

Abstract
Background and Purpose
Overactive lipolysis in adipose tissue contributes to the pathogenesis of alcoholic liver disease (ALD); however, the mechanisms involved have not been elucidated. We previously reported that chronic alcohol consumption produces a hypomethylation state in adipose tissue. In this study we investigated the role of hypomethylation in adipose tissue in alcohol-induced lipolysis and whether its correction contributes to the well-established hepatoprotective effect of betaine in ALD.
Experimental Approach
Male C57BL/6 mice were divided into four groups and started on one of four treatments for 5 weeks: isocaloric pair-fed (PF), alcohol-fed (AF), PF supplemented with betaine (BT/AF) and AF supplemented with betaine (BT/AF). Betaine, 0.5% (w v−1), was added to the liquid diet. Both primary adipocytes and mature 3T3-L1 adipocytes were exposed to demethylation reagents and their lipolytic responses determined.
Key Results
Betaine alleviated alcohol-induced pathological changes in the liver and rectified the impaired methylation status in adipose tissue, concomitant with attenuating lipolysis. In adipocytes, inducing hypomethylation activated lipolysis through a mechanism involving suppression of protein phosphatase 2A (PP2A), due to hypomethylation of its catalytic subunit, leading to increased activation of hormone-sensitive lipase (HSL). In line with in vitro observations, reduced PP2A catalytic subunit methylation and activity, and enhanced HSL activation, were observed in adipose tissue of alcohol-fed mice. Betaine attenuated this alcohol-induced PP2A suppression and HSL activation.
Conclusions and Implications
In adipose tissue, a hypomethylation state contributes to its alcohol-induced dysfunction and an improvement in its function may contribute to the hepatoprotective effects of betaine in ALD.
Keywords: methionine, S-adenosylmethionine, S-adenosylhomocysteine, PP2A, HSL
Introduction
Alcohol liver disease (ALD) covers a wide spectrum of liver diseases ranging from simple hepatic steatosis [accumulation of triglyceride (TG) inside hepatocytes] to steatohepatitis (necrosis and inflammation), with some people ultimately progressing to fibrosis/cirrhosis and liver failure. The disease ranks among the major causes of morbidity and mortality in the world, and affects millions of patients worldwide each year (O'Shea et al., 2010). Fatty liver is the most common and earliest response of the liver to chronic alcohol consumption. Although it can be a completely benign condition, excessive fat accumulation makes hepatocytes vulnerable to the ‘second hit’, mainly pro-inflammatory cytokines and oxidative stress, ultimately resulting in steatohepatitis (Stewart et al., 2001; Reddy and Rao, 2006).
Although much progress has been made in recent decades, the pathogenesis of ALD remains to be clearly defined. In general, excess fat accumulation in the liver results from an imbalance in the uptake, synthesis, export and oxidation of fatty acids. Chronic alcohol exposure, by up-regulating sterol regulatory element-binding protein (SREBP)-1c, a master transcription factor controlling de novo lipogenesis (You et al., 2002), results in increased hepatic TG biosynthesis. Specific knockout of SREBP-1c in the liver has been shown to protect mice from alcohol-induced liver damage (Ji et al., 2006), suggesting that up-regulation of hepatic de novo lipogenesis contributes to ALD. Moreover, chronic alcohol consumption has been demonstrated to impair hepatic fatty acid β-oxidation by suppressing AMPK and PPAR-α activation (Fischer et al., 2003; You et al., 2004). Furthermore, both increased hepatic fatty acid absorption (Zhong et al., 2012) and impaired hepatic very-low-density lipoprotein secretion (Sugimoto et al., 2002) were reported to contribute to the development of hepatic steatosis after chronic alcohol exposure.
Non-esterified free fatty acids (FFAs) in the form of TG are mainly stored in adipose tissue and are released into the circulation via a process called lipolysis. Uncontrolled lipolysis in adipose tissue, due to insulin resistance, leads to excessive exposure and uptake of FFA by the liver, which plays a causal role in the development of obesity-related non-alcoholic fatty liver disease (NAFLD) (Sanyal, 2005). There is also accumulating evidence that adipose tissue dysfunction plays a pivotal role in the pathogenesis of ALD. In the clinical setting, visceral fat accumulation is positively associated with the onset of alcoholic liver damage and body mass index represents an independent risk factor for fibrosis in alcoholic patients (Loomba et al., 2009; Hart et al., 2010; Shen et al., 2010; Tsai et al., 2011). Moreover, adipose tissue inflammation is correlated with the severity of pathological features in livers of patients with ALD (Souto et al., 2005). Experimentally, chronic alcohol consumption induces oxidative stress in adipose tissue, insulin resistance, inflammation, adipocyte cell death and reduced levels of adiponectin (Chen et al., 2007; Kang et al., 2007; Tang et al., 2007; Song et al., 2008; Sebastian et al., 2011). Moderate obesity and alcohol act synergistically to induce steatohepatitis (Xu et al., 2011). Moreover, chronic alcohol exposure evokes the mobilization of TG in adipose tissue, which results in increased plasma FFA concentrations (Kang et al., 2007). Importantly, both rosiglitazone (a PPAR-γ agonist mainly targeting adipocytes) and recombinant adiponectin (an adipokine exclusively produced by adipocytes) improve ALD (Xu et al., 2003; Sun et al., 2012), suggesting that improving adipose tissue function represents a potential therapeutic approach for ALD.
The mechanism underlying alcohol-induced adipose tissue dysfunction has not yet been clearly defined. We previously reported that chronic alcohol consumption resulted in impaired methionine metabolism in adipose tissue, characterized by homocysteine overproduction and a decreased ratio of S-adenosylmethionine:S-adenosylhomocysteine (SAM:SAH) (an indicator of cellular hypomethylation status) due to a reduction in SAM and increased generation of SAH (Song et al., 2008). We further reported that increased homocysteine accumulation in adipocytes contributes to the alcohol-induced decline in adiponectin and that homocysteine also suppresses adipogenesis by inhibiting the transactivation of PPAR-γ (Song et al., 2008; Wang et al., 2011). Collectively, these observations suggest that impaired methionine metabolism plays an important role in alcohol-triggered adipose tissue dysfunction and its rectification may therefore confer protection against alcohol-induced liver damage.
Hormone-sensitive lipase (HSL) is the major lipase in white adipose tissue responsible for lipolysis (Schweiger et al., 2006). Lipolytic hormones quickly activate HSL via cAMP/PKA-mediated phosphorylation (Anthonsen et al., 1998). In contrast, protein phosphatase 2A (PP2A) dephosphorylates HSL and inactivates it (Wood et al., 1993). PP2A is a highly regulated heterotrimeric protein phosphatase whose enzymatic activity requires leucine carboxyl methyltransferase-dependent methylation, which enhances PP2A enzymatic activity (Janssens and Goris, 2001). Therefore, PP2A represents a critical link between hypomethylation and lipolysis activation in adipose tissue.
Betaine (trimethylglycine) is a natural compound found in many foods, including wheat, shellfish, spinach and sugar beets. It is also a metabolite of choline and an essential biochemical component of the methionine-homocysteine cycle (Craig, 2004). Hepatoprotective effects of betaine were reported in a variety of experimental animal models of liver diseases, including ALD, bile acid-induced liver injury and NAFLD (Graf et al., 2002; Ji and Kaplowitz, 2003; Song et al., 2007; Wang et al., 2010). Although different mechanisms have been proposed, our studies showed that betaine supplementation was associated with an alleviation of the impairments in adipose tissue methionine and improved adipose tissue function (Song et al., 2007; Wang et al., 2010), suggesting that improved adipose tissue function may contribute to the hepatoprotective effects of betaine.
