

Abstract
Background and Purpose
The transcriptional co-activator with PDZ-binding motif (TAZ) is a key controller of mesenchymal stem cell differentiation through its nuclear localization and subsequent interaction with master transcription factors. In particular, TAZ directly associates with myoblast determining protein D (MyoD) and activates MyoD-induced myogenic gene expression, thereby enhancing myogenic differentiation. Here, we have synthesized and characterized low MW compounds modulating myogenic differentiation via induction of TAZ nuclear localization.
Experimental Approach
COS7 cells stably transfected with GFP-TAZ were used in a high content imaging screen for compounds specifically enhancing nuclear localization of TAZ. We then studied the effects of such TAZ modulators on myocyte differentiation of C2C12 cells and myogenic transdifferentiation of mouse embryonic fibroblast cells in vitro and muscle regeneration in vivo.
Key Results
We identified two TAZ modulators, TM-53, and its structural isomer, TM-54. Each compound strongly enhanced nuclear localization of TAZ by reducing S89-phosphorylation and dose-dependently augmented myogenic differentiation and MyoD-mediated myogenic transdifferentiation through an activation of MyoD–TAZ interaction. The myogenic stimulatory effects of TM-53 and TM-54 were impaired in the absence of TAZ, but retrieved by the restoration of TAZ. In addition, administration of TM-53 and TM-54 enhanced injury-induced muscle regeneration in vivo and attenuated myofiber injury in vitro.
Conclusions and Implications
The novel TAZ modulators TM-53 and TM-54 accelerated myogenic differentiation and improved muscle regeneration and function after injury, demonstrating that low MW compounds targeting the nuclear localization of TAZ have beneficial effects in skeletal muscle regeneration and in recovery from muscle degenerative diseases.
Keywords: TAZ modulators, MyoD, myogenic differentiation, muscle regeneration, muscle injury
Introduction
The transcriptional co-activator with PDZ-binding motif (TAZ) was isolated as a protein that interacted with the 14-3-3 protein in the cytosol (Kanai et al., 2000). TAZ is phosphorylated at residue S89 and is sequestered by 14-3-3 protein in the cytosol. Conversely, dephosphorylation of TAZ by protein phosphatase 1 leads to its nuclear localization and modulation of the activity of many transcription factors (Kanai et al., 2000; Lei et al., 2008; Liu et al., 2011; Hong and Guan, 2012). In addition, TAZ is phosphorylated at Y316 by hypertonic stimulation and mainly localized in the nucleus whereas, under hypotonic conditions, TAZ resides in the cytoplasm (Jang et al., 2012a). The WW domain within TAZ facilitates interaction with several PPXY motif-containing transcription factors, such as runt-related transcription factor 2 (RUNX2) and PPARγ, which, in turn, modulates their transcriptional activities (Cui et al., 2003; Hong and Yaffe, 2006). Accordingly, TAZ accelerates RUNX2-mediated osteogenic differentiation and suppresses PPARγ-induced adipogenesis driven from mesenchymal stem cells (Hong et al., 2005). Skeletal muscle differentiation of mesenchymal stem cells is also accelerated by TAZ through direct interaction with and transactivation of the myoblast determining protein D (MyoD) (Olson, 1992; Lassar et al., 1994; Arnold and Winter, 1998; Perry and Rudnick, 2000). TAZ deficiency blocks the induction of MyoD-mediated gene expression and myogenic differentiation. However, restoration of TAZ in TAZ knockout (KO) cells retrieves MyoD-dependent myogenic gene expression and also stimulates myogenic transdifferentiation of non-muscle cells (Jeong et al., 2010).
Skeletal myogenic differentiation of mesenchymal stem cells is tightly regulated through sequential activation of a variety of muscle regulatory factors (MRFs), such as MyoD, myocyte enhancing factor 2 (MEF2), myogenin (Myog), myogenic determination factor 5 (Myf5) and MRF4. MRFs promote myoblast formation, terminal differentiation into myocytes and myocyte fusion into multinucleated myofibers. Muscle marker genes, including myosin heavy chain (MyHC) and muscle creatine kinase (MCK), are strongly increased in differentiated myocytes and modulate muscle activity (Berkes and Tapscott, 2005; Le Grand and Rudnicki, 2007). Muscle injury normally induces myogenic differentiation and muscle regeneration through production of inflammatory cytokines such as TNF-α, IL-1β and IL-6, and the activation of myogenic factor Myog (Tidball, 2005). However, severe skeletal muscle loss is characterized by significant muscle protein degradation by the activation of two muscle-specific ubiquitin ligases, MAFbx (muscle atrophy F-box) and MuRF1 (muscle-specific RING finger protein 1) (Glass, 2003; 2005; Murton et al., 2008; Bonaldo and Sandri, 2013). Common environmental factors that contribute to this catabolic process include starvation, denervation and chronic inflammatory conditions (Bonetto et al., 2009; Kumar et al., 2012; Muscaritoli et al., 2013). Accelerated or exaggerated muscle loss increases morbidity and mortality associated with chronic inflammatory diseases (Wolfe, 2006; Workeneh and Mitch, 2010). Thus, enhancement of myogenic differentiation would protect against injury-induced severe skeletal muscle loss and improve the management of inflammatory diseases (Wang and Pessin, 2013).
The discovery of low MW compounds that promote myogenic differentiation would be an important step in the development of clinical approaches for stimulating myogenic differentiation in damaged muscle and attenuating muscle atrophy. Because TAZ functions as a molecular stimulator of myogenic differentiation, we reasoned that molecules that modulate TAZ activity may provide important lead compounds. Here, we report the isolation of TM-53 and TM-54 as low MW compounds that stimulated nuclear localization of TAZ. Furthermore, we describe the effects of these compounds on the process of myogenic differentiation.
Methods
High-throughput screening for isolation of TAZ modulators
COS7 cells were transfected with GFP-TAZ and RFP-H2B (red fluorescence protein-tagged histone 2B), followed by treatment with a range of low MW compounds, at a fixed concentration (10 μM), from the Korea Research Institute of Chemical Technology (KRICT) Chemical Library Bank (Daejeon, Korea) (Jang et al., 2012b). Live cells were monitored with a BD Pathway Bioimaging system in real time (BD Biosciences, San Jose, CA, USA). At least 500 cells of both green and red fluorescence cells were analysed. Fluorescence signals were captured in real time in living cells. Nuclear TAZ expression was calculated after normalization with the RFP-H2B signal (Jang et al., 2012b).
