

Abstract
Background and Purpose
Palmitoylethanolamide (PEA), a naturally occurring acylethanolamide chemically related to the endocannabinoid anandamide, interacts with targets that have been identified in peripheral nerves controlling gastrointestinal motility, such as cannabinoid CB1 and CB2 receptors, TRPV1 channels and PPARα. Here, we investigated the effect of PEA in a mouse model of functional accelerated transit which persists after the resolution of colonic inflammation (post-inflammatory irritable bowel syndrome).
Experimental Approach
Intestinal inflammation was induced by intracolonic administration of oil of mustard (OM). Mice were tested for motility and biochemical and molecular biology changes 4 weeks later. PEA, oleoylethanolamide and endocannabinoid levels were measured by liquid chromatography-mass spectrometry and receptor and enzyme mRNA expression by qRT-PCR.
Key Results
OM induced transient colitis and a functional post-inflammatory increase in upper gastrointestinal transit, associated with increased intestinal anandamide (but not 2-arachidonoylglycerol, PEA or oleoylethanolamide) levels and down-regulation of mRNA for TRPV1 channels. Exogenous PEA inhibited the OM-induced increase in transit and tended to increase anandamide levels. Palmitic acid had a weaker effect on transit. Inhibition of transit by PEA was blocked by rimonabant (CB1 receptor antagonist), further increased by 5′-iodoresiniferatoxin (TRPV1 antagonist) and not significantly modified by the PPARα antagonist GW6471.
Conclusions and Implications
Intestinal endocannabinoids and TRPV1 channel were dysregulated in a functional model of accelerated transit exhibiting aspects of post-inflammatory irritable bowel syndrome. PEA counteracted the accelerated transit, the effect being mediated by CB1 receptors (possibly via increased anandamide levels) and modulated by TRPV1 channels.
Table of Links
| TARGETS | LIGANDS |
| CB1 receptors | Anandamide, AEA |
| CB2 receptors | 2-Arachidonoylglycerol, 2-AG |
| TRPV1 channels | GW6471 |
| 5′-Iodoresiniferatoxin, I-RTX | |
| Oleoylethanolamide, OEA | |
| Rimonabant | |
| SR144528 |
This Table lists the protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013a, b2013b).
Introduction
Irritable bowel syndrome (IBS) is a very common condition (incidence: 10–15% of the population) defined by abdominal pain and abdominal discomfort, associated with changes in stool frequency and form, in the absence of organic disease that is likely to explain the symptoms (Camilleri, 2012; Fichna and Storr, 2012; Izzo, 2013). Post-inflammatory IBS represents a subset of IBS, which occurs after episodes of inflammation. Although the symptoms of IBS can be targeted with a variety of agents, there is a paucity of drugs specifically licensed for the treatment of IBS (Voß et al., 2012; De Ponti, 2013). Due to unsatisfactory results with conventional treatments, complementary and alternative medications, including food supplements and plant-derived products, are being increasingly used (Magge and Lembo, 2011; Rahimi and Abdollahi, 2012).
Palmitoylethanolamide (PEA) is an ubiquitous lipid in animals, with anti-inflammatory, analgesic and neuroprotective actions, abundant also in plants, particularly in seeds (Esposito and Cuzzocrea, 2013; Skaper et al., 2014). In some European countries (Italy, Spain, the Netherlands and Germany), PEA is available – as foods for special medical purposes (brand names: Pelvilen, Recoclix, Epitech Srl) – to alleviate bowel complaints (Skaper et al., 2014). PEA is also available as food supplement (Normalia, Innovet Italia Srl) in veterinary clinical practice.
Apart from being a dietary component, PEA is an endogenous mediator structurally related to the endocannabinoid anandamide and shares pathways for biosynthesis and breakdown with this mediator (Lambert and Di Marzo, 1999). However, unlike anandamide, PEA does not bind to cannabinoid (CB) receptors, although it can indirectly activate them via the so-called ‘entourage effect’, that is the augmentation of the endocannabinoid levels and/or actions at CB receptors (De Petrocellis et al., 2002; Smart et al., 2002; Ho et al., 2008; receptor nomenclature follows Alexander et al., 2013a). Additional and pharmacologically relevant targets of PEA action include PPARα (Lo Verme et al., 2005; O'Sullivan and Kendall, 2010) and transient receptor potential vanilloid type-1 (TRPV1) channels (De Petrocellis et al., 2001; Ambrosino et al., 2013; channel nomenclature follows Alexander et al., 2013b), which have been identified in peripheral nerves controlling intestinal motility (Boesmans et al., 2011; Holzer, 2011). PEA has been identified in the rodent (Capasso et al., 2001; Fu et al., 2007; Izzo et al., 2010; 2012; Diep et al., 2011; Balvers et al., 2013) and human (Darmani et al., 2005; D'Argenio et al., 2007; Zhang et al., 2014) digestive tract. When given exogenously, PEA reduces gastrointestinal transit and displays anti-inflammatory effects in the gut (Di Paola et al., 2012; Petrosino et al., 2013; Esposito et al., 2014).
In this study, we have evaluated the effect of PEA in a mouse model of accelerated transit that persists after the resolution of colonic inflammation (Kimball et al., 2005). The experimental model is generated by intracolonic administration of oil of mustard (OM), which induces transient colitis (1–5 days after OM), and in the longer term (i.e. 28 days after OM), a functional increase in gastrointestinal transit that is observed when there is no more inflammation (Kimball et al., 2005). We selected this model to study the effect of PEA in post-inflammatory conditions because OM induces pathophysiological perturbations resembling those found in IBS patients, such as altered motility (Kimball et al., 2005) and visceral hyperalgesia (Laird et al., 2001).
Methods
Animals
All animal care and experimental procedures complied with the principles of laboratory animal care (NIH publication no.86–23, revised 1985) and the Italian D.L. no.116 of 27 January 1992 and associated guidelines in the European Communities Council Directive of 24 November 1986 (86/609/ECC). All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). A total of 423 animals were used in the experiments described here.
Male ICR mice were purchased from Harlan Italy (Corezzana, Milan, Italy) and housed in polycarbonate cages in isolators under a 12 h light/12 h dark cycle, with controlled temperature 23 ± 2°C and humidity 60%. Mice were fed ad libitum with standard food, except for the 24 h period immediately preceding the administration of OM and for the 12 h period preceding the measurement of intestinal transit.
Induction of experimental colitis
Colitis was induced by the intracolonic administration of OM (Kimball et al., 2005). Briefly, mice (26–28 g) were anaesthetized with inhaled 5% isoflurane (Centro Agrovete Campania, Scafati, SA, Italy) and subsequently OM (50 μL of a solution of 0.5% OM in 30% ethanol) was inserted into the colon using a polyethylene catheter (1 mm in diameter) via the rectum (4.5 cm from the anus). Control mice received intracolonic vehicle (50 μL of 30% ethanol). The mice were allowed to recover from anaesthesia under a warming light, and then were maintained with normal food and water for 28 days at which time they were tested for biochemical, biology molecular and pharmacological (upper gastrointestinal transit) experiments. In order to assess colonic damage, in some experiments, mice were killed 3 and 7 days after OM, the abdomen was opened by a midline incision. The colon was removed, isolated from surrounding the tissues, opened along the antimesenteric border, rinsed, weighed and length measured [in order to determined the colon weight/colon length ratio (mg·cm−1) used as a marker of inflammation] (Borrelli et al., 2013).
Upper gastrointestinal transit
Upper gastrointestinal transit was measured in control and in mice treated with OM (28 days after its intracolonic administration). Mice were deprived of food overnight prior to experimentation, although water was provided ad libitum. Upper gastrointestinal transit was determined by identifying the leading front of an intragastrically administered charcoal meal marker (0.1 mL per 10 g·body weight of a 10% charcoal suspension in 5% gum Arabic) in the small intestine as previously described (Kimball et al., 2010; Forbes et al., 2012; Wade et al., 2012). Twenty minutes after charcoal administration, the mice were killed by asphyxiation with CO2 and the small intestine was isolated by cutting at the pyloric and ileocaecal junctions. The distance travelled by the marker was measured and expressed as a percentage of the total length of the small intestine from pylorus to caecum (Forbes et al., 2012; Wade et al., 2012).
Pharmacological treatment
PEA (1–10 mg·kg−1) or palmitic acid was administered intraperitoneally 30 min prior to charcoal administration (both in control and in OM-treated mice). In some experiments, the effect of PEA (10 mg·kg−1, i.p.) was evaluated in animals pretreated (i.p., 30 min before PEA) with the CB1 receptor antagonist rimonabant (0.1 mg·kg−1), the CB2 antagonist SR144528 (1 mg·kg−1), the PPARα antagonist GW6471 (1 mg·kg−1) or the TRPV1 channels antagonist 5′-iodoresiniferatoxin (I-RTX, 0.17 mg·kg−1). The doses of rimonabant, SR144528 and GW6471 were selected on the basis of earlier work on gastrointestinal pharmacology (Capasso et al., 2008; Abalo et al., 2010; Di Paola et al., 2012). The dose of I-RTX was selected on the basis of preliminary experiments showing that the antagonist, at 0.17 mg·kg−1, did not affect, per se, upper gastrointestinal transit. Higher doses of I-RTX (i.e. 0.35 and 0.70 mg·kg−1), when given alone, did reduce intestinal transit in OM-treated mice. In some experiments, PEA (1–10 mg·kg−1) was given orally (1 h before charcoal administration, intragastric administration with the aid of a stomach tube).