In the present study, using the Lieber-DeCarli alcohol-containing liquid feed mouse model of ALD, we aimed to (i) elucidate the mechanisms underlying alcohol-induced adipose tissue dysfunction, with a focus on whether and how impaired adipose tissue methionine metabolism contributes to alcohol-triggered lipolysis; (ii) evaluate the beneficial effects of betaine on adipose tissue function during chronic alcohol exposure; and (iii) investigate the mechanisms involved in the hepatoprotective effect of betaine in ALD. We demonstrated that the suppression of PP2A contributes to alcohol-induced overactivation of adipose tissue lipolytic response by unleashing HSL activation. Betaine corrected the adipose tissue hypomethylation status and protected against alcohol-induced fatty liver and liver injury.
Methods
Animals and treatments
All animal studies were approved by the Institutional Animal Care and Use Committee of the University of Illinois at Chicago, which is certified by the American Association of Accreditation of Laboratory Animal Care. Male C57BL/6 mice weighing 25 ± 0.5 g (means ± SD) were obtained from the Charles River Laboratories (Wilmington, MA, USA). All mice were initially housed in conventional conditions and fed standard diet and water ad libitum at the animal facility (Research Resource Facility) for 1 week before experiments began. Thereafter, animals were randomly assigned to four groups (n = 6–8 per group) and pair-fed a modified Lieber-DeCarli alcohol (AF) or isocaloric maltose dextrin control liquid diet (PF) for 5 weeks with a stepwise feeding procedure, as described previously (Song et al., 2008). The ethanol content started at 30% of total calories and gradually increased up to 36% in the last week. Betaine (anhydrous; Sigma, St. Louis, MO, USA) was administered as a supplement in the liquid diets (both PF and AF) at a concentration of 0.5% (w v−1), simultaneously with the alcohol. All animals had access to diet and water ad libitum. At the end of the experiment, the mice were killed after being deprived of food for 4 h, and the plasma, liver and epididymal fat pad samples were harvested for assays. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010).
Cell culture
Mouse embryo fibroblast 3T3-L1 cells and HepG2 cells, a human hepatoma cell line, were both obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). HepG2 cells were cultured in DMEM containing 10% (v v−1) FBS, 2 mM glutamine, 5 U·mL−1 penicillin and 50 μg·mL−1 streptomycin at 37°C in a humidified O2/CO2 (19:1) atmosphere. 3T3-L1 pre-adipocytes were grown in DMEM containing 10% FBS and 1% antibiotics (Cellgro, Manassas, VA, USA) and differentiated exactly as previously described (Zhang et al., 2013,2013). The cells were used 9–11 days after they had been induced to differentiate.
Primary adipocytes were isolated from epididymal fat pads from male C57BL/6 mice (8–9 weeks old), as described previously (Gu et al., 2013; Zhang et al., 2013,2013). Briefly, mice were anaesthetized with Avertin (300 mg/kg BW, ip) and killed via cervical dislocation. Epididymal fat pads were harvested, washed in PBS (pH 7.4) at room temperature and minced thoroughly (2–3 mm) in collagenase solution (0.2 mg·mL−1 collagenase A; 4 mL·g−1 of adipose tissue). This mixture was incubated at 37°C with shaking at 120 r.p.m. for 30 min. After digestion, the mixture was filtered through a 250 μm gauze mesh into a 50 mL conical polypropylene tube and allowed to stand for 2–3 min. The floating layer of adipocytes was washed three times and incubated at 37°C in DMEM containing 1% BSA.
Conditioned medium (CM) experiments
Fully differentiated 3T3-L1 adipocytes were treated with 3-deazaadenosine (DZA, 100 μM) or okadaic acid (OA, 50 nM) for 24 h. Medium (CM) was then transferred freshly onto HepG2 cells (2 mL per well in a 6-well plate). Twenty-four hours later, HepG2 cells were rinsed and intracellular TG levels were determined. To determine the potential direct effect of DZA or OA on intracellular TG accumulation in hepatocytes, in another set of experiments, oleic acid (0.2 mM) was directly added to the media of HepG2 cells after 2 h of pretreatment with either DZA or OA.
Determination of lipolysis in adipocytes
Glycerol and FFA contents in the culture medium of adipocytes were measured in both basal and isoproterenol-stimulated conditions, using commercially available colorimetric assay kits for both glycerol (Cayman Chemical Company, Ann Arbor, MI, USA) and FFA (BioVision, Mountain View, CA, USA).
Measurements of liver injury and hepatic fat content
Liver injury was determined by measuring plasma alanine aminotransferase (ALT) activities using a commercially available kit (Infinity, Thermo Electron, Melbourne, Australia). Hepatic fat accumulation was determined by measuring total hepatic TG content and by histopathological evaluations. For intrahepatic TG measurement, liver tissues were homogenized and total lipids were extracted and redissolved in 2% Triton X-100 in water. Hepatic TG content was determined by enzymatic colorimetric methods using a commercially available kit (Infinity, Thermo Electron).
Plasma biochemical assays
The plasma biochemical assays were performed with commercially available kits: glycerol (Cayman Chemical Company), FFAs (Waco Chemicals, Richmond, VA, USA). Blood alcohol level was determined in plasma using a NAD-alcohol dehydrogenase reagent (Sigma-Aldrich, St Louis, MO, USA).
Quantitative real-time RT-PCR
Total RNA from the epididymal fat pad was isolated with a phenol–chloroform extraction. For each sample, 1.0 μg of total RNA was reverse transcribed using a high-capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA, USA). The cDNA was amplified in MicroAmp Optical 96-well reaction plates with a SYBR Green PCR Master Mix (Applied Biosystems) on an Applied Biosystems Prism 7000 sequence detection system. Relative gene expression was calculated after normalization to a housekeeping gene (mouse or human 18S rRNA).
Cell lysates and Western blot analyses
Total proteins from either cultured adipocytes or epididymal fat pads were obtained using Western lysis buffer consisting of the following: 20 mM Tris–HCl (pH 7.4), 150 mM NaCl, 10% glycerol, 2% Nonidet P-40, 1 mM EDTA (pH 8.0), 20 mM sodium fluoride, 30 mM sodium pyrophosphate, 0.2% SDS, 0.5% sodium deoxycholate, 1 mM PMSF, 1 mM dithiothreitol, 1 mM sodium vanadate, 50 μM leupeptin and 5 μM aprotinin. Samples were incubated on ice with frequent vortexing for 15 min and centrifuged for 20 min at 18 000× g. The protein content of each supernatant was quantified via a protein assay reagent from Bio-Rad Laboratories (Hercules, CA, USA) in accordance with the manufacturer's instructions. Proteins were separated by SDS-PAGE and transferred to 0.45 μm Immobilin-P PVDF membranes (PerkinElmer Life Sciences, Waltham, MA, USA). After this transfer, membranes were blocked in 5% (w v−1) non-fat dry milk in PBS/0.1% Tween 20 and probed with primary antibodies, as indicated. Phospho-HSL (p-HSL) at Ser660 and HSL antibodies were purchased from Cell Signaling (Danvers, TX, USA). Methylated-PP2A/C subunit (04–1479) and PP2A/C (05–421) antibodies were obtained from Millipore (Billerica, MA, USA). Betaine-homocysteine methyltransferase (BHMT) antibody was purchased from Santa Cruz Biotechnology (Dallas, TX, USA). HRP-conjugated secondary antibodies (Sigma) and enhanced chemiluminescence substrate kit (PerkinElmer Life Science) were used in the detection of specific proteins.
Intracellular PP2A activity assay
The intracellular PP2A activities were determined by a commercially available elisa assay kit according to the manufacturer's instructions (R&D System, Minneapolis, MN, USA).
HPLC analysis
Cultured adipocytes or adipose tissue were homogenized and deproteinized in 4% metaphosphoric acid (1:8, w v−1). The homogenates were centrifuged at 15 000× g for 10 min. SAM, SAH and homocysteine levels were determined by HPLC using a 5 mm Hypersil C-18 column (250 × 4.6 mm). The mobile phase consisted of 40 mM ammonium phosphate, 8 mM heptane sulfonic acid [ion-pairing reagent (pH 5.0)] and 6% acetonitrile, and was delivered at a flow rate of 1.0 mL·min−1. SAM, SAH and homocysteine were detected using a Waters 740 UV detector (Milford, MA, USA) at 254 nm.
