

Abstract
Background and Purpose
Peptide welding technology (PWT) is a novel chemical strategy that allows the synthesis of multibranched peptides with high yield, purity and reproducibility. Using this technique, we have synthesized and pharmacologically characterized the tetrabranched derivatives of the tachykinins, substance P (SP), neurokinin A (NKA) and B (NKB).
Experimental Approach
The following in vitro assays were used: calcium mobilization in cells expressing human recombinant NK receptors, BRET studies of G-protein – NK1 receptor interaction, guinea pig ileum and rat urinary bladder bioassays. Nociceptive behavioural response experiments were performed in mice following intrathecal injection of PWT2-SP.
Key Results
In calcium mobilization studies, PWT tachykinin derivatives behaved as full agonists at NK receptors with a selectivity profile similar to that of the natural peptides. NK receptor antagonists display similar potency values when tested against PWT2 derivatives and natural peptides. In BRET and bioassay experiments PWT2-SP mimicked the effects of SP with similar potency, maximal effects and sensitivity to aprepitant. After intrathecal administration in mice, PWT2-SP mimicked the nociceptive effects of SP, but with higher potency and a longer-lasting action. Aprepitant counteracted the effects of PWT2-SP in vivo.
Conclusions and Implications
The present study has shown that the PWT technology can be successfully applied to the peptide sequence of tachykinins to generate tetrabranched derivatives characterized with a pharmacological profile similar to the native peptides. In vivo, PWT2-SP displayed higher potency and a marked prolongation of action, compared with SP.
Keywords: tachykinins, substance P, PWT2-SP, NK1 receptor, calcium mobilization, bioluminescence resonance energy transfer, guinea pig ileum, rat urinary bladder, nociceptive behaviour induced by spinal substance P, mice
Introduction
Most currently available pharmaceutical agent are orally available, low MW molecules. This formulation has undoubted pharmacokinetic advantages, but quite often suffer from limited target selectivity and consequent side effects. In contrast, peptides display extraordinarily high selectivity of action, but at the expense of a poor pharmacokinetic profile; including low bioavailability, poor barrier penetrance and high susceptibility to enzymatic degradation. One strategy to improve the pharmacokinetic properties of peptides, particularly their poor metabolic stability, is the use of multibranched peptides. However, the synthesis of these molecules is rather difficult, and in general, characterized by low yield and purity of the final product.
We have recently developed an innovative chemical approach, referred to as peptide welding technology (PWT), which allows the synthesis of multibranched peptides with excellent high yield, purity and reproducibility. With this approach, three different tetrabranched derivatives of the neuropeptide, nociceptin/orphanin FQ, have been synthesized and pharmacologically characterized in vitro and in vivo. In vitro, these compounds retained the biological activity of the natural peptide both at human recombinant and native receptors. In mice, these peptides showed higher potency and a marked prolongation of action (Rizzi et al., 2014). Based on these results, in the present study, we sought to investigate the pharmacological profile of PWT derivatives of tachykinins. Substance P (SP), neurokinin A (NKA) and neurokinin B (NKB) share a common C-terminal sequence Phe-X- Gly-Leu-Met that accounts for their biological activity. Tachykinins bind to and activate three different GPCRs: NK1, NK2 and NK3 receptors (Regoli et al., 1994; receptor nomenclature follows Alexander et al., 2013, see also Neubig et al., 2003). Tachykinins are neurotransmitters widely distributed both in the central and in the peripheral nervous systems where they control several biological functions, such as pain and migraine, nausea and vomiting, mood and anxiety, drug abuse and inflammatory conditions of the gastrointestinal tract (Douglas et al., 2014).
In the present study, PWT has been used to generate tetrabranched derivatives of the tachykinins: PWT2-SP, PWT2-NKA and PWT2-NKB. The chemical structure of PWT2-SP is shown in Figure 1. These compounds were evaluated pharmacologically for their ability to stimulate calcium mobilization in CHO cells stably expressing the human recombinant NK1, NK2 and NK3 receptors. Moreover PWT2-SP has been further characterized in vitro for its ability to promote the association of the NK1 receptor with G-protein in BRET experiments and to evoke contractions of the guinea pig ileum and rat urinary bladder in bioassay experiments. Finally, PWT2-SP has been assayed in vivo after intrathecal (i.t.) administration in the mouse for its ability to evoke a set of nociceptive behaviours such as scratching, biting and licking (SBL) responses.
Figure 1.
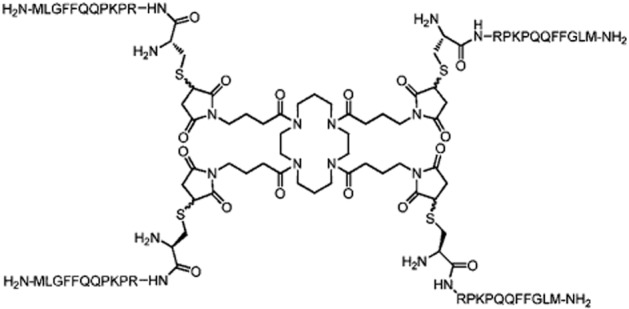
Chemical formula of PWT2-SP.
Methods
Synthesis of PWT tachykinin derivatives
These were prepared using a convergent synthetic approach. As an example, the synthesis of PWT2-SP is described. [Cys0]SP was synthesized using a solid phase methodology with an automatic solid phase peptide synthesizer Syro II (Biotage, Uppsala, Sweden) and Fmoc/tBu chemistry (Benoiton, 2005). The resin 4-(2′,4′-dimethoxyphenyl-Fmoc-aminomethyl)-phenoxyacetamido-norleucyl-MBHA (Rink amide MBHA resin) was used as a solid support. The resin was treated with 40% piperidine/dimethylformamide (DMF) and linked with Fmoc-Met-OH by using [O-(7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate] (HATU) as the coupling reagent. The following Fmoc amino acids were sequentially coupled to the growing peptide chain: Fmoc-Leu-OH, Fmoc-Gly-OH, Fmoc-Phe-OH, Fmoc-Phe-OH, Fmoc-Gln(Trt)-OH, Fmoc-Gln(Trt)-OH, Fmoc-Pro-OH, Fmoc-Lys(Boc)-OH, Fmoc-Pro-OH, Fmoc-Arg(Pmc)-OH, Fmoc-Cys(Trt)-OH. All the Fmoc amino acids (4 equiv) were coupled to the growing peptide chain using HATU (4 equiv) in DMF in the presence of an equimolar concentration of 4-methylmorpholine (NMM). The coupling reaction time was 1 h. To improve the analytical profile of the crude peptide, capping with acetic anhydride (0.5 M in DMF) in the presence of NMM (0.25 M in DMF) (3:1 v/v; 2 mL per 0.2 g of resin) was performed at any step. 40% piperidine in DMF was used to remove the Fmoc. The protected peptide-resin was treated with reagent B (Solé and Barany, 1992) [trifluoroacetic acid (TFA)/H2O/phenol/triisopropylsilane 88: 5:5:2; v/v; 10 mL 0.2·g−1 of resin] for 1.5 h at room temperature. After filtration of the resin, the solvent was concentrated in vacuo and the residue triturated with ether. Crude [Cys0]SP was purified by preparative reversed-phase HPLC using a Waters Delta Prep 3000 system (Milford, MA, USA) with a Jupiter C18 column (250 × 30 mm, 300 A, 15 μm spherical particle size). The column was perfused at a flow rate of 20 mL·min−1 with a mobile phase containing solvent A (5%, v/v, acetonitrile in 0.1% TFA), and a linear gradient from 0 to 70% of solvent B (60%, v/v, acetonitrile in 0.1% TFA) over 25 min for the elution of peptides. Purified [Cys0]SP was reacted in solution with PWT2 core in a classical thio-Michael reaction using experimental conditions previously optimized for the synthesis of nociceptin/orphanin FQ tetrabranched derivatives. The reaction proceeded rapidly at room temperature in acetonitrile/water and with the presence of a weak base (NaHCO3), it was complete in approximately 4 min, and displayed an impressive yield (virtually 100%). Analytical HPLC analyses were performed on a Beckman 116 liquid chromatograph equipped with a Beckman 166 diode array detector. Analytical purity of [Cys0]SP and PWT2-SP were determined using a Luna C18 column (4.6 × 100 mm, 3 μm particle size) with the solvent system mentioned earlier (solvents A and B) programmed at a flow rate of 0.5 mL·min−1 using a linear gradient from 0 to 80% B over 25 min. Final product showed ≥95% purity when monitored at 220 nm. Similar procedures were applied for the synthesis of PWT2-NKA and PWT2-NKB.
