

Abstract
Background and Purpose
Proteinase activated receptor 2 (PAR2) is a GPCR associated with inflammation, metabolism and disease. Clues to understanding how to block PAR2 signalling associated with disease without inhibiting PAR2 activation in normal physiology could be provided by studies of biased signalling.
Experimental Approach
PAR2 ligand GB88 was profiled for PAR2 agonist and antagonist properties by several functional assays associated with intracellular G-protein-coupled signalling in vitro in three cell types and with PAR2-induced rat paw oedema in vivo.
Key Results
In HT29 cells, GB88 was a PAR2 antagonist in terms of Ca2+ mobilization and PKC phosphorylation, but a PAR2 agonist in attenuating forskolin-induced cAMP accumulation, increasing ERK1/2 phosphorylation, RhoA activation, myosin phosphatase phosphorylation and actin filament rearrangement. In CHO-hPAR2 cells, GB88 inhibited Ca2+ release, but activated Gi/o and increased ERK1/2 phosphorylation. In human kidney tubule cells, GB88 inhibited cytokine secretion (IL6, IL8, GM-CSF, TNF-α) mediated by PAR2. A rat paw oedema induced by PAR2 agonists was also inhibited by orally administered GB88 and compared with effects of locally administered inhibitors of G-protein coupled pathways.
Conclusions and Implications
GB88 is a biased antagonist of PAR2 that selectively inhibits PAR2/Gq/11/Ca2+/PKC signalling, leading to anti-inflammatory activity in vivo, while being an agonist in activating three other PAR2-activated pathways (cAMP, ERK, Rho) in human cells. These findings highlight opportunities to design drugs to block specific PAR2-linked signalling pathways in disease, without blocking beneficial PAR2 signalling in normal physiology, and to dissect PAR2-associated mechanisms of disease in vivo.
Keywords: protease, protease inhibitor, peptide, proteinase activated receptor 2, antagonist, agonist, GPCR, inflammation, biased signalling, cell signalling
Introduction
Proteolytic enzymes (proteases) play important, though incompletely defined, roles in regulating cell signalling and mammalian physiology (Heutinck et al., 2010; Adams et al., 2011; Di Cera, 2011; Vergnolle and Chignard, 2011; Hollenberg et al., 2014). Inhibitors of specific proteases have successfully progressed through the clinic (Leung et al., 2000; Abbenante and Fairlie, 2005; Turk, 2006). However, many proteases are involved in most diseases and each protease usually has pleiotropic functions in both normal physiology and disease (Cudic and Fields, 2009; Pejler et al., 2010; Sharony et al., 2010; Moore and Crocker, 2012). A common cellular target of several proteases, as well as more selective modulation of the functions induced by proteases, may be necessary for more specific and more effective drugs. One such target is the proteinase activated receptor 2 (PAR2; nomenclature follows Alexander et al., 2013a), which is a membrane-spanning GPCR that is the target of a range of serine proteases (Adams et al., 2011; Ramachandran et al., 2012).
PAR2 has a unique activation mechanism. Its N-terminal domain is pruned by extracellular serine proteases, such as trypsin, tryptase, tissue factor TF-FVII-FXa, kallikreins and human leukocyte elastase (Barry et al., 2006; Ramachandran et al., 2012). If productively cleaved, the new N-terminus of PAR2 self-activates by inducing intracellular G-protein-coupled or β-arrestin-mediated signalling pathways, leading to a diverse range of intracellular and physiological responses. While activated PAR2 couples to Gq/11, Gs, Gi/o and G12/13 (Adams et al., 2011; Hirota et al., 2012), individual coupled pathways seem to be protease-, cell- and context-dependent, and there is conflicting evidence derived from different conditions. Furthermore, PAR2 can signal independently of G-proteins via ß-arrestin1/2 (Nichols et al., 2012). PAR2 is an important mediator in many animal models of inflammatory diseases, including asthma (Cocks et al., 1999), pancreatitis (Kawabata et al., 2006), irritable bowel syndrome (Cenac et al., 2007), colitis (Lohman et al., 2012b), arthritis (Lohman et al., 2012a), glomerulonephritis (Moussa et al., 2007), obesity and diabetes (Badeanlou et al., 2011; Lim et al., 2013).
Recently, we reported the discovery of a new, selective and orally active antagonist of PAR2, 5-isoxazoyl-Cha-Ile-spiroindene-1,4-piperidine (GB88; Barry et al., 2010; Suen et al., 2012). It binds at high nM to low μM concentrations to PAR2 on the cell surface and antagonizes intracellular calcium release induced in many human, rat and mouse cell types by either synthetic peptide (SLIGRL-NH2, SLIGKV-NH2, 2f-LIGRLO-NH2), peptidomimetic (GB110) or endogenous protease (trypsin, tryptase) agonists of PAR2. GB88 also inhibited inflammation in rat models of PAR2-induced paw oedema (Barry et al., 2010; Suen et al., 2012), PAR2 agonist- or TNBS-induced colitis (Lohman et al., 2012b), collagen-induced arthritis (Lohman et al., 2012a) and diet-induced obesity, adipose and macrophage inflammation, insulin sensitivity and cardiovascular remodeling (Lim et al., 2013).
The present study reports the surprising finding that the anti-inflammatory PAR2 antagonist GB88 activates a range of other PAR2-mediated signalling pathways. The study reports the regulation of PAR2-mediated intracellular signalling pathways by GB88, providing important new mechanistic insights to PAR2-dependent signalling and inflammatory activity in vivo and their potential for modulation by PAR2 ligands. Our goal was to understand the spectrum of effects of this new PAR2 ligand, which has important anti-inflammatory properties in vivo after oral administration, and to use GB88 to mechanistically dissect PAR2 signalling. The differential and unique spectrum of effects of GB88 on PAR2-mediated intracellular signalling pathways reveals potentially useful ligand-induced biased signalling that, in this particular paper, highlights the relevance of the Gq/11-Ca2+ signalling pathway in PAR2-mediated inflammation in human cells and in PAR2-induced rat paw oedema. The research uncovered a pathway-selective antagonist with potentially valuable uses as a new tool for selectively inhibiting Gq signalling in vitro and in vivo.
Methods
Cell culture
All cell culture reagents were purchased from Invitrogen (Carlsbad, CA, USA) and Sigma-Aldrich (St. Louis, MO, USA). Human colorectal carcinoma human colon adenocarcinoma grade II cell line (HT-29) and CHO cells were cultured in medium at 37°C and 5% CO2 according to American Tissue Culture Collection instructions. HT-29 cells were grown in DMEM and CHO cells were grown in F12 supplemented with 200 μg·mL−1 hygromycin B. All media were supplemented with 10% FBS, 100 units·per mL penicillin and 100 units·per mL streptomycin. During cell culture passage, cell dissociation solution (Sigma-Aldrich) was used to dissociate cells from the surface of culture flasks and cells were counted manually using a haemocytometer or automated cell counter.
Competitive binding assay
Assays were performed as described (Hoffman et al., 2012). Cells were seeded overnight in a 384-well plate at a density of 2500 cells per well. On the day of experiment, media was aspirated and cells were washed with PBS followed by 2% BSA blocking for 1 h at 37°C. After blocking, cells were simultaneously exposed to 2f-LIGRLO(diethylene triamine pentaacetic acid; dtpa)-NH2 (300 nM) and different concentrations of 2f-LIGRLO-NH2 for 15 min. GB88 was pre-incubated for 15 min prior to the addition of 2f-LIGRLO(dtpa)-NH2 (300 nM) due to its slow on-rate (Suen et al., 2012). Cells were then washed three times with PBS supplemented with 20 μM EDTA, 0.01% Tween and 0.2% BSA. After washings, cells were then incubated with 40 μL of DELFIA enhancement solution (Perkin Elmer, Santa Clara, CA, USA) for 90 min. Fluorescence was determined with terminal restriction fragment analysis (Pherastar FS, BMG Labtech, Ortenberg, Germany): 340 nm excitation followed by 400 μs delay before a 400 μs 615 nm emission.
Isolation and primary culture of human tubule epithelial cells (HTEC)
Use of human renal tissue for primary culture was reviewed and approved by the Princess Alexandra Hospital Research Ethics Committee. Informed consent was obtained prior to each operative procedure. Segments of macroscopically and histologically normal renal cortex (5–10 g) were obtained aseptically from the non-cancerous pole of adult human kidneys removed surgically because of small renal cancers. Patients were otherwise healthy.
The method for isolation and primary culture of HTEC is fully described elsewhere (Vesey et al., 2005; 2007; 2009; Qi et al., 2007). Briefly, the cortical tissue was minced finely, washed several times and agitated for 20 min at 37°C in a Krebs-Henseleit buffer (KHB) containing collagenase type II (1 mg·mL−1). Cold KHB was added and the solution passed through a 297 μm sieve (50 Mesh, Sigma). After washing three times, the tubular fragments were re-suspended in 45% Percoll – KHB and centrifuged at 20 000× g. A high-density band, previously shown to be tubule fragments, was removed and cultured in a serum free, hormonally defined DMEM/F12 media (containing 10 ng·mL−1 epidermal growth factor, 5 μg·mL−1 insulin, 5 μg·mL−1 transferrin, 50 nM hydrocortisone, 50 μM PGE1, 50 nM selenium and 5 pM triiodothyronine). All experiments were performed on confluent passage 2 HTEC made quiescent by two washes followed by incubation for 24 h in serum and growth factor free DMEM/F12 media.