Quantitative real-time (RT)-PCR analysis
The intestine (5cm length of jejunoileal tissues, starting 5 cm proximal of the caecum) from animals treated with vehicle (control group) or OM were removed (28 days after the administration of OM or vehicle), collected in RNA later (Invitrogen, Carlsbad, CA, USA) and homogenized by a rotorstator homogenizer in 1.5 mL of Trizol® (Invitrogen). Total RNA was purified, quantified, characterized and retro-transcribed as previously described (Grimaldi et al., 2009). For all samples tested, the RNA integrity number (Bionalyzer 2100, Agilent Technologies, Milan, Italy) was greater than 8 relative to a 0–10 scale. Quantitative RT-PCR was performed by an iCycler-iQ5® (Bio-Rad Laboratories, Milan, Italy) in a 20 μL reaction mixture as described. Assays were performed in quadruplicate (maximum ΔCt of replicate samples <0.5), and a standard curve from consecutive fivefold dilutions (100 to 0.16 ng) of a cDNA pool representative of all samples was included for PCR efficiency determination. Optimized primers for SYBR-green analysis and optimum annealing temperatures were designed by the Allele-Id software version 7.0 (Biosoft International, Palo Alto, CA, USA) and were synthesized (HPLC-purification grade) by Eurofins-MWG (Ebersberg, Germany). For each target, all mRNA sequences at http://www.ncbi.nlm.nih.gov/gene/ were aligned and common primers were designed (see Supporting Information Table S1 for primer sequences). Relative expression calculation, correct for PCR efficiency and normalized with respect to reference genes β-actin and hypoxanthine-guanine phosophoribosyltransferase (HPRT) was performed by the iQ5 software. Results are expressed as fold expression, compared with control (= 1) (Romano et al., 2013).
Identification and quantification of PEA, endocannabinoids and oleoylethanolamide
Full-thickness intestinal tissues (5cm length of jejunoileal tissues, starting 5 cm proximal of the caecum) from animals receiving OM or vehicle (with or without PEA 10 mg·kg−1, i.p.) were removed (28 days after OM administration), immediately immersed into liquid nitrogen and stored at –80°C until extraction of endocannabinoids [anandamide and 2-arachidonoylglycerol (2-AG)] and PEA. Tissues were extracted with chloroform/methanol (2:1, by volume) containing each 10 pmol of d8-anandamide, 50 pmol of d4-PEA and d5-2-AG, synthesized as described previously (for the former two compounds) (Di Marzo et al., 2008), or provided by Cayman Chemicals (for d5-2-AG, Ann Arbor, MI, USA). The lipid extracts were purified by silica column chromatography and the fractions containing anandamide, PEA, oleoylethanolamide and 2-AG were analysed by isotope dilution liquid chromatography–atmospheric pressure–chemical ionization mass spectrometry. Results were expressed as picomoles per milligram of tissue.
Data analysis
Data are expressed as the mean ± SEM of n experiments. To determine statistical significance, Student's t-test was used for comparing a single treatment mean with a control mean, and a one-way anova followed by a Tukey–Kramer multiple comparisons test was used for the analysis of multiple treatment means. Values of P less than 0.05 were considered significant.
Materials
Ultramicronized PEA (powder particle size <10 μm, with the following distribution: <6 μm, 99.9%; <2 μm, 59.6%; <1 μm, 14.7%; <0.6 μm, 2%, as described in patent EP 2475352 A1, with text from patent WO2011027373A1) was kindly provided by Epitech Group (Saccolongo, Italy). Ultramicronized PEA might differ, in terms of bioavailability, from non-ultramicronized PEA only when administered in an aqueous vehicle. Rimonabant and SR144528 (N-[(1S)-endo-1,3,3-trimethylbicyclo [2.2.1]heptan2-yl]-5-(4-chloro-3-methylphenyl)-1-[(4-methylphenyl)methyl]-1H-pyrazole-3-carboxamide) were provided by SANOFI Recherche, Montpellier, France. OM was purchased from Sigma Aldrich S.r.l. (Milan, Italy), 5′-Iodoresiniferatoxin (I-RTX), palmitic acid and GW6471 (N-[(2S)-2-[[(1Z)-1-methyl-3-oxo-3-[4-(trifluoromethyl)phenyl]-1-propen-1-yl]amino]-3-[4-[2-(5-methyl-2-phenyl-4-oxazolyl)ethoxy]phenyl]propyl]-propanamide) were supplied by Tocris (Bristol, UK). PEA and palmitic acid were dissolved in ethanol for i.p. injection (4 μL·per mouse) or suspended in carboxymethylcellulose (1.5%, 0.2 mL·per mouse) for oral administration. Rimonabant, SR144528, I-RTX and GW6471 were dissolved in DMSO (4 μL per·mouse). PEA, rimonabant, SR144528, GW6471 and I-RTX vehicles had no significant effects on the responses under study.
Results
OM induced transient colitis
OM administration evoked transient colitis. Distal colons from OM-treated mice showed shrinkage, thickening and severe erythema. When we assessed colonic inflammation by measuring the colon weight/colon length ratio (a simple and useful marker of intestinal inflammation), we found that colitis was transient, peaking at day 3 and absent at day 7 post-OM [colon weigh colon length−1 ratio, mg·cm−1: day 0 (control): 24.5 ± 0.9; day 3: 31.1 ± 1.6 (P < 0.01 vs. control); day 7: 26.6 ± 1.4, n = 5 for each experimental group]. Our data are in agreement with those of Kimball et al. who showed that the colitis induced by OM did not extend beyond 7 days (Kimball et al., 2005).
OM induced a post-inflammatory functional increase of upper gastrointestinal transit
In agreement with previous work (Kimball et al., 2005), OM induced a post-inflammatory accelerated upper gastrointestinal transit 28 days after its administration (Figure 1). Thus, OM increased transit 4 weeks after its administration in a site, the small intestine, distant from the site of acute inflammation, the colon.
Figure 1.
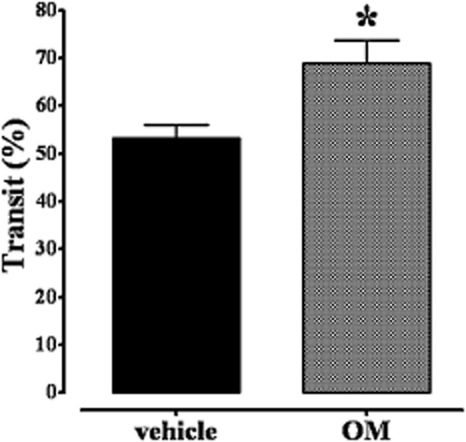
Effect of oil of mustard (OM) on upper gastrointestinal transit 28 days after its intracolonic administration (50 μL of a solution of 0.5% OM in 30% ethanol). Results are expressed as percentage of upper gastrointestinal transit (a low percentage indicates an anti-prokinetic effect). Bars represent the means ± SEM of 9–10 mice. *P < 0.05, significantly different from vehicle.
PEA reduced the post-inflammatory accelerated upper gastrointestinal transit induced by OM
Upper gastrointestinal transit studies were performed with PEA (single administration) given to mice 28 days after intracolonic vehicle (30% ethanol, control group) or OM. PEA (1–10 mg·kg−1, i.p.), given 30 min before the administration of the charcoal, reduced upper gastrointestinal transit both in control (Figure 2A) and in OM-treated mice (Figure 2B). However, while in control mice, the effect was significant at the highest dose tested (10 mg·kg−1), in OM-treated animals, PEA significantly reduced motility starting from the 2.5 mg·kg−1 dose. Analysis of the curves representing the percentage of inhibition of transit in control and OM-treated animals revealed that PEA preferentially decreased motility in OM-treated mice (Figure 2C).
Figure 2.