Synthesis of TM-53 and TM-54
The 2-butyl-5-methoxycarbonyl-6-bromobenzimidazole was prepared in four steps from 2-bromo-5-nitrobenzoic acid. Reaction with biphenyltetrazole group gave a N-alkylated regioisomer mixture at 1 or 3 position of benzimidazole, which were separated by column chromatography. Subsequent Suzuki reaction with trans-2-phenylvinylboronic acid followed by acidic deprotection yielded the target compounds TM-53 (2-butyl-6-styryl-1-[2′-(1H-tetrazol-5-yl)-biphenyl-4-ylmethyl]-1H-benzoimidazole-5-carboxylic acid methyl ester) and TM-54 (2-butyl-5-styryl-1-[2′-(1H-tetrazol-5-yl)-biphenyl-4-ylmethyl]-1H-benzoimidazole-6-carboxylic acid methyl ester). TM-53 (MW = 568) and its structural isomer, TM-54, were further purified by HPLC to >98% purity at KRICT and dissolved in dimethyl sulfoxide (10 mg·mL−1).
In vitro myogenic differentiation and transdifferentiation
Mouse-derived C2C12 myoblasts (ATCC, Manassas, VA, USA) were maintained in DMEM supplemented with 10% FBS (HyClone, Logan, UT, USA) and grown to 100% confluence for inducing myogenic differentiation. For myogenic differentiation, C2C12 cells were replaced with DMEM supplemented with 2% horse serum every 2 days and were additionally treated with vehicle, TM-53 or TM-54 as indicated. For myogenic transdifferentiation of non-muscle cells, mouse embryonic fibroblasts (MEFs) of wild-type (WT) and TAZ KO MEFs (Hong et al., 2005) were infected with viruses expressing MyoD and/or TAZ and subsequently cultured under myogenic differentiation conditions (Jeong et al., 2010) in the presence of either TM-53 or TM-54.
Myogenic cell fusion index
C2C12 cells were induced to differentiate into myocytes and harvested for determining myogenic fusion index. Cells were rinsed and subjected to immunostaining using anti- MyHC (MF-20; DSHB, Iowa City, IA, USA). Stained sections were observed under a microscope. Myogenic fusion index was determined by dividing cell number of nuclei in MyHC-expressing multinucleated myotubes by the total number of myogenic nuclei, in a given microscopic field (Sabourin et al., 1999; Jankowski et al., 2002). Data are given as mean ± SEM (n = 10) from three independent cultures.
Retroviral infection
Platinum E (plat E) cells (from (ATCC) were transiently transfected with retroviral vectors; mock, MyoD or TAZ expression vector and pSRP (coni) and pSRP-shTAZ (TAZi), and cells were refreshed and transferred to a 32°C incubator. Viruses were concentrated using PEG Virus Precipitation Kit (BioVision Inc., Milpitas, CA, USA) and used for infection of C2C12 cells and MEFs in the presence of polybrene (8 μg·mL−1; Sigma-Aldrich, St. Louis, MO, USA) for 24 h (Jang et al., 2012a). Cell clones were selected in the presence of puromycin (2.5 μg·mL−1; Sigma-Aldrich) for 7 days and used for the myogenic differentiation.
Immunoprecipitation and immunoblotting
Cell extracts were prepared and resolved by SDS-PAGE, followed by incubation with Ab against MyHC (MF20), Myog, MyoD, β-actin (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA), and TAZ (Cell Signaling Technology, Inc., Danvers, MA, USA). For immunoprecipitation, cell extracts were incubated with Flag-M2 agarose beads (Sigma-Aldrich) overnight. Immune complexes were washed and subjected to immunoblotting analysis. Each experiment was repeated at least three times.
Reverse transcription and real-time PCR
Total RNA was isolated using TRIzol reagent and used for reverse transcription using the Superscript II kit (Invitrogen, Carlsbad, CA, USA). Quantitative real-time PCR was performed using SYBR Green PCR Master Mix with an ABI 7300 real-time PCR system (Applied Biosystems, Foster City, CA, USA). The specific primers used were as follows: β-actin, 5′-agagggaaatcgtgcgtgac-3′, 5′-caatagtgatgacctggccgt-3′; MCK 5′-cacctccacagcacagacag-3′, 5′-accttggccatgtgattgtt-3′; MyoD, 5′-tgggatatggagcttctatcgc-3′, 5′-ggtgagtcgaaacacggatcat-3′; myogenin 5′-caaccaggaggagcgagacctccg-3, 5′-aggcgctgtgggatatgcattcact-3′; and TAZ 5′-gtcaccaacagtagctcagatc-3′, 5′-agtgattacagccaggttagaaag-3′.
Immunocytochemistry and immunohistochemistry
Cultured cells were fixed with 4% paraformaldehyde, permeabilized and incubated with Ab against TAZ and MyHC and Alexa Fluor 594-conjugated secondary Ab (Molecular Probes, Invitrogen, Carlsbad, CA, USA). Nuclei were counterstained using DAPI and cells were visualized under fluorescence microscopy. Tibialis anterior (TA) muscles were fixed and embedded in paraffin, followed by section and stain with haematoxylin and eosin (Sigma-Aldrich). Tissue sections were incubated with anti-TAZ Ab and subsequently incubated with HRP using diaminobenzidine as a substrate, followed by observation under the light microscope.
In vivo muscle regeneration after injury
All animal care and experimental procedures complied with the institutional guidelines and were approved by the IACUC (IACUC-2011-01-027, 2012-01-035 and 2013-01-074). All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). A total of 36 animals were used in the experiments described here.
C57BL/6 WT mice were purchased from the Jackson Laboratories (Bar Harbor, ME, USA) and housed in an animal facility of Ewha Womans University. Mice (12–16 weeks of age, male, 25–30 g) were used for sciatic nerve injury-induced muscle regeneration. The nerve was isolated by making a small incision below the hip bone and tightly ligated with 6-0 silk suture thread (Abbadie et al., 2003). For sham operation in mice, the sciatic nerve was exposed but not ligated. The mice were additionally treated with TM-53 and TM-54 for 7 days and were killed, followed by analyses of the TA (Muller et al., 2007).