Statistical analysis
All data are expressed as means ± SD. Statistical analysis was performed using a one-way anova and was analysed further by Newman–Keuls test for statistical difference. Differences between treatments were considered to be statistically significant at P < 0.05.
Results
Betaine supplementation alleviates alcohol-induced fatty liver and liver injury
Chronic alcohol feeding for 5 weeks resulted in a significant increase in liver-to-body weight ratio, which was attenuated by betaine supplementation (Figure 1A). The pathological alterations in the livers from different groups were evaluated by measuring hepatic TG contents and circulating liver enzyme levels. As shown in Figure 1B–D, chronic alcohol exposure was associated with fatty liver and liver injury in mice, manifested as a significantly increased hepatic TG content as well as augmented plasma ALT and AST levels respectively. A massive accumulation of fat in the liver of mice chronically exposed to the alcohol-containing diet was also observed via H&E staining (Figure 1E). When betaine was added to the alcohol-containing liquid diet, both hepatic TG accumulation and liver injury were significantly reduced (Figure 1B–E).
Figure 1.
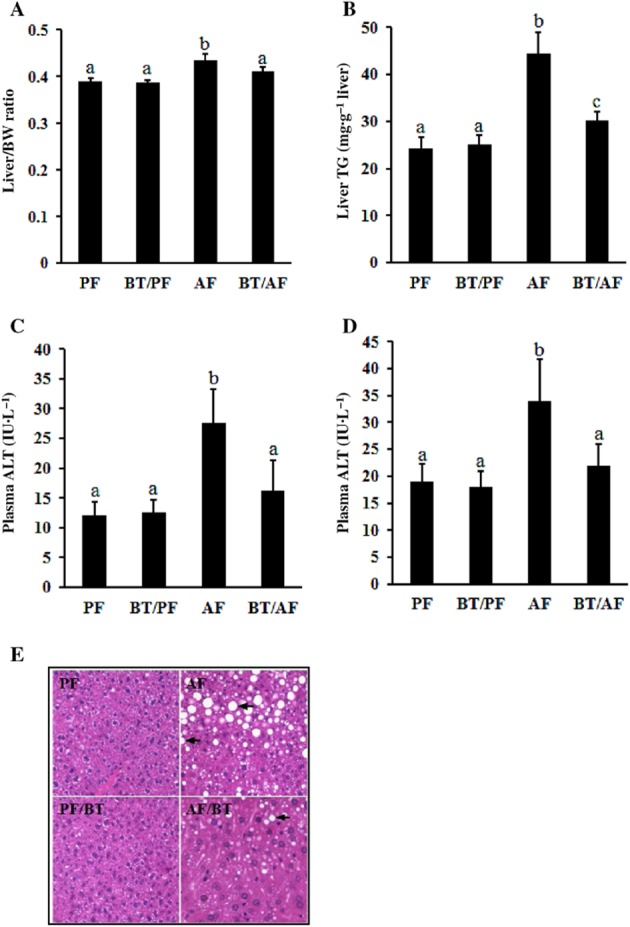
Betaine supplementation alleviates alcohol-induced fatty liver and liver injury. C57BL/6 mice were fed with control or alcohol-containing diet with/without betaine supplementation (0.5%, w v−1) for 5 weeks. (A) Changes in liver-to-body weight (BW). (B) Changes in the liver triglyceride content. (C) Changes in plasma ALT levels. (D) Changes in plasma AST levels. Data are means ± SD (n = 8 for PF, BT/PF and BT/AF; n = 6 for AF). Bars with different letters differ significantly (P < 0.05). (E) H&E staining of liver tissue shows that betaine supplementation significantly decreased hepatic fat accumulation induced by chronic alcohol consumption. AF, alcohol-fed; BT, betaine supplementation; PF, pair-fed.
Betaine supplementation rectifies alcohol-induced adipose tissue dysfunction
Betaine supplementation attenuated epididymal fat pad mass loss induced by chronic alcohol feeding (Figure 2A). Light microscopy examination revealed that alcohol exposure led to a reduction in the size of adipocytes in the epididymal fat pad, which was also rectified by betaine supplementation (Figure 2B). Moreover, chronic alcohol exposure was associated with enhanced lipolysis in adipose tissue, as indicated by the significantly higher fasting plasma glycerol (Figure 2C) and FFA (Figure 2D) concentrations in alcohol-fed mice when compared with pair-fed counterparts.
Figure 2.
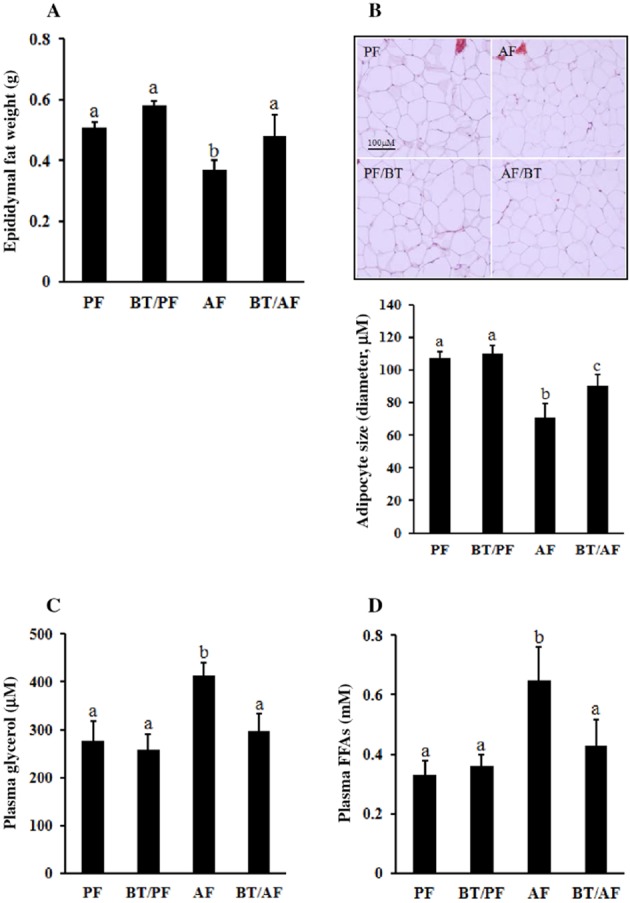
Betaine supplementation rectifies alcohol-induced adipose tissue dysfunction. C57BL/6 mice were fed a control or alcohol-containing diet with/without betaine supplementation (0.5%, w v−1) for 5 weeks. (A) Epididymal fat pad weights. (B) H&E staining of epididymal adipose tissue shows that betaine supplementation prevents the reduction in adipocyte size induced by chronic alcohol exposure. (C) Plasma glycerol levels. (D) Plasma non-esterified fatty acid levels. Data are means ± SD (n = 8 for PF, BT/PF and BT/AF; n = 6 for AF). Bars with different letters differ significantly (P < 0.05). AF, alcohol-fed; BT, betaine supplementation; PF, pair-fed.