Calcium mobilization assay
Cells stably expressing the human NK1 receptor were a generous gift from the laboratories of Prof. T. Costa (ISS, Rome, Italy), while cells stably expressing the human NK2 and NK3 receptors were a gift from Prof. T.W. Schwartz (University of Copenhagen, Denmark). CHONK1, CHONK2 and CHONK3 cells were maintained in DEMEM/F-12 medium supplemented with 10% FBS, 100 U·mL−1 penicillin and 100 μg·mL−1 streptomycin and 200 μg·mL−1 G418, and cultured at 37°C in 5% CO2 humidified air. Cells were seeded at a density of 50 000 cells per well into 96-well black, clear-bottom plates. The following day, the cells were incubated with medium supplemented with 2.5 mM probenecid, 3 μM of the calcium sensitive fluorescent dye Fluo-4 AM and 0.01% pluronic acid, for 30 min at 37°C. After that time the loading solution was aspirated and 100 μL·well−1 of assay buffer (HBSS) supplemented with 20 mM HEPES, 2.5 mM probenecid and 500 μM Brilliant Black (Sigma Aldrich, St. Louis, MO, USA) was added. Concentrated solutions (1 mM) of SP and PWT2-SP were made in bi-distilled water and stored at −20°C. NKA, PWT2-NKA, NKB and PWT2-NKB were solubilized in 0.1 M Na2CO3 at a final concentration of 1 mM. Aprepitant, GR159897 and SB222200 (10 mM) were dissolved in DMSO. Stock solutions were kept at −20°C until use. Serial dilutions were made in HBSS/HEPES (20 mM) buffer (containing 0.02% BSA fraction V). After placing both plates (cell culture and master plate) into the fluorometric imaging plate reader FlexStation II (Molecular Devices, Sunnyvale, CA, USA), fluorescence changes were measured. Online additions were carried out in a volume of 50 μL per well. To facilitate drug diffusion into the wells, the present studies were performed at 37°C, and in antagonism experiments, three cycles of mixing (25 μL from each well moved up and down three times) were performed immediately after antagonist injection to the wells. Antagonists were injected into the wells 24 min before adding agonists. Maximum change in fluorescence, expressed as percentage over the baseline fluorescence, was used to determine agonist response.
BRET assay
Mouse fibroblasts MB19tsA cells, 2B2 clone from the laboratory of Prof. O.H. Onaran (Ankara University, Ankara, Turkey), were cultured in a 50% mixture of DMEM and F12, containing 10% FBS in a humidified atmosphere of 5% CO2 at 37°C. Cell lines with stable co-expression of Renilla luciferase tagged human NK1 receptor and the Renilla green fluorescent protein fused Gβ1 were obtained by infecting cells sequentially with retroviruses encoding each single fusion protein followed by selection with G418 (500 μg·mL−1) in combination with hygromicin B (100 μg·mL−1). The BRET assay of G-protein coupling was performed using enriched membranes from transfected cells, prepared by differential centrifugation (Vachon et al., 1987) and stored in aliquots at −80°C before use. Membranes (5 μg·100 μL−1) in PBS, MgCl2 5 mM, were first preincubated with 500 nM coelenterazine for 10 min in white 96-well Optiplates (Perkin-Elmer, Waltham, MA, USA). Next, increasing concentrations of agonists were added, and the incubation proceeded for an additional 5 min. The plate was loaded into a a luminometer (VICTOR light, Perkin-Elmer) and the BRET signals were measured and analysed as described in Molinari et al. (2010). Results are expressed as BRET ratio (cps at 510 nm·cps−1 at 475 nm).
Isolated tissue bioassays
All animal care and experimental procedures complied with the European Communities Council directives (86/609/EEC) and national regulations (D.L. 116/92) and were approved by the Ethical Committee of the University of Ferrara. All experimental procedures adopted for in vivo and ex vivo studies were as humane as possible, complied with the, and have been reported according to ARRIVE guidelines (Kilkenny et al., 2010; McGrath et al., 2010). The total number of animals used in the experiments described in this study was 8 guinea pigs, 8 rats and 60 mice.
Male albino guinea pigs (300–350 g, Pamapaloni, Italy) were housed in 300 × 570 × 155 mm cages (Tecniplast, MN, Italy), under standard conditions (22°C, 55% humidity, 12 h light–dark cycle, lights on 07:00 h) with food (4RF, Mucedola, Italy) and water ad libitum for at least 7 days before the experiment. After killing the animal via isoflurane overdose, ileum segments were taken and suspended in 5 mL organ baths containing Krebs solution (composition in mM: NaCl 118.5, KCl 4.7, MgSO4 1.2, KH2PO4 1.2, NaHCO3 25, CaCl2 2.5, glucose 10, hexamethonium bromide 2.2 μM and benadril 0.3 μM) oxygenated with 95% O2 and 5% CO2. The temperature was set at 37°C. A resting tension of 1 g was applied to the tissues. Tissue contractions were measured isometrically with a force transducer (FT03, Grass Instruments, Warwick, RI, USA) and recorded on a multichannel chart recorder (Linseis Model L2005, Linseis Messgeraete GmbH, Selb, Germany). After an equilibration period of about 60 min, to check the responsiveness of the preparation, the contractile effect of carbachol 100 μM was recorded. After approximately 20 min, atropine 1 μM was added to the medium to prevent the effects of ACh release induced by activating presynaptic NK3 receptors (Laufer et al., 1985) and 20 min later, a cumulative concentration–response curve to SP or PWT2-SP were constructed. In antagonism experiments the concentration–response curve to SP or PWT2-SP were tested in the absence or presence of aprepitant (1 nM) with 15 min pre-incubation time.
Male Sprague-Dawley rats (Charles River, Calco, Italy) weighing 280–330 g were housed in 425 × 266 × 155 mm cages (Tecniplast, MN, Italy), under standard conditions (22°C, 55% humidity, 12 h light–dark cycle, lights on 07:00 h) with food (4RF) and water ad libitum for at least 7 days before the experiment. These experiments were performed following the protocol described by Meini and Maggi (2010). Briefly, after killing the animal via isoflurane overdose, the bladder was taken and dissected longitudinally in two parts, and then suspended in 5 mL organ baths containing Krebs solution oxygenated with 95% O2 and 5% CO2 at 37°C. A resting tension of 0.5 g was applied to the tissues and bath solution was replaced every 20 min. Tissue contractions were measured isotonically by means of Basile strain gauge transducers and recorded with a PC-based acquisition system (Power Lab, 4/25, ADInstruments, NSW, Australia). After an equilibration period of about 60 min, 100 μM carbachol was added to the bath solution to induce the maximal contractile response for use as a reference value. Subsequently, cumulative concentration–response curves (0.5 log unit steps) to SP and PWT2-SP were constructed. All experiments were performed in the presence of the NK2 receptor antagonist SR 48968 (1 μM). All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010).