MCK activity assay
C2C12 stable cells (coni and TAZi) were induced to differentiate into myocytes for 2, 4 and 6 days in the presence of TM-53 or TM-54 and harvested for the creatine kinase assay. Briefly, cell extracts were incubated with creatine kinase substrate according to the manufacturer's instruction (BioVision Inc.) and absorbance at 450 nm was measured by spectrophotometer. Creatine kinase activity was calculated from standard curve of NADH amount to the optical density and is given as nmol·min−1·mL−1.
Reporter gene assay
C2C12 or MEF cells were transiently transfected with MCK promoter-linked reporter gene (pMCK-Luc) and MyoD expression vector (Jeong et al., 2010). The pCMVβ-gal (Promega, Madison, WI, USA) was also transfected into the cells as a control for transfection efficiency. Cells were then treated with TM-53 and TM-54 for an additional 24 h and harvested for luciferase assay (Promega). The β-galactosidase activity from pCMVβ-gal was also assayed using the Glacto-Light (Applied Biosystems, Bedford, MA, USA) and used for normalization of transfection efficiency.
Data analysis
Data are presented as mean ± SEM of at least three independent experiments and were analysed using one-way anova and unpaired Student's t-test. P < 0.05 was considered statistically significant.
Results
TM-53 is a novel low MW compound enhancing nuclear localization of TAZ
We monitored the subcellular localization of GFP-TAZ following treatment with a panel of low MW compounds from the Chemical Bank at KRICT and identified TM-53, a compound that stimulated TAZ nuclear localization. Treatment across a broad concentration range (1–100 μM) further confirmed that TM-53 increased the nuclear localization of TAZ under both hypotonic and isotonic conditions in a dose-dependent manner (Figure 1A and B). TM-53 contains a carboxylic acid methyl ester and a styryl group at the 5- and 6-positions, whereas its structural isomer, TM-54, has these groups in the reverse orientation (Figure 1C). These compounds were synthesized to a purity of >98% prior to use in the subsequent in vitro and in vivo experiments.
Figure 1.
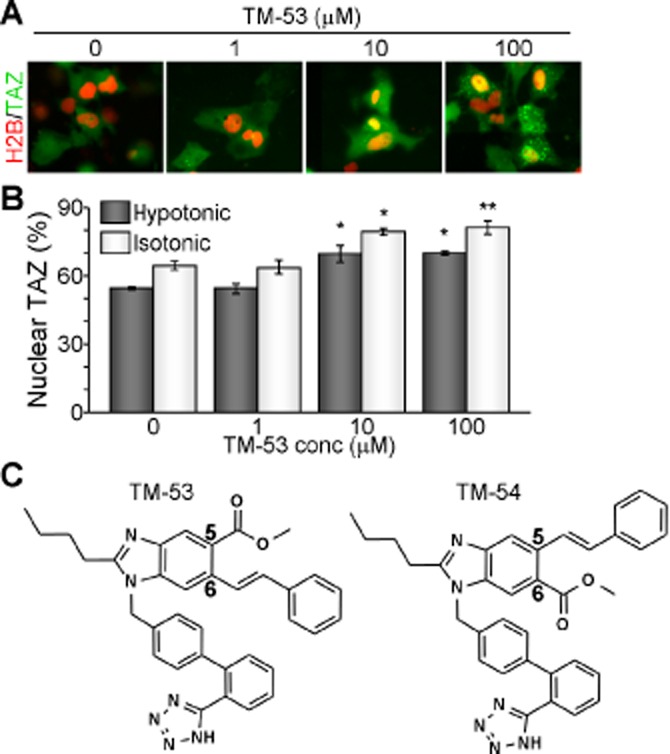
Isolation of the novel TAZ modulators, TM-53 and TM-54. (A, B) COS7 cells were transfected with GFP-TAZ and RFP-H2B and subsequently treated with different doses of TM-53 for 2 h under normal (isotonic) and low salt (hypotonic) conditions. Representative cell images were captured on a BD Pathway Bioimager system (A). The percentage of cells expressing TAZ in the nucleus was determined by counting at least 500 GFP-positive and RFP-positive cells from three independent experiments (B). (C) Chemical structures of TM-53 and TM-54.
TM-53 and TM-54 strongly enhance the nuclear localization of TAZ protein
As C2C12 cells serve as an excellent in vitro model system for skeletal muscle differentiation and TAZ is known to stimulate myogenic differentiation of C2C12 cells, we first examined the cytotoxicity of TM-53 and TM-54 in this system. Neither compound was cytotoxic between 1 and 20 μM, although TM-53 and TM-54 both decreased cell viability at 50 and 100 μM (Figure 2A). We thus treated C2C12 cells with doses below 20 μM for all subsequent experiments. Quantitative analysis of nuclear TAZ followed by immunofluorescent TAZ staining revealed that TM-53 and TM-54 significantly increased the proportion of cells with nuclear TAZ, at 10 and 20 μM (Figure 2B). This can also be observed in representative images taken after treatment with TM-53 and TM-54 (Figure 2C). Additional quantitative analysis of subcellular distribution of TAZ demonstrated that treatment with TM-53 and TM-54 induced nuclear TAZ level and concomitantly decreased cytoplasmic TAZ (Figure 2D). Furthermore, fractionation of nuclear and cytoplasmic proteins confirmed that TM-53 and TM-54 enhanced nuclear translocation of TAZ (Figure 2E). We then examined whether TM-53 and TM-54 affected S89 phosphorylation of TAZ as this modification leads to 14-3-3-dependent sequestration of TAZ in the cytoplasm (Kanai et al., 2000). Interestingly, phosphorylation of S89 was slightly but reproducibly attenuated by treatment with TM-53 and was more significantly decreased by TM-54 treatment (Figure 2F).
Figure 2.
