

Abstract
We generated homozygous knockin ES cells expressing a form of 3-phosphoinositide-dependent protein kinase-1 (PDK1) with a mutation in its pleckstrin homology (PH) domain that abolishes phosphatidylinositol 3,4,5-tris-phosphate (PtdIns(3,4,5)P3) binding, without affecting catalytic activity. In the knockin cells, protein kinase B (PKB) was not activated by IGF1, whereas ribosomal S6 kinase (RSK) was activated normally, indicating that PtdIns(3,4,5)P3 binding to PDK1 is required for PKB but not RSK activation. Interestingly, amino acids and Rheb, but not IGF1, activated S6K in the knockin cells, supporting the idea that PtdIns(3,4,5)P3 stimulates S6K through PKB-mediated activation of Rheb. Employing PDK1 knockin cells in which either the PtdIns(3,4,5)P3 binding or substrate-docking ‘PIF pocket' was disrupted, we established the roles that these domains play in regulating phosphorylation and stabilisation of protein kinase C isoforms. Moreover, mouse PDK1 knockin embryos in which either the PH domain or PIF pocket was disrupted died displaying differing phenotypes between E10.5 and E11.5. Although PDK1 plays roles in regulating cell size, cells derived from PH domain or PIF pocket knockin embryos were of normal size. These experiments establish the roles of the PDK1 regulatory domains and illustrate the power of knockin technology to probe the physiological function of protein–lipid and protein–protein interactions.
Keywords: mTOR, PI 3-kinase, PKB/Akt, PKC, RSK
Introduction
The 3-phosphoinositide-dependent protein kinase-1 (PDK1) phosphorylates the T loop and activates a group of related protein kinases belonging to the AGC family (Mora et al, 2004). These include isoforms of protein kinase B (PKB) (Brazil and Hemmings, 2001), p70 ribosomal S6 kinase (S6K) (Volarevic and Thomas, 2001), protein kinase C (PKC) (Newton, 2003) and p90 ribosomal S6 kinase (RSK) (Frodin and Gammeltoft, 1999). Once activated, these enzymes mediate diverse responses exerted by insulin and growth factors.
Much work has focused on understanding how phosphorylation of AGC kinases by PDK1 is regulated. This has revealed that PDK1 is constitutively active and that regulation involves the conversion of substrates to forms that can be phosphorylated by PDK1 in agonist-treated cells. For example, the phosphorylation of PKB by PDK1 is dependent on prior activation of the phosphoinositide 3-kinase (PI 3-kinase) and the production of the second messenger, phosphatidylinositol 3,4,5-trisphosphate (PtdIns(3,4,5)P3). This binds to the pleckstrin homology (PH) domain of PKB and is thought to induce a conformational change that enables PDK1 to phosphorylate PKB (Alessi et al, 1997; Stokoe et al, 1997; Milburn et al, 2003). PDK1 also contains a PH domain that binds PtdIns(3,4,5)P3 with high affinity. This interaction does not affect the catalytic activity of PDK1 (Currie et al, 1999), but may enhance its ability to activate PKB by colocalising PDK1 and PKB at the plasma membrane. However, this hypothesis is unproven because it has never been tested in a physiological setting.
Unlike PKB, other PDK1 substrates, including isoforms of S6K, PKC and RSK, do not bind PtdIns(3,4,5)P3 as they lack PH domains, nor is their phosphorylation by PDK1 stimulated by PtdIns(3,4,5)P3. However, recent work indicates that the ability of PDK1 to phosphorylate S6K and RSK is dependent on the phosphorylation of these enzymes at a C-terminal Ser/Thr residue located in a region known as the hydrophobic motif (reviewed in Mora et al, 2004). This phosphorylation allows PDK1 to interact with these enzymes through a specific substrate-docking site termed the ‘PIF pocket', which is situated in the small lobe of the catalytic domain of PDK1 (Biondi et al, 2000, 2001). In the case of S6K, the PI 3-kinase pathway controls phosphorylation of the hydrophobic motif. Recent studies performed by many groups indicate that activation of PKB leading to phosphorylation of TSC2 and subsequent activation of the Rheb GTPase stimulates the protein kinase mTOR, allowing it to phosphorylate the hydrophobic motif of S6K (reviewed in Manning and Cantley, 2003; Li et al, 2004). Nutrients such as amino acids also regulate the activity of mTOR through a pathway independent of PI 3-kinase and PKB. In contrast, the isoforms of RSK possess their own hydrophobic motif kinase as a second catalytic domain located in their C-terminal domains (Frodin and Gammeltoft, 1999). This C-terminal hydrophobic motif kinase of RSK is activated by the MAP kinases ERK1 and ERK2, triggering phosphorylation of the hydrophobic motif located on the N-terminal catalytic domain. This allows RSK to dock with the PIF pocket of PDK1, which then phosphorylates the T loop of the N-terminal AGC kinase domain of RSK resulting in activation (Frodin et al, 2000, 2002). The PDK1 PIF pocket is also required for the activation of atypical and related PKC isoforms (Balendran et al, 2000a). The hydrophobic motif of conventional PKC isoforms is required for their activation by PDK1, implying a role for the PDK1 PIF pocket in regulating interaction of PDK1 with conventional PKC isoforms (Gao et al, 2001).
We have recently examined the in vivo importance of the PIF-binding pocket of PDK1 by generating a homozygous knockin mutation in embryonic stem (ES) cells. In these cells, Leu155, which is required for the functional integrity of the PIF pocket, was changed to Glu, in order to disrupt the ability of the PIF pocket of PDK1 to interact with its substrates (Collins et al, 2003). In the knockin cells, PKB was activated normally, but S6K and RSK could not be activated, demonstrating the crucial role that the PIF pocket of PDK1 plays in enabling the activation of PDK1 substrates lacking a PH domain. In this study, we use knockin technology to examine the roles of PtdIns(3,4,5)P3 binding to the PH domain of PDK1.
Results
Generation of PDK1(PHKI/PHKI) knockin ES cells
We originally reported that mutation of three consecutive Arg residues (472–474) to Leu in the PH domain of PDK1 abolished the ability of PDK1 to interact with PtdIns(3,4,5)P3 and the plasma membrane (Currie et al, 1999). As the mutation of Arg474 alone had been suggested by others to inhibit PtdIns(3,4,5)P3 binding (Anderson et al, 1998), we compared the ability of both mutants to bind PtdIns(3,4,5)P3 in side-by-side assays. We found that the PDK1[R472L] mutant possessed weak, but nevertheless measurable binding to PtdIns(3,4,5)P3, compared to the PDK1[RRR472-474LLL] mutant (EJ McManus, data not shown). This prompted us to generate the PDK1[RRR472-474LLL] knockin, in order to abolish PtdIns(3,4,5)P3 binding. This was achieved using methodology described in Materials and methods and Figure 1. Homozygous knockin PDK1(PHKI/PHKI) and wild-type PDK1(+/+) ES cell lines were selected and employed in the studies described below. All experimental findings were confirmed using two different PDK1(PHKI/PHKI) ES cell clones.
Figure 1.