Nociceptive behaviours induced by i.t. PWT2-SP in mice
Male CD-1 mice (weight 20–25 g, Harlan, Udine, Italy) were housed in 425 × 266 × 155 mm cages (Tecniplast), under standard conditions (22°C, 55% humidity, 12 h light–dark cycle, lights on 07:00 h) with food (4RF) and water ad libitum for at least 3 days before experiments began. Each animal was used only once. SP (100 pmol) and PWT2-SP (2.5–250 pmol) were given spinally. I.t. injections (5 μL per mouse) were performed according to the procedure described by Hylden and Wilcox (1980) and routinely used in our laboratory (Rizzi et al., 2006). Approximately, 45 min before i.t. injection, mice were adapted to an individual plastic cage, which served as the observation chamber. The animals were randomly assigned to treatments (saline, SP and PWT2-SP) and individually observed for 30 min. The total time (s) spent displaying the following behaviours was measured: hind limb scratching directed towards the flank; biting or licking of the fore and hind paws; biting or licking of the tail (Takahasi et al., 1987). Aprepitant (1 mg·kg−1, i.p.) was solubilized in 5% DMSO and administered 2 h before PWT2-SP (25 pmol, i.t.). All experiments started at 09:00 h.
Data analysis
Data from in vitro experiments are expressed as means ± SEM of n experiments. Non-linear regression analysis using GraphPad Prism software (v.4.0; GraphPad Software, Inc, La Jolla, CA, USA) allowed logistic iterative fitting of the resultant responses and the calculation of agonist potencies and maximal effects. Agonist potencies were expressed as pEC50. In inhibition response experiments (i.e. increasing concentrations of antagonist vs. a fixed concentration of agonist) the antagonist potency was expressed as pIC50. A fixed concentration of aprepitant was also challenged against the agonist concentration–response curve. In calcium mobilization and guinea pig ileum experiments the antagonist displayed insurmountable behaviour, thus according to Gaddum et al. (1955), the following equation was used to derive antagonist potency:
In practice, equiactive concentrations of the agonist in the absence and presence of an insurmountable antagonist [ (B) ] are compared in a double reciprocal plot describing a straight line. In contrast, in rat urinary bladder experiments aprepitant displayed a competitive behaviour and its antagonist potency was derived from following equation:
where CR is the ratio of potency between the agonist EC50 in the presence and absence of antagonist and (B) is the antagonist molar concentration.
In vivo data are expressed as mean ± SEM of n animals. Data were analysed using Student's t-test for unpaired data or one-way anova followed by Dunnett's post hoc test. Differences were considered statistically significant when P < 0.05.
Materials
GR159897, SB222200 atropine and carbachol were purchased from Sigma Chemical Co. (St Louis, MO, USA). NKA and NKB were purchased from Tocris Biosciences (Bristol, UK). SP was synthesized and purified in our laboratories. Aprepitant was a generous gift from Dr C Pietra (Helsinn, Lugano, CH).
Results
Calcium mobilization studies
In a first series of experiments the effects of PWT2-SP, PWT2-NKA and PWT2-NKB were assessed in CHO cells expressing the human NK1, NK2 and NK3 receptors and compared with those elicited by the natural tachykinins SP, NKA and NKB. In addition the actions of the peptides used in the synthesis of PWT molecules, that is [Cys0]SP, [Cys0]NKA and [Cys0]NKB, were also evaluated in parallel experiments. The effects of [Cys0] derivatives of tachykinins were always identical to those of the natural peptides and, for the sake of clarity, these results have not been not displayed in the Figures. As shown in Figure 2, in CHONK1 cells, SP stimulated calcium mobilization in a concentration-dependent manner with high potency (pEC50 = 10.40) and an Emax value of 109 ± 8% over the basal levels. NKA and NKB mimicked SP action with similar maximal effects, but lower potency (by approximately 10- and 100-fold respectively). PWT2-SP and PWT2-NKA stimulated intracellular calcium mobilization producing similar maximal effects as the natural peptides, but were approximately 30-fold less potent. In contrast, the concentration–response curves to NKB and PWT2-NKB were virtually superimposable (Figure 2, panels A, B and C). In CHONK2 cells NKA increased intracellular calcium levels in a concentration-dependent manner, with a pEC50 value of 9.32 and an Emax value of 174 ± 11% over the basal. SP and NKB produced maximal effects similar to those of NKA, but were approximately 100-fold less potent. PWT2-NKA elicited similar maximal effects as the natural peptide, but was 30-fold less potent. Similar results were obtained with PWT2-SP and PWT2-NKB, but the reduction in potency for these compounds in comparison with the natural peptides was limited to 10- and threefold respectively (Figure 2, panels D, E and F). In CHONK3 cells, NKB increased intracellular calcium levels in a concentration-dependent manner with a pEC50 value of 7.96 and an Emax value of 207 ± 18%. SP and NKA evoked similar maximal effects as NKB, but were significantly less potent. In this cell line, PWT tachykinin derivatives elicited concentration–response curves superimposable to those of the natural peptides with the exception of PWT2-NKA that was sixfold less potent than NKA (Figure 2, panels G, H and I). Results obtained in this series of experiments have been summarized in Table 1.
Figure 2.

Calcium mobilization assay performed in CHO cells expressing NK1 (panels A, B and C), NK2 (panels D, E and F) and NK3 (panels G, H and I) receptors. Concentration–response curve to SP and PWT2-SP (panels A, D and G), NKA and PWT2-NKA (panels B, E and H), and NKB and PWT2-NKB (panels C, F and I). Data are mean ± SEM of four experiments performed in duplicate.
Table 1.
Potencies and maximal effects of natural tachykinins and their PWT derivatives in calcium mobilization experiments performed in CHO cells expressing human NK receptors
| NK1 | NK2 | NK3 | ||||
|---|---|---|---|---|---|---|
| pEC50 (CL95%) | Emax | pEC50 (CL95%) | Emax | pEC50 (CL95%) | Emax | |
| SP | 10.40 (9.93–10.87) | 109 ± 8% | 7.18 (6.99–7.37) | 178 ± 19% | 7.15 (7.01–7.29) | 217 ± 30% |
| PWT2-SP | 9.02 (8.85–9.19) | 95 ± 2% | 6.17 (5.62–6.72) | 168 ± 11% | 7.15 (6.90–7.45) | 190 ± 21% |
| NKA | 9.70 (9.32–10.08) | 108 ± 10% | 9.32 (9.05–9.58) | 174 ± 11% | 7.39 (6.81–7.97) | 222 ± 21% |
| PWT2-NKA | 8.44 (7.79–9.09) | 93 ± 10% | 7.70 (7.06–8.36) | 183 ± 10% | 6.63 (6.03–7.23) | 204 ± 18% |
| NKB | 8.34 (8.13–8.55) | 99 ± 10% | 7.15 (6.81–7.49) | 177 ± 9% | 7.96 (7.62–8.30) | 207 ± 18% |
| PWT2-NKB | 8.03 (7.82–8.24) | 88 ± 11% | 6.73 (6.22–7.24) | 176 ± 13% | 7.82 (7.25–8.39) | 199 ± 17% |
Data are mean ± SEM of at least four experiments performed in duplicate. CL95%, 95% confidence limits.