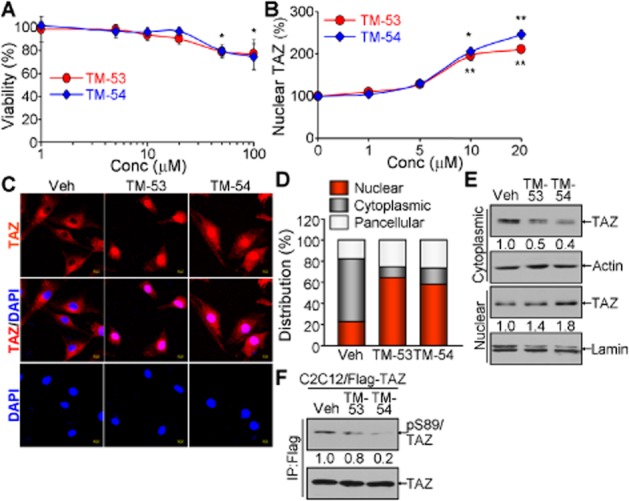
Enhancement of TAZ nuclear translocation by TM-53 and TM-54. (A) C2C12 cells were treated with TM-53 and TM-54 (1, 5, 10, 20, 50 and 100 μM) for 24 h and subjected to the cytotoxicity assay. Cell viability was calculated by measuring optical density at 450 nm after staining and is given as mean ± SEM for three experiments. The viability of cells treated with vehicle was set as 100%. (B) Cells were treated with TM-53 and TM-54 (0, 1, 5, 10 and 20 μM) and stained with anti-TAZ antibody. The location of the nuclei was indicated by DAPI staining. Nuclear TAZ was calculated by dividing nuclear TAZ-expressing cells by the total number of TAZ-expressing cells (100 cells for each counting, five times). Nuclear TAZ expression in vehicle-treated cells was regarded as 100% and is expressed as mean ± SEM. (C, D) C2C12 cells were treated with 10 μM TM-53 and TM-54 for 24 h. Cells were subjected to immunofluorescence staining with anti-TAZ antibody. Representative images of three independent experiments are shown (C). Percentages of nuclear, cytoplasmic and pan-cellular TAZ distribution were calculated by counting 80–150 cells in multiple fields of view (n = 5 per sample) (D). (E, F) C2C12 cells stably expressing Flag-TAZ were incubated with TM-53 (10 μM) and TM-54 (10 μM) for 24 h. Cells were harvested and subjected to fractionation for nuclear and cytoplasmic protein, followed by immunoblotting analysis (E). Total protein extract was used for immunoprecipitation with anti-Flag antibody and subsequently analysed by SDS-PAGE and immunoblotting with anti-pS89 TAZ antibody.
MyHC expression is enhanced by TM-53 and TM-54 during myogenic differentiation
We then examined the effects of TM-53 and TM-54 on myogenic differentiation in C2C12 cells. Myocyte morphological characteristics and MyHC expression were both remarkably increased in C2C12 cells after treatment with TM-53 and TM-54 (Figure 3A). This was confirmed by a quantitative analysis of the fraction of MyHC-expressing cells in the presence of TM-53 and TM-54 compared with vehicle-treated cells (Figure 3B). In addition, there was an increase in the cell fusion index in myocytes treated with TM-53 and TM-54 (Figure 3C). Muscle marker genes such as Myog, MyHC and MCK were significantly up-regulated by treatment with TM-53 and TM-54 at various time points (Figure 3D and E). These results indicate that TM-53 and TM-54 may enhance myogenic differentiation and muscle activity.
Figure 3.
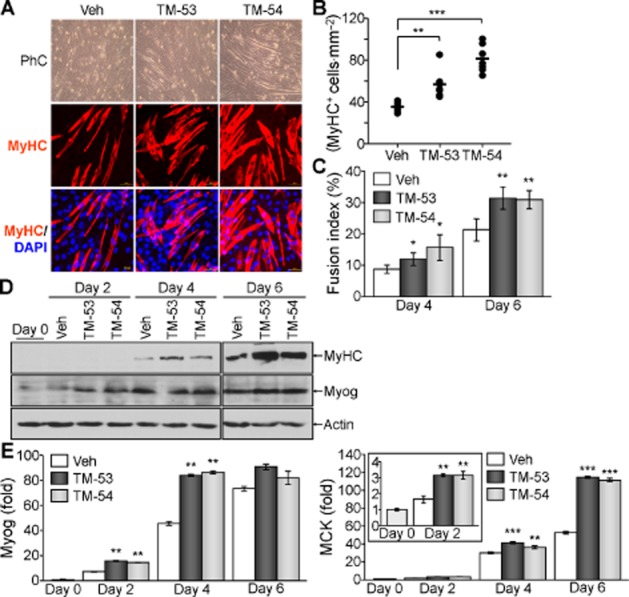
Stimulation of myogenic differentiation by TM-53 and TM-54. C2C12 cells were induced to differentiate into mature myocytes in the presence of vehicle, TM-53 (10 μM) or TM-54 (10 μM) for 6 days. (A) Differentiated myocytes were fixed and incubated with MyHC Ab, followed by counterstaining with DAPI and observation under a fluorescence microscope. (B) For quantitative analysis, the MyHC-expressing cells were counted from six different regions of the three independent culture experiments. (C) Multinucleated MyHC+ cells (between 50 and 100 cells counted) were scored in 5 different fields of view per sample. Fusion index (>2 nuclei per cell) was calculated by dividing the number of multinucleated cells by the number of MyHC+ cells from three independent experiments. (D) Total cell extracts were harvested at the indicated time points and subjected to immunoblotting with anti-MCK Ab. (E) Total RNA was prepared from differentiating cells and the relative expression of MyHC was determined by real-time PCR analysis. The results of (D) and (E) are given as mean ± SEM for three experiments. (F) Cells were harvested at day 5 and incubated with Fura-2AM for 1 h. Cells were washed with calcium-free buffer and measured at 340 and 380 nm. The ratio of calcium influx was calculated at the indicated times. This experiment was repeated three times and a representative graph is shown. *P < 0.05; **P < 0.005; ***P < 0.0005; significantly different from vehicle.
TM-53 and TM-54 are equipotent and induce the expression of myogenic markers in a dose-dependent manner
We further analysed the expression of myogenic markers to assure the myogenic stimulatory effects of TM-53 and TM-54. TM-53 and TM-54 dose-dependently increased the levels of MyHC and Myog during myogenic differentiation, as evidenced by immunoblotting and quantitative analysis (Figure 4A). Concomitantly, the relative transcript levels of MCK and Myog were augmented by treatment with TM-53 and TM-54 in a dose-dependent manner (Figure 4B). On the contrary, the expression of MyoD and TAZ was not affected by either TM-53 or TM-54 (Figure 4C).
Figure 4.