Betaine supplementation corrects the abnormalities in methionine metabolism in adipose tissue induced by chronic alcohol exposure
Betaine is an intermediate in methionine metabolic pathway and a homocysteine-lowering agent (Figure 3A) via a reaction catalysed by BHMT. To determine whether the beneficial effect of betaine supplementation on adipose tissue function in the setting of chronic alcohol consumption involves the correction of adipose tissue methionine metabolism, we examined SAM, SAH and homocysteine concentrations in adipose tissues obtained from different treatments. As shown in Figure 3B–D, alcohol consumption resulted in a significant reduction in SAM in adipose tissue (Figure 3B), but significantly increased the SAH level (Figure 3C), so reducing the SAM:SAH ratio (Figure 3D), a strong indicator of cellular hypomethylation state. In keeping with our previous findings, the epididymal homocysteine levels were significantly increased by chronic alcohol feeding (Figure 3E). Betaine supplementation prevented the depletion of SAM and reduced the increase in SAH, thereby improving the methylation state of the adipose tissue (Figure 3B–D). As expected, betaine supplementation also attenuated adipose tissue homocysteine accumulation in alcohol-fed mice (Figure 3E). BHMT, the enzyme that integrates betaine into the methionine/homocysteine cycle, is known to be abundantly expressed in the liver. However, its expression in adipose tissue has never been reported. We therefore examined BHMT expression in epididymal fat pads obtained from different groups. As shown in Figure 3F, the basal BHMT expression can be detected in adipose tissue. Chronic alcohol exposure had a tendency to increase BHMT gene expression; however, the increase did not reach statistical significance. Interestingly, betaine supplementation increased the expression of BHMT, at both the mRNA and protein levels.
Figure 3.

Betaine supplementation corrects the abnormal methionine metabolism in adipose tissue induced by chronic alcohol exposure. C57BL/6 mice were fed a control or alcohol-containing diet with/without betaine supplementation (0.5%, w v−1) for 5 weeks. (A) Intracellular methionine metabolic pathway. (B) SAM levels in epididymal fat pads. (C) SAH levels in epididymal fat pads. (D) Epididymal adipose tissue SAM:SAH ratio. (E) Homocysteine levels in epididymal fat pads. (F) BHMT gene expression and protein in epididymal fat pads. Data are means ± SD (n = 8 for PF, BT/PF and BT/AF; n = 6 for AF). Bars with different letters differ significantly (P < 0.05). AF, alcohol-fed; BHMT: betaine homocysteine methyltransferase; BT, betaine supplementation; PF, pair-fed; SAHH, SAH hydrolase.
A reduction in intracellular SAM:SAH ratio (hypomethylation) contributes to the enhanced lipolytic response in adipocytes
Cell culture experiments were subsequently conducted to determine whether the cellular hypomethylation status in adipocytes contributes to the enhanced lipolytic response. Primary mouse adipocytes were treated with DZA, a potent inhibitor of SAH hydrolase (SAHH), the enzyme involved in SAH degradation (see Figure 3A), for 6 h, and the levels of intracellular SAM and SAH were measured and SAM:SAH ratios calculated. As shown in Figure 4A,B, DZA increased the cellular hypomethylation status, observed as a marked reduction in the SAM:SAH ratio in DZA-treated adipocytes; this resulted from both a reduction in SAM and an increase in SAH. To determine the effect of DZA on the lipolytic response, primary mouse adipocytes were pretreated with DZA for 6 h before stimulation with 10 μM isoprenaline. Glycerol release was measured after 1 h. As shown in Figure 4C, DZA promoted glycerol release from adipocytes in both basal and isoprenaline-stimulated conditions. To further validate this observation, we conducted similar experiments in fully differentiated 3T3-L1 adipocytes transfected with the siRNA for SAHH overnight. As shown in Figure 4D–F, similar to DZA, SAHH knockdown significantly reduced the cellular SAM:SAH ratio, and this was associated with an enhanced lipolytic response in both basal and isoprenaline-stimulated adipocytes. To exclude the potential cytotoxic effects of DZA or SAHH siRNA transfection in adipocytes, we also monitored LDH release in the culture medium. No differences in LDH release were found between the control groups and the treated groups (data not shown).
Figure 4.

A reduction in intracellular SAM:SAH (hypomethylation) enhances the lipolytic response in adipocytes. Primary adipocytes isolated from mouse epididymal fat pad were cultured in a 24-well plate (1 × 106 cells·mL−1). Adipocytes were treated with DZA for 6 h followed by the stimulation with 10 μM isoprenaline (iso) for 1 h. (A) Intracellular SAM and SAH concentrations. (B) Intracellular SAM:SAH ratio. (C) Glycerol levels in the culture media. All values are denoted as means ± SD from three or more independent studies. *P < 0.05 compared to corresponding control. Bars with different letters differ significantly (P < 0.05). Fully differentiated 3T3-L1 adipocytes were transfected with siRNA for SAH hydrolase (SAHH) overnight, followed by stimulation with isoprenaline for 1 h. (D) Intracellular SAM and SAH concentrations. (E) Intracellular SAM:SAH ratio. (F) Glycerol levels in culture media. All values are denoted as means ± SD from three or more independent studies. *P < 0.05 versus control (scramble siRNA). Bars with different letters differ significantly (P < 0.05).
Activation of HSL due to PP2A suppression contributes to the hypomethylation-triggered lipolytic response in adipocytes
HSL activation has a central role in controlling the lipolytic response of adipocytes. In response to lipolytic hormone stimulation, several serine residues of HSL, including Ser660, are phosphorylated by PKA, which leads to HSL activation. In contrast, HSL activity is also regulated by PP2A, which deactivates HSL by dephosphorylation, and the methylation state of PP2A affects its enzymatic activity (Janssens and Goris, 2001) (Figure 5A). Catalytic (C) subunit methylation is required for the binding of the regulatory subunit (B) to facilitate the assembly of PP2A holoenzyme trimers and subsequent activation (Stanevich et al., 2011). To determine whether the lowered intracellular SAM:SAH ratio is associated with an altered methylation state and activity of PP2A, primary adipocytes were treated with DZA for 16 h. As shown in Figure 5B, DZA reduced the PP2A C subunit methylation state, without affecting the total amount of C subunit protein. Hence the activity of PP2A was significantly decreased by DZA (Figure 5C). Furthermore, DZA enhanced the lipolytic activity of HSL by markedly increasing the phosphorylation of HSL at Ser660 (Figure 5D). To further confirm that the suppression of PP2A is involved in the hypomethylation-triggered activation of HSL and increased lipolysis, primary adipocytes were treated with OA, a potent inhibitor of PP2A (Haystead et al., 1989) for 16 h. OA inhibited PP2A activity (Figure 5C), increased HSL activation (Figure 5D) and significantly increased the release of glycerol release (Figure 5E).
Figure 5.
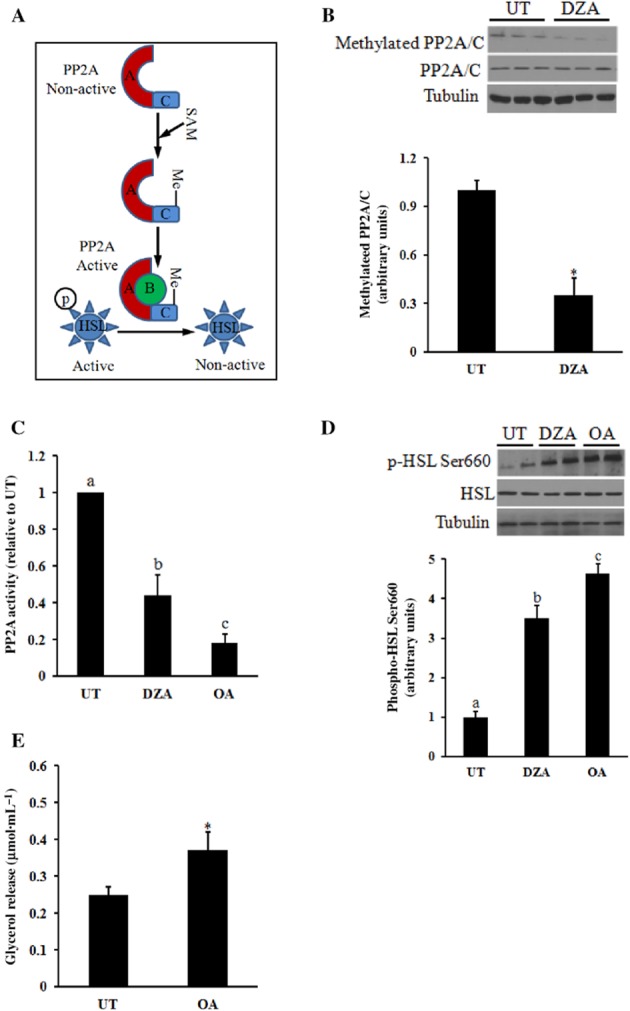
Activation of HSL induced by PP2A suppression contributes to hypomethylation-triggered lipolytic response in adipocytes. Primary adipocytes isolated from mouse epididymal fat pad were cultured in a 6-well plate (1 × 106 cells·mL−1). Adipocytes were treated with DZA or OA for the indicated time periods. (A) Catalytic (C) subunit methylation status regulates PP2A activity. (B) DZA, a hypomethylation inducer, reduces PP2A C subunit methylation. (C) DZA suppresses PP2A activation. (D) Suppression of PP2A activation enhances the activity of HSL. (E) OA, a PP2A inhibitor, enhances lipolysis in adipocytes. All values are denoted as means ± SD from three or more independent studies. *P < 0.05 vs. corresponding control. Bars with different letters differ significantly (P < 0.05).
