.948. [DOI] [PubMed] [Google Scholar]
- 5.Rodriguez AL, T A, Collins L, Katzenellenbogen JA. J Med Chem. 2004;47:600. doi: 10.1021/jm030404c. [DOI] [PubMed] [Google Scholar]
- 6.Scott JE. Biochem. 1987;12:318. [Google Scholar]
- 7.Nolan WP, R BS, Rees DC. Tet Lett. 1992;33:6879. [Google Scholar]
- 8.Yin HL, L G, Sedey KA, Kutzki O, Park HS, Orner BP, Ernst JT, Wang HG, Sebti SM, Hamilton AD. J Am Chem Soc. 2006;127:10191. doi: 10.1021/ja050122x. [DOI] [PubMed] [Google Scholar]
- 9.Parent AAG, G JR, Katzenellenbogen JA. J Med Chem. 2009;51:6512. doi: 10.1021/jm800698b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Caboni L, Lloyd DG. Beyond the Ligand-Binding Pocket: Targeting Alternate Sites in Nuclear Receptors. Medicinal Research Reviews. 2013;33 doi: 10.1002/med.21275. [DOI] [PubMed] [Google Scholar]
- 11.Williams AB, Weiser PT, Hanson RN, Gunther JR, Katzenellenbogen JA. Synthesis of Biphenyl Proteomimetics as Estrogen Receptor-α Coactivator Binding Inhibitors. Org Lett. 2009;11:5370. doi: 10.1021/ol901999f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Savkur RS, B TP. J Peptide Res. 2004;63:207. doi: 10.1111/j.1399-3011.2004.00126.x. [DOI] [PubMed] [Google Scholar]
- 13.Weiser PT, Williams AB, Chang CY, McDonnell DP, Hanson RN. 3,3′-Disubstituted bipolar biphenyls as inhibitors of nuclear receptor coactivator binding. Bioorganic & Medicinal Chemistry Letters. 2012;22:6587. doi: 10.1016/j.bmcl.2012.09.007. [DOI] [PubMed] [Google Scholar]
- 14.Ishar MPS, S G, Signh S, Sreenivasan KK, Sign G. Bioorg Med Chem Lett. 2006;16:1366. doi: 10.1016/j.bmcl.2005.11.044. [DOI] [PubMed] [Google Scholar]
- 15.Beaulieu PL, Gillard J, Bykowski D, Brochu C, Dansereau N, Duceppe J, Hache B, Jakalian A, Lagace L, LaPlante S, McKercher G, Moreau E, Perreault S, Stammers T, Thauvette L, Warrington J, Kukolj G. Improved replicon cellular activity of non-nucleoside allosteric inhibitors of HCV NS5B polymerase: From benzimidazole to indole scaffolds. Bioorganic & Medicinal Chemistry Letters. 2006;16:4987. doi: 10.1016/j.bmcl.2006.07.074. [DOI] [PubMed] [Google Scholar]
- 16.Tang G, Ding K, Nikolovska-Coleska Z, Yang C, Qui S, Shangary S, Wang R, Guo J, Gao W, Meagher J, Stuckey J, Krajewski K, Jiang S, Roller PP, Wang S. Structure-Based Design of Flavonoid Compounds As a New Class of Small-Molecule Inhibitors of the Anti-apoptotic Bcl-2 Proteins. J Med Chem. 2007;50:3163. doi: 10.1021/jm070383c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Khodaei MM, Bahrami K, Shahbazi F. An efficient method for aromatic Friedel-Crafts acylation reactions. Chem Lett. 2008;37:844. [Google Scholar]
- 18.Pallavicini M, Budriesi R, Fumagalli L, Ioan P, Chiarini A, Bolchi C, Ugenti MP, Colleoni S, Gobbi M, Valoti E. WB4101-Related Compounds: New Subtype-Selective 1-Adrenoreceptor Antagonists (or Inverse Agonists?) J Med Chem. 2006;49:7140. doi: 10.1021/jm060358r. [DOI] [PubMed] [Google Scholar]
- 19.Valoti E, Pallavicini M, Villa L, Pezzetta D. Synthesis of Homochiral 5- and 8- Substituted2-[((2-(2,6-Dimethoxyphenoxy)-ethyl)amino)methyl]-1,4-benzodioxanes and Electrophoretic Determination of Their Enantiomeric Excess. J Org Chem. 2001;66:1018. doi: 10.1021/jo0008340. [DOI] [PubMed] [Google Scholar]
- 20.Agai B, Proszenyak A, Tarkanyi G, Vida L, Faigl F. Convenient, benign and scalable synthesis of 2- and 4-substituted benzylpiperidines. Eur J Org Chem. 2004:3623. [Google Scholar]
- 21.Hummel CW, Geiser AG, Bryant HU, Cohen IR, Dally RD, Fong KC, Frank SA, Hinklin R, Jones SA, Lewis G, McCann DJ, Rudmann DG, Shepherd TA, Tian H, Wallace OB, Wang M, Wang Y, Dodge JA. A Selective Estrogen Receptor Modulator Designed for the Treatment of Uterine Leiomyoma with Unique Tissue Specificity for Uterus and Ovaries in Rats. J Med Chem. 2005;48:6772. doi: 10.1021/jm050723z. [DOI] [PubMed] [Google Scholar]
- 22.Punna S, Meunier S, Finn MG. A Hierarchy of Aryloxide Deprotection by Boron Tribromide. Org Lett. 2004;6:2777. doi: 10.1021/ol0489898. [DOI] [PubMed] [Google Scholar]
- 23.Ren Y, Himmeldirk Klaus, Chen Xiaozhuo. Synthesis and Structure-Activity Relationshiop Study of Antidiabetic Penta-O-galloyl-D-Glucopyranose and Its Analogues. Journal of Medicinal Chemistry. 2006;49:2829. doi: 10.1021/jm060087k. [DOI] [PubMed] [Google Scholar]
- 24.Ishiyama T, Murata Miki, Miyaura Norio. Palladium(0)-Catalyzied Cross-Coupling Reaction of Alkoxydiboron with Haloarenes: A Direct Procedure for Arylboronic Esters. Journal of Organic Chemistry. 1995;60:7508. [Google Scholar]
- 25.Murata Miki, W S, Masuda Yuzuru. Novel Palladium(0)-Catalyzed Coupling Reaction of Dialkoxyborane with Aryl Halides: Convenient Synthetic Route to Arylboronates. J Org Chem. 1997;62:6458. [Google Scholar]
- 26.Chang CY, F D, Gron H, Hamilton PT, Kenan DJ, McDonnell DP, Norris JD, Paige LA. Dissection of the LXXLL Nuclear Receptor-Coactivator Interaction Motif Using Combinatorial Peptide Libraries: Discovery of Peptide Antagonists of Estrogen Receptors α and β. Mol Cell Biol. 1999;19:8226. doi: 10.1128/mcb.19.12.8226. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.