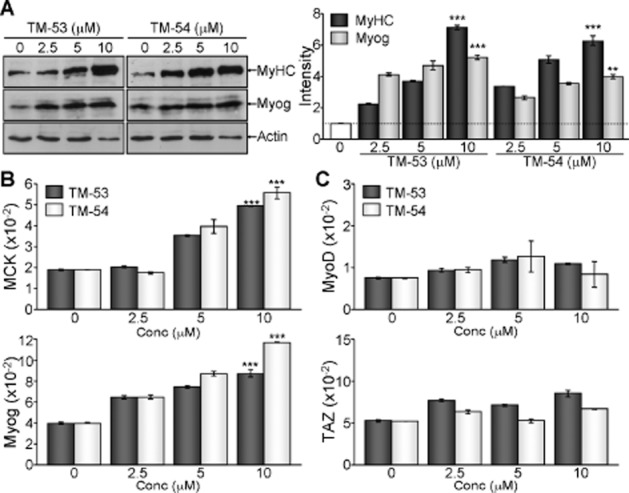
Augmentation of myogenic gene expression by treatment with TM-53 and TM-54. C2C12 myoblasts were treated with different amounts (0, 2.5, 5 and 10 μM) of TM-53 and TM-54 for 4 days during myogenic differentiation. (A) Protein extracts were resolved by SDS-PAGE, followed by immunoblotting analysis with Ab against MyHC, Myog and actin. The bands of MyHC and Myog were quantitatively analysed by densitometry. Representative immunoblotting images are shown and protein band intensity is mean ± SEM for three independent experiments after normalization to the actin level. (B, C) Total RNA was harvested using TRIzol and reverse-transcribed for quantitative real-time PCR analysis. Relative expression level of MCK and Myog (B), MyoD and TAZ (C) was determined after normalization to β-actin levels. **P < 0.005; ***P < 0.0005; significantly different from control.
Myogenic stimulatory effects of TM-53 and TM-54 are dependent upon TAZ expression
To confirm the importance of TAZ in myogenic stimulatory activities by TM-53 and TM-54, we established TAZ knockdown myoblasts (TAZi) and induced myogenic differentiation in the presence of TM-53 and TM-54. As previously reported (Jeong et al., 2010), myogenic differentiation was impaired in TAZi cells compared with control cells, as evidenced by diminished MyHC expression in TAZi cells. In addition, TM-53 and TM-54 strongly augmented the expression of MyHC in control cells, but this effect was absent in TAZi cells (Figure 5A). Accordingly, MCK and Myog were also significantly induced in response to TM-53 and TM-54 treatment in control cells, but failed to increase in TAZi cells (Figure 5B). In addition, MCK activity increased time-dependently increased during the course of myogenesis and was enhanced following treatment with TM-53 and TM-54 in control cells; this activity was unchanged in response to TM-53 and TM-54 in TAZi cells (Figure 5C). TAZ is critical for the regulation of MyoD-induced gene expression through a direct interaction with MyoD, and seemed to be prerequisite for the activities of TM-53 and TM-54. We thus inquired whether TM-53 and TM-54 affected dimerization between MyoD and TAZ, and found that both TM-53 and TM-54 enhanced the interaction between two molecules (Figure 5D). We also examined the effects of TM-53 and TM-54 on MyoD-induced gene expression. Treatment with TM-53 and TM-54 enhanced Myog and MCK promoter activities, but this effect was abrogated in the absence of TAZ (Figure 5E). These results suggest that TM-53 and TM-54 stimulate myogenic gene expression through the activation of MyoD–TAZ interaction.
Figure 5.

TAZ-dependent stimulatory effect of TM-53 and TM-54 on myogenic differentiation. (A–C) Control (Coni) and TAZ knockdown (TAZi) C2C12 cells were established and differentiated into myocytes in the presence of TM-53 or TM-54. Cell extracts were harvested at day 6 and subjected to immunoblotting with anti-MyHC antibody. MyHC expression level was analysed using densitometry (A). Total RNA was collected at day 6 and used for reverse transcription and real-time PCR analysis. Relative expression level of MCK, Myog and TAZ was calculated after normalization to β-actin levels. *P < 0.05; **P < 0.005; ***P < 0.0005; significantly different from vehicle-treated group; #P < 0.01; ##P < 0.001; significantly different from coni group (B). Cells were harvested at different time points and used for MCK activity assay using MCK colorimetric assay kit (BioVision Inc.). MCK activity was determined as nmoL·min−1·mL−1 (C). (D) Highly transfectable 293T cells were transfected with Flag-TAZ and MyoD expression vector and were treated with TM-53 (10 μM) and TM-54 (10 μM) for 24 h. Cell extracts were subjected to immunoprecipitation and immunoblotting. (E) WT and KO MEF cells were transfected with MyoD expression vector together with either pMyog-luc or pMCK-luc. Cells were then treated with TM-53 (10 μM) and TM-54 (10 μM) for 24 h and collected for reporter gene assay. The promoter activity was expressed as fold change after normalization with β-galactosidase activity. The MyoD-induced promoter activity in vehicle-treated cells was set as 1. All experiments in Figure 5 were repeated at least three times and data are given as mean ± SEM. Representative image from three blots was shown in Figure 5A and D.
MyoD-mediated myogenic transdifferentiation is enhanced by TM-53 and TM-54 in a TAZ-dependent manner
As TM-53 and TM-54 enhanced MyoD-induced MCK promoter activity in the presence of TAZ, we investigated the effects of TM-53 and TM-54 on MyoD-induced myogenic transdifferentiation of non-muscle cell type, MEF cells. Retroviral infection of WT MEFs with MyoD substantially increased MyHC expression; this effect was lost when MyoD was introduced into TAZ KO MEFs (Figure 6A). Contemporarily, the expression of MCK and Myog was robust in WT MEFs that overexpressed MyoD, but was significantly attenuated in KO cells (Figure 6B). Comparable MyoD expression in WT and KO cells and TAZ deficiency in KO cells were also confirmed (Figure 6B). Additional treatment of MyoD-transduced WT MEFs with TM-53 and TM-54 substantially elevated the expression of MCK and Myog. However, the stimulatory effects of TM-53 and TM-54 were abolished in the absence of TAZ (Figure 6C). Interestingly, restoration of TAZ expression together with MyoD rescued the attenuated expression of MyHC and Myog in KO cells. This induction was further augmented by treatment with TM-53 and TM-54 (Figure 6D). In addition, the robust expression of MyHC and Myog was prominently observed in TAZ-reconstituted KO cells treated with TM-53 and TM-54 (Figure 6E). These results suggest that the restoration of TAZ acts together with MyoD in TAZ KO MEF cells to elicit myogenic differentiation.