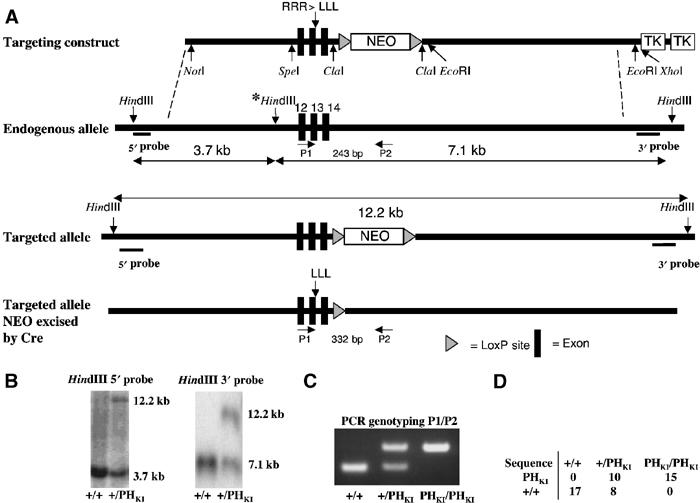
PDK1(PHKI/PHKI) knockin strategy. (A) Diagram depicting the knockin construct, the endogenous PDK1 allele containing exons 12–14, the targeted allele with the neomycin selection cassette still present, and the targeted allele with the neomycin cassette removed by Cre recombinase. The black boxes represent exons and the grey triangles represent LoxP sites. The three HindIII restriction sites present in the endogenous allele are shown, the middle site (marked with an asterisk), being removed upon targeting of the allele. (B) After positive and negative selection of cell lines (see Materials and methods), genomic DNA from the indicated cell lines was purified and digested overnight using HindIII, and the digested DNA was electrophoresed on a 1% agarose gel and transferred to nitrocellulose. The membrane was then incubated with a 32P-labelled 5′ or 3′ probe. In the case of the 5′ probe, the wild-type allele generates a 3.7 kb fragment whereas the knockin allele produces a 12.2 kb fragment. Similarly, the 3′ probe detects a fragment of 7.1 kb from the wild-type allele and 12.2 kb from the targeted knockin allele. (C) The indicated ES cell lines derived from E3 murine blastocysts (see Materials and methods) were genotyped using PCR primers P1 and P2. The wild-type allele generates a 243 bp product, while the knockin allele generates a 332 bp product due to the presence of the LoxP site and flanking region, which remains in an intronic region following Cre-mediated excision of the neomycin selection cassette. (D) Genomic DNA purified from the indicated ES cell lines was subjected to PCR to generate a product that encompasses the knockin mutation region. The resultant PCR products were ligated into the pCR-Topo 2.1 vector, transformed into Escherichia coli and clones sequenced. The numbers of the wild-type and knockin sequences obtained for each cell line are indicated.
Initial characterisation of PDK1(PHKI/PHKI) ES cells
We first compared the activity and expression of PDK1 protein in wild-type PDK1(+/+) and PDK1(PHKI/PHKI) knockin ES cells (Figure 2). These studies revealed that the level of expression of PDK1 in the knockin ES cells was only 20% that of the wild-type cells. These results could be explained if binding of PDK1 to phosphoinositides was required to stabilise the protein and/or if the PDK1 mutant was rendered less stable by the knockin mutation. In order to generate a control ES cell line that expresses similar levels of nonmutated PDK1, we employed a homozygous targeted ES cell line in which a neomycin cassette was inserted between exons 2 and 3 of both PDK1 alleles, which we had previously found to reduce the level of expression of PDK1 (Lawlor et al, 2002). The resulting hypomorphic PDK1(fl/fl) ES cell line expressed only 10% of the PDK1 activity compared to the wild-type PDK1(+/+) cells (Figure 2), and was employed as a control throughout this study. In this manner, any observed effect could be attributed to the lack of binding of PDK1 to 3-phosphoinositides rather than the lower level of expression of PDK1. As a further control, we employed the PDK1(−/−) ES cell line that does not express PDK1 at all (Figure 2; Williams et al, 2000).
Figure 2.
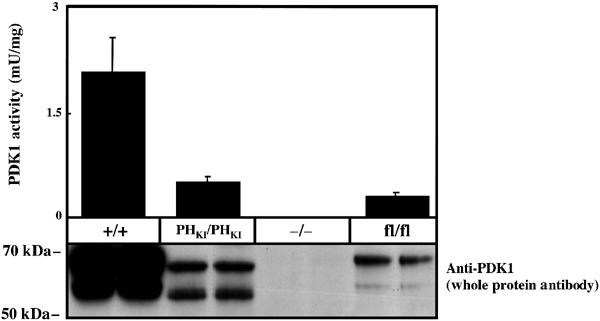
Expression and kinase activity of PDK1 in knockin ES cells. The indicated ES cell lines were grown and lysed. The activity of PDK1 was measured following immunoprecipitation employing the PDKtide substrate peptide as described in Materials and methods. The results shown are the mean±s.d. for three dishes of cells each assayed in duplicate. In order to quantify PDK1 expression in cell lysates, PDK1 was affinity purified on Sepharose conjugated to PIF as described in the Materials and methods and resin was immunoblotted for PDK1. Similar results were obtained in three separate experiments. It should be noted that PDK1 in ES cells, as observed in other cell lines, is detected as two bands on immunoblot analysis, but the reasons for this are unknown (Balendran et al, 1999; Williams et al, 2000).
Normal activation of RSK in PDK1(PHKI/PHKI) knockin ES cells
As discussed in Introduction, RSK is activated following its phosphorylation by PDK1 and ERK1/ERK2. The role played by the PH domain of PDK1 in regulating RSK activation is unknown. We therefore tested whether RSK could be activated in the PDK1(PHKI/PHKI) knockin ES cells in response to the phorbol ester TPA (Figure 3A) or serum (Figure 3B). These studies revealed that TPA or serum induced a similar activation of RSK in wild-type PDK1(+/+), PDK1(PHKI/PHKI) and PDK1(fl/fl) ES cells (Figure 3). Consistent with this, TPA and serum induced comparable phosphorylation of three of the activating residues in RSK, including Ser227, the site of PDK1 phosphorylation, as well as phosphorylation of GSK3α/GSK3β, a physiological substrate of RSK (Frame and Cohen, 2001). As expected, PD 184352, an inhibitor of MKK1 (Sebolt-Leopold et al, 1999), prevented TPA- or serum-induced phosphorylation of ERK and the activation of RSK. No detectable RSK activity or phosphorylation of RSK at Ser227 was observed in TPA- or serum-stimulated PDK1(−/−) ES cells. As a further control we demonstrate that the PI 3-kinase inhibitor wortmannin failed to inhibit serum-induced activation of RSK (Figure 3B), under conditions in which it prevented phosphorylation of PKBα at Thr308, a process mediated by PI 3-kinase (data not shown). These results demonstrate that binding of phosphoinositides to the PH domain of PDK1 is not required for ERK-mediated RSK activation, and demonstrate that the reduced levels of PDK1 in the PDK1(PHKI/PHKI) knockin and PDK1(fl/fl) hypomorphic ES cells do not affect the activation of RSK.
Figure 3.