The hypomethylation state in adipocytes contributes to TG accumulation in hepatocytes in vitro
CM experiments were conducted to directly test whether the hypomethylation state or PP2A inhibition in adipocytes is capable of inducing TG accumulation in hepatocytes. Fully differentiated 3T3-L1 adipocytes were treated with either DZA or OA for 24 h, after which the supernatant (CM) was collected and applied to HepG2 cells. The intracellular TG content of HepG2 cells was determined 24 h later. As shown in Figure 6A, the TG content of HepG2 cells cultured in CM from either DZA- or OA-treated adipocytes was significantly higher than that in the cells cultured in CM from untreated (UT) adipocytes. The LDH release assay indicated that the CM had no effect on hepatocyte cell death (Figure 6B). To exclude any potential direct effect of DZA or OA on hepatocytes, HepG2 cells were pretreated with either DZA or OA, at the same concentrations as used for adipocytes, for 2 h, followed by the addition of 0.2 mM oleic acid. Intracellular TG contents were determined 24 h later. As shown in Figure 6C, neither DZA nor OA pretreatment resulted in a further elevation of intracellular TG accumulation.
Figure 6.
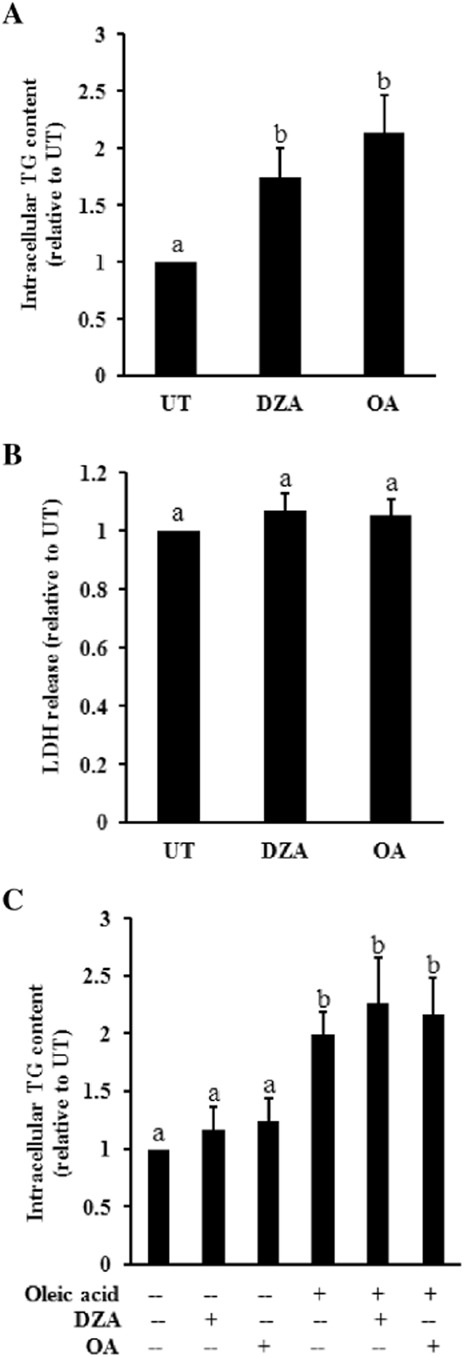
Conditioned medium (CM) from adipocytes treated with either DZA or OA induces triglyceride accumulation in hepatocytes in vitro. Fully differentiated 3T3-L1 adipocytes were treated with either DZA or OA for 24 h, after which the supernatant (CM) was collected and applied to HepG2 cells. Intracellular triglyceride content in HepG2 cells and LDH level in culture medium were determined 24 h later. (A) Biochemical assay of intercellular triglyceride concentration. (B) LDH release assay. HepG2 cells were pretreated with DZA or OA for 2 h. Oleic acid (0.2 mM) was then added into the culture medium. Intracellular triglyceride content in HepG2 cells were determined 24 h later. All values are denoted as means ± SD from three or more independent studies. Bars with different letters differ significantly (P < 0.05).
Betaine supplementation alleviates alcohol-induced PP2A inhibition and HSL activation in adipose tissue
To test the relevance of our mechanistic observations from cell culture studies in vivo, we measured PP2A activity, PP2A C subunit methylation state, as well as HSL Ser660 phosphorylation in epididymal fat pads obtained from alcohol-fed mice. As shown in Figure 7, chronic alcohol exposure decreased PP2A activity in adipose tissue, in parallel with a reduced C subunit methylation state (Figure 7A,B). Accordingly, chronic alcohol enhanced HSL activity in adipose tissues, observed as increased HSL Ser660 phosphorylation (Figure 7C). Importantly, all these alterations were attenuated by betaine (Figure 7A–C).
Figure 7.
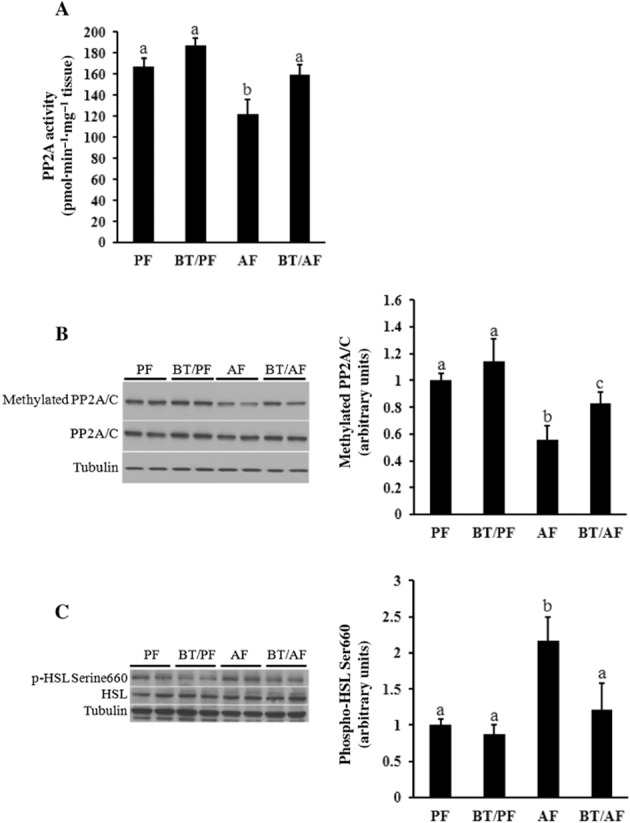
Betaine supplementation alleviates alcohol-induced inhibition of PP2A and activation of HSL in adipose tissue. C57BL/6 mice were fed a control or alcohol-containing diet with/without betaine supplementation (0.5%, w v−1) for 5 weeks. Total protein extracts from epididymal fat pads were prepared thereafter. Forty micrograms of protein was subjected to Western blot analysis for (methylated) PP2A C subunit and HSL phosphorylation using specific antibodies. (A) PP2A activity in epididymal fat pad. (B) Epididymal adipose tissue PP2A C subunit methylation. (C) Epididymal adipose tissue HSL activity. Data are means ± SD (n = 8 for PF, BT/PF and BT/AF; n = 6 for AF). Bars with different letters differ significantly (P < 0.05). AF, alcohol-fed; BT, betaine supplementation; PF, pair-fed.