Figure 6.

Stimulation of myogenic transdifferentiation of non-muscle cells by TM-53 and TM-54. (A, B) TAZ WT and KO MEFs were infected with viruses expressing mock vector (RV) or MyoD (RV-MyoD) and were subjected to conditions that induce myogenic differentiation. Morphological changes were observed under a microscope. (A). Total RNA was harvested and subjected to reverse transcription and real-time PCR analysis. Relative levels of MCK, Myog and MyoD transcripts were determined after normalization to β-actin levels (B) *P < 0.05; ***P < 0.0005; significantly different from WT. In (C), WT and KO MEFs infected with MyoD-expressing viruses were treated with TM-53 or TM-54 under myogenic differentiation conditions. Total RNA was prepared and subjected to reverse transcription and real-time PCR. Relative transcript levels of MCK and Myog were determined after normalization to β-actin level and expressed as fold change after normalization to MyoD. Data are shown as mean ± SEM from three independent experiments. (D, E) TAZ KO MEFs were transfected with MyoD alone or with TAZ via retroviral infection, and additionally treated with TM-53 and TM-54 during myogenic differentiation. Relative transcript levels of MCK and Myog were determined by real-time PCR (D). Protein extracts were analysed by immunoblotting. Band intensity in TAZ-transduced cells was analysed by densitometry (E). *P < 0.05; **P < 0.005; ***P < 0.0005; significantly different from vehicle.
TM-53 and TM-54 stimulate muscle regeneration in vivo
In order to examine the effects of TM-53 and TM-54 on muscle regeneration in vivo, we examined the response of mice to induction of sciatic nerve injury. These mice were unable to run and showed an increased number of tibial shocks during treadmill running. However, administration of TM-53 and TM-54 improved muscular activity during running after injury, as shown by a decrease in shock number during running (Figure 7A). Histological analysis revealed that the myofibers ruptured and necrotized in the group with nerve injury. Interestingly, TM-53 and TM-54 treatment increased the centrally nucleated myofibers in muscle, indicating an improved regeneration of myofibers in response to TM-53 and TM-54 treatment (Figure 7B). Furthermore, the reduction of MyHC expression after injury was blocked by administration of TM-53 and TM-54 (Figure 7C). Interestingly, TAZ expression was mainly observed in the cytoplasm of the injured muscle, whereas TM-53 and TM-54 treatment increased nuclear localization of TAZ in the nucleus of regenerating myofibers (Figure 7D). TM-induced blockade of MyHC reduction was also confirmed in differentiated myocytes in vitro. MyHC level was substantially decreased in differentiated myocytes after treatment with dexamethasone (DEX). However, the DEX-induced decrease of MyHC protein was abolished by TM-53 and TM-54 treatment (Figure 7E). These results imply that TM-53 and TM-54 accelerate injury-induced muscle regeneration and protect against muscle damage in vitro and in vivo.
Figure 7.
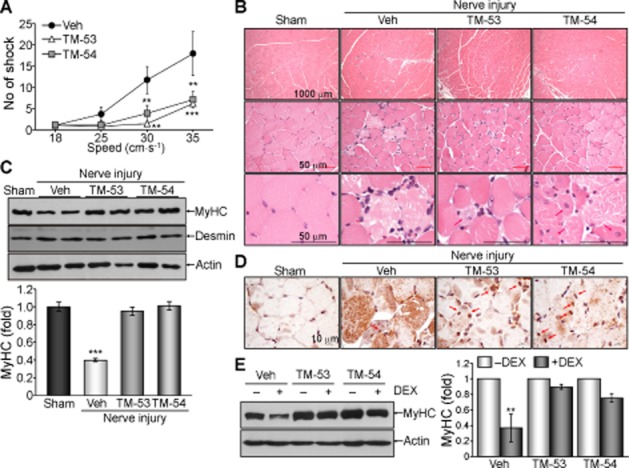
Improvement of muscle regeneration and physical activity after injury upon administration of TM-53 and TM-54. (A–D) WT mice were subjected to sciatic nerve injury and were administered daily with either vehicle (Veh, n = 6), TM-53 (10 mg·kg−1, n = 6) or TM-54 (10 mg·kg−1, n = 6) by i.p. injection for 7 days. Mice (n = 6) were subjected to a treadmill running test at days 5, 6 and 7. The number of electric shocks experienced at day 7 in response to the running speed of treadmill is given as mean ± SEM; **P < 0.005; ***P < 0.0005; significantly different from vehicle (A). TA muscle tissue was sectioned and subjected to haematoxylin and eosin staining. Representative images are shown from six different staining results for each group. Scale bars indicate 1000 and 50 μm. Red arrows indicate central nuclei in regenerating myofibers (B). Protein extracts were isolated from TA muscle and were subsequently analysed by immunoblotting. Representative images for two mice are shown and band intensity was calculated from three independent blots (n = 6); ***P < 0.0005; significantly different from sham (C). TA muscle tissue was sectioned and stained with anti-TAZ Ab. Representative images from six different sections were shown (D). (E) C2C12 cells were induced to undergo myogenic differentiation in the presence of TM-53 or TM-54 for 6 days. Cells were treated with DEX (500 μM) for 24 h and harvested. Protein extracts were analysed by immunoblotting. The change in MyHC expression is expressed as mean ± SEM from three independent immunoblottings and subsequent densitometric analysis; **P < 0.005; significantly different from vehicle.
Discussion
Our results demonstrated that a novel TAZ modulator, TM-53, and its isomer, TM-54, strongly promoted nuclear localization of TAZ, and further augmented MyoD-induced myogenic gene transcription, leading to an acceleration of myogenic differentiation and transdifferentiation. Interestingly, the loss of myogenic stimulatory activity of both TM-53 and TM-54 in TAZ-deficient cells was restored after overexpression of TAZ. TM-53 and TM-54 substantially enhanced muscle regeneration and improved muscle activity after injury in vivo, suggesting a beneficial role of TM-53 and TM-54 in treatment of muscular dysfunction.