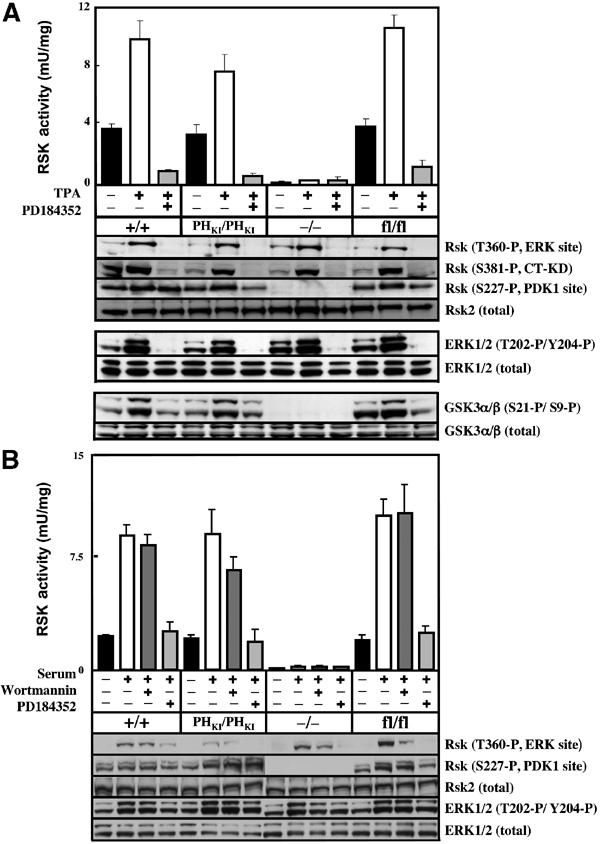
RSK isoforms are activated normally in PDK1(PHKI/PHKI) cells. (A) The indicated ES cell lines were grown to 80% confluence, serum starved for 3 h, incubated with or without 2 μM PD184352 for 1 h and then either left unstimulated or stimulated with 0.4 μg/ml TPA for 15 min. The cells were then lysed and the activity of RSK was measured after immunoprecipitation with an antibody that recognises all RSK isoforms. The results shown are mean±s.d. for three dishes of cells each assayed in duplicate. Lysates from the ES cells were also immunoblotted using the indicated antibodies. Thr360 is phosphorylated by ERK, Ser381 is phosphorylated by the C-terminal kinase domain of RSK (CT-KD) and Ser227 is the site phosphorylated by PDK1 (Frodin et al, 2000). (B) As above, except that the serum-starved cells were incubated with or without 2 μM PD184352 for 1 h or 100 nM wortmannin for 10 min and stimulated with 10% (v/v) serum for 15 min. Similar results were obtained in three different experiments.
Lack of PKB activation in the PDK1(PHKI/PHKI) ES cells
In order to assess PKBα activity, the different ES cell lines were stimulated with IGF1 in the presence or absence of the PI 3-kinase inhibitor wortmannin and PKBα immunoprecipitated and assayed. For both PDK1(+/+) and PDK1(fl/fl) ES cells, a similar low basal PKBα activity was detected in unstimulated cells, while IGF1 induced a robust ∼6-fold activation in both cell lines, which was inhibited by wortmannin (Figure 4). In unstimulated PDK1(PHKI/PHKI) knockin ES cells, PKBα activity was undetectable and IGF1 only increased PKBα activity to a low basal level, similar to that found in unstimulated PDK1(+/+) and PDK1(fl/fl) ES cells (Figure 4). The small increase in PKBα activity in the PDK1(PHKI/PHKI) cells was prevented by wortmannin. As expected, PKBα was not activated by IGF1 in PDK1(−/−) ES cells.
Figure 4.
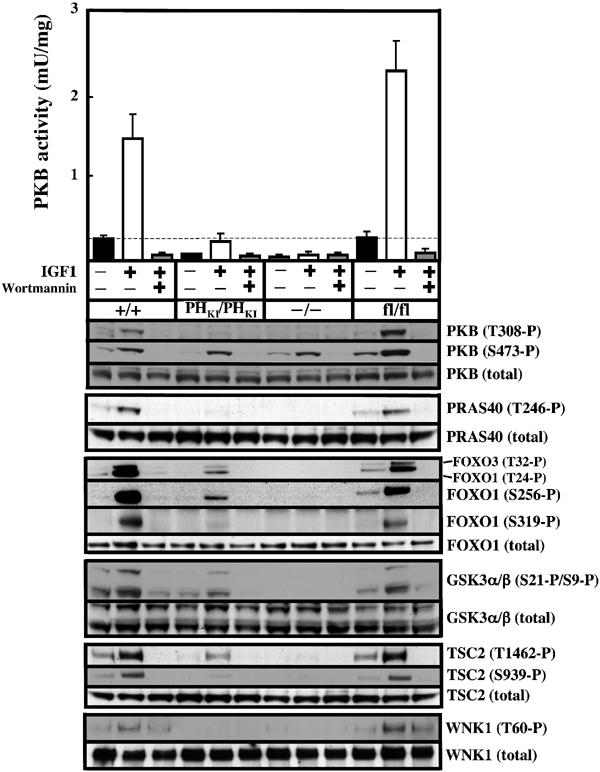
Activation of PKBα is severely impaired in PDK1(PHKI/PHKI) cells. The indicated ES cell lines were grown to 80% confluence and serum starved for 4 h, incubated with or without 100 nM wortmannin for 10 min and then either left unstimulated or stimulated with 20 ng/ml IGF1 for 15 min. The cells were lysed and the activity of PKBα was measured after immunoprecipitation. The results shown are mean±s.d. for three dishes of cells each assayed in duplicate. The dotted line represents the basal activity of PKBα in unstimulated PDK1(+/+) cells. The activation of PKB and the phosphorylation of the indicated substrates were also assessed using the appropriate antibodies (see Materials and methods). Similar results were obtained in three different experiments.
We also assessed the state of the PDK1 phosphorylation site in PKBα (Thr308), as well as the hydrophobic motif (Ser473), employing phosphospecific antibodies. In both wild-type PDK1(+/+) and PDK1(fl/fl) ES cells, but not PDK1(PHKI/PHKI) or PDK1(−/−) ES cells, IGF1 induced a marked phosphorylation of PKB at Thr308. In all cell types, IGF1 induced a wortmannin-sensitive phosphorylation of PKB at Ser473. This is consistent with previous studies indicating that a protein kinase distinct from PDK1 mediates this phosphorylation (reviewed in Leslie et al, 2001). We also assessed the phosphorylation of 10 established sites of PKB phosphorylation in six different substrates (PRAS40[Thr246] (Kovacina et al, 2003), Foxo1[Thr24, Ser256, Ser319] and Foxo3[Thr32] (Brunet et al, 1999; Rena et al, 1999), GSK3α[Ser21], GSK3β[Ser9] (Cross et al, 1995), TSC2[Ser939, Thr1462] (Manning et al, 2002)) and WNK1[Thr60] (Vitari et al, 2004). In the wild-type PDK1(+/+) and PDK1(fl/fl) ES cells, IGF1 induced a marked increase in the phosphorylation of these substrates, which was prevented by wortmannin. In contrast, in the PDK1(PHKI/PHKI) cells, IGF1 induced either no detectable phosphorylation or only a slight phosphorylation comparable to the levels observed in unstimulated PDK1(+/+) or PDK1(fl/fl) ES cells. This correlates with the levels of PKBα activity that are observed in the different PDK1 ES cell lines. As expected, no significant phosphorylation of any PKB substrate was observed in the PDK1(−/−) ES cells.