The stimulatory effects elicited by PWT tachykinin derivatives were challenged with selective NK receptor antagonists; aprepitant (Kramer et al., 1998) for NK1, GR159897 (Beresford et al., 1995) for NK2, and SB222200 (Sarau et al., 2000) for the NK3 receptors. Antagonists were evaluated in inhibition response experiments, where a fixed concentration of agonist, approximately corresponding to its EC80, was challenged against increasing concentrations of antagonist. In CHONK1 cells, aprepitant produced a concentration-dependent inhibition of the stimulatory actions of both SP (1 nM) and PWT2-SP (10 nM) with similar potency values (pIC50 = 8.57 and 8.53, respectively, Figure 3, panels A and B). GR159897 and SB222200 up to 1 μM did not affect PWT2-SP responses. In CHONK2 cells, GR159897 blocked the stimulatory effects of NKA (1 nM) and PWT2-NKA (100 nM) with pIC50 values of 8.15 and 8.11 respectively (Figure 3, panels C and D). Aprepitant and SB222200 up to 1 μM did not affect PWT2-NKA responses. Finally, in CHONK3 cells SB222200 blocked the stimulatory effects of NKB and PWT2-NKB (both at 100 nM) with pIC50 values of 7.01 and 7.00, respectively (Figure 3, panels E and F), while aprepitant and GR159897 were inactive. The results of this series of antagonist experiments have been summarized in Table 2.
Figure 3.
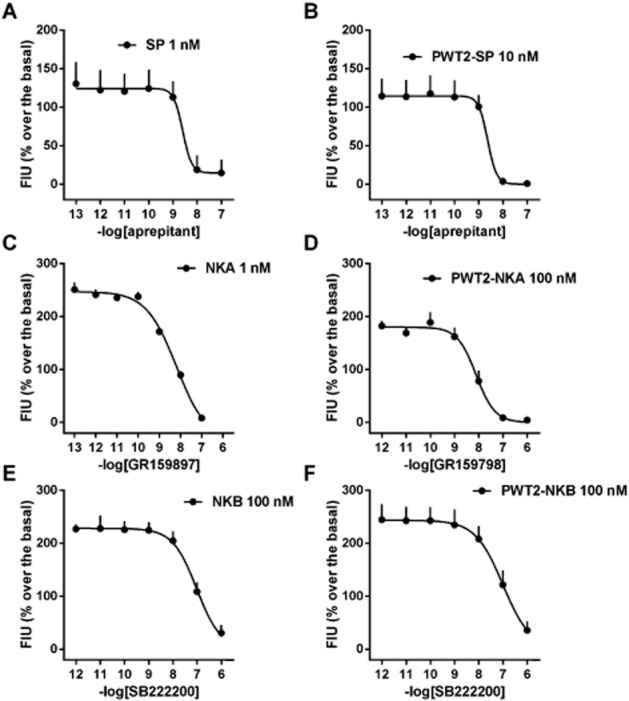
Calcium mobilization assay performed in CHO cells expressing NK1 (panels A and B), NK2 (panels C and D), and NK3 (panels E and F) receptors. Inhibition–response curve to NK receptor antagonists against the stimulatory effect of natural tachykinins (panels A, C and E) and their PWT derivatives (panels B, D and F). Data are mean ± SEM of four experiments performed in duplicate.
Table 2.
pIC50 values of NK receptor antagonists obtained in inhibition response experiments against PWT2-NK derivatives in calcium mobilization experiments performed in CHO cells expressing human NK receptors
| Receptor agonist | NK1 PWT2-SP | NK2 PWT2-NKA | NK3 PWT2-NKB |
|---|---|---|---|
| Aprepitant | 8.53 (8.16–8.90) | <6 | <6 |
| GR159897 | <6 | 8.11 (7.59–8.63) | <6 |
| SB222200 | <6 | <6 | 7.00 (6.60–7.40) |
Data are mean (CL95%) of at least four experiments performed in duplicate.
When the concentration–response curve to SP was carried out in the presence of a fixed concentration of aprepitant (1 nM), this antagonist produced a slight rightward shift of the curve associated with a large reduction in agonist maximal effects (Figure 4, panel A). A pKB value of 9.66 was derived from these experiments. Similarly, aprepitant slightly shifted to right the concentration–response curve to PWT2-SP, reducing its maximal effects and with a pKB value of 9.46 (Figure 4, panel B).
Figure 4.
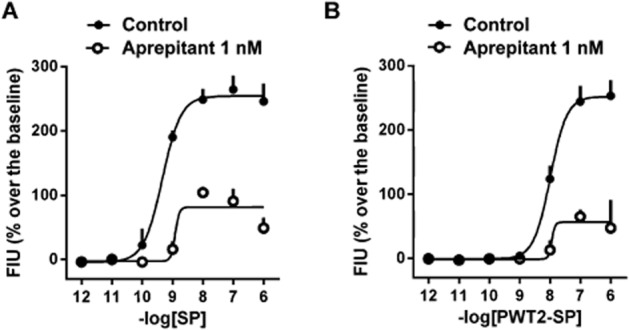
Calcium mobilization assay performed in CHO cells expressing NK1 receptors. Concentration–response curve to SP (panel A) and PWT2-SP (panel B) in absence and in presence of 1 nM aprepitant. Data are mean ± SEM of three experiments performed in duplicate.
BRET studies
In the BRET assay, SP produced a concentration-dependent stimulation of NK1 receptor–G-protein association with a maximal effect of 0.53 ± 0.01 and a pEC50 of 9.07 (CL95% 8.73–9.42). PWT2-SP mimicked the action of the natural peptide with a concentration–response curve not different from that of SP (Figure 5).
Figure 5.
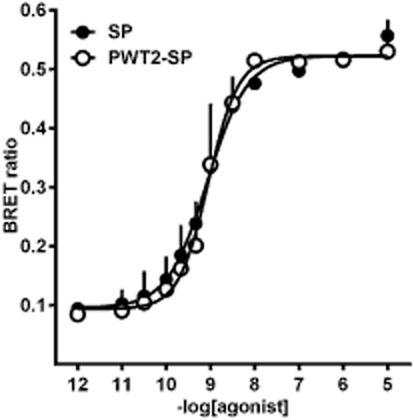
BRET assay performed in fibroblasts stably expressing Renilla luciferase tagged NK1 receptors and Renilla green fluorescent protein fused Gβ1 protein. Concentration–response curves to SP and PWT2-SP. Data are mean ± SEM of three experiments performed in duplicate.
Isolated tissue bioassays
In the guinea pig ileum, SP produced a concentration-dependent contraction with a pEC50 and Emax value of 8.50 (CL95% 8.19–8.81) and 3.25 ± 0.37 g respectively. PWT2-SP mimicked the contractile effects of SP with similar potency [8.64 (CL95% 8.48–8.78) ] and maximal effects (3.11 ± 0.16 g, Figure 6, panel A). Panel B of Figure 6 displays typical tracings of the contractile effects of SP and PWT2-SP in the guinea pig ileum. Immediately after agonist injection the tissue rapidly responded. The contractile responses induced by SP and PWT2-SP were very similar. In contrast, after washing, SP-induced contraction rapidly disappeared with only a few seconds needed to return to baseline, while the contractile effect evoked by PWT2-SP was less readily washed out and more than 1 min was required to return to baseline. In order to investigate the receptor involved in the contractile effects of SP and PWT2-SP in the guinea pig ileum their actions were challenged with the known NK1 receptor antagonist aprepitant. As shown in Figure 6, panels C and D, aprepitant (1 nM) produced a rightward shift in the concentration–response curves to SP and PWT2-SP along with a significant reduction in their maximal effects. From these experiments pKB values of 10.41 and 10.21 were derived for aprepitant versus SP and PWT2-SP respectively.
Figure 6.