The mechanisms underlying the increase TAZ nuclear localization by TM-53 and TM-54 are not clear. Nuclear translocation of TAZ can be regulated by post-translational modifications such as dephosphorylation of S89 or phosphorylation of Y316 upon hypertonic stimulation (Jang et al., 2012a). Although S89 phosphorylation of TAZ was attenuated by treatment with TM-53 and TM-54, it is not yet clear whether TM-53 and TM-54 affect tyrosine phosphorylation of TAZ during myogenic differentiation. It is thought that the changes in the phosphorylation status affect the structures of TAZ proteins. In particular, domains that participate in protein–protein interactions, such as the WW domain, coiled-coil domain, and PDZ-binding motif, undergo conformational changes that may alter interactions with partner proteins including 14-3-3; this may, in turn, facilitate nuclear translocation. In addition, several low MW compounds, including TM-53 and TM-54, may directly bind to TAZ protein. However, a detailed characterization and X-ray crystallographic structural study of TAZ in the presence and absence of compounds will be required in order to confirm this hypothesis.
TM-54 is a structural isomer of TM-53 that contains a carboxylic acid methyl ester and a styryl group at the 5- and 6-positions respectively. Despite being structural isomers, TM-53 and TM-54 were equally effective in enhancing nuclear localization of TAZ, induction of myocyte differentiation of myoblasts in vitro, and stimulation of muscle regeneration after injury in vivo. This suggests that the position of the styryl and carboxylic acid methyl ester groups was not a critical determinant of TAZ-modulatory activity and myogenic differentiation associated with these compounds. However, TM-54 did suppress S89-phosphorylation of TAZ more effectively than TM-53 and more efficiently enhanced myogenic differentiation of myoblasts. On the contrary, reduction of S89-phosphorylation by TM-53 was relatively moderate but significant, thus enhancing nuclear localization of TAZ. TM-53 was more potent with regard to stimulation of MyoD-induced myogenic transdifferentiation of non-muscle cells. This was probably due to significantly more robust induction of MyHC, MCK and Myog by TM-53, which was mediated through the activation of MyoD in non-muscle cells. These results indicate a potential difference in the mechanisms of action between TM-53 and TM-54.
Importantly, TM-53 and TM-54 dramatically improved physical performance and disability caused by injury, as shown by the decreased frequency of exercise-induced shock. Accordingly, histological and immunological analyses also confirmed that TM-53 and TM-54 attenuated nerve injury-induced muscle fibre damage and increased the number of regenerative muscle fibres with central nuclei in injured muscle. Furthermore, TM-53 and TM-54 intensified intracellular calcium transients and MCK activity in differentiated myocytes in vitro. These results together suggest that TM-53 and TM-54 may have beneficial roles in preventing and treating muscular injury.
In conclusion, the novel TAZ modulators, TM-53 and TM-54, augmented myogenic differentiation of C2C12 myoblasts and transdifferentiation of non-muscle cell type in a TAZ-dependent manner, by increasing the nuclear localization of TAZ. Activation of TAZ by TM-53 and TM-54 robustly increased the expression of muscle-specific marker genes, calcium transients and MCK activity. Furthermore, TM-53 and TM-54 accelerated injury-induced muscle regeneration and improved physical performance after injury in mice. Moving forward, it will be important to examine whether TM-53 and TM-54 are capable of delivering substantial clinical benefits.
Acknowledgments
This work was supported by the basic research program (2012R1A1B3000603) of the National Research Foundation funded by Ministry of Education, Science, and Technology and partly by the Ministry of Health & Welfare (AI21102 and AI20476).
Glossary
- DEX
dexamethasone
- KO
knockout
- KRICT
Korea Research Institute of Chemical Technology
- MAFbx
muscle atrophy F-box
- MCK
muscle creatine kinase
- MEF
mouse embryonic fibroblast
- MRF
muscle regulatory factor
- MuRF1
muscle-specific RING finger protein 1
- MYF5
myogenic determination factor 5
- MyHC
myosin heavy chain
- MyoD
myoblast determining protein D
- Myog
myogenine
- RFP-H2B
red fluorescence protein-tagged histone 2B
- TAZ
transcriptional co-activator with PDZ-binding motif
- TM
TAZ modulator
- WT
wild type
Author contributions
G. H. P., H. J., M-G. J. and E. J. J. conducted all experiments and analyses; M. A. B., Y-L. L. and N. J. K. synthesized TM-53 and TM-54 and characterized their characteristics and purity; J-H. H. generated the expression vectors and stable cell lines; and E. S. H. designed all experiments and wrote the paper.
Conflict of interest
No potential conflicts of interest were disclosed.
References
- Abbadie C, Lindia JA, Cumiskey AM, Peterson LB, Mudgett JS, Bayne EK, et al. Impaired neuropathic pain responses in mice lacking the chemokine receptor CCR2. Proc Natl Acad Sci U S A. 2003;100:7947–7952. doi: 10.1073/pnas.1331358100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold HH, Winter B. Muscle differentiation: more complexity to the network of myogenic regulators. Curr Opin Genet Dev. 1998;8:539–544. doi: 10.1016/s0959-437x(98)80008-7. [DOI] [PubMed] [Google Scholar]
- Berkes CA, Tapscott SJ. MyoD and the transcriptional control of myogenesis. Semin Cell Dev Biol. 2005;16:585–595. doi: 10.1016/j.semcdb.2005.07.006. [DOI] [PubMed] [Google Scholar]
- Bonaldo P, Sandri M. Cellular and molecular mechanisms of muscle atrophy. Dis Model Mech. 2013;6:25–39. doi: 10.1242/dmm.010389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonetto A, Penna F, Muscaritoli M, Minero VG, Rossi Fanelli F, Baccino FM, et al. Are antioxidants useful for treating skeletal muscle atrophy? Free Radic Biol Med. 2009;47:906–916. doi: 10.1016/j.freeradbiomed.2009.07.002. [DOI] [PubMed] [Google Scholar]