Intracellular calcium mobilization
Cells were grown to 80% confluence. Prior to experiment, cells were seeded overnight in 96-well black wall, clear bottom, plates at approximately 5 × 104 cells per well. On the day of experiment, supernatant was removed and cells were incubated in dye loading buffer (HBSS with 4 μM Fluo-3, 0.04% pluronic acid, 1% FBS and 2.5 mM probenecid) for 1 h at 37°C. Cells were washed twice with HBSS and transferred to a FLIPR Tetra instrument (Molecular Device, Sunnyvale CA, USA) for agonist injection and fluorescence measurements. PAR2 agonists were added 10 s after reading commenced at various concentrations and fluorescence was measured in real time using excitation at 480 nm and emission at 520 nm. HBSS was prepared in-house, while all other reagents were purchased from Invitrogen. Plates were supplied by DKSH (Zurich, Switzerland). Calcimycin (A23187, Invitrogen) was used to measure maximum fluorescence, with individual results normalized accordingly.
cAMP accumulation
LANCE Ultra cAMP assay was performed in accordance with the manufacturer's instructions (Perkin Elmer). In brief, cells were dissociated from flasks by Versene (Invitrogen) on the day of experiment. Cells (5 μL, 4 × 105 cells·per mL) were transferred to a 384-well proxiplate (Perkin Elmer) and incubated with various concentrations of GB88 (2.5 μL) for 20 min at room temperature. Forskolin (2.5 μL, 120 nM) was then added into each well and incubated for a further 10 min at room temperature. Finally, europium (Eu)-cAMP tracer (5 μL) and ULight™-anti-cAMP (5 μL; Perkin Elmer, Waltham, MA, USA) were added to each well and incubated for 1 h at room temperature. The plate was read using a Pherastar FS fluorimeter (BMG Labtech).
ERK1/2 phosphorylation
SureFire phospho-ERK1/2 assay was performed in accordance with the manufacturer's instructions (Perkin Elmer). In brief, cells were seeded overnight in 96-well tissue culture plate (∼5 × 104 cells per well). On the day of experiment, cells were treated with various concentrations of compounds dissolved in serum-free medium and incubated for 10 min at 37°C. Supernatant was removed and cells were lysed with cell lysis buffer provided by the kit. Cell lysate (4 μL) was transferred to a 384-well proxiplate (Perkin Elmer) and incubated with reaction mixture (7 μL) for 2 h at room temperature before plate reading.
Western blot analysis
HT-29 cells were seeded at a density of 5 × 105 cells·mL−1 and allowed to adhere overnight. Cells were then serum-starved up to 8 h followed by treatment with 2f-LIGRLO-NH2 and GB88 at specific concentrations. Cells were lysed in Tris buffer (50 mM, pH 7.4) containing SDS (1%) and protease inhibitor cocktail (Roche Applied Science, Penzberg, Germany). Equal amounts of proteins were separated by SDS-PAGE and transferred onto polyvinylidene fluoride membrane. Proteins were detected using specific antibodies targeting a protein of interest: phospho-myosin phosphatase, (MYPT1; Thr696), PKC pan, phospho-PKC, (Thr638/641), phospho-PKC antibody sampler kit (Cell Signaling Technology, Danvers, MA, USA), GAPDH (Sigma-Aldrich) and PKC epsilon (Ser729; Abcam, Cambridge, UK). Relative densitometry analysis on protein bands was performed using ImageJ 1.40e software (U.S. National Institutes of Health, Bethesda, MD, USA). Results were normalized against control bands.
G-LISA Ras homologue gene family, member A (RhoA) activation assay
HT-29 cells were seeded at 30% confluency and serum-starved for 2 days prior to experiment. Cells were treated with various agents, and cell lysates were collected by lysis buffer provided by kit. RhoA activation was examined using G-LISA kit (Cytoskelelon Inc., Denver, CO, USA; BK124). Briefly, cell lysates were added 1:1 v/v to binding buffer and triplicate assays were performed. Samples were incubated (30 min), washed (3 times) with washing buffer, antigen-presenting buffer was added (2 min), then incubated with anti-RhoA antibody (45 min), washed three more times, then incubated with secondary antibodies (45 min, room temperature). HRP detection reagent was added and signal was read by measuring absorbance at 490 nm after stopping the reaction with 2 M sulfuric acid. Total RhoA in cell lysates was measured by RhoA elisa kit (Cytoskeleton Inc., BK150).
Confocal microscopy of HT-29 cells
Cells were seeded on sterile glass coverslips and allowed to adhere overnight. The cells were incubated in medium containing 0.5% serum for 24 h followed by serum-starved for another 24 h. Cells were treated with PBS, calpeptin, 2f-LIGRLO-NH2 or GB88 and fixed with 4% formaldehyde for 10 min. Cells were then permeabilized with 0.5% Triton for 5 min and incubated with fluorescein-phalloidin for 30 min at room temperature. The slides were mounted with Prolong Gold, and examined by an LSM-510 META inverted microscope (Zeiss, North Ryde, NSW, Australia).
Elisa
Cells in 6-, 12- or 48-well plates at 80% confluence were allowed to adhere overnight. On the day of experiment, cells were treated with test agents for 24 h. Supernatants were then collected and examined using BD Pharmingen elisa set. Briefly, the plate was coated with capturing antibody overnight at 4°C overnight. The plate was then blocked by 10% serum for 1 h at room temperature. Samples were added to each well, along with standards, and allowed to incubate for 2 h at room temperature. Afterwards, HRP-conjugated antibody was added and incubated for a further 1 h at room temperature. K-blue substrate (Elisa Systems, Brisbane, Australia) was allowed to develop for 30 min in the dark, stopped by sulfuric acid (50 μL, 2 M), and absorbance was measured.
Animals
All animal care and experimental procedures were approved by the animal ethics committee of The University of Queensland. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). A total of 43 animals were used in the experiments described here.
Male Wistar rats (8–9 weeks, 250 ± 20 g) were bred at the Australia Animal Resource Centre (Canning Vale, WA, Australia). Following Australian ethical standard animal air transport, animals were housed at The University of Queensland Biological Resources (UQBR) Animal Facility at The Australian Institute for Bioengineering and Nanotechnology at The University of Queensland, Australia. Animals were housed in the appropriate temperature/pressure environment in a 12 h light/dark cycle, according to the standards of the accredited holding facility, with food and water provided ad libitum. At least 48 h habituation in the UQBR facility was provided prior to any experimental intervention. After experimentation, animals were humanely killed by CO2 inhalation as stipulated by approved ethical agreements.
PAR2-induced paw oedema
Male Wistar rats (8–9 weeks) were injected with 2f-LIGRLO-NH2 (350 μg in 100 μL isotonic saline) or trypsin (20 μg) into the plantar surface of both hind paw pads using a 30G needle. Inhibitors at various concentrations in saline were injected into the plantar surface of the right hind paw pad, 30 min before agonist, while the left hind paw pad received saline only by injection. Paw thickness and width were measured using digital calipers (WPI) at 0, 1, 2 and 24 h after PAR2 agonist administration. Hind paw size is expressed as % change in area after 1 h from baseline, right (inhibitor) paw compared with left (control) paw, and then normalized against maximum swelling induced by agonist alone.
Data analysis
Data were analysed in GraphPad Prism (GraphPad Software, San Diego, CA, USA) using anova or Student's t-test, with values expressed as mean ± SEM (n ≥ 3). Data are presented as the mean value of the entire data set. Significance was determined as P < 0.05. Concentration-response curves were fitted in GraphPad Prism with a standard Hill slope of 1 (three-parameter fit).
Materials
PAR2 activating peptide agonist (2f-LIGRLO-NH2), non-peptide agonist (GB110) and non-peptide antagonist (GB88) were synthesized in-house as described (Barry et al., 2010; Suen et al., 2012). elisa sets were purchased from BD Pharmingen (San Jose, CA, USA). SureFire phosphor-ERK1/2 kit and LANCE Ultra cAMP kit were purchased from Perkin Elmer. G-LISA kit was purchased from Cytoskeleton Inc. Pertussis toxin (PTX) was purchased from Abacus ALS (Brisbane, Australia). Y-27632, Gö6983 and FITC-labelled phalloidin were purchased from Sigma-Aldrich. The CHOLERA toxin used in the study was purchased from Sigma-Aldrich. U0126 was purchased from Merck (White House Station, NJ, USA). Prolong Gold was purchased from Invitrogen.