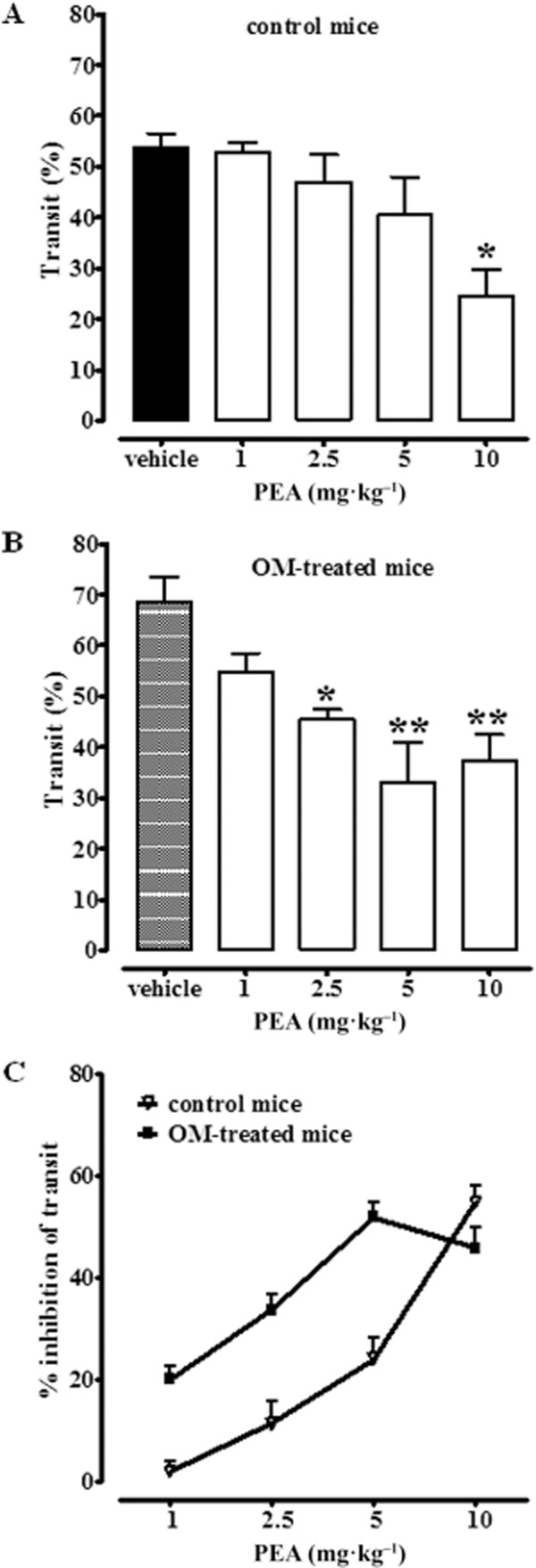
Inhibitory effect of PEA (1–10 mg·kg−1, i.p.) on upper gastrointestinal transit in control mice (A) and (B) in mice treated with OM. Transit was measured 28 days after OM or vehicle (30% ethanol) administration. Results (the means ± SEM of 9–10 mice for each experimental group) are expressed as a percentage of upper gastrointestinal transit. *P < 0.05, **P < 0.01, significantly different from vehicle. In (C), the effect of PEA (1–10 mg·kg−1, i.p.) on upper gastrointestinal transit is expressed as % of inhibition of corresponding control values. A statistically significant difference (P < 0.01) was observed between the two dose-response curves shown in (C). Note that in (A) the term ‘vehicle’ refers to the vehicle used to dissolve PEA, while in (B) the term ‘vehicle’ refers to the vehicle used to dissolve OM.
Palmitic acid had a little inhibitory effect on post-inflammatory accelerated upper gastrointestinal transit induced by OM
Because PEA may be hydrolyzed to palmitic acid, we tested the effects of this fatty acid (1–10 mg·kg−1, i.p.) and found no significant effects on upper gastrointestinal transit in control animals (Figure 3A). In OM-treated mice, palmitic acid significantly inhibited transit only at the highest dose (10 mg·kg−1) tested (Figure 3B).
Figure 3.
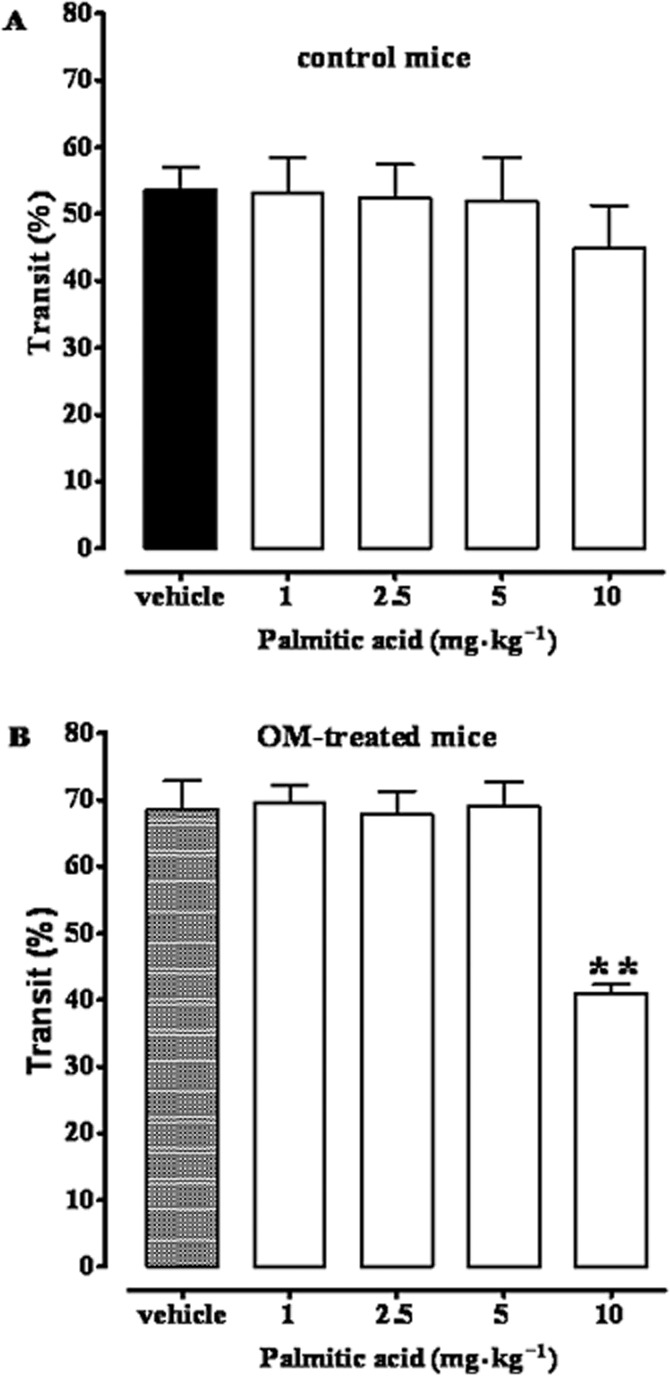
Effect of palmitic acid (1–10 mg·kg−1, i.p.) on upper gastrointestinal transit in control mice (A) and in OM-treated mice (B). Transit was measured 28 days after OM or vehicle (30% ethanol) administration. Results (the means ± SEM of 9–10 mice for each experimental group) are expressed as a percentage of upper gastrointestinal transit. **P < 0.01, significantly different from vehicle. Note that in (A) the term ‘vehicle’ refers to the vehicle used to dissolve palmitic acid, while in (B) the term ‘vehicle’ refers to the vehicle used to dissolve OM.
The CB1 receptor antagonist rimonabant blocked the inhibitory effect of PEA on upper gastrointestinal transit in OM-treated but not in control mice
Because PEA might indirectly activate CB receptors via the so-called entourage effect (De Petrocellis et al., 2002), we investigated the effect of PEA on upper gastrointestinal transit, both in control and OM-treated mice, in the presence of selective cannabinoid CB1 and CB2 receptor antagonists. Both rimonabant (CB1 antagonist, 0.1 mg·kg−1, i.p.) and SR144528 (CB2 antagonist, 1 mg·kg−1, i.p.) did not modify the inhibitory effect of PEA on upper gastrointestinal transit in control mice (Figure 4A). However, rimonabant, but not SR144528, attenuated the decrease in upper gastrointestinal transit induced by PEA in mice treated with OM (28 days after its administration) (Figure 4B).
Figure 4.