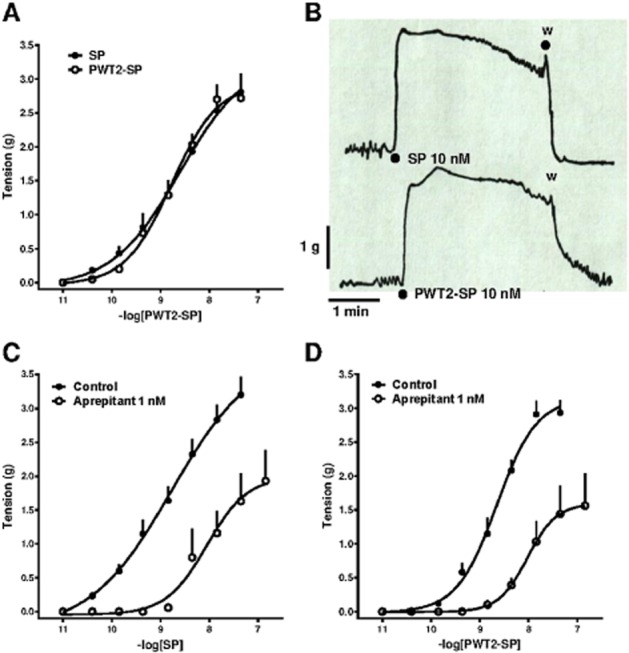
Guinea pig ileum bioassay. Concentration–response curves to SP and PWT2-SP (panel A) and representative tracings of tissue contraction in response to SP and PWT2-SP (panel B). Concentration–response curve to SP (panel C) and PWT2-SP (panel D) in absence and in presence of aprepitant 1 nM. Data are mean ± SEM of four experiments performed in duplicate.
In the rat urinary bladder, SP and PWT2-SP produced superimposable concentration–response curves with similar potencies and maximal effects (Figure 7, panel A). Panel B of Figure 7 displays typical tracings of the contractile effects of SP and PWT2-SP in the rat urinary bladder. Immediately after SP injection, the tissue rapidly contracted reaching the plateau in less than 2 min. After washing, this rapidly returned to baseline. In contrast, the contraction elicited by PWT2-SP was slower to reach a plateau, taking more than 6 min, and essentially resistant to washing; Aprepitant (1 μM) did not modify the contractile effect of SP and PWT2-SP in the rat urinary bladder (data not shown). At 10 μM, the antagonist produced a slight rightward shift in the concentration–response curves to SP and PWT2-SP without significantly modifying their maximal effects. From these experiments, pKB values of 5.42 and 5.31 were derived for aprepitant versus SP and PWT2-SP (Figure 7, panels C and D).
Figure 7.
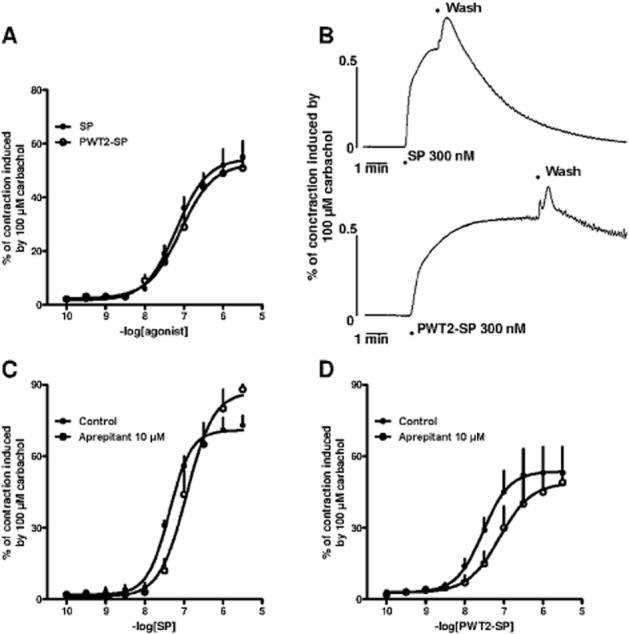
Rat urinary bladder bioassay. Concentration–response curves to SP and PWT2-SP (panel A) and representative tracings of tissue contraction in response to SP and PWT2-SP (panel B). Concentration–response curve to SP (panel C) and PWT2-SP (panel D) in absence and in presence of aprepitant 10 μM. Data are mean ± SEM of four experiments performed in duplicate.
Nociceptive behaviours induced by i.t. PWT2-SP in mice
Previous studies (Hylden and Wilcox, 1981) recently replicated in our laboratories (Rizzi et al., 2012) demonstrated that the i.t. injection of SP in mice elicits a typical nociceptive behaviour consisting of scratching (S), biting (B) and licking (L). In line with these findings, in the present experiments SP (100 pmol; i.t.) elicited a typical SBL response. The effect of SP peaked immediately after injection and rapidly disappeared after 10 min (Figure 8, panel A). Under the same experimental conditions, PWT2-SP (2.5–250 pmol) dose-dependently elicited the SBL response mimicking the effect of SP. PWT2-SP was more potent and efficacious and produced longer-lasting effects. Comparing the effects of 100 pmol SP and 25 pmol PWT2-SP (these doses contain the same number of peptide sequences) indicates the effect elicited by PWT2-SP is approximately sixfold larger than that of the natural peptide. Moreover the action of SP was no longer evident 10 min after injection while that of PWT2-SP lasted for the entire duration of the experiment, 30 min (Figure 8, panel A). In order to investigate the receptor involved in the nociceptive effect of PWT2-SP, a series of experiments were performed in mice treated with vehicle or 1 mg·kg−1 aprepitant. At this dose the antagonist did not produce, per se, any nociceptive behaviour (data not shown). However, when aprepitant was tested against 25 pmol PWT2-SP, it significantly inhibited agonist-induced nociceptive behaviour (Figure 8, panels C and D).
Figure 8.
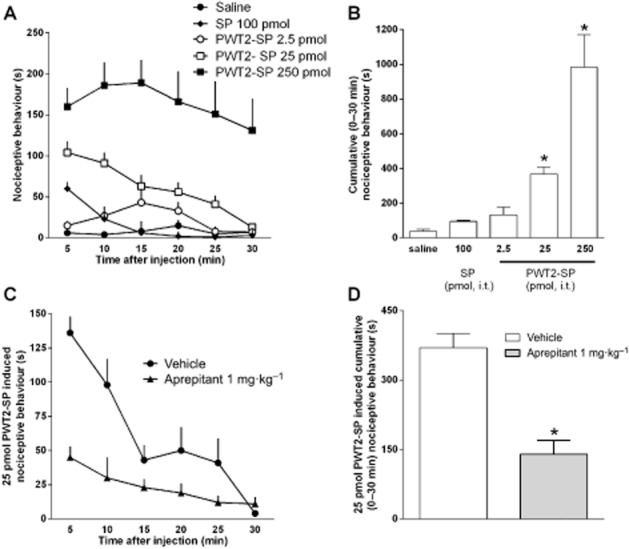
SBL test in mice. Time course of the behaviour response to i.t. SP (100 pmol) and PWT2-SP (2.5–250 pmol, panel A); the same results are shown as cumulative nociceptive behaviour displayed over the 30 min period in the panel B. One-way anova followed by the Dunnett's post hoc test revealed a significant effect of PWT2-SP [F(3,31) = 19.87]. Effects of pretreatment with aprepitant (1 mg·kg−1, for 2 h) on nociceptive behaviour induced by PWT2-SP (25 pmol). The time-course is shown in panel C while the same results are shown as cumulative nociceptive behaviour displayed over the 30 min period in the panel D. Data are mean ± SEM of at least eight mice per group. *P < 0.05, significantly different from saline or vehicle; unpaired t-test .(t = 5.33, d.f. = 14).