Results
GB88 is a PAR2 antagonist of Ca2+ release and PKC phosphorylation
Recently, we discovered an orally active PAR2 antagonist (GB88) that inhibited PAR2-, but not PAR1-, induced intracellular calcium mobilization in multiple human cell types treated with peptide, non-peptide or protease agonists of PAR2 (Barry et al., 2010; Suen et al., 2012). Here, we use a fluorescence-based binding assay (Hoffman et al., 2012) to show that GB88 directly competes with a Eu-tagged peptide agonist of PAR2, namely 2f-LIGRLO-NH2 (Figure 1A and 1B). CHO cells transfected with human PAR2 were used because a high level of PAR2 expression is required to allow binding measurements. On human non-transfected colorectal HT-29 cells, GB88 behaved as a full antagonist in inhibiting Ca2+ release induced by the PAR2 agonist 2f-LIGRLO-NH2 (Figure 1C and 1D). GB88 was also a PAR2 antagonist in inhibiting Ca2+ release in CHO-hPAR2 cells (Supporting Information Fig. S1), but did not bind to untransfected CHO cells or induce Ca2+ release in HT-29 cells at concentrations where it was an antagonist. In terms of Ca2+ release, GB88 behaved as a PAR2-specific antagonist with no agonist activity up to 10 μM concentration (Supporting Information Figs S2 and S3) in HT-29 cells. GB88 inhibition of 2f-LIGRLO-NH2-induced calcium release was not Gs-dependent, as cholera toxin (CTX) did not decrease the response induced by the agonist (Supporting Information Fig. S4). PAR2 agonists activated various subtypes of PKC (Reibman et al., 2000; Amadesi et al., 2009; Chen et al., 2011; Huang et al., 2012). When HT-29 cells were treated here with 2f-LIGRLO-NH2, there was an increase in phosphorylated PKCα/β (Figure 1E and Supporting Information Fig. S5) that was prevented by GB88 treatment (Figure 1F). These results indicate that GB88 is a full antagonist in inhibiting the PAR2-Gq/11-Ca2+-PKC signalling axis in vitro in cells expressing human PAR2.
Figure 1.
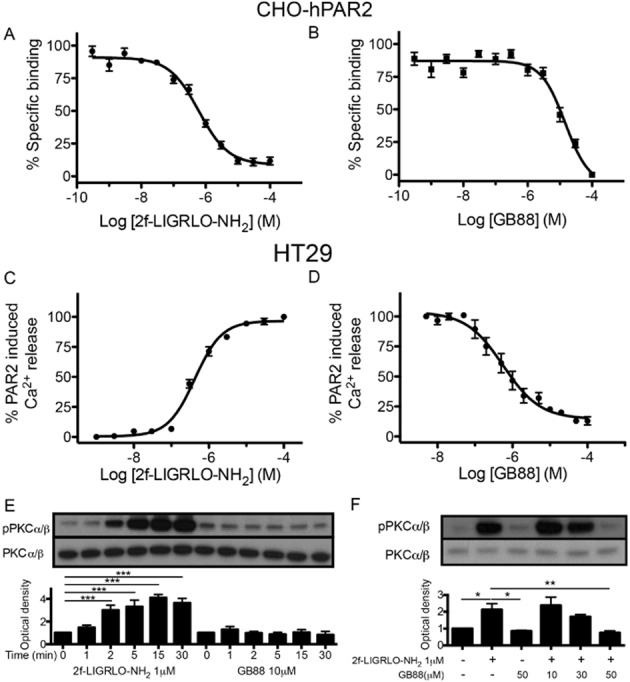
GB88 is an antagonist of the PAR2-Ca2+-PKC signalling axis. (A) 2f-LIGRLO-NH2 competed in a concentration-dependent manner with 300 nM Eu-tagged 2f-LIGRLO-NH2 (KD 240 nM, n = 6) in a receptor binding assay in CHO-hPAR2. (B) GB88 competed in a concentration-dependent manner with 300 nM Eu-tagged 2f-LIGRLO-NH2 (Ki 7.7 μM, n = 3) in CHO-hPAR2 cells. (C) 2f-LIGRLO-NH2 induced intracellular calcium release (EC50 340 nM, n = 12) in a concentration-dependent manner in HT-29 cells. (D) GB88 inhibited 2f-LIGRLO-NH2 (1 μM) induced intracellular calcium release (IC50 560 nM, n = 3) in HT-29 cells. (E) Time-course of PKC phosphorylation by 2f-LIGRLO-NH2 and GB88 (n = 6) in HT-29 cells. (F) GB88 blocked PKC phosphorylation induced by PAR2 in HT-29 cells (n = 6). (E, F) One representative Western blot is shown, bar chart results are for six independent experiments (n = 6). Data shown are means ± SEM. n, number of independent experiments, **P < 0.01, ***P < 0.001; significant differences as indicated.
GB88 inhibits PAR2-mediated cytokine release
PAR2 activation has previously been associated with inflammatory cytokine production, so we investigated whether GB88 was able to antagonize cytokine release. In HT-29 cells, GB88 blocked the secretion of IL-8 induced by a PAR2 agonist (Supporting Information Fig. S6). Effects of PAR2 agonist and antagonist were also examined in primary HTEC of the kidney, as these cells have been previously reported to respond strongly to PAR2 activation by releasing some cytokines (Vesey et al., 2007). Indeed, GB88 (10 μM) did significantly reduce production of IL-8, IL-6, TNF-α and GM-CSF induced by 2f-LIGRLO-NH2 (1 μM) as measured by (Figure 2). For comparison, cytokine production was abolished by blocking the activity of PKC using the commercially available PKC inhibitor, Gö6983. GB88 alone (10 μM) did not cause any significant increase in cytokine secretion.
Figure 2.
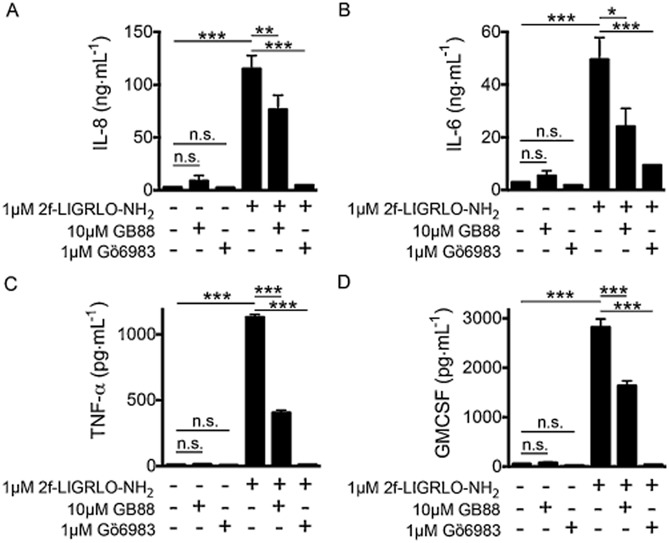
GB88 blocks PAR2 induced cytokine release in HTEC kidney cells. GB88 (10 μM, 30 min) inhibited >50% of IL-8 (A), IL-6 (B), TNF-α (C) and GM-CSF (D) secretion induced by PAR2 activation (2f-LIGRLO-NH2 1 μM, 24 h). Inhibition of PKC by Gö6983 (1 μM, 30 min) also showed decreased secretion of these cytokines. Treatment with GB88 or Gö6983 alone failed to change cytokine release. Data shown are means ± SEM of three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001; significant differences as indicated.
GB88 attenuates forskolin-induced cAMP accumulation via PAR2-Gi/o
GB88 was examined for effects in other PAR2-mediated signalling pathways. It induced a similar response to the PAR2 agonist 2f-LIGRLO-NH2 in down-regulating forskolin-induced cAMP levels in CHO-hPAR2 cells (Figure 3A and 3B) and in HT-29 cells (Figure 3C and 3D). Neither GB88 nor 2f-LIGRLO-NH2 had any effect on cAMP in untransfected CHO cells (Figure 3A and 3B). In HT-29 cells, this PAR2-mediated response induced by either 2f-LIGRLO-NH2 or GB88 was abolished by pretreatment of the same cells with PTX (200 ng·mL−1, 24 h), indicating that the response was Gi/o dependent (Figure 3C and 3D). The results reveal that GB88 behaves like other PAR2 agonists in reducing forskolin-induced cAMP accumulation via activating the PAR2-Gi/o signalling pathway.
Figure 3.