Discussion
In the present study, we provided evidence that improving adipose tissue function contributed to the beneficial effect of betaine in ALD. We demonstrated for the first time that the hypomethylation status of adipose tissue, indicated by the significant reduction in cellular SAM:SAH ratio, contributes to the enhanced lipolytic response observed in adipose tissue of mice chronically exposed to an alcohol-containing diet. Specifically, we revealed that the reduction in intracellular SAM:SAH ratio in adipocytes was associated with a hypomethylated PP2A C subunit and suppressed enzymatic activity, which leads to uncontrolled activation of HSL. Betaine supplementation rectified this methionine metabolism abnormality in adipose tissue and prevented alcohol-induced PP2A suppression and consequently HSL overactivation.
Increased adipose tissue TG mobilization contributes to alcohol-induced fatty liver and liver injury (Kang et al., 2007; Sun et al., 2012; Zhong et al., 2012); however, the exact mechanism underlying alcohol-triggered overactivation of lipolysis remains elusive. We previously reported that chronic alcohol feeding resulted in abnormal methionine metabolism in adipose tissue, and increased homocysteine levels in adipose tissue contribute to the decreased adiponectin response induced by chronic alcohol exposure (Song et al., 2008). In keeping with our previous findings, in this study we also observed that methionine metabolism was impaired in adipose tissue of mice chronically exposed to alcohol. In addition to the significantly increased levels of homocysteine, the alcohol also enhanced the hypomethylation status in adipose tissue, manifested as a significant reduction in SAM:SAH ratio, which was a consequence of both a decrease in SAM and an elevation of SAH. Betaine rectified the SAM:SAH ratio in adipose tissue and concomitantly attenuated the enhanced lipolytic response, suggesting that the hypomethylation state in adipocytes may play a mechanistic role in alcohol-induced lipolysis. This notion was indeed confirmed by the subsequent cell culture studies, in which a lowered intracellular SAM:SAH ratio, induced either by pharmacological or genetic approaches, led to an enhanced lipolytic response of both basal and isoprenaline-stimulated adipocytes. Importantly, our CM experiments further supported the possibility that the hypomethylation status in adipocytes plays an important role in the development of hepatocyte steatosis.
Although adipocyte lipolysis is regulated by a variety of proteins exhibiting lipase activity (Birner-Gruenberger et al., 2005), HSL plays an important role in this process and, together with ATGL, is responsible for more than 95% of lipase activity in murine white adipose tissue (Schweiger et al., 2006). A recent study from Zhou's group elegantly showed that chronic alcohol feeding was associated with increased ATGL expression and HSL activation in adipose tissue (Sun et al., 2012). In the present study, our data also indicate that increased plasma glycerol and FFA concentrations in response to chronic alcohol exposure are closely assocaited with the HSL Ser660 phosphorylation levels in epididymal fat pads. Since betain corrected the alcohol-induced changes in adipose tissue methylation status and concomitantly attenuated HSL activation, it is likely that intracellular methylation status in adipocytes may play a regulatory role in HSL phosphorylation. Therefore, further investigation into whether and how hypomethylation results in HSL activation will provide useful information regarding mechanisms underlying alcohol-induced lipolysis.
Upon stimulation with lipolytic hormones, such as catecholamines, HSL is quickly activated by cAMP/PKA-mediated phosphorylation of several serine residues (Anthonsen et al., 1998). In contrast, PP2A dephosphorylates HSL at some serine residues, leading to its inactivation (Wood et al., 1993). PP2A is a highly regulated heterotrimeric protein phosphatase, composed of a catalytic (C) subunit and a structural (A) subunit, in association with a third variable regulatory (B) subunit (Perrotti and Neviani, 2013). It is now emerging as an important regulator of many cellular processes involving protein phosphorylation. Interestingly, the methylation status of subunit C of PP2A is positively associated with its enzymatic activity. Subunit C methylation dramatically enhances the binding of B regulatory subunit to AC complex and facilitates the assembly of PP2A holoenzyme trimers (Janssens and Goris, 2001). Employing both a pharmacological and genetic approach, we revealed that PP2A represents a critical link between adipose tissue hypomethylation and lipolysis activation. We demonstrated that the reduction in SAM:SAH ratio in adipocytes lowered PP2 subunit C methylation, leading to a reduction in its enzymatic activity, which was associated with an up-regulation of HSL Ser660 phosphorylation and increased glycerol release. This notion was further supported by the observation that OA, a potent inhibitor of PP2A, enhanced lipolysis in cultured adipocytes, which was associated with increased HSL Ser660 phosphorylation. Taken together, these observations suggest that, by enhancing the activity of HSL, PP2A suppression contributes to the increased lipolysis induced by the decreased intracellular methylation status. It is noteworthy that, although the hypomethylation status induced by SAHH suppression in our cell culture experiments does not mimic that observed in the adipose tissue of alcohol-fed mice in that SAHH inhibition is potentially associated with decreased homocysteine generation, it represents an ideal approach to differentiate the effect of hypomethylation and hyperhomocysteinaemia on adipocyte physiology.
Betaine is a metabolite in the choline metabolism pathway and is an important participant in methionine metabolism. Betaine converts homocysteine to methionine, by a BHMT-catalysed reaction, thereby preventing the accumulation of homocysteine (hyperhomocysteinaemia) (Craig, 2004). The beneficial effect of betaine supplementation on ALD is well recognized and has been extensively investigated. However, the exact mechanisms are still elusive. Since hyperhomocysteinaemia, resulting from dys-regulated hepatic methionine metabolism, is one of major metabolic abnormalities induced by chronic alcohol consumption and represents a pathological factor involved in the pathogenesis of ALD, the rectification of hepatic methionine metabolism was considered to be a major mechanism accounting for the beneficial effects of betaine. Using a mouse model of ALD, it was reported that betaine supplementation prevented fatty liver and liver injury induced by chronic alcohol consumption by attenuating hyperhomocysteinemia and subsequent ER stress in the liver (Ji and Kaplowitz, 2003). Similarly, betaine supplementation alleviated high-fat diet-induced NAFLD in rats by correcting impaired sulfur-amino acid metabolism and oxidative stress in the liver (Kwon do et al., 2009). However, several recent reports suggested that in addition to serving as a homocysteine-reducing agent, other mechanisms exist. In high-fat diet-fed rats, betaine was found to protect against fatty liver and liver injury by reducing hepatic high-mobility group box 1 and toll-like receptor 4 expression (Zhang et al., 2013,2013). Using a high-sucrose diet model of NAFLD, we demonstrated that betaine directly activates the AMPK system in hepatocytes, leading to improved liver function (Song et al., 2007). Moreover, betaine improves insulin resistance in HepG2 cells triggered by high-glucose exposure and alleviates hepatic insulin resistance induced by a high-fat diet in mice (Kathirvel et al., 2010). A recent study in our group indicated that in addition to directly acting on the liver, betaine also improves adipose tissue dysfunction induced by a high-fat diet in mice, which accounts for its hepatoprotective effect in NAFLD (Wang et al., 2011). Furthermore, betaine was reported to prevent the blood alcohol cycle in alcohol-fed rats (Li et al., 2011). Altogether these results suggest that the mechanisms underlying the beneficial effects of betaine on liver diseases are multifactorial and involve different organs/tissue. Employing both in vitro and in vivo experimental approaches, the present work not only confirmed the beneficial effect of betaine on adipose tissue function but also provided a mechanistic explanation for its hepatoprotective effect in ALD. We clearly demonstrated that adipose tissue hypomethylation status contributes to the development of alcoholic liver disease. Betaine supplementation may, therefore, have a hepatoprotective effect by improving adipose tissue function during chronic alcohol exposure.