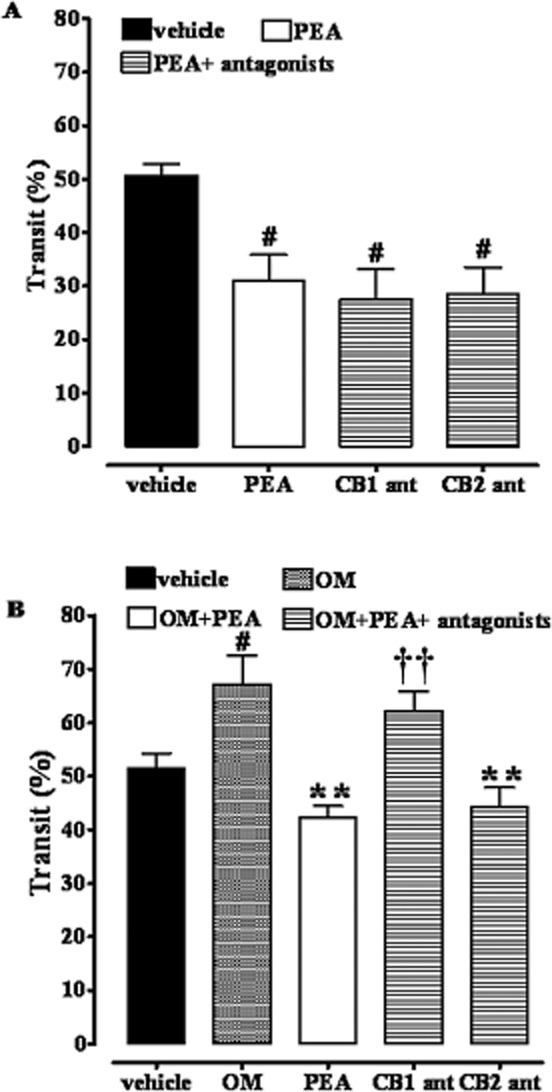
Inhibitory effect of PEA (10 mg·kg−1, i.p.) on upper gastrointestinal transit in control mice (A) and in OM-treated mice (B) in the presence of rimonabant (CB1 ant, 0.1 mg·kg−1, i.p.) or SR144528 (CB2 ant, 1 mg·kg−1, i.p.). Transit was measured 28 days after OM or vehicle (30% ethanol) administration. Results (the means ± SEM of six to eight mice for each experimental group) are expressed as a percentage of upper gastrointestinal transit. #P < 0.05, significantly different from vehicle; **P < 0.01, significantly different from OM; ††P < 0.01, significantly different from PEA. Note that in (A) the term ‘vehicle’ refers to the vehicle used to dissolve PEA, while in (B) the term ‘vehicle’ refers to the vehicle used to dissolve OM.
The TRPV1 channel antagonist I-RTX increased the inhibitory effect of PEA on upper gastrointestinal transit in OM-treated mice
PEA is able to activate and desensitize TRPV1 channels (Ambrosino et al., 2013). Thus, we evaluated the effect of PEA in the presence of the TRPV1 channel antagonist I-RTX. A dose of I-RTX (0.17 mg·kg−1), which was not effective when given alone, did increase the inhibitory effect of PEA on upper gastrointestinal transit 28 days after OM administration (Figure 5B). Higher doses of I-RTX (i.e. 0.35 mg·kg−1 and 0.75 mg·kg−1), given alone, decreased transit in OM-treated mice [percentage of transit: vehicle 49.2 ± 1.3; OM 63.3 ± 2.1 (P < 0.05 vs. vehicle); OM + I-RTX 0.35 mg·kg−1: 45.3 ± 2.4 (P < 0.05 vs. OM); OM + I-RTX 0.75 mg·kg−1: 41.5 ± 1.9 (P < 0.001 vs. OM); n = 6 for each experimental group].
Figure 5.
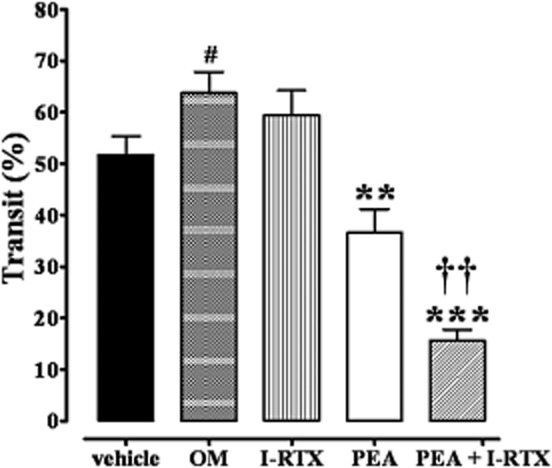
Inhibitory effect of PEA (10 mg·kg−1, i.p.) on upper gastrointestinal transit in OM-treated mice or in the presence of the TRPV1 channel antagonist 5′-iodoresiniferatoxin (I-RTX, 0.17 mg·kg−1, i.p.). Transit was measured 28 days after OM or vehicle (30% ethanol) administration. Results (the means ± SEM of six to eight mice for each experimental group) are expressed as a percentage of upper gastrointestinal transit. #P < 0.05, significantly different from vehicle, **P < 0.01, ***P < 0.001 significantly different from OM; ††P < 0.01, significantly different from PEA.
Effects of the PPARα antagonist GW6471 on upper gastrointestinal transit 28 days post-intracolonic OM challenge
This set of experiments was performed because PPARα has been proposed as one of the main molecular targets of PEA actions (Lo Verme et al., 2005; Esposito and Cuzzocrea, 2013). The PPARα antagonist GW6471 (1 mg·kg−1, i.p.) did not modify the inhibitory effect of PEA on upper gastrointestinal transit 28 days after the intracolonic administration of OM (Figure 6), although the effect of PEA was lost in GW6471-treated animals.
Figure 6.

Inhibitory effect of PEA (10 mg·kg−1, i.p.) on upper gastrointestinal transit in OM-treated mice alone or in the presence of PPARα antagonist GW6471 (1 mg·kg−1, i.p.). Transit was measured 28 days after OM or vehicle (30% ethanol) administration. Results (the means ± SEM of six to eight mice for each experimental group) are expressed as a percentage of upper gastrointestinal transit. #P < 0.05, significantly different from vehicle; **P < 0.01, significantly different from OM.
TRPV1 channels, but not PPARα, were down-regulated in the small intestine 28 days after intracolonic OM challenge
Kimball et al. showed hyperexpression of both CB1 and CB2 receptors in jejunoileal tissues, 28 days after intracolonic OM (Kimball et al., 2010). We thus evaluated, by RT-PCR, the mRNA expression of other targets of PEA, namely TRPV1 channels and PPARα. Results, illustrated in Figure 7, showed a down-regulation of TRPV1 channels in the small intestine, with no changes in PPARα expression (Figure 7).
Figure 7.
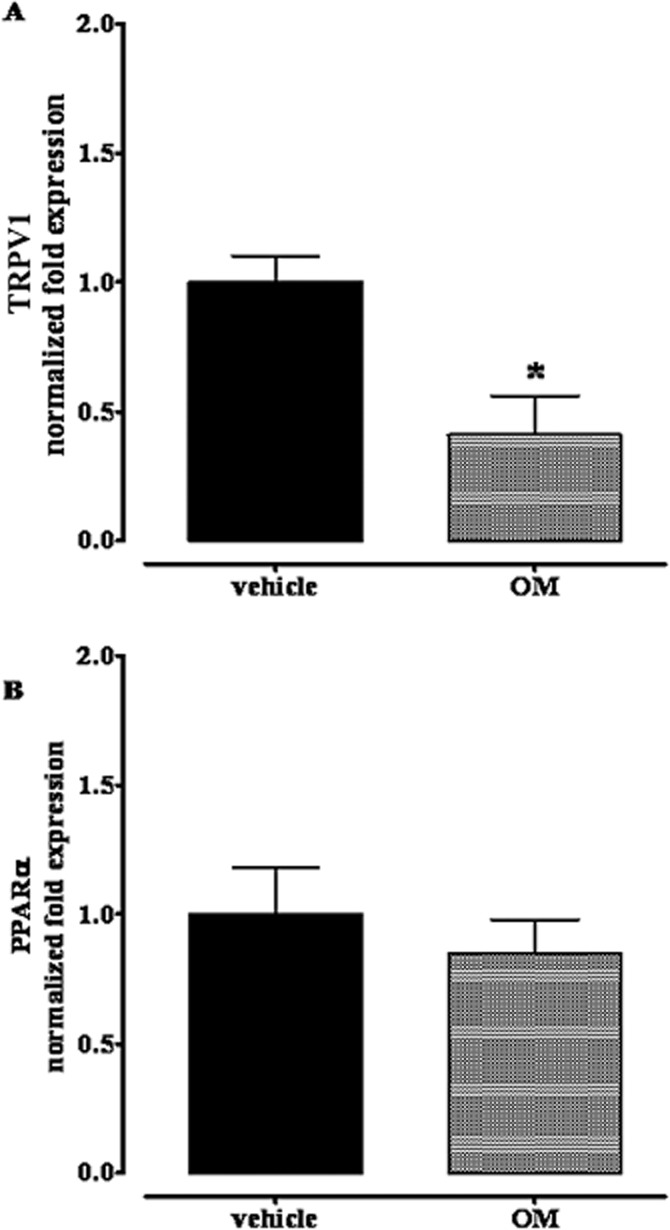
Expression of mRNA for TRPV1 (A) and PPAR-α (B) in jejunoileal segments of in mice treated with OM or vehicle. Tissues were analysed 28 days after OM or vehicle administration. Results are the means ± SEM of three experiments. RT-PCR analysis was performed as described in methods, for statistical significance analysis Cq data were also analysed by the RESTR 2009 software(Pfaffl M.W., Nucleic Acid Research 2002, 30, E-36). *P < 0.05, significantly different from vehicle.
Intracolonic OM treatment did not change PEA levels 28 days later
Isotope GC-MS analysis of lipid extracts from the small intestine of control and OM-treated mice showed no changes in endogenous PEA levels (Figure 8). Furthermore, the mRNA expression of N-acylethanolamine-hydrolyzing acid amidase (NAAA; a key specific enzyme involved in PEA degradation) showed no significant changes between samples of small intestine from control and OM-treated mice (insert to Figure 8).