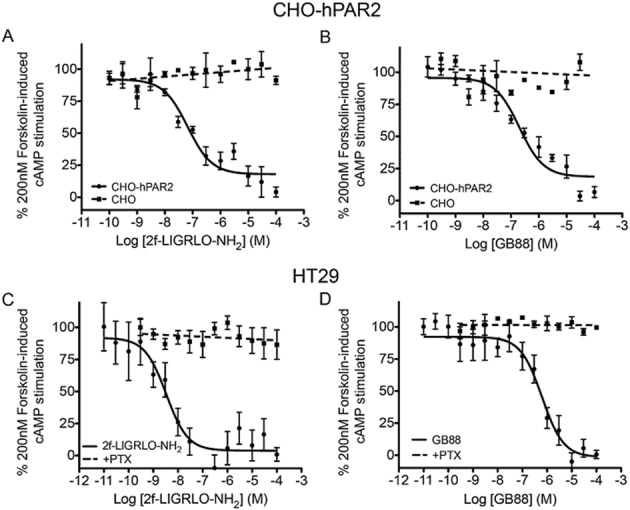
GB88 down-regulates cAMP via PAR2-Gi/o. (A) 2f-LIGRLO-NH2 dose dependently down-regulated forskolin-stimulated cAMP in CHO cells transfected with human PAR2 (EC50 70 nM, n = 3), but was inactive in untransfected CHO cells (dotted line). (B) GB88 dose-dependently down-regulated cAMP in CHO-hPAR2 cells as did the PAR2 agonist 2f-LIGRLO-NH2 (EC50 120 nM, n = 3), but was inactive in untransfected CHO cells. Thus, the response was PAR2 dependent. (C–D) Dose-dependent down-regulation of forskolin-stimulated cAMP by 2f-LIGRLO-NH2 (C; EC50 4 nM) and GB88 (D; EC50 600 nM) in HT-29 cells (n = 12), both inhibited with PTX (200 ng·mL−1, 24 h, n = 3). This indicates that down-regulation of cAMP was via Gi/o activation. Data shown are means ± SEM, n, number of independent experiments.
GB88 stimulates RhoA activity
With respect to the G12/13 pathway, HT-29 cells were treated with either 2f-LIGRLO-NH2 or GB88 and examined for formation of the characteristic actin stress fibres that result from RhoA activation. Calpeptin (CN01), a known RhoA activator, was used as positive control. Under confocal microscopy, cells treated with 2f-LIGRLO-NH2 or GB88 both showed rearrangement of actin filaments to the same extent as the positive control (Figure 4A). This induction of RhoA activity by both treatments was confirmed, using G-LISA, to directly measure production of RhoA protein. Both PAR2 ligands were able to stimulate RhoA release (Figure 4B). Downstream phosphorylation of MYPT, known to be a messenger associated with RhoA and Rho-associated kinase (ROCK), was also observed. Both PAR2 ligands increased MYPT phosphorylation (Figure 4C and 4D). Thus, GB88 behaved like other PAR2 agonists in up-regulating G12/13 signalling with RhoA activation.
Figure 4.
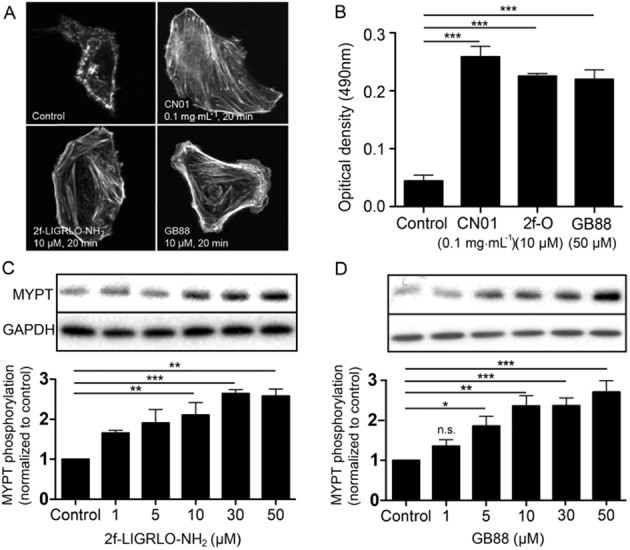
GB88 activates RhoA via PAR2. (A) Confocal microscopy images of HT-29 cells treated with various agents [control; Calpeptin (CN01), 0.1 mg·mL−1, 20 min; 2f-LIGRLO-NH2, 10 μM, 20 min; GB88, 50 μM, 20 min]. Actin filament was stained with FITC-phalloidin (200 nM, 30 min). (B) RhoA activation in HT-29 cells was measured by G-LISA. Serum-free medium was used as negative control and calpeptin (CN01, 0.1 mg·mL−1, 20 min) was used as positive control. Both 2f-LIGRLO-NH2 (10 μM, 20 min) and GB88 (50 μM, 20 min) up-regulated RhoA activition (n = 3). (C–D) Phosphorylation of MYPT by (C) 2f-LIGRLO-NH2 (10 μM) and (D) GB88 (50 μM) in HT29 cells, indicating an increase in activity of ROCK. One representative Western blot is shown, bar chart results are for three independent experiments (n = 3). Optical densities were normalized against controls of individual blots. Data shown are means ± SEM. n, number of independent experiments; n.s. not statistically significant, *P < 0.05, **P < 0.01, ***P < 0.001; significant differences as indicated.
GB88 phosphorylates ERK1/2 via PAR2
Both 2f-LIGRLO-NH2 and GB88 induced ERK1/2 phosphorylation in CHO-hPAR2 cells and in HT-29 cells, whereas no effect was detected in untransfected CHO cells (Figure 5A and 5B). This is consistent with GB88 being a PAR2-selective agonist via this pathway. However, unlike the other agonist-like effects of GB88 above, the GB88-induced response was only ∼50% of the magnitude of the response to 2f-LIGRLO-NH2. In order to delineate the signalling pathways associated with ERK1/2 phosphorylation, HT-29 cells were treated with inhibitors of various intracellular messengers (Figure 5C–F). The mitogen-activated kinase (MEK)1/2 inhibitor, U0126, completely abolished the effects of both PAR2 ligands, while treatment with RhoA inhibitor (Y-27632) resulted in slight changes in compound potencies. PTX was inactive in regulating levels of pERK1/2, consistent with the Gi/o coupling not being involved in this event. The PKC inhibitor, Gö 6983, failed to cause any effect on ERK1/2 phosphorylation induced by GB88. However, it did significantly reduce the 2f-LIGRLO-NH2-induced response by ∼50%, bringing it down to a level similar to GB88. Of the four commercial inhibitors tested, only the PKC inhibitor showed differential effects on the two PAR2 ligands examined.
Figure 5.
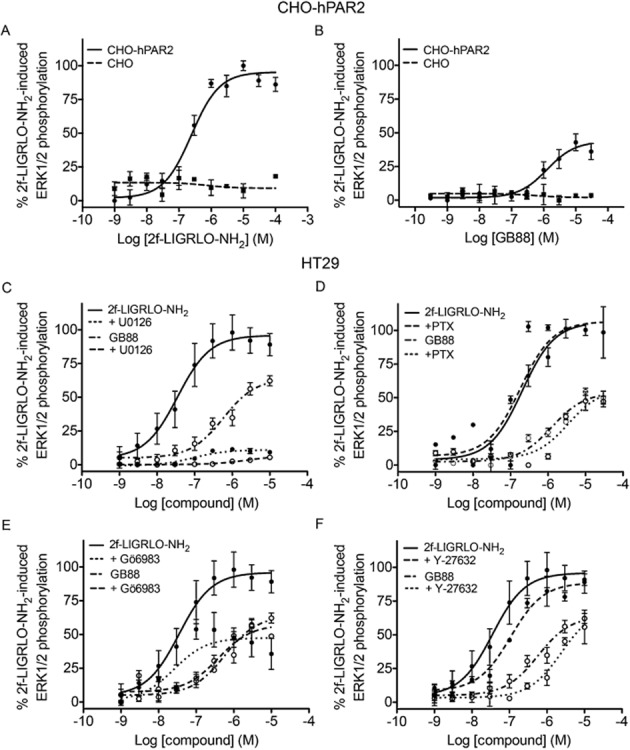
GB88 phosphorylates ERK1/2 via PAR2. (A–B), Concentration-dependent phosphorylation of ERK1/2 by 2f-LIGRLO-NH2 (A; EC50 250 nM) or GB88 (B; EC50 1.3 μM) in CHO-hPAR2 cells (solid lines), compared with no activity in untransfected CHO cells (dotted lines). This suggests a PAR2-dependent effect. (C) ERK1/2 phosphorylation of both PAR2 ligands in HT29 cells was abolished by the MEK1/2 inhibitor, U0126 (10 μM, 30 min). (D) Gi/o inhibitor, PTX (200 ng·mL−1, 24 h) failed to have any significant effects on ERK1/2 phosphorylation induced in HT29 cells by both PAR2 ligands. (E) 2f-LIGRLO-NH2-induced ERK1/2 phosphorylation in HT29 cells was significantly reduced by PKC inhibitor, Gö6983 (1 μM, 30 min). The reduced level was similar for GB88. Gö6983 was inactive against GB88-induced response. (F) ERK1/2 phosphorylation in HT29 cells in the presence of ROCK inhibitor, Y-27632 (1 μM, 2 h). Data shown are means ± SEM of three independent experiments.