Discussion and conclusions
In the present study, the pharmacological profile of tachykinin tetrabranched derivatives produced using PWT technology have been investigated. In vitro, in the calcium mobilization assay, PWT2-SP, PWT2-NKA and PWT2-NKB behaved as full agonists at human recombinant NK receptors maintaining a profile of selectivity in common with the natural peptides. The potency of PWT derivatives compared with natural tachykinins was similar or reduced depending on both peptide sequence and receptor. In BRET experiments investigating NK1 receptor / G-protein interactions, both PWT2-SP and SP produced results that were superimposable in terms of maximal effects and potency. Moreover, similar findings were obtained in bioassay experiments at native NK1 receptors expressed in the guinea pig ileum and the rat urinary bladder. Finally, in vivo after i.t. injection in mice, PWT2-SP mimicked the nociceptive effect of SP, but with higher potency and effectiveness along with a prolonged duration of action. The in vitro and in vivo effects of PWT2-SP were sensitive to the NK1-selective antagonist aprepitant. Thus, the present study demonstrated that the PWT technology can be successfully applied to tachykinin peptide sequences to obtain novel ligands that display an in vitro pharmacological profile similar to natural tachykinins, but display higher potency and longer-lasting effects in vivo.
PWT technology has recently been developed and validated by generating tetrabranched derivatives of the opioid-like neuropeptide N/OFQ, using three different maleimide functionalized cores: (Lys)2-Lys-NH2 for PWT1-N/OFQ, Cyclam for PWT2-N/OFQ, and (Lys)2-ethylendiamine for PWT3-N/OFQ. In in vitro and in vivo pharmacological studies PWT2-N/OFQ generated the best results in terms of maintaining the selectivity of the natural peptide sequence along with higher in vivo potency and longer-lasting effects (Rizzi et al., 2014). Based on these results, the PWT2 core was selected for generating PWT tachykinin derivatives. In addition to the core used for the design of multivalent ligands another crucial issue is the selection of the attachment point (Shonberg et al., 2011). As noted earlier, tachykinins share a common C-terminal sequence Phe-X-Gly-Leu-Met that accounts for their biological activity (Regoli et al., 1994; Douglas et al., 2014). Thus, we decided to use the N terminal of the peptide sequence as attachment point for the Cys residue crucial for PWT reaction. Therefore [Cys0]SP, [Cys0]NKA, and [Cys0]NKB were synthesized. A large body of evidence suggests that the N terminal of SP can be utilized to generate conjugated molecules without losing the ability of the peptide to bind and activate the NK1 receptor. This strategy has been successfully used for delivery of several different toxins including saporin (Mantyh et al., 1997), diphtheria (Benoliel et al., 1999), cholera (Caudle et al., 2007) and, more recently, botulinum toxin (Mustafa et al., 2013) into cells expressing NK1 receptors. In line with these findings the present results obtained in calcium mobilization experiments demonstrated that the pharmacological profile of [Cys0]SP, [Cys0]NKA, and [Cys0]NKB are identical to those for the natural tachykinins in terms of efficacy, potency, and selectivity of action. Thus [Cys0]tachykinins were linked together with the PWT2 core using a thio-Michael reaction. As in our results obtained using the N/OFQ sequence, the reaction proceeded in a few minutes and was characterized by an extremely high yield and purity of the final products: PWT2-SP, PWT2-NKA and PWT2-NKB.
In the calcium mobilization assay performed with CHO cells stably transfected with human recombinant NK receptors, the natural tachykinins SP, NKA and NKB behaved as full agonists, with the following rank–order potency: SP > NKA > NKB, NKA > SP = NKB, NKB > NKA > SP at NK1, NK2 and NK3 receptors respectively. These findings are fully consistent with results obtained using bioassay techniques and animal tissues (Regoli et al.,1988; 1994) and cells expressing recombinant human receptors (Rizzi et al., 2012). PWT2 tachykinin derivatives mimicked the stimulatory effects of the natural peptides with similar maximal effects in the three cell lines. Thus all PWT2 tachykinins behaved as full agonists. This mirrors our previous work with N/OFQ PWT derivatives (Rizzi et al., 2014). Based on these findings, we can suggest that PWT technology used for generating these compounds has negligible impact on the ability of the peptide sequence to adopt the biologically active conformation responsible for full agonist activity.
With respect to selectivity of action, the following rank–order of potency has been measured for PWT2 tachykinins PWT2-SP > PWT2-NKA > PWT2-NKB, PWT2-NKA > PWT2-NKB > PWT2-SP, PWT2-NKB > PWT2-SP > PWT2-NKA at NK1, NK2 and NK3 receptors respectively. This rank–order (PWT2 tachykinins compared with natural tachykinins) demonstrated that PWT technology did not affect the selectivity of action of the natural peptide sequence. This finding is in line with previous results obtained with N/OFQ PWT derivatives that maintain an extremely high selectivity of action over classical opioid receptors, typical of the natural peptide N/OFQ (Rizzi et al., 2014).
Considering potency, comparison of each PWT2 derivative with the native peptide sequence suggests differences depending on both the peptide sequence and the receptor under study. In particular, PWT2-NKB always displayed similar potency to NKB, and PWT derivatives displayed similar potency to the natural peptides at NK3 receptors. In contrast, PWT2-SP and PWT2-NKA displayed lower potency compared with SP and NKA at NK1 and NK2 receptors. We cannot explain those differences, but the present results indicate that agonist potency may or may not be affected by the application of PWT technology depending on the peptide sequence and the receptor interacting with the ligand. Independently of agonist potency, the interaction of PWT derivatives with NK receptors seems to mirror that of the natural sequences as the NK receptor antagonists (aprepitant, GR159897, SB222200) displayed inhibitor potencies against PWT2-SP, PWT2-NKA and PWT2-NKB, close to those obtained in earlier studies against SP, NKA and NKB (Rizzi et al., 2012). In addition, aprepitant behaved as an insurmountable antagonist against both PWT2-SP and SP with similar potency. Collectively, calcium mobilization studies demonstrated that tachykinin PWT derivatives behave as full agonists at human recombinant NK receptors with a selectivity profile superimposable to that of the parent peptides.
We further characterized the effects of PWT2-SP in vitro for its ability to promote the interaction of the NK1 receptor with G-protein in BRET experiments and to evoke contractions of the guinea pig ileum and rat urinary bladder. In the BRET assay SP produced a concentration-dependent stimulation of NK1 receptor–G-protein association with high potency. PWT2-SP mimicked the action of SP and no differences were found between SP and PWT2-SP potency in this assay. To evaluate the action of PWT2-SP at native receptors the guinea pig ileum and rat urinary bladder were used. Guinea pig ileum studies were performed in the presence of atropine in order to block the effects of prejunctional NK3 receptors, whose activation elicits ACh release (Laufer et al., 1985). Under these experimental conditions, SP produced a concentration-dependent contraction with potency and maximal effects similar to those obtained in previous studies (Rizzi et al., 2012). The effects of SP were mimicked by PWT2-SP with a concentration–response curve identical to that of the naturally occurring peptide. Experiments with the selective NK1 receptor antagonist aprepitant demonstrated that the biological effects of PWT2-SP (and SP) are solely due to activation of NK1 receptors. Very similar results have been obtained in the rat urinary bladder. The studies were performed in the presence of 1 μM SR 48968 to exclude the interference of the NK2 receptor (Meini and Maggi, 2010). Under these experimental conditions, SP produced a concentration-dependent contraction with potency and maximal effects similar to those reported by Meini and Maggi (2010). The contractile action of SP was mimicked by PWT2-SP with similar potency and maximal effects. The exclusive involvement of the NK1 receptor in the contractile effect of both SP and PWT2-SP was demonstrated by aprepitant sensitivity. Of note, the pharmacological features of aprepitant are very different at the rat NK1 receptor (low potency, competitive behaviour) compared with the guinea pig or human receptor isoforms (very high potency, insurmountable behaviour). Identical results were previously obtained by us (Rizzi et al., 2012) and others (Leffler et al., 2009) investigating the effects of aprepitant at the rat and human recombinant NK1 receptors. The reader is referred to Rizzi et al. (2012) for a detailed discussion of the mechanisms underlying the insurmountable antagonist behaviour of aprepitant. Collectively, bioassay studies at native receptors confirmed that PWT technology did not affect the pharmacological activity, high potency and selectivity of action of the natural peptide SP.