Figure 8.
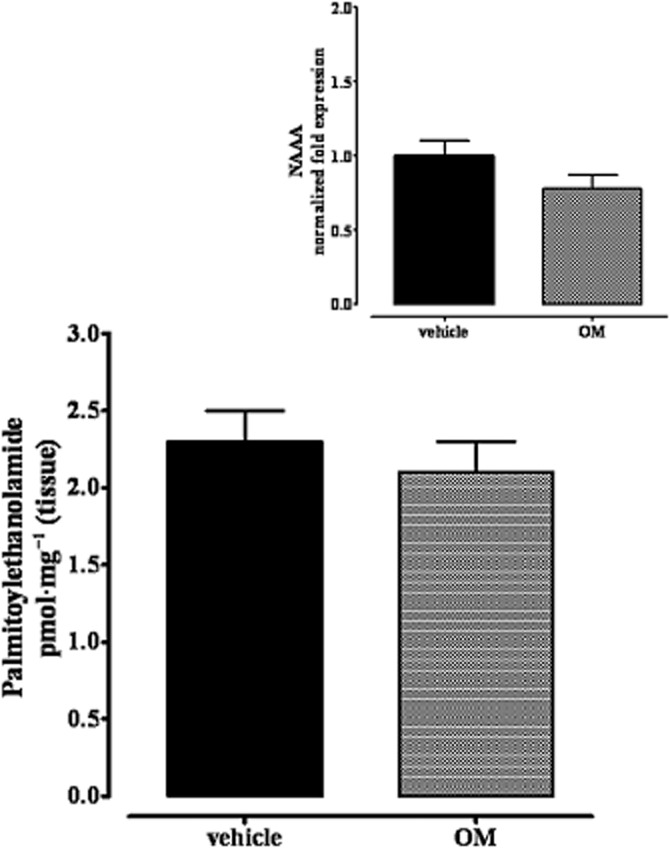
Levels of PEA in jejunoileal segments of mice treated with OM or vehicle. The insert shows the mRNA expression of N-acylethanolamine acid amidase (NAAA, a specific enzyme involved in palmitoylethanolamide degradation) in control and OM-treated mice. Tissues were analysed 28 days after OM or vehicle administration. Data are the means ± SEM of five mice (n = 3 for the data contained in the insert). No significant differences were found.
Intracolonic OM increased anandamide, but not 2-AG or oleoylethanolamide, levels
This set of experiments was performed because there is evidence in the literature that PEA can inhibit anandamide inactivation (Bisogno et al., 1997; Di Marzo et al., 2001) and thus indirectly activate endocannabinoid targets such as CB receptors and TRPV1 channels. The levels of anandamide, but not of 2-AG, were clearly increased, compared with control tissues, in the small intestine 28 days after OM. These levels also showed a strong trend towards further increase (P = 0.057) following treatment with exogenous PEA (10 mg·kg−1, i.p., Figure 9). Also, OM treatment did not change the small intestinal levels of oleoylethanolamide (pmol mg tissue–1: vehicle 0.22 ± 2.04; OM 0.18 ± 0.03; OM + PEA 0.12 ± 0.02, n = 6 for each experimental group).
Figure 9.
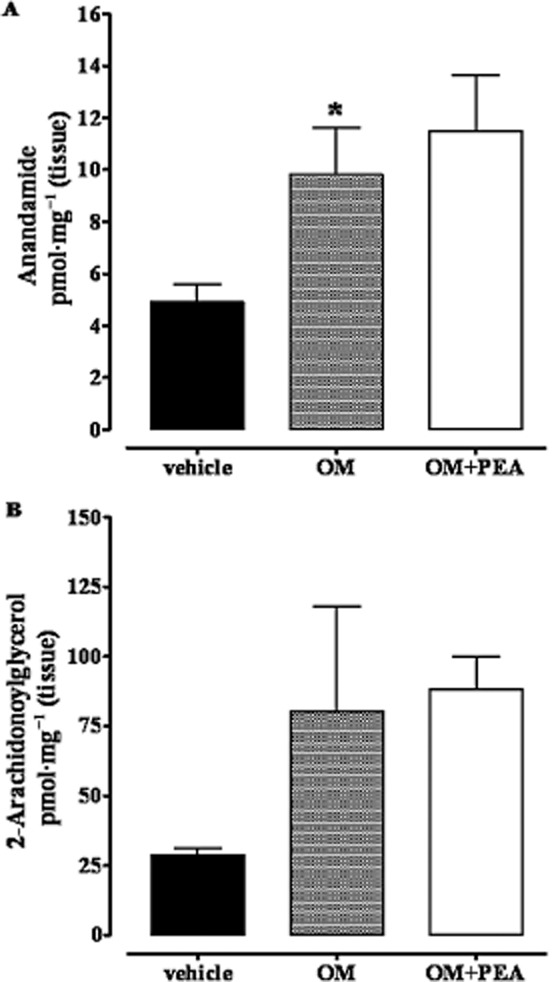
Anandamide (A) and 2-arachidonoylglycerol (B) levels in the small intestine of mice treated with OM or vehicle: effect of exogenous PEA. Tissues were analysed 28 days after OM or vehicle administration. PEA (10 mg·kg−1, i.p.) was administered 30 min before the assay. Data are the means ± SEM of five mice. *P < 0.05, significantly different from vehicle (A: OM vs. OM plus PEA, P = 0.057; B: OM vs. vehicle, P = 0.245).
Oral PEA reduced the post-inflammatory accelerated upper gastrointestinal transit induced by OM
Because preparations of PEA are marketed for the treatment of functional intestinal complaints in humans, we investigated its possible efficacy after oral administration to mice. PEA (1–10 mg·kg−1, p.o.) inhibited upper gastrointestinal transit only at 5 mg·kg−1, at 28 days after OM (Figure 10).
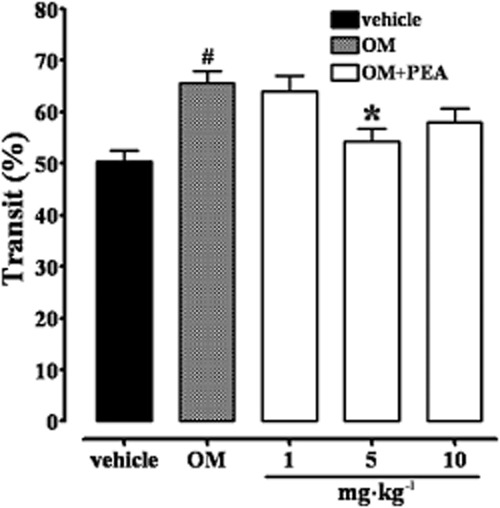
Inhibitory effect of PEA (1–10 mg·kg−1, oral) on upper gastrointestinal transit in mice treated with OM. Transit was measured 28 days after OM administration. Results (the means ± SEM of 12–15 mice for each experimental group) are expressed as a percentage of upper gastrointestinal transit. #P < 0.05, significantly different from vehicle; *P < 0.05, significantly different from OM.
Discussion
Gut inflammation is now considered as a predisposing event in a significant proportion of patients who experience IBS-like symptoms, without signs of co-existent inflammation. In some patients, functional bowel disorders are the result of an earlier infection or exposure to allergen that provoked gut inflammation (Spiller, 2004). In the present study, we showed that PEA normalized the accelerated upper gastrointestinal transit in a murine model of post-inflammatory condition of the gut, exhibiting some aspects of post-inflammatory IBS.
As shown already (Kimball et al., 2005; 2010), in the present study, OM induced transient colitis – which resolved 7 days after its intracolonic administration – and a functional increase in upper gastrointestinal transit that was observed after the recovery from the colonic inflammation, i.e. at 28 days after OM. As in the original study by Kimball et al. (2005), we used a method of measuring motility that reflects a combination of gastric emptying and small intestinal transit. So, even if our results do not distinguish between a gastric or intestinal site of action for PEA, but rather reflect a combination of both, they clearly show that exogenously administered PEA preferentially inhibited transit in mice with a functional increase of gastrointestinal motility. It is likely that PEA was acting in the small intestine rather than in the stomach, as we have previously shown that PEA inhibits upper gastrointestinal transit (Capasso et al., 2008) without affecting gastric emptying (Aviello et al., 2008). The functional increase in intestinal motility was not accompanied by changes in endogenous PEA levels nor in the mRNA expression of NAAA, a key enzyme of PEA catabolism. These results rule against a possible anti-prokinetic effect of endogenous PEA following the experimental post-inflammatory functional increase in intestinal motility. Recently, increased PEA levels in the plasma (Fichna et al., 2013) and in the colon (Zhang et al., 2014) of IBS patients have been reported.