GB88 inhibits PAR2-induced paw oedema
To correlate with the above in vitro findings, the properties of GB88 were compared with those of the commercially available signalling pathway inhibitors above in a PAR2-induced paw oedema in Wistar rats, an acute inflammation model in vivo. The PAR2 agonists, 2f-LIGRLO-NH2 (350 μg) or trypsin (20 μg), were given by intra-plantar (i.pl.) injection to rat paws to induce an acute paw oedema in vivo. Oral administration of GB88 (10 mg·kg−1) 3 h prior to induction of paw oedema, commensurate with its Tmax of 4 h (Lohman et al., 2012a; Suen et al., 2012), reduced (>50%) the swelling mediated by PAR2 activation after 30 min to 2 h (Figure 6A and 6B). Elsewhere, we have reported that GB88 does not inhibit rat paw oedema induced by PAR1 agonists thrombin and the pentapeptide TFLLR-NH2 (Suen et al., 2012). As GB88 was an antagonist of PAR2-induced paw oedema, but behaved similarly to PAR2 agonists in the in vitro assays except for those associated with intracellular calcium and PKC phosphorylation, we also examined the effect of the PKC inhibitor (Gö6983, 1 μM and 10 μM) in the same in vivo rat model of acute inflammation. This inhibitor was locally injected into the hind paw pads 30 min prior to i.pl. injection of either 2f-LIGRLO-NH2 or trypsin, resulting in significant reduction in PAR2-induced paw oedema (up to 90%; Figure 6C and 6D). Similar experiments were performed with inhibitors of other secondary messengers of PAR2 signalling pathways, including U73122 (PLCβ), U0126 (MEK1) and PTX (Gi/o), and all except the PLCβ inhibitor failed to reduce PAR2-mediated paw oedema (Supporting Information Fig. S7). Thus, orally delivered systemic GB88 had a similar anti-inflammatory effect in rat paws as a PKC inhibitor (and possibly any PAR2-Gq/11 pathway inhibitor) locally administered to rat paws in this model of paw oedema. This directly contrasts with the pro-inflammatory effects of the PAR2 agonists 2f-LIGRLO-NH2 and trypsin (Figure 6), as well as tryptase, which all induce rat paw oedema in this inflammatory model (Lohman et al., 2012b; Suen et al., 2012).
Figure 6.
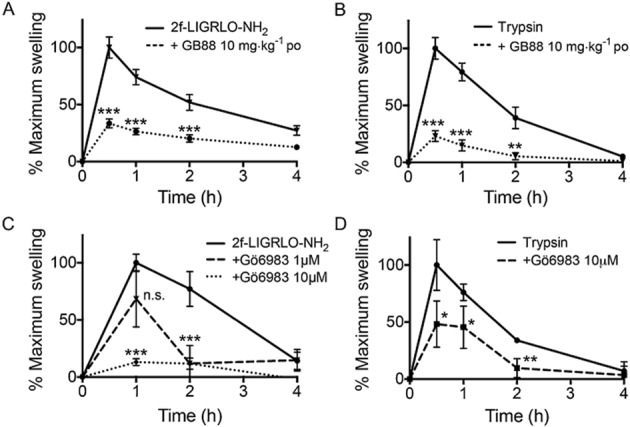
GB88 blocks PAR2-induced paw oedema in rats. (A–B) GB88 (10 mg·kg−1 p.o., 3 h prior) inhibited rat paw oedema induced by i.pl. injection of PAR2 agonist, 2f-LIGRLO-NH2 (A, 350 μg) or trypsin (B, 20 μg). (C–D) PKC inhibitor, Gö6983 (1 and 10 μM, intraplanar), inhibited Wistar rat paw oedema induced by i.pl. injection of PAR2 agonist 2f-LIGRLO-NH2 (C, 350 μg) or trypsin (D, 20 μg). Samples were compared with 2f-LIGRLO-NH2 or trypsin-treated controls. Data shown are means ± SEM; n.s. not statistically significant, *P < 0.05, **P < 0.01, ***P < 0.001; significant differences as indicated. Number of rats: A, 2F (n = 9), GB88 (n = 5); B, tryp (n = 9), GB88 (n = 5); C, 2F (n = 3), Go 1 µM (n = 3) and 10 µM (n = 3); D, tryp (n = 3), Go 10 µM (n = 3).
Thus, GB88 has dual functions mediated through PAR2 (Figure 7). It was a selective antagonist of the PAR2-Gq/11-Ca2+-PKC signalling pathway (Figure 1), exerting anti-inflammatory activity like Gö6983 both in vivo and in vitro, as shown by inhibiting PAR2-induced paw oedema in rats and by inhibiting secretion of inflammatory cytokines from human cells (IL8, IL6, TNF-α, GMCSF) stimulated with the PAR2 agonist 2f-LIGRLO-NH2 (Figure 2). However, GB88 was a PAR2 agonist in other pathways in vitro, behaving like 2f-LIGRLO-NH2 in activating Gi/o (Figure 3), G12/13 (Figure 4) and ERK1/2 (Figure 5) signalling pathways. By contrast, PAR2 agonists like trypsin, SLIGRL-NH2, 2f-LIGRLO-NH2 all show pro-inflammatory effects in vitro and in vivo in the assays described, unlike the anti-inflammatory effects of GB88.
Figure 7.
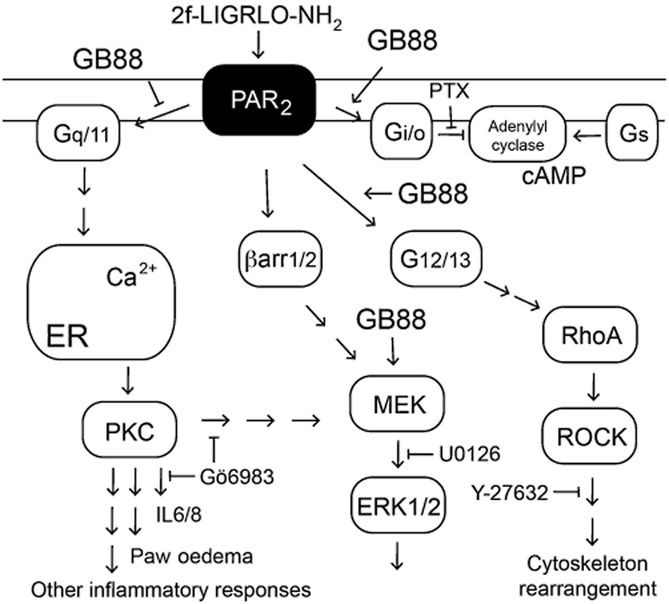
Schematic diagram of effects of GB88 on PAR2-mediated signalling. GB88 activates (arrows) three of four PAR2-coupled pathways shown, and selectively inhibits (T arrow) the PAR2-Gq/11-Ca2+-PKC pathway.
Discussion and conclusions
Protein and peptide-activated GPCRs are embedded in the plasma membrane of cells and are pivotal mediators in disease (Tyndall et al., 2005; Blakeney et al., 2007). About 30% of all pharmaceutical agents activate (agonist) or inhibit (antagonist) GPCRs. Many GPCR ligands were discovered by screening chemical libraries for GPCR binding, followed by optimizing for higher affinity, selectivity and functional potency in one or two in vitro assays (Klabunde and Hessler, 2002; Blakeney et al., 2007). Compounds assigned as agonists or antagonists have historically been assumed to be so across all functions mediated by that receptor. However, this is now known not to be always true, due to ligand-induced biased signalling (Shukla et al., 2008; Rajagopal et al., 2010; Kenakin, 2011; 2012). Two structurally similar agonists, even for the same GPCR subtype, can activate different intracellular signalling cascades due to subtle differences in interactions with the GPCR (Quirk et al., 2007; Shukla et al., 2008; Rajagopal et al., 2010; Kenakin, 2012), resulting in different ligand-bound receptor conformations. In turn, intracellular G protein coupling to a GPCR can also influence extracellular ligand binding (Kobilka and Deupi, 2007; Kobilka and Schertler, 2008; Kobilka, 2011). A biased ligand differentially affects certain signal transduction pathways, and can potentially exert more refined control over GPCR-mediated cell signalling by activating or inhibiting a particular signalling pathway. To date, most GPCR research on biased signalling has focused on distinguishing G-protein-dependent from independent (e.g. ß-arrestin) signalling. A role for ß-arrestin was recently identified in PAR2-induced responses in lung tissues, with an opposing physiological effect between ß-arrestin and G-protein heterotrimers (Nichols et al., 2012). However, in principle, biased signalling can refer to differences between any signalling pathways and may profoundly influence whether ‘agonist’ or ‘antagonist’ properties measured in vitro translate into different in vivo properties.
In this study, different G-protein-coupled PAR2-dependent pathways were assessed for effects of an orally active ligand known as GB88. Previously, we reported that GB88 was an antagonist of PAR2-activating peptide agonists (2f-LIGLRO-NH2, SLIGKV-NH2, SLIGRL-NH2), non-peptide agonists (GB110), and protease agonists (trypsin, tryptase), as measured by intracellular calcium mobilization in different human cell types (Lohman et al., 2012a; Suen et al., 2012). In that assay, GB88 had no agonist activity and was selective for PAR2 over PAR1 agonists thrombin or pentapeptide TFLLR, or the PAR4 agonist hexapeptide AYPGKF, in human cells and in a PAR2-induced rat paw oedema model. In the present study, GB88 was found to compete with 2f-LIGRLO-NH2 in a competitive ligand-binding assay measuring hPAR2 affinity on CHO-hPAR2 and to antagonize 2f-LIGRLO-NH2-induced Ca2+ release; yet it behaved like PAR2 agonists in attenuating forskolin-induced cAMP accumulation and inducing ERK1/2 phosphorylation. In HT-29 cells, GB88 is an antagonist of PAR2 by blocking Ca2+ release, PKC phosphorylation and IL-8 secretion, but it is an agonist in activating Gi/o, increasing pERK1/2, and increasing RhoA activity and MYPT phosphorylation. In primary human kidney cells, GB88 reduced secretion of several inflammatory cytokines induced by PAR2 agonist 2f-LIGRLO-NH2, and in rats it blocked PAR2-mediated paw oedema. These differential effects of GB88 indicate pathway-selective modulation of PAR2 (Figure 7), with agonist or antagonist activity dependent on the signal transduction being examined but not on the cell type, at least for these cells examined here. Elsewhere, GB88 alone reportedly recruited PAR2-dependent ß-arrestin-1/2 to the plasma membrane but did not either induce or inhibit receptor internalization (Adams et al., 2011; Hollenberg et al., 2014).