Interestingly, the potency of PWT2-SP at NK1 receptors in BRET and bioassay experiments was identical to that of SP while. in calcium mobilization studies. PWT2-SP displayed an almost 30-fold lower potency. Previous studies demonstrated that PWT derivatives of N/OFQ displayed a reduced potency in calcium mobilization studies while being more potent than the parent peptide in [35S]GTPγS binding and bioassay studies (Rizzi et al., 2014). In isolated tissues, PWT-N/OFQ derivatives displayed slow onset of action and their effects were resistant to washing, while the action of N/OFQ occurred immediately after adding the peptide to the bath and was immediately reversible after washing. Thus, the relatively long time needed to obtain full activation of the N/OFQ receptor by PWT-N/OFQ might not be compatible with the rapid and transient nature of the calcium response. Thus, according to Charlton and Vauquelin (2010), we suggest that the reversal of the rank–order of agonist potency measured in the calcium assay compared with [35S]GTPγS binding and bioassay studies may derive from kinetic artefacts because of the hemiequilibrium conditions that characterize the calcium assay. The present bioassay studies, particularly the rat urinary bladder, also demonstrated kinetic differences between PWT2-SP and SP, namely slower onset of action and lower sensitivity to wash for PWT2-SP. Thus, similar kinetic artefacts may account for the discrepant results we obtained with PWT2-SP in the calcium mobilization studies where SP > PWT2-SP and in BRET and bioassay studies, where SP = PWT2-SP.
Collectively the in vitro studies demonstrated that the PWT technology can be applied to tachykinins for the generation of potent full NK receptor agonists that maintain a selectivity of action typical of the parent peptides. Based on these encouraging data, we further characterized the effects of PWT2-SP in vivo.
Previous experiments performed in our laboratories (Rizzi et al., 2012) confirmed that i.t. injection of SP in mice elicited the characteristic SBL response. The action of SP was dose-dependent (10–1000 pmol) and short lasting. This was confirmed in the present experiments with 100 pmol SP. PWT2-SP in the dose range 2.5–25 pmol mimicked the effects of the natural peptide. However, PWT2-SP was more potent, produced more intense and longer-lasting nociceptive effects compared with the natural peptide. These results are similar to those obtained with N/OFQ and PWT-N/OFQ. Indeed, after supraspinal injection, in mice PWT-N/OFQ inhibited locomotor activity similar to N/OFQ; however, PWT-N/OFQ was more potent, produced more pronounced and longer-lasting effects (Rizzi et al., 2014). Thus, high in vivo potency and long-lasting actions seem to be a common feature of PWT derivatives. These features could derive from the lower susceptibility to cleavage by peptidases, reported for multibranched peptides compared with free peptide sequences. This has been demonstrated for different peptide sequences including enkephalins, neurotensin and N/OFQ (Bracci et al., 2003). A structure-based hypothesis of branched peptide resistance to proteolysis has been proposed by the same group (Falciani et al., 2007). Peptide metabolism is likely to be more relevant in vivo than in vitro. Thus, it is reasonable to propose that the increase in agonist potency and duration of action displayed by multibranched peptides in vivo is largely due to their lower susceptibility to peptidase action. Another in vitro feature common to PWT derivatives of N/OFQ and SP is reduced sensitivity to washing, in organ bath experiments. This suggests longer-lasting target binding compared with native peptides. Several mechanisms have been proposed to explain the mode of action of multivalent ligands such as PWT peptides. Receptor clustering, cooperative binding, rebinding and subsite binding (Gestwicki et al., 2002) may be relevant. Longer lasting target binding might be important for the prolongation of in vivo drug action (for a detailed analysis of this topic see Vauquelin and Charlton, 2010). Thus different mechanisms (lower susceptibility to peptidases and longer-lasting target binding) alone or in combination may explain the high potency and long-lasting effects displayed by PWT derivatives in vivo.
Finally, the effects of PWT2-SP in the mouse SBL assay were sensitive to the NK1 receptor antagonist, aprepitant, demonstrating the exclusive involvement of spinal NK1 receptors. Identical results have been obtained previously by using this antagonist against the natural peptide SP (Rizzi et al., 2012). These results demonstrate that while the PWT modification does increase potency and duration of action, it does so without affecting peptide selectivity in vivo.
In conclusion, the present study has shown that PWT technology can be successfully applied to the tachykinin peptide sequence to generate tetrabranched derivatives with an in vitro pharmacological profile similar to the native peptides. In addition, PWT2-SP demonstrated higher in vivo potency and a marked prolongation of action compared with SP. This NK1 receptor-preferring ligand is therefore proposed as a novel research tool particularly useful to investigate those conditions in which a prolonged activation of the NK1 receptor is desirable. More generally, this study confirms and extends previous findings demonstrating that the PWT strategy may be easily used to generate innovative and interesting ligands for peptidergic GPCRs.
Acknowledgments
This work was supported by funds from UFPeptides s.r.l. We thank T.W. Schwartz (University of Copenhagen, Denmark) for the generous gift of CHO NK2 and NK3 cells, Dr. M. Bastepe (Harvard Medical School, Boston, MA, USA) and Dr O.H. Onaran (Ankara University, Ankara, Turkey) for 2B2 cells, and Dr. C Pietra (Helsinn, Lugano, CH) for Aprepitant.
Glossary
- DMF
dimethylformamide
- HATU
[O-(7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate
- NKA
neurokinin A
- NKB
neurokinin B
- NMM
4-methylmorpholine
- PWT
peptide welding technology
- SBL
scratching, biting and licking
- SP
substance P
Author contributions
CR and AR performed the in vivo experiments; CR, DM, MCC, FF, CA and CG performed the in vitro experiments; EM, RG and SS synthesized and purified the tachykinin tetrabranched derivatives; and GC wrote the first draft of the paper that has been finalized with the contributions of CR, TC and RG.