Because PEA is readily hydrolyzed to palmitic acid, we tested the effects of this fatty acid on intestinal motility. We found that palmitic acid was much less active than PEA in reducing intestinal motility, 28 days after OM treatment (a small inhibitory effect was observed only at the highest dose tested), suggesting that palmitic acid would contribute only for a minimal part of the pharmacological effect of exogenous PEA.
It is well established that many of the pharmacological effects of PEA involve indirect activation of CB receptors or TRPV1 channels (via the so-called ‘entourage effect’), activation of PPARα as well as activation and/or desensitization of TRPV1 channels (De Petrocellis et al., 2002; Smart et al., 2002; Lo Verme et al., 2005; D'Agostino et al., 2007; Ambrosino et al., 2013; Skaper et al., 2014). Thus, in order to investigate the mode of action of PEA on intestinal transit, we considered the possible involvement of such receptors. The discussion of this part of our results is detailed below.
Activation of CB1 and CB2 receptor is known to reduce intestinal transit in pathophysiological states (Izzo and Coutts, 2005; Storr et al., 2008). Both CB1 and CB2 receptor agonists inhibit upper gastrointestinal transit in the experimental model of post-inflammatory accelerated transit induced by OM (Kimball et al., 2010). In physiological states, anandamide inhibits transit via activation of CB1 receptors (Calignano et al., 1997; Izzo et al., 2001), while PEA inhibits transit via CB-independent mechanisms (Capasso et al., 2001). In the present study, the inhibitory effect of PEA on motility was significantly attenuated by a CB1, but not a CB2 receptor, antagonist. A direct activation of cannabinoid CB1 receptors by PEA is unlikely as this acylethanolamide does not bind to CB1 and CB2 receptors (Lambert and Di Marzo, 1999). We therefore suggest that PEA might indirectly activate cannabinoid CB1 receptors and thus reduce motility via the so-called ‘entourage effect’, that is the augmentation of the endocannabinoid levels and/or modulatory actions at CB receptors (De Petrocellis et al., 2001; 2002; Smart et al., 2002). Consistent with our suggestion, we showed, in the present study, an increase of anandamide levels in the small intestine after all overt signs of inflammation had resolved (i.e. 28 days after OM) in mice that exhibit increased upper gastrointestinal transit and, more importantly, that exogenous PEA further augmented endogenous anandamide levels (although a conventional statistical difference was not achieved). Furthermore, a more intense CB1 receptor immunostaining in myenteric neurons 28 days after OM treatment was previously observed (Kimball et al., 2010), suggesting that hyperactivation of the endogenous CB system is present in post-inflammatory states, even in the absence of inflammation. Importantly, reversal by rimonabant was specific for the pathophysiological condition as the antagonist did not change the inhibitory effects of PEA, on transit in control mice. Collectively, these results suggest that PEA could indirectly activate CB1 receptors, only when endogenous anandamide and/or CB1 receptors were up-regulated and not in the presence of ‘low’ basal levels of endocannabinoids and CB1 receptors. On the other hand, it is unlikely that PEA might exert anti-transit effects via the CB2 receptor, as the selective CB2 receptor antagonist SR144528, at a dose previously shown to counteract the inhibitory effect of the selective CB2 receptor antagonist JWH015 on intestinal transit, did not change the effect of PEA. Others have found that the effect of PEA might exert pharmacological effects via putative CB2-like receptors (Calignano et al., 1998; Farquhar-Smith et al., 2002).
In the alimentary canal, TRPV1 channels are present in extrinsic sensory neurons and in intrinsic myenteric neurons (Holzer, 2011). Animal experiments have demonstrated a key role for these ion channels in the modulation of mechanosensitivity, chemosensitivity and pain in IBS (Boesmans et al., 2011). Increased numbers of TRPV1 channels in nerve fibres have been observed in IBS patients and these are believed to contribute to visceral hypersensitivity and pain in IBS (Akbar et al., 2008). In the present study, we have shown that 28 days after the acute colitis, two changes were observed. (i) TRPV1 channels were down-regulated in the small intestine and (ii) a dose of I-RTX which was in effective by itself, further increased the anti-transit effect of PEA. Such data suggest that the effect of PEA on transit is negatively modulated by TRPV1 channels. Consistent with our data, De Novellis et al. (2012) have recently reported that PEA decreased the burst and increased the latency of tail flick-evoked onset of ‘ON cell’ activity in the rostral ventromedial medulla and such effects were enhanced by I-RTX. Furthermore, we have recently shown that the intestinal anti-inflammatory effect of PEA was augmented by capsazepine, another TRPV1 channel antagonist (F. Borrelli, unpublished).
Another mediator of the pharmacological actions of PEA, such as the analgesic, antiepileptic and anti-inflammatory effects, is the nuclear receptor PPARα (Lo Verme et al., 2005; Citraro et al., 2013; Esposito et al., 2014; Okine et al., 2014). We thus investigated the possible involvement of this receptor by measuring its intestinal mRNA expression 28 days after OM administration and by evaluating the effect of PEA in the presence of GW6471, a PPARα receptor antagonist. Although PPARα immunoreactivity has been detected in myenteric neurons throughout the mouse gastrointestinal tract (Cluny et al., 2009), the involvement of this nuclear receptor in the control of intestinal motility has not been demonstrated to date. In the present study, we showed that GW6471 did not significantly counteract the effect of PEA on intestinal motility, although the effect of PEA was lost in GW6471-treated rats. The dose of GW6471 used in the present study has been previously shown to reveal PPARα-mediated anti-inflammatory effects of simvastatin (Esposito et al., 2012).
Finally, because the naturally occurring lipid PEA is available for human use as food for medical purposes, promoted for the treatment of functional disturbances of the gastrointestinal tract, we tested the efficacy of PEA after oral administration. We observed that PEA significantly – and only at the 5 mg·kg−1 dose – reduced intestinal motility at day 28 post-OM treatment. Thus, PEA was less active when given orally than when given intraperitoneally, a result which can be explained by the presence, in the digestive tract, of NAAA and other amidases able to metabolize PEA (Tsuboi et al., 2007; Borrelli and Izzo, 2009).
In conclusion, by using a functional model of accelerated gastrointestinal transit, exhibiting some aspects of post-inflammatory IBS, we have shown that levels of endogenous anandamide and TRPV1 channels were altered, after an initial inflammatory period, thus suggesting their involvement in the underlying pathophysiology of functional post-inflammatory disturbances of gastrointestinal motility. Importantly from a pharmacological viewpoint, PEA normalized the functional post-inflammatory accelerated intestinal transit, an effect which probably involves indirect activation of CB1 receptors and is modulated by TRPV1 channels. In view of its safety, PEA might be considered for clinical use in disorders of intestinal motility, such as post-inflammatory IBS.
Acknowledgments
This study was partly supported by funds from PON_BIAM-EPI ‘Research and Development of bioregulators active on epigenetic mechanisms of inflammatory processes in chronic and degenerative diseases’.
Glossary
- 2-AG
2-arachidonylglycerol
- CB
cannabinoid
- IBS
irritable bowel syndrome
- HPRT
hypoxanthine-guanine phosophoribosyltransferase
- I-RTX, NAAA
N-acylethanolamine-hydrolyzing acid amidase
- OM
oil of mustard
- PEA
palmitoylethanolamide
- TRPV1
transient receptor potential vanilloid type-1
Author contributions
A. A. I. conceived and coordinated the study and wrote the manuscript. V. D. M. and P. O. reviewed and provided scientific input to the manuscript. R. C., E. P., F. B., L. B., T. A. planned and executed the experimental work.
Conflict of interest
VDM is co-inventor of patents claiming the use of palmitoylethanolamide against inflammatory conditions, and receives research support from Epitech Italia S.r.l. which markets palmitoylethanolamide. The other authors declare that they have no conflict of interest.
Supporting Information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:http://dx.doi.org/10.1111/bph.12759
Primer sequences.