In summary, our data suggest that overactivation of lipolysis in adipose tissue plays an important role in the pathogenesis of ALD and adipose tissue hypomethylation status in response to chronic alcohol exposure represents a novel mechanism in this process. A reduced intracellular SAM:SAH ratio in adipose tissue induced by chronic alcohol exposure results in compromised PP2A activation, leading to constant HSL activation, increased FFA release and the resultant fatty liver and liver injury. Our results provide evidence that rectification of this methionine metabolism abnormality in adipose tissue, such as that inuced by betaine supplementation, represents a potential and novel therapeutic target for the treatment of ALD. Betaine is a natural component of many foods and safe in doses ranging from 3 to 30 g·day–1; therefore, betaine represents an ideal therapeutic agent for ALD. Our present study provides strong evidence for further evaluation of the potential therapeutic role of betaine in ALD or other types of liver disease in which a dysfunction in adipose tissue is mechanistically involved.
Acknowledgments
This study was supported by grants from Public Health Department of Zhejiang Province Project (China) 2013KYA138 (X. D.), the National Basic Research Program of China (973 Program) 2014 CB543001 (X. D.), NSFC 81241145 (X. D.) and the National Institutes of Health NIAAA grants R01 AA017442 (Z. S.).
Glossary
- ALD
alcoholic liver disease
- ALT
alanine aminotransferase
- BHMT
betaine homocysteine methyltransferase
- HSL
hormone-sensitive lipase
- NEFAs
non-esterified free fatty acids
- PP2A
protein phosphatase 2A
- SAH
S-adenosylhomocysteine
- SAM
S-adenosylmethionine
Author contributions
X. D. contributed to the design and conduct of the study, data collection and analysis, data interpretation and manuscript writing. Y. X. contributed to data collection and analysis, data interpretation and manuscript writing. J. C., Y. Q., S. L. and X. Z. contributed to data collection and analysis. Z. S. contributed to data interpretation and manuscript editing. All authors approved the final version of the manuscript.
Conflict of interest
None declared.
References
- Anthonsen MW, Rönnstrand L, Wernstedt C, Degerman E, Holm C. Identification of novel phosphorylation sites in hormone-sensitive lipase that are phosphorylated in response to isoproterenol and govern activation properties in vitro. J Biol Chem. 1998;273:215–221. doi: 10.1074/jbc.273.1.215. [DOI] [PubMed] [Google Scholar]
- Birner-Gruenberger R, Susani-Etzerodt H, Waldhuber M, Riesenhuber G, Schmidinger H, Rechberger G, et al. The lipolytic proteome of mouse adipose tissue. Mol Cell Proteomics. 2005;4:1710–1717. doi: 10.1074/mcp.M500062-MCP200. [DOI] [PubMed] [Google Scholar]
- Chen X, Sebastian BM, Nagy LE. Chronic ethanol feeding to rats decreases adiponectin secretion by subcutaneous adipocytes. Am J Physiol Endocrinol Metab. 2007;292:E621–E628. doi: 10.1152/ajpendo.00387.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig SA. Betaine in human nutrition. Am J Clin Nutr. 2004;80:539–549. doi: 10.1093/ajcn/80.3.539. [DOI] [PubMed] [Google Scholar]
- Fischer M, You M, Matsumoto M, Crabb DW. Peroxisome proliferator-activated receptor alpha (PPARalpha) agonist treatment reverses PPARalpha dysfunction and abnormalities in hepatic lipid metabolism in ethanol-fed mice. J Biol Chem. 2003;278:27997–28004. doi: 10.1074/jbc.M302140200. [DOI] [PubMed] [Google Scholar]
- Graf D, Kurz AK, Reinehr R, Fischer R, Kircheis G, Haussinger D. Prevention of bile acid-induced apoptosis by betaine in rat liver. Hepatology. 2002;36:829–839. doi: 10.1053/jhep.2002.35536. [DOI] [PubMed] [Google Scholar]
- Gu D, Wang Z, Dou X, Zhang X, Li S, Vu L, et al. Inhibition of ERK1/2 pathway suppresses adiponectin secretion via accelerating protein degradation by Ubiquitin-proteasome system: relevance to obesity-related adiponectin decline. Metabolism. 2013;62:1137–1148. doi: 10.1016/j.metabol.2013.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart CL, Morrison DS, Batty GD, Mitchell RJ, Davey Smith G. Effect of body mass index and alcohol consumption on liver disease: analysis of data from two prospective cohort studies. BMJ. 2010;340:c1240. doi: 10.1136/bmj.c1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haystead TA, Sim AT, Carling D, Honnor RC, Tsukitani Y, Cohen P, et al. Effects of the tumour promoter okadaic acid on intracellular protein phosphorylation and metabolism. Nature. 1989;337:78–781. doi: 10.1038/337078a0. [DOI] [PubMed] [Google Scholar]
- Janssens V, Goris J. Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem J. 2001;353:417–439. doi: 10.1042/0264-6021:3530417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji C, Kaplowitz N. Betaine decreases hyperhomocysteinemia, endoplasmic reticulum stress, and liver injury in alcohol-fed mice. Gastroenterology. 2003;124:1488–1499. doi: 10.1016/s0016-5085(03)00276-2. [DOI] [PubMed] [Google Scholar]
- Ji C, Chan C, Kaplowitz N. Predominant role of sterol response element binding proteins (SREBP) lipogenic pathways in hepatic steatosis in the murine intragastric ethanol feeding model. J Hepatol. 2006;45:717–724. doi: 10.1016/j.jhep.2006.05.009. [DOI] [PubMed] [Google Scholar]
- Kang L, Chen X, Sebastian BM, Pratt BT, Bederman IR, Alexander JC, et al. Chronic ethanol and triglyceride turnover in white adipose tissue in rats: inhibition of the anti-lipolytic action of insulin after chronic ethanol contributes to increased triglyceride degradation. J Biol Chem. 2007;282:28465–28473. doi: 10.1074/jbc.M705503200. [DOI] [PubMed] [Google Scholar]
- Kathirvel E, Morgan K, Nandgiri G, Sandoval BC, Caudill MA, Bottiglieri T, et al. Betaine improves nonalcoholic fatty liver and associated hepatic insulin resistance: a potential mechanism for hepatoprotection by betaine. Am J Physiol Gastrointest Liver Physiol. 2010;299:G1068–G1077. doi: 10.1152/ajpgi.00249.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon do Y, Jung YS, Kim SJ, Park HK, Park JH, Kim YC. Impaired sulfur-amino acid metabolism and oxidative stress in nonalcoholic fatty liver are alleviated by betaine supplementation in rats. J Nutr. 2009;139:63–68. doi: 10.3945/jn.108.094771. [DOI] [PubMed] [Google Scholar]
- Li J, Li XM, Caudill M, Malysheva O, Bardag-Gorce F, Oliva J, et al. Betaine feeding prevents the blood alcohol cycle in rats fed alcohol continuously for 1 month using the rat intragastric tube feeding model. Exp Mol Pathol. 2011;91:540–547. doi: 10.1016/j.yexmp.2011.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loomba R, Bettencourt R, BarrettConnor E. Synergistic association between alcohol intake and body mass index with serum alanine and aspartate aminotransferase levels in older adults: the Rancho Bernardo Study. Aliment Pharmacol Ther. 2009;30:1137–1149. doi: 10.1111/j.1365-2036.2009.04141.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Drummond GB, McLachlan EM, Kilkenny C, Wainwright CL. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Shea RS, Dasarathy S, McCullough AJ Practice Guideline Committee of the American Association for the Study of Liver Diseases; Practice Parameters Committee of the American College of Gastroenterology. Alcoholic liver disease. Hepatology. 2010;51:307–328. doi: 10.1002/hep.23258. [DOI] [PubMed] [Google Scholar]