- Cui CB, Cooper LF, Yang X, Karsenty G, Aukhil I. Transcriptional coactivation of bone-specific transcription factor Cbfa1 by TAZ. Mol Cell Biol. 2003;23:1004–1013. doi: 10.1128/MCB.23.3.1004-1013.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass DJ. Signalling pathways that mediate skeletal muscle hypertrophy and atrophy. Nat Cell Biol. 2003;5:87–90. doi: 10.1038/ncb0203-87. [DOI] [PubMed] [Google Scholar]
- Glass DJ. Skeletal muscle hypertrophy and atrophy signaling pathways. Int J Biochem Cell Biol. 2005;37:1974–1984. doi: 10.1016/j.biocel.2005.04.018. [DOI] [PubMed] [Google Scholar]
- Hong JH, Yaffe MB. TAZ: a beta-catenin-like molecule that regulates mesenchymal stem cell differentiation. Cell Cycle. 2006;5:176–179. doi: 10.4161/cc.5.2.2362. [DOI] [PubMed] [Google Scholar]
- Hong JH, Hwang ES, McManus MT, Amsterdam A, Tian Y, Kalmukova R, et al. TAZ, a transcriptional modulator of mesenchymal stem cell differentiation. Science. 2005;309:1074–1078. doi: 10.1126/science.1110955. [DOI] [PubMed] [Google Scholar]
- Hong W, Guan KL. The YAP and TAZ transcription co-activators: key downstream effectors of the mammalian Hippo pathway. Semin Cell Dev Biol. 2012;23:785–793. doi: 10.1016/j.semcdb.2012.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang EJ, Jeong H, Han KH, Kwon HM, Hong JH, Hwang ES. TAZ suppresses NFAT5 activity through tyrosine phosphorylation. Mol Cell Biol. 2012a;32:4925–4932. doi: 10.1128/MCB.00392-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang EJ, Jeong H, Kang JO, Kim NJ, Kim MS, Choi SH, et al. TM-25659 enhances osteogenic differentiation and suppresses adipogenic differentiation by modulating the transcriptional co-activator TAZ. Br J Pharmacol. 2012b;165:1584–1594. doi: 10.1111/j.1476-5381.2011.01664.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowski RJ, Deasy BM, Cao B, Gates C, Huard J. The role of CD34 expression and cellular fusion in the regeneration capacity of myogenic progenitor cells. J Cell Sci. 2002;115:4361–4374. doi: 10.1242/jcs.00110. [DOI] [PubMed] [Google Scholar]
- Jeong H, Bae S, An SY, Byun MR, Hwang JH, Yaffe MB, et al. TAZ as a novel enhancer of MyoD-mediated myogenic differentiation. FASEB J. 2010;24:3310–3320. doi: 10.1096/fj.09-151324. [DOI] [PubMed] [Google Scholar]
- Kanai F, Marignani PA, Sarbassova D, Yagi R, Hall RA, Donowitz M, et al. TAZ: a novel transcriptional co-activator regulated by interactions with 14-3-3 and PDZ domain proteins. EMBO J. 2000;19:6778–6791. doi: 10.1093/emboj/19.24.6778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Bhatnagar S, Paul PK. TWEAK and TRAF6 regulate skeletal muscle atrophy. Curr Opin Clin Nutr Metab Care. 2012;15:233–239. doi: 10.1097/MCO.0b013e328351c3fc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassar AB, Skapek SX, Novitch B. Regulatory mechanisms that coordinate skeletal muscle differentiation and cell cycle withdrawal. Curr Opin Cell Biol. 1994;6:788–794. doi: 10.1016/0955-0674(94)90046-9. [DOI] [PubMed] [Google Scholar]
- Le Grand F, Rudnicki MA. Skeletal muscle satellite cells and adult myogenesis. Curr Opin Cell Biol. 2007;19:628–633. doi: 10.1016/j.ceb.2007.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei QY, Zhang H, Zhao B, Zha ZY, Bai F, Pei XH, et al. TAZ promotes cell proliferation and epithelial-mesenchymal transition and is inhibited by the hippo pathway. Mol Cell Biol. 2008;28:2426–2436. doi: 10.1128/MCB.01874-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CY, Lv X, Li T, Xu Y, Zhou X, Zhao S, et al. PP1 cooperates with ASPP2 to dephosphorylate and activate TAZ. J Biol Chem. 2011;286:5558–5566. doi: 10.1074/jbc.M110.194019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Drummond GB, McLachlan EM, Kilkenny C, Wainwright CL. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller FL, Song W, Jang YC, Liu Y, Sabia M, Richardson A, et al. Denervation-induced skeletal muscle atrophy is associated with increased mitochondrial ROS production. Am J Physiol Regul Integr Comp Physiol. 2007;293:R1159–R1168. doi: 10.1152/ajpregu.00767.2006. [DOI] [PubMed] [Google Scholar]
- Murton AJ, Constantin D, Greenhaff PL. The involvement of the ubiquitin proteasome system in human skeletal muscle remodelling and atrophy. Biochim Biophys Acta. 2008;1782:730–743. doi: 10.1016/j.bbadis.2008.10.011. [DOI] [PubMed] [Google Scholar]
- Muscaritoli M, Lucia S, Molfino A, Cederholm T, Rossi Fanelli F. Muscle atrophy in aging and chronic diseases: is it sarcopenia or cachexia? Intern Emerg Med. 2013;8:553–560. doi: 10.1007/s11739-012-0807-8. [DOI] [PubMed] [Google Scholar]
- Olson EN. Interplay between proliferation and differentiation within the myogenic lineage. Dev Biol. 1992;154:261–272. doi: 10.1016/0012-1606(92)90066-p. [DOI] [PubMed] [Google Scholar]
- Perry RL, Rudnick MA. Molecular mechanisms regulating myogenic determination and differentiation. Front Biosci. 2000;5:D750–D767. doi: 10.2741/perry. [DOI] [PubMed] [Google Scholar]
- Sabourin LA, Girgis-Gabardo A, Seale P, Asakura A, Rudnicki MA. Reduced differentiation potential of primary MyoD−/− myogenic cells derived from adult skeletal muscle. J Cell Biol. 1999;144:631–643. doi: 10.1083/jcb.144.4.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tidball JG. Inflammatory processes in muscle injury and repair. Am J Physiol Regul Integr Comp Physiol. 2005;288:R345–R353. doi: 10.1152/ajpregu.00454.2004. [DOI] [PubMed] [Google Scholar]
- Wang Y, Pessin JE. Mechanisms for fiber-type specificity of skeletal muscle atrophy. Curr Opin Clin Nutr Metab Care. 2013;16:243–250. doi: 10.1097/MCO.0b013e328360272d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe RR. The underappreciated role of muscle in health and disease. Am J Clin Nutr. 2006;84:475–482. doi: 10.1093/ajcn/84.3.475. [DOI] [PubMed] [Google Scholar]
- Workeneh BT, Mitch WE. Review of muscle wasting associated with chronic kidney disease. Am J Clin Nutr. 2010;91:1128S–1132S. doi: 10.3945/ajcn.2010.28608B. [DOI] [PubMed] [Google Scholar]