GB88 behaved like 2f-LIGRLO-NH2 in inducing ERK1/2 phosphorylation. Increased pERK1/2 was observed for cells treated with either 2f-LIGRLO-NH2 or GB88. However, unlike the comparable levels of cAMP reduction or RhoA activation by both compounds, GB88 only induced 50% of the increase in pERK1/2 that was caused by 2f-LIGRLO-NH2. Only 2f-LIGRLO-NH2-induced pERK1/2 was sensitive to PKC inhibition, and there was no significant difference in pERK1/2 between GB88 and 2f-LIGRLO-NH2 in cells pretreated with PKC inhibitor. As inhibitors of various G-protein-coupled pathways were ineffective against GB88-induced ERK1/2 phosphorylation, it is likely that this proceeds via a G-protein-independent pathway, possibly via β-arrestin1/2.
In summary (Figure 7), if we had not assessed antagonism of Ca2+ and PKC signalling by GB88, this compound would have been considered a weak agonist in the other pathways examined herein and its biased signalling properties and anti-inflammatory properties would have been missed. Could the antagonist properties in various animal models be explained by GB88 being a pathway-selective PAR2 agonist that simply fails to recruit Gq/11? If so, why does it inhibit other agonists only in this pathway. Is it possible that GB88 antagonizes PAR2-mediated Ca2+ release, cytokine production and paw oedema by biasing the receptor toward G12/13, Gi/o or even β-arrestin1/2 activation, thereby preventing a typical agonist (such as 2f-LIGRLO-NH2) from activating the Gq/11 pathway? If so, why don't other agonists that activate these pathways behave similarly. Finally, we have shown elsewhere that GB88 is a competitive antagonist of 2f-LIGRLO-NH2 (Suen et al., 2012), so why should it be viewed as an agonist. While the precise mechanism of antagonism remains uncertain, it is clear that GB88 has beneficial inhibitory properties of PAR2 activation in a key inflammatory pathway. This is evidenced here by GB88 inhibiting (i) PAR2-Gq-Ca2+-PKC-cytokine signalling in human cells, (ii) PAR2-induced inflammatory rat paw oedema, and (iii) in our recent in vivo studies, PAR2- and TNBS-induced experimental colitis in rats (Lohman et al., 2012b), collagen-induced arthritis in rats (Lohman et al., 2012a), and diet-induced metabolic dysfunction, adiposity, cardiac fibrosis and remodelling in rats (Lim et al., 2013). Thus, at least in the context of the various cells and the disease model examined here and those in our recent publications, GB88 can be considered as a valuable pathway-selective antagonist of PAR2, an antagonist with a biased signalling profile (Figure 7).
As GB88 is an antagonist of PAR2-mediated intracellular calcium release and PKC phosphorylation, we investigated whether a PKC inhibitor could also mimic properties of GB88. PAR2-mediated PKC activation has been reported to be essential in GM-CSF production in PAR2-activated airway epithelial cells, and PAR2-PKC activation has been associated with neurogenic inflammation and neuropathic pain, possibly through increased PKA activity and sensitization of TRPV1, TRPV4 and TRPA1 channels (Reibman et al., 2000; Amadesi et al., 2009; Chen et al., 2011; Huang et al., 2012; channels nomenclature follows Alexander et al., 2013b). In this study, PAR2-mediated secretion of proinflammatory cytokines and PAR2-induced rat paw oedema were both inhibited by the PKC inhibitor Gö6983, as well as by GB88. This supported our findings that GB88 inhibits the PAR2-Gq-Ca2+-PKC axis, whereas its agonist properties have no role in its anti-inflammatory effects in the rat paw oedema model.
PAR2 activation is known to activate many different genes, physiological processes and metabolic pathways (Barry et al., 2006; Suen et al., 2010; Yau et al., 2013), so the prospect of using rationally designed PAR2-binding ligands to inhibit only one PAR2-mediated signalling pathway associated with a particular diseased state is an exciting opportunity that may also be possible for other GPCRs. There have been seemingly paradoxical reports of PAR2 being both pro- and anti-inflammatory (see Barry et al., 2006; Yau et al., 2013), depending upon the context of cell or animal experiments conducted or disease models studied. The new findings here provide a possible explanation for such opposing actions, suggesting that it may be possible for some ligands to activate certain PAR2-mediated pathways while blocking others, enabling PAR2-mediated disease intervention without affecting beneficial or protective PAR2-mediated physiological responses. Importantly, this potentially useful ligand-induced fine-tuning control over PAR2-mediated cellular responses cannot be predicted or interrogated by receptor knockouts, knockdowns or phenotypic screens and this highlights the value of a biased ligand like GB88 in offering unique mechanistic insights to relationships between extracellular ligand binding, intracellular signalling, physiological responses and disease modulation.
The general prospect of ligand-induced biased signalling via GPCRs opens a new dimension for drug design, with the novel possibility of exerting a new level of fine-tuning control and specificity over intracellular regulation that could be of considerable pharmacological significance and therapeutic potential. Extracellular ligands that differentially regulate downstream intracellular signalling through protein receptors located in the plasma membrane, activating or inhibiting selective signalling pathways associated with specific diseases, may present new opportunities for drug design. In this study, we have identified biased signalling by the PAR2 ligand GB88 in human cells. In HT-29 cells, we discovered that GB88 was a biased antagonist of PAR2-Gq/11-dependent Ca2+ release and PKC activation, as well as blocking secretion of several cytokines in human kidney cells. On the other hand, GB88 behaved as a PAR2 agonist in activating PAR2-mediated Gi/o-dependent cAMP down-regulation, PAR2-mediated G12/13-dependent RhoA activation, and PAR2-mediated ERK phosphorylation. These novel findings reveal new opportunities to selectively modulate the PAR2-Gq/11 pathway for therapeutic benefit in inflammatory diseases. There are few known specific inhibitors of the Gq/11 signalling pathway (Nishimura et al., 2010) and the properties of GB88 in human cells and rat inflammation contribute to our mechanistic understanding of PAR2 signalling and of Gq/11-Ca2+-PKC signalling in inflammation. An exciting implication is the possibility of harnessing pathway-selective antagonism without preventing activation of other, potentially protective or beneficial, physiological effects mediated by other PAR2-coupled signalling pathways. We expect that this work will stimulate further searches for, and mechanistic studies of, biased ligands of PAR2 and other GPCRs, for which ligand-induced biased signalling is not fully explored.
Acknowledgments
We thank Drs John Hooper and Mark Adams (Mater Medical Research Institute, Brisbane) for CHO-hPAR2 cells; the Australian Cancer Research Foundation (ACRF) Cancer Biology Imaging Facility (Brisbane, Qld, Australia) for use of confocal microscopes; the National Health and Medical Research Council for grants APP1000745 and 569595); the Australian Research Council for grant DP1093245 and for a Centre of Excellence in Advanced Molecular Imaging (CE140100011); and the Queensland Government for a CIF grant. D. P. F. acknowledges ARC Federation (FF668733) and NHMRC SPRF (1027369) fellowships.
Glossary
- dtpa
diethylene triamine pentaacetic acid
- FITC
fluorescein isothiocyanate
- GB88
5-isoxazoyl-Cha-Ile-spiroindene-1,4- piperidine
- HT-29
human colon adenocarcinoma grade II cell line
- HTEC
human tubule epithelial cells
- i.pl
intra-plantar
- MEK
mitogen-activated kinase
- MYPT
myosin phosphatase
- PAR2
proteinase activated receptor 2
- PTX
Pertussis toxin
- RhoA
Ras homologue gene family, member A
- ROCK
Rho-associated protein kinase
Author contributions
J. S. and D. F. wrote the manuscript; J. S., D. F., D. V., R. L. designed experiments; L. L., M. Y., D. F. developed and produced compounds; J. S., A. C., R. L., A. H., J. L., D. V. performed experiments, M. C. and D. V. contributed editorial assistance.
Conflict of interest
J. S., R. L., J. L., M. Y., L. L. and D. F. are named inventors on patent applications involving PAR2 agonists and antagonists owned by University of Queensland. No other competing interests.