References
- Abalo R, Cabezos PA, Vera G, Fernández-Pujol R, Martín MI. The cannabinoid antagonist SR144528 enhances the acute effect of WIN 55,212-2 on gastrointestinal motility in the rat. Neurogastroenterol Motil. 2010;22:694. doi: 10.1111/j.1365-2982.2009.01466.x. e206. [DOI] [PubMed] [Google Scholar]
- Akbar A, Yiangou Y, Facer P, Walters JR, Anand P, Ghosh S. Increased capsaicin receptor TRPV1-expressing sensory fibres in irritable bowel syndrome and their correlation with abdominal pain. Gut. 2008;57:923–929. doi: 10.1136/gut.2007.138982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: G Protein-Coupled Receptors. Br J Pharmacol. 2013a;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Catterall WA, et al. The Concise Guide to PHARMACOLOGY 2013/14: Ion Channels. Br J Pharmacol. 2013b;170:1607–1651. doi: 10.1111/bph.12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambrosino P, Soldovieri MV, Russo C, Taglialatela M. Activation and desensitization of TRPV1 channels in sensory neurons by the PPARα agonist palmitoylethanolamide. Br J Pharmacol. 2013;168:1430–1444. doi: 10.1111/bph.12029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aviello G, Matias I, Capasso R, Petrosino S, Borrelli F, Orlando P, et al. Inhibitory effect of the anorexic compound oleoylethanolamide on gastric emptying in control and overweight mice. J Mol Med (Berl) 2008;86:413–422. doi: 10.1007/s00109-008-0305-7. [DOI] [PubMed] [Google Scholar]
- Balvers MG, Verhoeckx KC, Meijerink J, Wortelboer HM, Witkamp RF. Measurement of palmitoylethanolamide and other N-acylethanolamines during physiological and pathological conditions. CNS Neurol Disord Drug Targets. 2013;12:23–33. doi: 10.2174/1871527311312010007. [DOI] [PubMed] [Google Scholar]
- Bisogno T, Maurelli S, Melck D, De Petrocellis L, Di Marzo V. Biosynthesis, uptake, and degradation of anandamide and palmitoylethanolamide in leukocytes. J Biol Chem. 1997;272:3315–3323. doi: 10.1074/jbc.272.6.3315. [DOI] [PubMed] [Google Scholar]
- Boesmans W, Owsianik G, Tack J, Voets T, Vanden Berghe P. TRP channels in neurogastroenterology: opportunities for therapeutic intervention. Br J Pharmacol. 2011;162:18–37. doi: 10.1111/j.1476-5381.2010.01009.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrelli F, Izzo AA. Role of acylethanolamides in the gastrointestinal tract with special reference to food intake and energy balance. Best Pract Res Clin Endocrinol Metab. 2009;23:33–49. doi: 10.1016/j.beem.2008.10.003. [DOI] [PubMed] [Google Scholar]
- Borrelli F, Fasolino I, Romano B, Capasso R, Maiello F, Coppola D, et al. Beneficial effect of the non-psychotropic plant cannabinoid cannabigerol on experimental inflammatory bowel disease. Biochem Pharmacol. 2013;85:1306–1316. doi: 10.1016/j.bcp.2013.01.017. [DOI] [PubMed] [Google Scholar]
- Calignano A, La Rana G, Makriyannis A, Lin SY, Beltramo M, Piomelli D. Inhibition of intestinal motility by anandamide, an endogenous cannabinoid. Eur J Pharmacol. 1997;340:R7–R8. [PubMed] [Google Scholar]
- Calignano A, La Rana G, Giuffrida A, Piomelli D. Control of pain initiation by endogenous cannabinoids. Nature. 1998;394:277–281. doi: 10.1038/28393. [DOI] [PubMed] [Google Scholar]
- Camilleri M. Peripheral mechanisms in irritable bowel syndrome. N Engl J Med. 2012;367:1626–1635. doi: 10.1056/NEJMra1207068. [DOI] [PubMed] [Google Scholar]
- Capasso R, Izzo AA, Fezza F, Pinto A, Capasso F, Mascolo N, et al. Inhibitory effect of palmitoylethanolamide on gastrointestinal motility in mice. Br J Pharmacol. 2001;134:945–950. doi: 10.1038/sj.bjp.0704339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capasso R, Borrelli F, Cascio MG, Aviello G, Huben K, Zjawiony JK, et al. Inhibitory effect of salvinorin A, from Salvia divinorum, on ileitis-induced hypermotility: cross-talk between kappa-opioid and cannabinoid CB(1) receptors. Br J Pharmacol. 2008;155:681–689. doi: 10.1038/bjp.2008.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citraro R, Russo E, Scicchitano F, van Rijn CM, Cosco D, Avagliano C, et al. Antiepileptic action of N-palmitoylethanolamine t hrough CB1 and PPAR-α receptor activation in a genetic model of absence epilepsy. Neuropharmacology. 2013;69:115–126. doi: 10.1016/j.neuropharm.2012.11.017. [DOI] [PubMed] [Google Scholar]
- Cluny NL, Keenan CM, Lutz B, Piomelli D, Sharkey KA. The identification of peroxisome proliferator-activated receptor alpha-independent effects of oleoylethanolamide on intestinal transit in mice. Neurogastroenterol Motil. 2009;21:420–429. doi: 10.1111/j.1365-2982.2008.01248.x. [DOI] [PubMed] [Google Scholar]
- Darmani NA, Izzo AA, Degenhardt B, Valenti M, Scaglione G, Capasso R, et al. Involvement of the cannabimimetic compound, N-palmitoyl-ethanolamine, in inflammatory and neuropathic conditions: review of the available pre-clinical data, and first human studies. Neuropharmacology. 2005;48:1154–1163. doi: 10.1016/j.neuropharm.2005.01.001. [DOI] [PubMed] [Google Scholar]
- D'Agostino G, La Rana G, Russo R, Sasso O, Iacono A, Esposito E, et al. Acute intracerebroventricular administration of palmitoylethanolamide, an endogenous peroxisome proliferator-activated receptor-alpha agonist, modulates carrageenan-induced paw edema in mice. J Pharmacol Exp Ther. 2007;322:1137–1143. doi: 10.1124/jpet.107.123265. [DOI] [PubMed] [Google Scholar]
- D'Argenio G, Petrosino S, Gianfrani C, Valenti M, Scaglione G, Grandone I, et al. Overactivity of the intestinal endocannabinoid system in celiac disease and in methotrexate-treated rats. J Mol Med (Berl) 2007;85:523–530. doi: 10.1007/s00109-007-0192-3. [DOI] [PubMed] [Google Scholar]
- De Novellis V, Luongo L, Guida F, Cristino L, Palazzo E, Russo R, et al. Effects of intra-ventrolateral periaqueductal grey palmitoylethanolamide on thermoceptive threshold and rostral ventromedial medulla cell activity. Eur J Pharmacol. 2012;676:41–50. doi: 10.1016/j.ejphar.2011.11.034. [DOI] [PubMed] [Google Scholar]
- De Petrocellis L, Davis JB, Di Marzo V. Palmitoylethanolamide enhances anandamide stimulation of human vanilloid VR1 receptors. FEBS Lett. 2001;506:253–256. doi: 10.1016/s0014-5793(01)02934-9. [DOI] [PubMed] [Google Scholar]
- De Petrocellis L, Bisogno T, Ligresti A, Bifulco M, Melck D, Di Marzo V. Effect on cancer cell proliferation of palmitoylethanolamide, a fatty acid amide interacting with both the cannabinoid and vanilloid signalling systems. Fundam Clin Pharmacol. 2002;16:297–302. doi: 10.1046/j.1472-8206.2002.00094.x. [DOI] [PubMed] [Google Scholar]
- De Ponti F. Drug development for the irritable bowel syndrome: current challenges and future perspectives. Front Pharmacol. 2013;4:7. doi: 10.3389/fphar.2013.00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Marzo V, Melck D, Orlando P, Bisogno T, Zagoory O, Bifulco M, et al. Palmitoylethanolamide inhibits the expression of fatty acid amide hydrolase and enhances the anti-proliferative effect of anandamide in human breast cancer cells. Biochem J. 2001;358:249–255. doi: 10.1042/0264-6021:3580249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Marzo V, Capasso R, Matias I, Aviello G, Petrosino S, Borrelli F, et al. The role of endocannabinoids in the regulation of gastric emptying: alterations in mice fed a high-fat diet. Br J Pharmacol. 2008;153:1272–1280. doi: 10.1038/sj.bjp.0707682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Paola R, Impellizzeri D, Torre A, Mazzon E, Cappellani A, Faggio C, et al. Effects of palmitoylethanolamide on intestinal injury and inflammation caused by ischemia-reperfusion in mice. J Leukoc Biol. 2012;91:911–920. doi: 10.1189/jlb.0911485. [DOI] [PubMed] [Google Scholar]
- Diep TA, Madsen AN, Holst B, Kristiansen MM, Wellner N, Hansen SH, et al. Dietary fat decreases intestinal levels of the anorectic lipids through a fat sensor. FASEB J. 2011;25:765–774. doi: 10.1096/fj.10-166595. [DOI] [PubMed] [Google Scholar]
- Esposito E, Cuzzocrea S. Palmitoylethanolamide in homeostatic and traumatic central nervous system injuries. CNS Neurol Disord Drug Targets. 2013;12:55–61. doi: 10.2174/1871527311312010010. [DOI] [PubMed] [Google Scholar]
- Esposito E, Rinaldi B, Mazzon E, Donniacuo M, Impellizzeri D, Paterniti I, et al. Anti-inflammatory effect of simvastatin in an experimental model of spinal cord trauma: involvement of PPAR-α. J Neuroinflammation. 2012;9:81. doi: 10.1186/1742-2094-9-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito G, Capoccia E, Turco F, Palumbo I, Lu J, Steardo A, et al. Palmitoylethanolamide improves colon inflammation through an enteric glia/toll like receptor 4-dependent PPAR-α activation. Gut. 2014;63:1300–1312. doi: 10.1136/gutjnl-2013-305005. [DOI] [PubMed] [Google Scholar]