S6K activity in the PDK1(PHKI/PHKI) knockin ES cell lines
To assess the activation of S6K1, ES cells were stimulated with IGF1 in the presence or absence of the mTOR inhibitor rapamycin, which abolishes S6K activation in vivo. As noted previously with PDK1(+/+) ES cells (Williams et al, 2000; Collins et al, 2003), a significant basal activity of S6K1 was observed in wild-type PDK1(+/+) ES cells, which was increased a further 2.5-fold following stimulation with IGF1 and reduced to undetectable levels by rapamycin (Figure 5A). Similar results were obtained with PDK1(fl/fl) hypomorphic cells, indicating that reducing the levels of PDK1 do not affect the activation of S6K. Interestingly, in unstimulated PDK1(PHKI/PHKI) ES cells, the basal S6K1 activity was similar to that found in PDK1(+/+) and PDK1(fl/fl) ES cells; however, stimulation with IGF1 did not increase S6K1 activity further (Figure 5A). As expected, no S6K1 activity was observed in PDK1(−/−) ES cells. We also assessed S6K1 activity using phosphospecific antibodies that recognise the phosphorylated hydrophobic motif of S6K1 (Thr389) and C-terminal residues (Thr421 and Ser424). We found that phosphorylation of the hydrophobic motif of S6K1 in the different ES cells lines correlated with the activation state of S6K1. In PDK1(−/−) and the other ES cell lines, phosphorylation of the Thr421 and Ser424 is high and reduced to basal levels by rapamycin, indicating that these phosphorylation events are regulated by mTOR but not PDK1 (Figure 5A). We also found that the relative activation of S6K2 compared to S6K1 was similar in the different PDK1 ES cell lines studied, with the PDK1(PHKI/PHKI) ES cells displaying a significant basal S6K2 activity, which was not stimulated further by IGF1 (Figure 5A).
Figure 5.
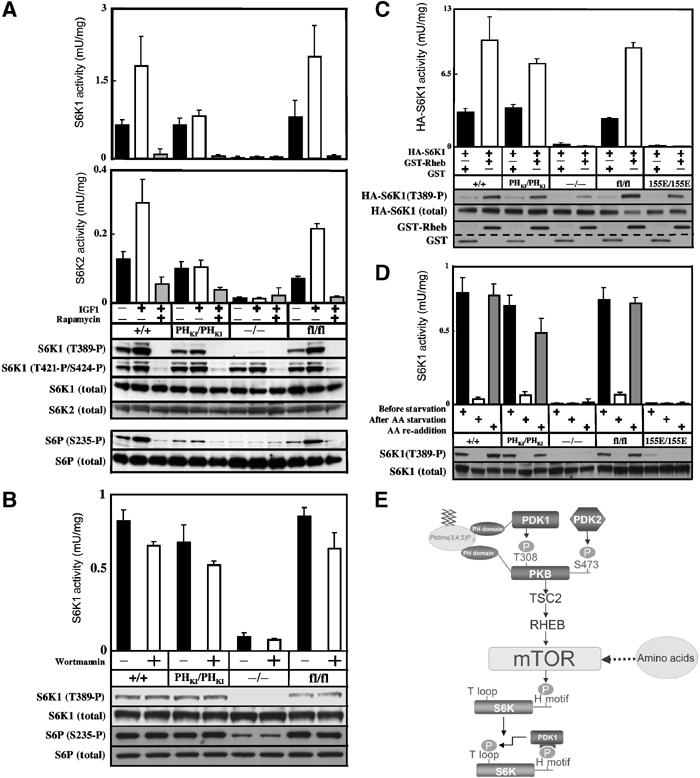
Regulation of S6K in PDK1(PHKI/PHKI) cells. (A) The indicated ES cell lines were grown to 80% confluence and serum starved for 3.5 h, incubated with or without 100 nM rapamycin for 30 min and then either left unstimulated or stimulated with 20 ng/ml IGF1 for 15 min. The cells were lysed and the activity of S6K1 (upper panel) or S6K2 (lower panel) was measured in independent experiments after immunoprecipitation using antibodies specific for either isoform. The results shown are mean±s.d. for three dishes of cells each assayed in duplicate. The activation of S6K and phosphorylation of its substrates were assessed using the indicated phosphospecific antibodies. (B) As above, except that serum-deprived cells were incubated in the presence or absence of 100 nM wortmannin for 10 min. (C) ES cells were transfected with HA epitope-tagged S6K1 (HA-S6K1) in the presence of either GST-Rheb or GST alone. At 24 h post-transfection, cells were serum starved for 4 h and HA-S6K1 immunoprecipitated with anti-HA antibody. The immunoprecipitates were either assayed for S6K1 activity or immunoblotted with the indicated antibodies. The levels of GST-Rheb and GST were determined by immunoblotting cell lysates with anti-GST. The dotted line emphasizes the molecular weight difference between GST-Rheb and GST. (D) ES cell lines were grown to 80% confluence and deprived of serum but not amino acids for 2.5 h. The cells were then either maintained in the presence of amino acids for 90 min (before starvation) or deprived of amino acids for 90 min (AA starvation) or deprived of amino acids for 90 min followed by the re-addition of amino acids for 30 min (AA re-addition). S6K activity and phosphorylation were assessed as above. Similar results were obtained in at least two separate experiments. (E) Model summarising the mechanism of activation of S6K by the PI 3-kinase and amino acids that is discussed in the main text.
To confirm the lack of effect of IGF1 stimulation on S6K activity in the PDK1(PHKI/PHKI) ES cells, we also employed phosphospecific antibodies recognising Ser235 of ribosomal protein S6, a site phosphorylated by S6K in vivo. In PDK1(+/+) and PDK1(fl/fl) ES cells, significant levels of phosphorylated S6 protein were observed in unstimulated cells, with IGF1 increasing phosphorylation ∼3-fold and rapamycin reducing phosphorylation to undetectable levels (Figure 5A). In PDK1(PHKI/PHKI) ES cells, the S6 protein was phosphorylated to the same extent as found in unstimulated PDK1(+/+) cells, but its phosphorylation was not increased further in IGF1-stimulated cells.
As the basal S6K activity in serum-starved PDK1(PHKI/PHKI) ES cells was normal, this indicated that PI 3-kinase might not regulate this state of S6K activity. Consistent with this idea, we show that wortmannin had no significant effect on the basal S6K1 activity in the PDK1(PHKI/PHKI) and control ES cell lines (Figure 5B). The lack of activation of S6K1 and S6K2 in PDK1(PHKI/PHKI) cells by IGF1 might be explained by the lack of phosphorylation of TSC2 by PKB, resulting in the Rheb GTPase not being activated. To explore this possibility, we tested whether overexpression of Rheb was able to stimulate S6K in the different ES cell lines. We found that Rheb stimulated S6K activity 2- to 3-fold in the PDK1(PHKI/PHKI) knockin ES cells to the same extent as in PDK1(+/+) and PDK1(fl/fl) cells (Figure 5C). We had previously generated PDK1(155E/155E) knockin ES cell lines in which the PIF pocket of PDK1 was disrupted (Collins et al, 2003) and found that Rheb was not able to stimulate S6K1 activity in these cells (Figure 5C). Rheb also failed to stimulate S6K activity in PDK1(−/−) ES cells. As outlined in Introduction, mTOR and hence S6K are also regulated by a nutrient-dependent pathway and we therefore tested how amino acids regulated S6K activity (Figure 5D). Withdrawal of amino acids from the PDK1(PHKI/PHKI) cells abolished basal S6K activity and phosphorylation at Thr389. Subsequent addition of amino acids stimulated S6K activity and phosphorylation to the level found in non-amino-acid-deprived cells similar to the level found in PDK1(+/+) and PDK1(fl/fl) cells. S6K activity was undetectable and was not stimulated by amino acids in PDK1(155E/155E) or PDK1(−/−) ES cells (Figure 5D).