Supporting Information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Figure S1 GB88 blocked 2f-LIGRLO-NH2-induced intracellular calcium release in CHO-hPAR2. Cells were incubated with various concentrations of GB88 for 15 min, then treated with 2f-LIGRLO-NH2 (1 μM) and monitored for intracellular calcium release.
Figure S2 GB88 induced intracellular calcium release at high concentration in HT-29 cells.
Figure S3 Intracellular calcium release by PAR2 ligands in HT-29 cells. Calcimycin was used as control. Cells were treated with 2f-LIGRLO-NH2 (1 μM) and GB88 (10 μM) and fluorescence measured for 30 min after compound addition. 2f-LIGRLO-NH2 showed similar maxima as calcimycin and GB88 showed no significant difference from buffer alone.
Figure S4 2f-LIGLRO-NH2 increased intracellular calcium release independent of Gs in HT-29 cells. HT-29 cells were treated with cholera toxin (200 ng·mL−1, 24 h) and then with 2f-LIGRLO-NH2 or forskolin for calcium release. Forskolin failed to induce intracellular calcium release and CTX had no effect on PAR2-mediated Ca2+ response.
Figure S5 2f-LIGRLO-NH2 and GB88 failed to increase phosphorylation of PKC subtypes in HT-29 cells. Cells were treated with 2f-LIGRLO-NH2 (1 μM) or GB88 (10 μM) for various durations and examined by Western blot using phosphor-specific antibodies.
Figure S6 GB88 blocked IL-8 secretion in HT-29 cells. Cells were treated with 2f-LIGRLO-NH2 (1 μM, 24 h) with various concentrations of GB88. Supernatants or cell lysates were collected and analysed by elisa. GB88 was able to block 2f-LIGRLO-NH2-induced IL-8 secretion and no significant changes in level of IL-8 in cells.
Figure S7 Effects of inhibitors on PAR2-induced paw oedema in rat. PLCβ inhibitor (100 μM, 30 min) inhibited Wistar rat paw oedema induced by intraplantar injection of PAR2 agonists, 2f-LIGRLO-NH2 (A, 350 μg) or trypsin (B, 20 μg). Other inhibitors [MEK1/2 (U0126, 10 μM) or Gi/o (PTX, 5 μg)] failed to cause any significant changes. n = 3, ***P < 0.001.
References
- Abbenante G, Fairlie DP. Protease inhibitors in the clinic. Med Chem. 2005;1:71–104. doi: 10.2174/1573406053402569. [DOI] [PubMed] [Google Scholar]
- Adams MN, Ramachandran R, Yau MK, Suen JY, Fairlie DP, Hollenberg MD, et al. Structure, function and pathophysiology of protease activated receptors. Pharmacol Ther. 2011;130:248–282. doi: 10.1016/j.pharmthera.2011.01.003. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The concise guide to PHARMACOLOGY 2013/14: G protein-coupled receptors. Br J Pharmacol. 2013a;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Catterall WA, et al. The Concise Guide to PHARMACOLOGY 2013/14: Ion Channels. Br J Pharmacol. 2013b;170:1607–1651. doi: 10.1111/bph.12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amadesi S, Grant AD, Cottrell GS, Vaksman N, Poole DP, Rozengurt E, et al. Protein kinase D isoforms are expressed in rat and mouse primary sensory neurons and are activated by agonists of protease-activated receptor 2. J Comp Neurol. 2009;516:141–156. doi: 10.1002/cne.22104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badeanlou L, Furlan-Freguia C, Yang G, Ruf W, Samad F. Tissue factor-protease-activated receptor 2 signaling promotes diet-induced obesity and adipose inflammation. Nat Med. 2011;17:1490–1497. doi: 10.1038/nm.2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry GD, Le GT, Fairlie DP. Agonists and antagonists of protease activated receptors (PARs) Curr Med Chem. 2006;13:243–265. doi: 10.2174/092986706775476070. [DOI] [PubMed] [Google Scholar]
- Barry GD, Suen JY, Le GT, Cotterell A, Reid RC, Fairlie DP. Novel agonists and antagonists for human protease activated receptor 2. J Med Chem. 2010;53:7428–7440. doi: 10.1021/jm100984y. [DOI] [PubMed] [Google Scholar]
- Blakeney JS, Reid RC, Le GT, Fairlie DP. Nonpeptidic ligands for peptide-activated G protein-coupled receptors. Chem Rev. 2007;107:2960–3041. doi: 10.1021/cr050984g. [DOI] [PubMed] [Google Scholar]
- Cenac N, Andrews CN, Holzhausen M, Chapman K, Cottrell G, Andrade-Gordon P, et al. Role for protease activity in visceral pain in irritable bowel syndrome. J Clin Invest. 2007;117:636–647. doi: 10.1172/JCI29255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Yang C, Wang ZJ. Proteinase-activated receptor 2 sensitizes transient receptor potential vanilloid 1, transient receptor potential vanilloid 4, and transient receptor potential ankyrin 1 in paclitaxel-induced neuropathic pain. Neuroscience. 2011;193:440–451. doi: 10.1016/j.neuroscience.2011.06.085. [DOI] [PubMed] [Google Scholar]
- Cocks TM, Fong B, Chow JM, Anderson GP, Frauman AG, Goldie RG, et al. A protective role for protease-activated recetpors in the airways. Nature. 1999;398:156–160. doi: 10.1038/18223. [DOI] [PubMed] [Google Scholar]
- Cudic M, Fields GB. Extracellular proteases as targets for drug development. Curr Protein Pept Sci. 2009;10:297–307. doi: 10.2174/138920309788922207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Cera E, editor. Proteases in Health and Disease in Progress in Nucleic Acid Research, Progress in Molecular Biology and Translational Science. Vol. 99. London: Academic Press; 2011. Vol. [Google Scholar]
- Heutinck KM, ten Berge IJ, Hack CE, Hamann J, Rowshani AT. Serine proteases of the human immune system in health and disease. Mol Immunol. 2010;47:1943–1955. doi: 10.1016/j.molimm.2010.04.020. [DOI] [PubMed] [Google Scholar]
- Hirota CL, Moreau F, Iablokov V, Dicay M, Renaux B, Hollenberg MD, et al. Epidermal growth factor receptor transactivation is required for proteinase-activated receptor-2-induced COX-2 expression in intestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2012;303:G111–G119. doi: 10.1152/ajpgi.00358.2011. [DOI] [PubMed] [Google Scholar]
- Hoffman J, Flynn AN, Tillu DV, Zhang Z, Patek R, Price TJ, et al. Lanthanide labeling of a potent protease activated receptor-2 agonist for time-resolved fluorescence analysis. Bioconjug Chem. 2012;23:2098–2104. doi: 10.1021/bc300300q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenberg MD, Mihara K, Polley D, Suen JY, Han A, Fairlie DP, et al. Biased signaling and proteinase-activated receptors (PARs): targeting inflammatory disease. Review. Br J Pharmacol. 2014;171:1180–1194. doi: 10.1111/bph.12544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang ZJ, Li HC, Cowan AA, Liu S, Zhang YK, Song XJ. Chronic compression or acute dissociation of dorsal root ganglion induces cAMP-dependent neuronal hyperexcitability through activation of PAR2. Pain. 2012;153:1426–1437. doi: 10.1016/j.pain.2012.03.025. [DOI] [PubMed] [Google Scholar]
- Kawabata A, Matsunami M, Tsutsumi M, Ishiki T, Fukushima O, Sekiguchi F, et al. Suppression of pancreatitis-related allodynia/hyperalgesia by proteinase-activated receptor-2 in mice. Br J Pharmacol. 2006;148:54–60. doi: 10.1038/sj.bjp.0706708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T. Functional selectivity and biased receptor signaling. J Pharmacol Exp Ther. 2011;336:296–302. doi: 10.1124/jpet.110.173948. [DOI] [PubMed] [Google Scholar]
- Kenakin TP. Biased signalling and allosteric machines: new vistas and challenges for drug discovery. Br J Pharmacol. 2012;165:1659–1669. doi: 10.1111/j.1476-5381.2011.01749.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: Reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klabunde T, Hessler G. Drug design strategies for targeting G-protein-coupled receptors. Chembiochem. 2002;3:928–944. doi: 10.1002/1439-7633(20021004)3:10<928::AID-CBIC928>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Kobilka B, Schertler GF. New G-protein-coupled receptor crystal structures: insights and limitations. Trends Pharmacol Sci. 2008;29:79–83. doi: 10.1016/j.tips.2007.11.009. [DOI] [PubMed] [Google Scholar]
- Kobilka BK. Structural insights into adrenergic receptor function and pharmacology. Trends Pharmacol Sci. 2011;32:213–218. doi: 10.1016/j.tips.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobilka BK, Deupi X. Conformational complexity of G-protein-coupled receptors. Trends Pharmacol Sci. 2007;28:397–406. doi: 10.1016/j.tips.2007.06.003. [DOI] [PubMed] [Google Scholar]