- Perrotti D, Neviani P. Protein phosphatase 2A: a target for anticancer therapy. Lancet Oncol. 2013;14:e229–e238. doi: 10.1016/S1470-2045(12)70558-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy JK, Rao MS. Lipid metabolism and liver inflammation. II. Fatty liver disease and fatty acid oxidation. Am J Physiol Gastrointest Liver Physiol. 2006;290:G852–G858. doi: 10.1152/ajpgi.00521.2005. [DOI] [PubMed] [Google Scholar]
- Sanyal AJ. Mechanisms of disease: pathogenesis of nonalcoholic fatty liver disease. Nat Clin Pract Gastroenterol Hepatol. 2005;2:46–53. doi: 10.1038/ncpgasthep0084. [DOI] [PubMed] [Google Scholar]
- Schweiger M, Schreiber R, Haemmerle G, Lass A, Fledelius C, Jacobsen P, et al. Adipose triglyceride lipase and hormone-sensitive lipase are the major enzymes in adipose tissue triacylglycerol catabolism. J Biol Chem. 2006;281:40236–40241. doi: 10.1074/jbc.M608048200. [DOI] [PubMed] [Google Scholar]
- Sebastian BM, Roychowdhury S, Tang H, Hillian AD, Feldstein AE, Stahl GL, et al. Identification of a cytochrome P4502E1/Bid/C1q-dependent axis mediating inflammation in adipose tissue after chronic ethanol feeding to mice. J Biol Chem. 2011;286:35989–35997. doi: 10.1074/jbc.M111.254201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Z, Li Y, Yu C, Shen Y, Xu L, Xu C, et al. A cohort study of the effect of alcohol consumption and obesity on serum liver enzyme levels. Eur J Gastroenterol Hepatol. 2010;22:820–825. doi: 10.1097/MEG.0b013e3283328b86. [DOI] [PubMed] [Google Scholar]
- Song Z, Deaciuc I, Zhou Z, Song M, Chen T, Hill D, et al. Involvement of AMP-activated protein kinase in beneficial effects of betaine on high-sucrose diet-induced hepatic steatosis. Am J Physiol Gastrointest Liver Physiol. 2007;293:G894–G902. doi: 10.1152/ajpgi.00133.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Z, Zhou Z, Deaciuc I, Chen T, McClain CJ. Inhibition of adiponectin production by homocysteine: a potential mechanism for alcoholic liver disease. Hepatology. 2008;47:867–879. doi: 10.1002/hep.22074. [DOI] [PubMed] [Google Scholar]
- Souto JC, Blanco-Vaca F, Soria JM, Buil A, Almasy L, Ordoñez-Llanos J, et al. A genomewide exploration suggests a new candidate gene at chromosome 11q23 as the major determinant of plasma homocysteine levels: results from the GAIT project. Am J Hum Genet. 2005;76:925–933. doi: 10.1086/430409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanevich V, Jiang L, Satyshur KA, Li Y, Jeffrey PD, Li Z, et al. The structural basis for tight control of PP2A methylation and function by LCMT-1. Mol Cell. 2011;41:331–342. doi: 10.1016/j.molcel.2010.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart S, Jones D, Day CP. Alcoholic liver disease: new insights into mechanisms and preventative strategies. Trends Mol Med. 2001;7:408–413. doi: 10.1016/s1471-4914(01)02096-2. [DOI] [PubMed] [Google Scholar]
- Sugimoto T, Yamashita S, Ishigami M, Sakai N, Hirano K, Tahara M, et al. Decreased microsomal triglyceride transfer protein activity contributes to initiation of alcoholic liver steatosis in rats. J Hepatol. 2002;36:157–162. doi: 10.1016/s0168-8278(01)00263-x. [DOI] [PubMed] [Google Scholar]
- Sun X, Tang Y, Tan X, Li Q, Zhong W, Sun X, et al. Activation of peroxisome proliferator-activated receptor-γ by rosiglitazone improves lipid homeostasis at the adipose tissue-liver axis in ethanol-fed mice. Am J Physiol Gastrointest Liver Physiol. 2012;302:G548–G557. doi: 10.1152/ajpgi.00342.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H, Sebastian BM, Axhemi A, Chen X, Hillian AD, Jacobsen DW, et al. Ethanol-induced oxidative stress via the CYP2E1 pathway disrupts adiponectin secretion from adipocytes. Alcohol Clin Exp Res. 2007;36:214–222. doi: 10.1111/j.1530-0277.2011.01607.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai J, Ford ES, Zhao G, Li C, Greenlund KJ, Croft JB. Co-occurrence of obesity and patterns of alcohol use associated with elevated serum hepatic enzymes in US adults. J Behav Med. 2011;35:200–210. doi: 10.1007/s10865-011-9353-5. [DOI] [PubMed] [Google Scholar]
- Wang Z, Yao T, Pini M, Zhou Z, Fantuzzi G, Song Z. Betaine improved adipose tissue function in mice fed a high-fat diet: a mechanism for hepatoprotective effect of betaine in nonalcoholic fatty liver disease. Am J Physiol Gastrointest Liver Physiol. 2010;298:G634–G642. doi: 10.1152/ajpgi.00249.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Dou X, Yao T, Song Z. Homocysteine inhibits adipogenesis in 3T3-L1 preadipocytes. Exp Biol Med (Maywood) 2011;236:1379–1388. doi: 10.1258/ebm.2011.011234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood SL, Emmison N, Borthwick AC, Yeaman SJ. The protein phosphatases responsible for dephosphorylation of hormone-sensitive lipase in isolated rat adipocytes. Biochem J. 1993;295:531–535. doi: 10.1042/bj2950531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu A, Wang Y, Keshaw H, Xu LY, Lam KS, Cooper GJ. The fat-derived hormone adiponectin alleviates alcoholic and nonalcoholic fatty liver diseases in mice. J Clin Invest. 2003;112:91–100. doi: 10.1172/JCI17797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Lai KK, Verlinsky A, Lugea A, French SW, Cooper MP, et al. Synergistic steatohepatitis by moderate obesity and alcohol in mice despite increased adiponectin and p-AMPK. J Hepatol. 2011;55:673–682. doi: 10.1016/j.jhep.2010.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You M, Fischer M, Deeg MA, Crabb DW. Ethanol induces fatty acid synthesis pathways by activation of sterol regulatory element-binding protein (SREBP) J Biol Chem. 2002;277:29342–29347. doi: 10.1074/jbc.M202411200. [DOI] [PubMed] [Google Scholar]
- You M, Matsumoto M, Pacold CM, Cho WK, Crabb DW. The role of AMP-activated protein kinase in the action of ethanol in the liver. Gastroenterology. 2004;127:1798–8084. doi: 10.1053/j.gastro.2004.09.049. [DOI] [PubMed] [Google Scholar]
- Zhang W, Wang LW, Wang LK, Li X, Zhang H, Luo LP, et al. Betaine protects against high-fat-diet-induced liver injury by inhibition of high-mobility group box 1 and toll-like receptor 4 expression in rats. Dig Dis Sci. 2013;58:3198–3206. doi: 10.1007/s10620-013-2775-x. [DOI] [PubMed] [Google Scholar]
- Zhang X, Wang Z, Li J, Gu D, Li S, Shen C, et al. Increased 4-hydroxynonenal formation contributes to obesity-related lipolytic activation in adipocytes. PLoS ONE. 2013;8:e70663. doi: 10.1371/journal.pone.0070663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong W, Zhao Y, Tang Y, Wei X, Shi X, Sun W, et al. Chronic alcohol exposure stimulates adipose tissue lipolysis in mice: role of reverse triglyceride transport in the pathogenesis of alcoholic steatosis. Am J Pathol. 2012;180:998–1007. doi: 10.1016/j.ajpath.2011.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]