Levels of PKC isoforms in the PDK1(PHKI/PHKI) knockin ES cell lines
Phosphorylation of PKC isoforms by PDK1 plays an important role in stabilising these enzymes (Newton, 2003). Consistent with this, the levels of many PKC isoforms were previously found to be markedly reduced in PDK1(−/−) ES cells (Balendran et al, 2000b). To study the importance of the PDK1 PH domain and PIF pocket in stabilising PKC isoforms, we assessed the levels of these enzymes in the PDK1(PHKI/PHKI) and PDK1(155E/155E) knockin ES cell lines. This was performed by immunoblotting cell extracts with PKC isoform-specific antibodies and quantitating the blots employing an infrared imaging system described in Materials and methods. In the PDK1(PHKI/PHKI) cells, the levels of most PKC isoforms were within 25% of those in the PDK1(+/+) and PDK1(fl/fl) controls (Figure 6A). In the case of PKCβII, the levels were reduced by 50% in the PDK1(PHKI/PHKI) knockin ES cells, indicating that PtdIns(3,4,5)P3 binding to the PH domain of PDK1 may play a role in regulating this isoform. Although the levels of PKCγ are reduced by 40% in the PDK1(PHKI/PHKI) knockin, this may be due to the lower levels of PDK1 in these cells as PKCγ levels are similarly reduced in the PDK1(fl/fl) cells (Figure 6A). In the PDK1(155E/155E) knockin ES cells, the levels of PKCα, PKBβII, PKCγ, PKCδ, PKCɛ and PRK1 were markedly reduced to a low level similar to that found in the PDK1(−/−) ES cells. The levels of PKCζ and PRK2 were reduced by 20–30% in the PDK1(155E/155E) knockin ES cells.
Figure 6.
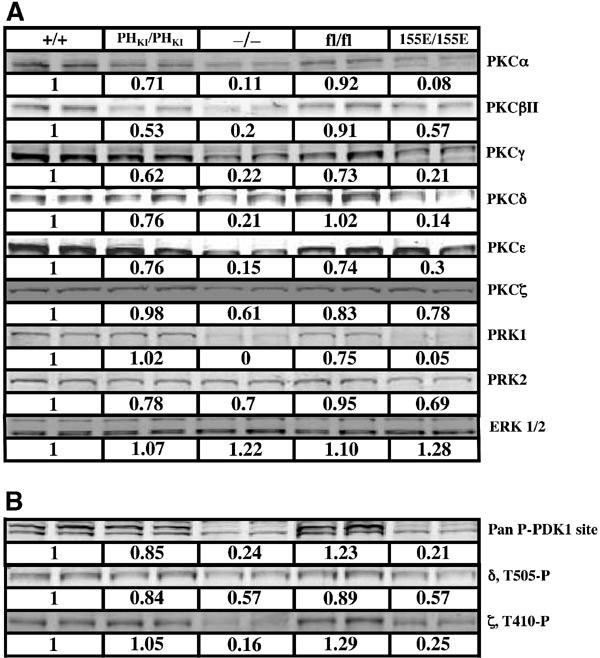
Immunoblotting of PKC isoforms in ES cells. The indicated ES cells cultured in the presence of 10% serum were lysed and aliquots (20 μg of protein) were subjected to SDS–polyacrylamide gel electrophoresis and immunoblotted with the indicated total PKC isoform antibodies (A) or phosphospecific T-loop PKC antibodies (B). Quantitation of the immunoblots, indicated immediately below each blot, was performed using LI-COR Odyssey infrared imaging system as described in Materials and methods. The value of expression of each PKC isoform detected in PDK1(+/+) cells is taken as 1.0 and the expression of the isoforms in other cells expressed relative to this. As a control, the cell lysates were immunoblotted for ERK1 and ERK2 whose levels are not regulated by PDK1. Similar results were obtained in at least three independent experiments.
The phosphorylation status of the T loop of PKC isoforms was also assessed using commercially available antibodies recognising the phosphorylated T loop of PKCγ that crossreacts with several conventional PKC isoforms (Pan PDK1 site), as well as with PKCδ and PKCζ phosphospecific T-loop antibodies (Figure 6B). T-loop phosphorylation measured using all three antibodies was reduced by 50–85% in the PDK1(−/−) and PDK1(155E/155E) cells, but not significantly lowered in PDK1(PHKI/PHKI) or PDK1(fl/fl) cells.
Embryonic lethality of PDK1(PHKI/PHKI) knockin mice
The PDK1(+/PHKI) heterozygous mice were healthy, of normal size and displayed no obvious phenotypes. In an attempt to generate complete PDK1 knockin mice, matings were set up between heterozygous PDK1(+/PHKI) mice (Table I). No PDK1(PHKI/PHKI) postnatal mice were ever recovered, indicating that this genotype resulted in embryonic lethality. We next analysed the genotype of embryos from days E7.0 to E11.5 of development derived from PDK1(+/PHKI) matings, which revealed the presence of PDK1(PHKI/PHKI) embryos at near Mendelian distribution between E7.5 and E10.5 (Table I). We were never able to isolate PDK1(PHKI/PHKI) embryos at E11.5 or later times, indicating that the mutant embryos had died and were reabsorbed after E11.0. Previous work has shown that PDK1 mRNA is expressed ubiquitously in all embryonic and extraembryonic tissues during this period of development (Lawlor et al, 2002).
Table 1.