- Leung D, Abbenante G, Fairlie DP. Protease inhibitors: current status and future prospects. J Med Chem. 2000;43:305–341. doi: 10.1021/jm990412m. [DOI] [PubMed] [Google Scholar]
- Lim J, Iyer A, Liu L, Suen JY, Lohman RJ, Seow V, et al. Diet-induced obesity, adipose inflammation, and metabolic dysfunction correlating with PAR2 expression are attenuated by PAR2 antagonism. FASEB J. 2013;27:4757–4767. doi: 10.1096/fj.13-232702. [DOI] [PubMed] [Google Scholar]
- Lohman RJ, Cotterell AJ, Barry GD, Liu L, Suen JY, Vesey DA, et al. An antagonist of human protease activated receptor-2 attenuates PAR2 signaling, macrophage activation, mast cell degranulation, and collagen-induced arthritis in rats. FASEB J. 2012a;26:2877–2887. doi: 10.1096/fj.11-201004. [DOI] [PubMed] [Google Scholar]
- Lohman RJ, Cotterell AJ, Suen J, Liu L, Do AT, Vesey DA, et al. Antagonism of protease-activated receptor 2 protects against experimental colitis. J Pharmacol Exp Ther. 2012b;340:256–265. doi: 10.1124/jpet.111.187062. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore CS, Crocker SJ. An alternate perspective on the roles of TIMPs and MMPs in pathology. Am J Pathol. 2012;180:12–16. doi: 10.1016/j.ajpath.2011.09.008. [DOI] [PubMed] [Google Scholar]
- Moussa L, Apostolopoulos J, Davenport P, Tchonque J, Tipping PG. Protease-activated receptor-2 augments experimental crescentic glomerulonephritis. Am J Pathol. 2007;171:800–808. doi: 10.2353/ajpath.2007.061155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols HL, Saffeddine M, Theriot BS, Hegde A, Polley D, El-Mays T, et al. beta-Arrestin-2 mediates the proinflammatory effects of proteinase-activated receptor-2 in the airway. Proc Natl Acad Sci U S A. 2012;109:16660–16665. doi: 10.1073/pnas.1208881109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura A, Kitano K, Takasaki J, Taniguchi M, Mizuno N, Tago K, et al. Structural basis for the specific inhibition of heterotrimeric Gq protein by a small molecule. Proc Natl Acad Sci U S A. 2010;107:13666–13671. doi: 10.1073/pnas.1003553107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pejler G, Ronnberg E, Waern I, Wernersson S. Mast cell proteases: multifaceted regulators of inflammatory disease. Blood. 2010;115:4981–4990. doi: 10.1182/blood-2010-01-257287. [DOI] [PubMed] [Google Scholar]
- Qi W, Johnson DW, Vesey DA, Pollock CA, Chen X. Isolation, propagation and characterization of primary tubule cell culture from human kidney. Nephrology (Carlton) 2007;12:155–159. doi: 10.1111/j.1440-1797.2007.00779.x. [DOI] [PubMed] [Google Scholar]
- Quirk K, Roberts DJ, Strange PG. Mechanisms of G protein activation via the D2 dopamine receptor: evidence for persistent receptor/G protein interaction after agonist stimulation. Br J Pharmacol. 2007;151:144–152. doi: 10.1038/sj.bjp.0707197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopal S, Rajagopal K, Lefkowitz RJ. Teaching old receptors new tricks: biasing seven-transmembrane receptors. Nat Rev Drug Discov. 2010;9:373–386. doi: 10.1038/nrd3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran R, Noorbakhsh F, Defea K, Hollenberg MD. Targeting proteinase-activated receptors: therapeutic potential and challenges. Nat Rev Drug Discov. 2012;11:69–86. doi: 10.1038/nrd3615. [DOI] [PubMed] [Google Scholar]
- Reibman J, Talbot AT, Hsu Y, Ou G, Jover J, Nilsen D, et al. Regulation of expression of granulocyte-macrophage colony-stimulating factor in human bronchial epithelial cells: roles of protein kinase C and mitogen-activated protein kinases. J Immunol. 2000;165:1618–1625. doi: 10.4049/jimmunol.165.3.1618. [DOI] [PubMed] [Google Scholar]
- Sharony R, Yu PJ, Park J, Galloway AC, Mignatti P, Pintucci G. Protein targets of inflammatory serine proteases and cardiovascular disease. J Inflamm. 2010;7:45. doi: 10.1186/1476-9255-7-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla AK, Violin JD, Whalen EJ, Gesty-Palmer D, Shenoy SK, Lefkowitz R. Distinct conformational changes in beta-arrestin report biased agonism at seven-transmembrane receptors. Proc Natl Acad Sci U S A. 2008;105:9988–9993. doi: 10.1073/pnas.0804246105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suen JY, Gardiner B, Grimmond S, Fairlie DP. Profiling gene expression induced by protease-activated receptor 2 (PAR2) activation in human kidney cells. PLoS ONE. 2010;5:e13809. doi: 10.1371/journal.pone.0013809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suen JY, Barry GD, Lohman RJ, Halili MA, Cotterell AJ, Le GT, et al. Modulating human proteinase activated receptor 2 with a novel antagonist (GB88) and agonist (GB110) Br J Pharmacol. 2012;165:1413–1423. doi: 10.1111/j.1476-5381.2011.01610.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turk B. Targeting proteases: successes, failures and future prospects. Nat Rev Drug Discov. 2006;5:785–799. doi: 10.1038/nrd2092. [DOI] [PubMed] [Google Scholar]
- Tyndall JDA, Pfeiffer B, Abbenante G, Fairlie DP. Over one hundred peptide-activated G protein-coupled receptors recognize ligands with turn structure. Chem Rev. 2005;105:793–826. doi: 10.1021/cr040689g. [DOI] [PubMed] [Google Scholar]
- Vergnolle N, Chignard M, editors. Proteases and Their Receptors in Inflammation, Progress in Inflammation Research. Basel AG: Springer; 2011. [Google Scholar]
- Vesey DA, Cheung CW, Kruger WA, Poronnik P, Gobe G, Johnson DW. Thrombin stimulates proinflammatory and proliferative responses in primary cultures of human proximal tubule cells. Kidney Int. 2005;67:1315–1329. doi: 10.1111/j.1523-1755.2005.00209.x. [DOI] [PubMed] [Google Scholar]
- Vesey DA, Kruger WA, Poronnik P, Gobe GC, Johnson DW. Proinflammatory and proliferative responses of human proximal tubule cells to PAR-2 activation. Am J Physiol Renal Physiol. 2007;293:F1441–F1449. doi: 10.1152/ajprenal.00088.2007. [DOI] [PubMed] [Google Scholar]
- Vesey DA, Qi W, Chen X, Pollock CA, Johnson DW. Isolation and primary culture of human proximal tubule cells. Methods Mol Biol. 2009;466:19–24. doi: 10.1007/978-1-59745-352-3_2. [DOI] [PubMed] [Google Scholar]
- Yau MK, Liu L, Fairlie DP. Toward drugs for protease-activated receptor 2 (PAR2) J Med Chem. 2013;56:7477–7497. doi: 10.1021/jm400638v. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 GB88 blocked 2f-LIGRLO-NH2-induced intracellular calcium release in CHO-hPAR2. Cells were incubated with various concentrations of GB88 for 15 min, then treated with 2f-LIGRLO-NH2 (1 μM) and monitored for intracellular calcium release.
Figure S2 GB88 induced intracellular calcium release at high concentration in HT-29 cells.
Figure S3 Intracellular calcium release by PAR2 ligands in HT-29 cells. Calcimycin was used as control. Cells were treated with 2f-LIGRLO-NH2 (1 μM) and GB88 (10 μM) and fluorescence measured for 30 min after compound addition. 2f-LIGRLO-NH2 showed similar maxima as calcimycin and GB88 showed no significant difference from buffer alone.
Figure S4 2f-LIGLRO-NH2 increased intracellular calcium release independent of Gs in HT-29 cells. HT-29 cells were treated with cholera toxin (200 ng·mL−1, 24 h) and then with 2f-LIGRLO-NH2 or forskolin for calcium release. Forskolin failed to induce intracellular calcium release and CTX had no effect on PAR2-mediated Ca2+ response.
Figure S5 2f-LIGRLO-NH2 and GB88 failed to increase phosphorylation of PKC subtypes in HT-29 cells. Cells were treated with 2f-LIGRLO-NH2 (1 μM) or GB88 (10 μM) for various durations and examined by Western blot using phosphor-specific antibodies.
Figure S6 GB88 blocked IL-8 secretion in HT-29 cells. Cells were treated with 2f-LIGRLO-NH2 (1 μM, 24 h) with various concentrations of GB88. Supernatants or cell lysates were collected and analysed by elisa. GB88 was able to block 2f-LIGRLO-NH2-induced IL-8 secretion and no significant changes in level of IL-8 in cells.
Figure S7 Effects of inhibitors on PAR2-induced paw oedema in rat. PLCβ inhibitor (100 μM, 30 min) inhibited Wistar rat paw oedema induced by intraplantar injection of PAR2 agonists, 2f-LIGRLO-NH2 (A, 350 μg) or trypsin (B, 20 μg). Other inhibitors [MEK1/2 (U0126, 10 μM) or Gi/o (PTX, 5 μg)] failed to cause any significant changes. n = 3, ***P < 0.001.