- Farquhar-Smith WP, Jaggar SI, Rice AS. Attenuation of nerve growth factor-induced visceral hyperalgesia via cannabinoid CB(1) and CB(2)-like receptors. Pain. 2002;97:11–21. doi: 10.1016/s0304-3959(01)00419-5. [DOI] [PubMed] [Google Scholar]
- Fichna J, Storr MA. Brain-gut interactions in IBS. Front Pharmacol. 2012;3:127. doi: 10.3389/fphar.2012.00127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fichna J, Wood JT, Papanastasiou M, Vadivel SK, Oprocha P, Salaga M, et al. Endocannabinoid and cannabinoid-like fatty acid amide levels correlate with pain-related symptoms in patients with IBS-D and IBS-C: a pilot study. PLoS ONE. 2013;8:e85073. doi: 10.1371/journal.pone.0085073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes S, Herzog H, Cox HM. A role for neuropeptide Y in the gender-specific gastrointestinal, corticosterone and feeding responses to stress. Br J Pharmacol. 2012;166:2307–2316. doi: 10.1111/j.1476-5381.2012.01939.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu J, Astarita G, Gaetani S, Kim J, Cravatt BF, Mackie K, et al. Food intake regulates oleoylethanolamide formation and degradation in the proximal small intestine. J Biol Chem. 2007;282:1518–1528. doi: 10.1074/jbc.M607809200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimaldi P, Orlando P, Di Siena S, Lolicato F, Petrosino S, Bisogno T, et al. The endocannabinoid system and pivotal role of the CB2 receptor in mouse spermatogenesis. Proc Natl Acad Sci U S A. 2009;106:11131–11136. doi: 10.1073/pnas.0812789106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho WS, Barrett DA, Randall MD. Entourage' effects of N-palmitoylethanolamide and N-oleoylethanolamide on vasorelaxation to anandamide occur through TRPV1 receptors. Br J Pharmacol. 2008;155:837–846. doi: 10.1038/bjp.2008.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzer P. Transient receptor potential (TRP) channels as drug targets for diseases of the digestive system. Pharmacol Ther. 2011;131:142–170. doi: 10.1016/j.pharmthera.2011.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izzo AA. Novel insights which may translate into treatments for irritable bowel syndrome. Front Pharmacol. 2013;4:160. doi: 10.3389/fphar.2013.00160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izzo AA, Coutts AA. Cannabinoids and the digestive tract. Handb Exp Pharmacol. 2005;168:573–598. doi: 10.1007/3-540-26573-2_19. [DOI] [PubMed] [Google Scholar]
- Izzo AA, Capasso R, Pinto L, Di Carlo G, Mascolo N, Capasso F. Effect of vanilloid drugs on gastrointestinal transit in mice. Br J Pharmacol. 2001;132:1411–1416. doi: 10.1038/sj.bjp.0703975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izzo AA, Piscitelli F, Capasso R, Marini P, Cristino L, Petrosino S, et al. Basal and fasting/refeeding-regulated tissue levels of endogenous PPAR-alpha ligands in Zucker rats. Obesity (Silver Spring) 2010;18:55–62. doi: 10.1038/oby.2009.186. [DOI] [PubMed] [Google Scholar]
- Izzo AA, Capasso R, Aviello G, Borrelli F, Romano B, Piscitelli F, et al. Inhibitory effect of cannabichromene, a major non-psychotropic cannabinoid extracted from Cannabis sativa, on inflammation-induced hypermotility in mice. Br J Pharmacol. 2012;166:1444–1460. doi: 10.1111/j.1476-5381.2012.01879.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimball ES, Palmer JM, D'Andrea MR, Hornby PJ, Wade PR. Acute colitis induction by oil of mustard results in later development of an IBS-like accelerated upper GI transit in mice. Am J Physiol Gastrointest Liver Physiol. 2005;288:G1266–G1273. doi: 10.1152/ajpgi.00444.2004. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: Reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimball ES, Wallace NH, Schneider CR, D'Andrea MR, Hornby PJ. Small intestinal cannabinoid receptor changes following a single colonic insult with oil of mustard in mice. Front Pharmacol. 2010;1:132. doi: 10.3389/fphar.2010.00132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird JM, Martinez-Caro L, Garcia-Nicas E, Cervero F. A new model of visceral pain and referred hyperalgesia in the mouse. Pain. 2001;92:335–342. doi: 10.1016/S0304-3959(01)00275-5. [DOI] [PubMed] [Google Scholar]
- Lambert DM, Di Marzo V. The palmitoylethanolamide and oleamide enigmas: are these two fatty acid amides cannabimimetic? Curr Med Chem. 1999;6:757–773. [PubMed] [Google Scholar]
- Lo Verme J, Fu J, Astarita G, La Rana G, Russo R, Calignano A, et al. The nuclear receptor peroxisome proliferator-activated receptor-alpha mediates the anti-inflammatory actions of palmitoylethanolamide. Mol Pharmacol. 2005;67:15–19. doi: 10.1124/mol.104.006353. [DOI] [PubMed] [Google Scholar]
- Magge S, Lembo A. Complementary and alternative medicine for the irritable bowel syndrome. Gastroenterol Clin North Am. 2011;40:245–253. doi: 10.1016/j.gtc.2010.12.005. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okine BN, Rea K, Olango WM, Price J, Herdman S, Madasu MK, et al. A role for PPAR-α in the medial prefrontal cortex in formalin-evoked nociceptive responding in rats. Br J Pharmacol. 2014;171:1462–1471. doi: 10.1111/bph.12540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Sullivan SE, Kendall DA. Cannabinoid activation of peroxisome proliferator-activated receptors: potential for modulation of inflammatory disease. Immunobiology. 2010;215:611–616. doi: 10.1016/j.imbio.2009.09.007. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledge base of drug targets and their ligands. Nucleic Acids Research. 2014;42(Database Issue):D1098–1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrosino S, Borrelli F, Pagano E, Di Marzo V, Izzo AA. 2013. Effect of palmitoylethanolamide in a murine model of colitis. XXXVI National Congress of the Italian Pharmacological Society; Turin (Italy)
- Rahimi R, Abdollahi M. Herbal medicines for the management of irritable bowel syndrome: a comprehensive review. World J Gastroenterol. 2012;18:589–600. doi: 10.3748/wjg.v18.i7.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romano B, Borrelli F, Fasolino I, Capasso R, Piscitelli F, Cascio M, et al. The cannabinoid TRPA1 agonist cannabichromene inhibits nitric oxide production in macrophages and ameliorates murine colitis. Br J Pharmacol. 2013;169:213–229. doi: 10.1111/bph.12120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaper SD, Facci L, Fusco M, Della Valle MF, Zusso M, Costa B, et al. Palmitoylethanolamide, a naturally occurring disease-modifying agent in neuropathic pain. Inflammopharmacology. 2014;22:79–94. doi: 10.1007/s10787-013-0191-7. [DOI] [PubMed] [Google Scholar]
- Smart D, Jonsson KO, Vandevoorde S, Lambert DM, Fowler CJ. ‘Entourage’ effects of N-acyl ethanolamines at human vanilloid receptors. Comparison of effects upon anandamide-induced vanilloid receptor activation and upon anandamide metabolism. Br J Pharmacol. 2002;136:452–458. doi: 10.1038/sj.bjp.0704732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiller R. Infection, immune function, and functional gut disorders. Clin Gastroenterol. 2004;2:445–455. doi: 10.1016/s1542-3565(04)00159-4. [DOI] [PubMed] [Google Scholar]
- Storr MA, Yüce B, Andrews CN, Sharkey KA. The role of the endocannabinoid system in the pathophysiology and treatment of irritable bowel syndrome. Neurogastroenterol Motil. 2008;20:857–868. doi: 10.1111/j.1365-2982.2008.01175.x. [DOI] [PubMed] [Google Scholar]
- Tsuboi K, Takezaki N, Ueda N. The N-acylethanolamine-hydrolyzing acid amidase (NAAA) Chem Biodivers. 2007;4:1914–1925. doi: 10.1002/cbdv.200790159. [DOI] [PubMed] [Google Scholar]
- Voß U, Lewerenz A, Nieber K. Treatment of irritable bowel syndrome: sex and gender specific aspects. Handb Exp Pharmacol. 2012;214:473–497. doi: 10.1007/978-3-642-30726-3_21. [DOI] [PubMed] [Google Scholar]
- Wade PR, Palmer JM, McKenney S, Kenigs V, Chevalier K, Moore BA, et al. Modulation of gastrointestinal function by MuDelta, a mixed μ opioid receptor agonist/μ opioid receptor antagonist. Br J Pharmacol. 2012;67:1111–1125. doi: 10.1111/j.1476-5381.2012.02068.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SC, Wang WL, Su PJ, Jiang KL, Yuan ZW. Decreased enteric fatty acid amide hydrolase activity is associated with colonic inertia in slow transit constipation. J Gastroenterol Hepatol. 2014;29:276–283. doi: 10.1111/jgh.12346. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Primer sequences.