Mice matings reported in this study
| Cross | Stage | Genotype | Total |
|---|---|---|---|
| PDK1(+/PHKI) × PDK1(+/PHKI) | E8 | PDK1(+/+) (29%) PDK1(+/PHKI) (24%) PDK1(PHKI/PHKI) (47%) | 42 |
| PDK1(+/PHKI) × PDK1(+I/PHKI) | E8.5 | PDK1(+/+) (21%) PDK1(+/PHKI) (58%) PDK1(PHKI/PHKI) (21%) | 24 |
| PDK1(+/PHKI) × PDK1(+I/PHKI) | E9.5 | PDK1(+/+) (32%) PDK1(+/PHKI) (49%) PDK1(PHKI/PHKI) (19%) | 114 |
| PDK1(+/PHKI) × PDK1(+I/PHKI) | E10.5 | PDK1(+/+) (26%) PDK1(+/PHKI) (59%) PDK1(PHKI/PHKI) (15%) | 54 |
| PDK1(+/PHKI) × PDK1(+I/PHKI) | Weaning | PDK1(+/+) (30%) PDK1(+/PHKI) (70%) PDK1(PHKI/PHKI) (0%) | 165 |
| PDK1(+/155E) × PDK1(+/155E) | E8 | PDK1(+/+) (50%) PDK1(+/155E) (20%) PDK1(155E/155E) (30%) | 10 |
| PDK1(+/155E) × PDK1(+/155E) | E8.5 | PDK1(+/+) (55%) PDK1(+/155E) (22%) PDK1(155E/155E) (23%) | 9 |
| PDK1(+/155E) × PDK1(+/155E) | E9.5 | PDK1(+/+) (14%) PDK1(+/155E) (45%) PDK1(155E/155E) (41%) | 22 |
| PDK1(+/155E) × PDK1(+/155E) | E10.5 | PDK1(+/+) (23%) PDK1(+/155E) (45%) PDK1(155E/155E) (32%) | 22 |
| PDK1(+/155E) × PDK1(+/155E) | E11.5 | PDK1(+/+) (32%) PDK1(+/155E) (58%) PDK1(155E/155E) (10%) | 57 |
| PDK1(+/155E) × PDK1(+/155E) |
Weaning |
PDK1(+/+) (51%) PDK1(+/155E) (49%) PDK1(155E/155E) (0%) |
172 |
| The indicated matings were set up and the progeny genotyped as described in Materials and methods. The percentage of each genotype observed is indicated in parentheses. Total indicates the number of progeny analysed. | |||
Analysis of the PDK1(PHKI/PHKI) phenotype
At day E8.0 (4–6 somites), PDK1(PHKI/PHKI) embryos are of similar size to their PDK1(+/PHKI) or PDK1(+/+) littermates (Figure 7A and B). They do not appear retarded, possess the same headfolds, posterior mesoderm, allantois and somites, and also possess a heart, which could be seen beating following dissection. However, at E8.5 (8–10 somites), the first phenotypic differences between the PDK1(PHKI/PHKI) and PDK1(+/PHKI) embryos become apparent (Figure 7C and D). Overall, the embryos are slightly retarded and blood islands begin to be observed from around the otic vesicle through to the forebrain. At day E9.5, both the PDK1(PHKI/PHKI) and the PDK1(+/PHKI) embryos have turned (Figure 7E and F). However, the PDK1(PHKI/PHKI) embryos are more obviously retarded in development, and further blood islands in various regions of the head are apparent. Moreover, there is also a change in the shape of the forebrain, which appears smaller with the maxillary portion of the first branchial arch coming to partially cover the eye. The frontonasal process is also smaller. Sectioning of E9.5 embryos also revealed the absence of dorsal root ganglia in PDK1(PHKI/PHKI) embryos (data not shown). The E9.5 PDK1(PHKI/PHKI) embryos have developed a placenta with umbilical vessels. However, these appear less developed than in the PDK1(+/+) embryos (Supplementary Figure 1). At E10.5, the last day that the knockin embryos are seen, development has not progressed significantly from E9.5 (Figure 7G and H). A greater number of blood islands are visible in the head, and tissues are becoming opaque indicating necrosis. Moreover, no visible blood vessels or blood islands are observed in the extraembryonic membranes of PDK1(PHKI/PHKI) E10.5 embryos (Figure 7I and J). Importantly, at E10.5, the PDK1(+/+) placenta has increased in both depth and complexity, while the PDK1(PHKI/PHKI) placenta did not noticeably develop from E9.5 (Supplementary Figure 1). The cause of death of the PDK1(PHKI/PHKI) embryos is most likely due to blood vessel defects in the head coupled with the lack of placental development.
Figure 7.
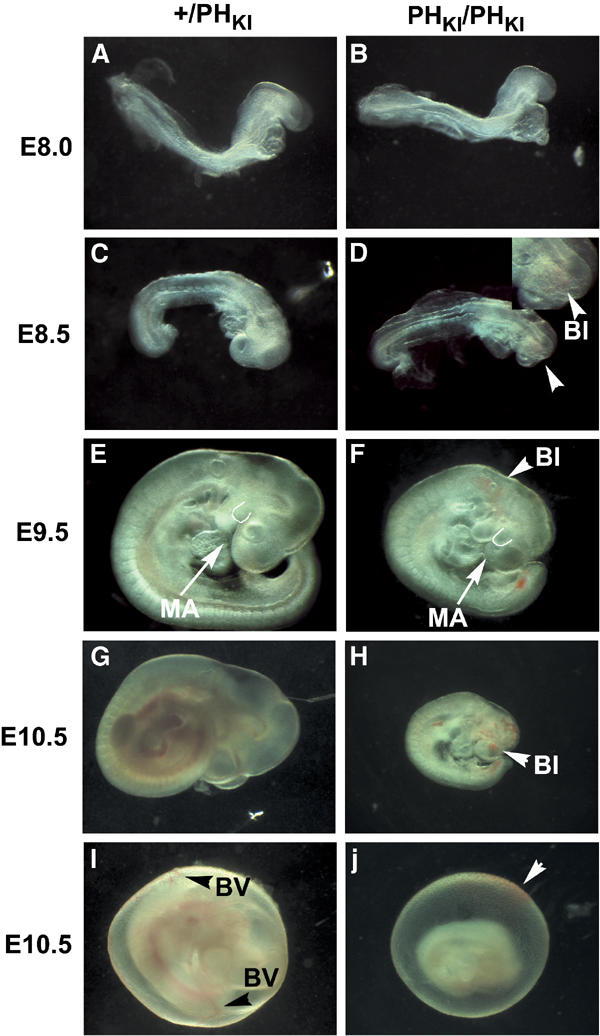
Phenotypes of PDK1(PHKI/PHKI) embryos. (A–J) PDK1(+/PHKI) or PDK1(PHKI/PHKI) embryos at the indicated embryonic stage were dissected in PBS and imaged on a Leica M275 microscope and whole-mount photographs were taken. BI, blood island; MA, maxillary arch; BV, blood vessel. In (D), an enlargement of the region of the blood island is shown. In (E, F), the first part of the maxillary arch, which covers the eye, is outlined. Black arrowheads in (I) indicate yolk sac blood vessels (BV). The white arrowhead in (J) indicates a dissection phenomenon, namely blood from the dissection dish adhering to the outside of the sac. In all images, PDK1(PHKI/PHKI) embryos are shown at the same magnification as PDK1(+/PHKI) embryos. The results presented are representative of the number of embryos shown in Table I.
Embryonic lethality of PDK1(155E/155E) knockin mice
We previously generated PDK1(155E/155E) knockin ES cell lines in which the PIF pocket of PDK1 was disrupted to prevent activation of S6K and RSK but not PKB (Collins et al, 2003). In an attempt to generate PDK1(155E/155E) knockin mice, matings were set up between heterozygous PDK1(+/155E) mice that displayed no apparent phenotype and the resulting progeny genotyped (Table I). No PDK1(155E/155E) postnatal mice were ever recovered, indicating that this genotype results in an embryonic lethal phenotype. We next analysed the genotype of embryos from days E7.0 to E11.5 derived from PDK1(+/155E) matings, which revealed the presence of PDK1(155E/155E) embryos at near Mendelian distribution (Table I). We were never able to isolate PDK1(155E/155E) embryos at E12.5 or later, indicating that the mutant embryos died and were reabsorbed after E12.0. We also noticed that the PDK1(+/155E) heterozygous mice were born at about half of the expected Mendelian frequency (Table I), indicating that a proportion of these mice failed to develop to term. Until E11.5, the Mendelian proportion of PDK1(+/155E) knockin embryos was observed and these embryos were indistinguishable in phenotype from wild-type PDK1(+/+) embryos. This suggests that a fraction of the PDK1(+/155E) knockin embryos die later in development.