Conflicts of interest
The following authors Girolamo Calo', Severo Salvadori, and Remo Guerrini are inventors of the patent application (EP13162532.9) focused on PWT technology and are founders of the University of Ferrara spin off company UFPeptides s.r.l. the assignee of such patent application.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, M Spedding, et al. The Concise Guide to PHARMACOLOGY 2013/14: G Protein-Coupled Receptors. Br J Pharmacol. 2013;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benoiton NL. CRC Press. Chemistry of Peptide Synthesis. Boca Raton, FL: Taylor & Francis; 2005. Solid-phase synthesis; pp. 125–154. [Google Scholar]
- Benoliel R, Eliav E, Mannes AJ, Caudle RM, Leeman S, Iadarola MJ. Actions of intrathecal diphtheria toxin-substance P fusion protein on models of persistent pain. Pain. 1999;79:243–253. doi: 10.1016/s0304-3959(98)00170-5. [DOI] [PubMed] [Google Scholar]
- Beresford IJ, Sheldrick RL, Ball DI, Turpin MP, Walsh DM, Hawcock AB, et al. GR159897, a potent non-peptide antagonist at tachykinin NK2 receptors. Eur J Pharmacol. 1995;272:241–248. doi: 10.1016/0014-2999(94)00655-q. [DOI] [PubMed] [Google Scholar]
- Bracci L, Falciani C, Lelli B, Lozzi L, Runci Y, Pini A, et al. Synthetic peptides in the form of dendrimers become resistant to protease activity. J Biol Chem. 2003;278:46590–46595. doi: 10.1074/jbc.M308615200. [DOI] [PubMed] [Google Scholar]
- Caudle RM, Mannes AJ, Keller J, Perez FM, Suckow SK, Neubert JK. Sensitization of spinal cord nociceptive neurons with a conjugate of substance P and cholera toxin. BMC Neurosci. 2007;8:30. doi: 10.1186/1471-2202-8-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlton SJ, Vauquelin G. Elusive equilibrium: the challenge of interpreting receptor pharmacology using calcium assays. Br J Pharmacol. 2010;161:1250–1265. doi: 10.1111/j.1476-5381.2010.00863.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas SD, Leeman SE, Barrett J, Dombrowsky E, Heyward CY, Remeshwar P, et al. 2014. Tachykinin receptors. UPHAR database (IUPHAR-DB). Last modified on 17/03/2014. Available at: http://www.iuphar-db.org/DATABASE/FamilyMenuForward?familyId=62 (accessed on 19/05/2014)
- Falciani C, Lozzi L, Pini A, Corti F, Fabbrini M, Bernini A, et al. Molecular basis of branched peptides resistance to enzyme proteolysis. Chem Biol Drug Des. 2007;69:216–221. doi: 10.1111/j.1747-0285.2007.00487.x. [DOI] [PubMed] [Google Scholar]
- Gaddum JH, Hameed KA, Hathway DE, Stephens FF. Quantitative studies of antagonists for 5-hydroxytryptamine. Q J Exp Physiol Cogn Med Sci. 1955;40:49–74. doi: 10.1113/expphysiol.1955.sp001097. [DOI] [PubMed] [Google Scholar]
- Gestwicki JE, Cairo CW, Strong LE, Oetjen KA, Kiessling LL. Influencing receptor-ligand binding mechanisms with multivalent ligand architecture. J Am Chem Soc. 2002;124:14922–14933. doi: 10.1021/ja027184x. [DOI] [PubMed] [Google Scholar]
- Hylden JL, Wilcox GL. Intrathecal morphine in mice: a new technique. Eur J Pharmacol. 1980;17:313–316. doi: 10.1016/0014-2999(80)90515-4. [DOI] [PubMed] [Google Scholar]
- Hylden JL, Wilcox GL. Intrathecal substance P elicits a caudally-directed biting and scratching behavior in mice. Brain Res. 1981;217:212–215. doi: 10.1016/0006-8993(81)90203-1. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer MS, Cutler N, Feighner J, Shrivastava R, Carman J, Sramek JJ, et al. Distinct mechanism for antidepressant activity by blockade of central substance P receptors. Science. 1998;281:1640–1645. doi: 10.1126/science.281.5383.1640. [DOI] [PubMed] [Google Scholar]
- Laufer R, Wormser U, Friedman ZY, Gilon C, Chorev M, Selinger Z. Neurokinin B is a preferred agonist for a neuronal substance P receptor and its action is antagonized by enkephalin. Proc Natl Acad Sci U S A. 1985;82:7444–7448. doi: 10.1073/pnas.82.21.7444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leffler A, Ahlstedt I, Engberg S, Svensson A, Billger M, Oberg L, et al. Characterization of species-related differences in the pharmacology of tachykinin NK receptors 1, 2 and 3. Biochem Pharmacol. 2009;77:1522–1530. doi: 10.1016/j.bcp.2009.01.020. [DOI] [PubMed] [Google Scholar]
- Mantyh PW, Rogers SD, Honore P, Allen BJ, Ghilardi JR, Li J, et al. Inhibition of hyperalgesia by ablation of lamina I spinal neurons expressing the substance P receptor. Science. 1997;278:275–279. doi: 10.1126/science.278.5336.275. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meini S, Maggi CA. Tachykinin receptor assays. Curr Protoc Pharmacol. 2010;Chapter 4:Unit 4.10. doi: 10.1002/0471141755.ph0410s48. [DOI] [PubMed] [Google Scholar]
- Molinari P, Vezzi V, Sbraccia M, Gro C, Riitano D, Ambrosio C, et al. Morphine-like opiates selectively antagonize receptor-arrestin interactions. J Biol Chem. 2010;285:12522–12535. doi: 10.1074/jbc.M109.059410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustafa G, Anderson EM, Bokrand-Donatelli Y, Neubert JK, Caudle RM. Anti-nociceptive effect of a conjugate of substance P and light chain of botulinum neurotoxin type A. Pain. 2013;154:2547–2553. doi: 10.1016/j.pain.2013.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neubig RR, Spedding M, Kenakin T, Christopoulos A. International union of pharmacology committee on receptor nomenclature and drug classification. XXXVIII. Update on terms and symbols in quantitative pharmacology. Pharmacol Rev. 2003;55:597–606. doi: 10.1124/pr.55.4.4. [DOI] [PubMed] [Google Scholar]
- Regoli D, Drapeau G, Dion S, Couture R. New selective agonists for neurokinin receptors: pharmacological tools for receptor characterization. Trends Pharmacol Sci. 1988;9:290–295. doi: 10.1016/0165-6147(88)90013-2. [DOI] [PubMed] [Google Scholar]
- Regoli D, Boudon A, Fauchere JL. Receptors and antagonists for substance P and related peptides. Pharmacol Rev. 1994;46:551–599. [PubMed] [Google Scholar]
- Rizzi A, Nazzaro C, Marzola GG, Zucchini S, Trapella C, Guerrini R, et al. Endogenous nociceptin/orphanin FQ signalling produces opposite spinal antinociceptive and supraspinal pronociceptive effects in the mouse formalin test: pharmacological and genetic evidences. Pain. 2006;124:100–108. doi: 10.1016/j.pain.2006.03.021. [DOI] [PubMed] [Google Scholar]
- Rizzi A, Campi B, Camarda V, Molinari S, Cantoreggi S, Regoli D, et al. In vitro and in vivo pharmacological characterization of the novel NK1 receptor selective antagonist Netupitant. Peptides. 2012;37:86–97. doi: 10.1016/j.peptides.2012.06.010. [DOI] [PubMed] [Google Scholar]
- Rizzi A, Malfacini D, Cerlesi MC, Ruzza C, Marzola E, Bird MF, et al. In vitro and in vivo pharmacological characterization of nociceptin/orphanin FQ tetrabranched derivatives. Br J Pharmacol. 2014;171:4138–4153. doi: 10.1111/bph.12799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarau HM, Griswold DE, Bush B, Potts W, Sandhu P, Lundberg D, et al. Nonpeptide tachykinin receptor antagonists. II. Pharmacological and pharmacokinetic profile of SB-222200, a central nervous system penetrant, potent and selective NK-3 receptor antagonist. J Pharmacol Exp Ther. 2000;295:373–381. [PubMed] [Google Scholar]
- Shonberg J, Scammells PJ, Capuano B. Design strategies for bivalent ligands targeting GPCRs. ChemMedChem. 2011;6:963–974. doi: 10.1002/cmdc.201100101. [DOI] [PubMed] [Google Scholar]
- Solé NA, Barany G. Optimization of solid-phase synthesis of [Ala8]dynophin A. J Org Chem. 1992;57:5399–5403. [Google Scholar]
- Takahasi K, Sakurada T, Sakurada S, Kuwahara H, Yonezawa A, Ando R, et al. Behavioural characterization of substance P-induced nociceptive response in mice. Neuropharmacology. 1987;26:1289–1293. doi: 10.1016/0028-3908(87)90089-x. [DOI] [PubMed] [Google Scholar]
- Vachon L, Costa T, Herz A. Opioid receptor desensitization in NG 108–15 cells. Differential effects of a full and a partial agonist on the opioid-dependent GTPase. Biochem Pharmacol. 1987;36:2889–2897. doi: 10.1016/0006-2952(87)90199-7. [DOI] [PubMed] [Google Scholar]
- Vauquelin G, Charlton SJ. Long-lasting target binding and rebinding as mechanisms to prolong in vivo drug action. Br J Pharmacol. 2010;161:488–508. doi: 10.1111/j.1476-5381.2010.00936.x. [DOI] [PMC free article] [PubMed] [Google Scholar]