Analysis of PDK1(155E/155E) phenotype
Until E8.0, homozygous PDK1(155E/155E) embryos displayed no phenotype (Figure 8A and B). In analysing over 50 PDK1(155E/155E) embryos, a variation in phenotype was observed. Firstly, some PDK1(155E/155E) embryos are severely retarded relative to littermates from E8.5 onwards (Figure 8D, G, J and M). This retardation progresses until E11.5 when some embryos are retarded by at least 1 day (Figure 8M). Secondly, in some PDK1(155E/155E) embryos, the posterior portion of the body axis begins to twist abnormally around the dorso-ventral and left–right axes from day E9.5, so that on viewing the embryos from the dorsal part of the posterior trunk the mid-thoracic levels are visible (compare Figure 8L and N). In the PDK1(155E/155E) embryos that have this phenotype, the twist occurs only at the thoracic somites and not at other levels. Furthermore, once twisting has occurred, the body axis continues to grow along the new line with no further twisting. The third phenotype that is observed in some of the PDK1(155E/155E) embryos, which is most clearly seen at E10.5 and E11.5, is a major reduction in the forebrain size, with the forebrain vesicles being noticeably smaller resulting in a prominent indentation (compare Figure 8I and K). This defect is also apparent earlier at E8.5–E9.5, shown by the reduced distance from the eye to the anterior of the forebrain (compare Figure 8C and E). In PDK1(PHKI/PHKI) knockin embryos at E9.5, the maxillary portion of the first branchial arch was partially covering the eye (Figure 7F). However, in the PDK1(155E/155E) embryos this phenotype is not observed, indicating a different part of the forebrain is affected. Forebrain reduction and posterior twisting were found alone or together, in severely retarded or more normal stage homozygotes. Cross-sections of various E10.5 PDK1(155E/155E) embryos all revealed the presence of dorsal root ganglia (data not shown). In contrast to the PDK1(PHKI/PHKI) knockin embryos, the PDK1(155E/155E) embryos display no defects in blood vessel formation and possessed a normal placenta and extraembryonic membranes between E9.5 and E11.5 (data not shown). No live PDK1(155E/155E) embryos were ever recovered after E12.0. The cause of death in these embryos is unknown.
Figure 8.
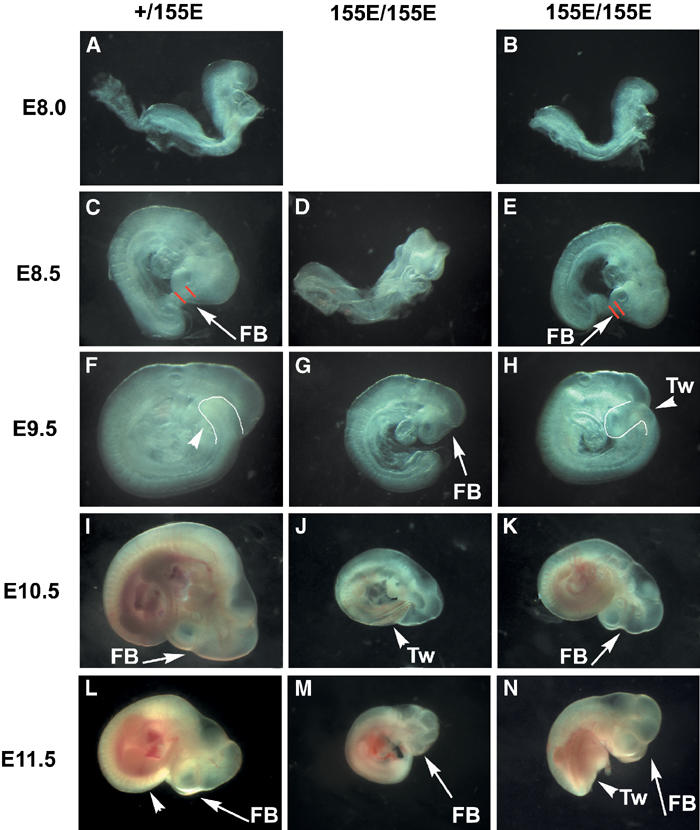
Phenotypes of PDK1 (155E/155E) embryos. (A–N) PDK1(+/155E) and PDK1(155E/155E) embryos were dissected in PBS and imaged on a Leica M275 microscope and whole-mount photographs were taken. FB, forebrain; Tw, twisting. In (C, D), the red lines represent the distance between the eye and the anterior of the forebrain. In (F, H), the outline represents twisting of the thoracic somites. In all images, PDK1(155E/155E) embryos are shown at the same magnification as PDK1(+/155E) embryos. The results presented are representative of the number of embryos shown in Table I.
Evidence that cell volume is normal in the PDK1 (PHKI/PHKI) and PDK1(155E/155E) embryos
Our previous analysis employing PDK1(−/−) cells indicated that one of the major effects of knocking out PDK1 is a marked reduction in cell volume. For example, E7.0 PDK1(−/−) embryos are markedly smaller than their PDK1(+/+) littermates (Lawlor et al, 2002). Employing a stereological approach called the dissector principle to sample the cells in an unbiased manner before estimating their volume (other nonstereological methods of volume estimation may yield biased estimates; Sterio, 1984; Gundersen, 1986), we reported previously that embryonic endoderm cells from PDK1(−/−) E7.0 embryos are 60% smaller than wild-type cells (Lawlor et al, 2002). In contrast, our findings showed that E7.0 PDK1(PHKI/PHKI) and PDK1(155E/155E) embryos were of similar size to their wild-type littermates (Figure 9A). Visual (Figure 9B) and quantitative stereological (Figure 9C) estimation of endoderm cell volumes revealed that neither the PDK1(PHKI/PHKI) nor PDK1(155E/155E) cells differed significantly in volume compared to their wild-type littermates. As a control we also reassessed the size of PDK1(−/−) endoderm cells and confirmed that these cells were reduced by 50% in volume compared to PDK1(+/+) cells (Figure 9).
Figure 9.
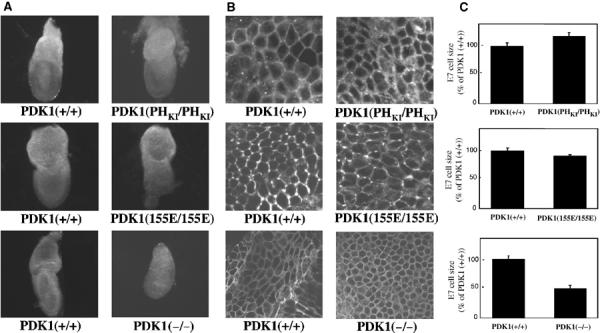
The PDK1(PHKI/PHKI) and PDK1(155E/155E) cells are of normal size. (A) The indicated E7.0 PDK1 knockin or knockout embryos were dissected in PBS and imaged on a Leica M275 microscope and whole-mount photographs were taken. Embryos are shown at the same magnification. In each case, the mutant PDK1 embryos are compared to their wild-type littermates. (B) Representative images of the indicated E7.0 embryonic endoderm cells stained with the lipid dye DilC16(3) using a Zeiss LSM510 microscope. (C) The size of the E7.0 embryonic endoderm cells was quantitated as described in Materials and methods. Three embryos of each genotype were analysed, with 120 cells in each embryo being measured and P<0.0004.
Discussion
In a previous study, we showed that PDK1(−/−) embryos died at E9.5 displaying multiple abnormalities including lack of forebrain, heart, dorsal root ganglia and somites. In addition, the PDK1(−/−) embryos did not undergo turning, possess a circulation or form a placenta. In marked contrast, both PDK1(PHKI/PHKI) and PDK1(155E/155E) embryos develop to a much greater extent and possessed a forebrain, heart, functioning circulation, somites, turn normally and form placenta. Interestingly, the PDK1(PHKI/PHKI) and PDK1(155E/155E) embryos display significantly different phenotypes. The distinctive features of the PDK1(PHKI/PHKI) embryos are formation of blood islands in the head, the lack of blood vessels in the extraembryonic membranes and lack of placental development after E9.5. In the PDK1(155E/155E) embryos, various combinations of retardation, twisting of the posterior portion of embryo and reduction in size of the forebrain vesicles were observed. The PDK1(PHKI/PHKI) mice possess normal anterior forebrain, but the posterior forebrain was reduced in size resulting in flexing of the head so that the maxillary portion of first branchial arch covers the eye.
The phenotype of the PDK1(PHKI/PHKI) embryos is unlikely to be caused by the reduction in PDK1 levels, as we previously demonstrated that hypomorphic PDK1(−/fl) embryos that possess 10-fold lower than normal amounts of PDK1 develop normally and are born at the expected Mendelian frequency. The only phenotype of these embryos and mice was a 40–50% reduction in overall size (Lawlor et al, 2002). Stereological analysis of cell size and number in a variety of organs in the hypomorphic PDK1(−/fl) mice, indicated that the reduction in mouse size and organ volume was due to a decrease in cell volume rather than a decrease in cell number. This was also confirmed by the finding that mice lacking PDK1 in striated muscle possessed smaller hearts, and that the reduction in size was caused by decreased cell volume rather than cell number (Mora et al, 2003). These results are consistent with previous genetic findings in Drosophila that PDK1 plays an important role in regulating cell volume (Cho et al, 2001; Rintelen et al, 2001).
Quantitative stereological analysis revealed that the PDK1(PHKI/PHKI) or PDK1(155E/155E) knockin cells did not differ significantly in volume compared to wild-type cells (Figure 9). This suggests that specific disruption of the PKB/S6K pathways in the PDK1(PHKI/PHKI) embryos or the S6K/RSK pathways in the PDK1(155E/155E) embryos is not sufficient to affect cell size. This is also supported by the finding that E7.0 and E8.0 PDK1(PHKI/PHKI) or PDK1(155E/155E) embryos are of normal size and display no obvious abnormalities (Figures 7 and 8). A key question concerns the mechanism by which PDK1 is regulating cell size. A striking result was that in hypomorphic PDK1(−/fl) mice, a reduction of PDK1 levels, which markedly reduced cell size, was insufficient to affect activation of PKB, S6K or RSK in response to an insulin injection in a variety of tissues (Lawlor et al, 2002). Taken together, these findings indicate that PDK1 may regulate mammalian cell size independently of the ability of growth factors to activate PKB, S6K or RSK, perhaps involving the phosphorylation of another substrate or binding partner. However, we cannot rule out the possibility that compensation for the deficient activation of an AGC kinase pathway occurs in the knockin cells, resulting in a normal cell size.
We found that RSK was activated normally in the PDK1(PHKI/PHKI) ES cells in response to TPA or serum stimulation (Figure 3). In ES cells, as in most cell lines, TPA does not trigger the activation of PI 3-kinase or PKB and inhibitors of PI 3-kinase do not affect TPA-induced activation of ERK/RSK (data not shown). This could explain why RSK activation was normal in PDK1(PHKI/PHKI) ES cells in response to TPA. We therefore also utilised a more physiological activator of RSK such as serum that induces activation of ERK and PKB. Even under these conditions, RSK activation was normal in the PDK1(PHKI/PHKI) ES cells and not affected by inhibiting PI 3-kinase with wortmannin (Figure 3B). This results establish that binding of the PH domain to PtdIns(3,4,5)P3 is not required for activation of RSK.
Much work indicates that phosphorylation of PKC isoforms by PDK1 plays an important role in phosphorylating and stabilising these enzymes (reviewed in Newton, 2003). Studies by Newton and colleagues (Gao et al, 2001) also showed that the hydrophobic motif of PKCβII is important for the activation of this enzyme by PDK1, indicating a role for the PIF pocket of PDK1 in regulating PKC isoforms. Our analysis confirms that the PIF pocket of PDK1 plays a significant role in stabilising PKCα, PKCβII, PKCγ, PKCδ, PKCɛ and PRK1, as the levels of these enzymes in PDK1(155E/155E) knockin ES cell lines is reduced to the same level as the PDK1(−/−) knockout cells (Figure 6). PKCζ and PRK2 expression may not be so dependent upon PDK1, as the levels of these enzymes are near normal in PDK1(155E/155E) and PDK1(−/−) cells. Previous in vitro and overexpression studies have indicated that PtdIns(3,4,5)P3 enhances the phosphorylation of several PKC isoforms by PDK1 (reviewed by Newton, 2003). However, with the exception of PKCβII, the levels of PKC isoforms in the PDK1(PHKI/PHKI) cells were within 70% of that of the control cells, indicating that binding of PDK1 to phosphoinositides may not be crucial for PDK1 to regulate the stability of most PKC isoforms in vivo. It is therefore likely that the effects of PI 3-kinase on PKC isoforms are mediated independently of PtdIns(3,4,5)P3 binding to PDK1.
A key result in this study is that PKB can only be activated to a low basal level in PDK1(PHKI/PHKI) ES cells, providing the first genetic evidence that the PH domain of PDK1 plays a crucial role in enabling PDK1 to activate PKB in vivo. This most likely results from PDK1 being colocalised with PKB at cell membranes of agonist-treated cells through its PH domain-mediated interaction with PtdIns(3,4,5)P3. The basal level of PKB activation observed in IGF1-stimulated PDK1(PHKI/PHKI) ES cells might be mediated through small amounts of PDK1 being randomly localised at the cell membrane or low levels of PDK1 that are associated with membranes through a non-PtdIns(3,4,5)P3-mediated mechanism.
Recent studies indicate that mTOR functions as a crucial regulator of S6K by controlling the phosphorylation of the hydrophobic motif (see Introduction and Figure 5E). mTOR activity is known to be regulated through two different pathways, one involving a PI 3-kinase and the other a nutrient sensing pathway that is independent of PI 3-kinase. As summarised in Figure 5E, recent work indicates that the PI 3-kinase-regulated input of mTOR is exerted through the PKB-mediated phosphorylation of TSC2, which then allows the activation of mTOR through a pathway involving the small Rheb GTPase (reviewed in Manning and Cantley, 2003; Shamji et al, 2003; Li et al, 2004). In serum-starved ES cells (in which PI 3-kinase and PKB are minimally active), there is a significant basal S6K activity, which is not significantly affected by wortmannin (Figure 5B). As found in all other cell lines studied, this activity is reduced to undetectable levels when amino acids are withdrawn from the medium (Figure 5D). This indicates that the basal level of S6K activity seen in serum-starved ES cells is maintained through the non-PI 3-kinase, nutrient sensing input to mTOR. Our results indicate that PtdIns(3,4,5)P3 binding to PDK1 is not required for nutrient-dependent activation of S6K, as it is activated normally by amino acids in PDK1(PHKI/PHKI) ES cells (Figure 5D). However, employing PDK1(155E/155E) ES cells, we demonstrate that the PIF pocket of PDK1 is required for amino-acid-induced activation, as S6K activity was not stimulated by amino acids in these cells (Figure 5D). This is not unexpected, as the PDK1[155E] mutant is unable to interact with S6K phosphorylated at its hydrophobic motif. Interestingly, in unstimulated PDK1(PHKI/PHKI) ES cells, the basal activity of S6K was normal, but not increased further with IGF1. This result could be explained by the low level of IGF1-induced PKB activation in the PDK1(PHKI/PHKI) knockin cells, resulting in the marked reduction of the phosphorylation of TSC2 at Ser939 and Thr1462 (Figure 4). As recent evidence supports the notion that phosphorylation of TSC2 by PKB is likely to stimulate mTOR through activation of Rheb (Manning and Cantley, 2003; Shamji et al, 2003; Li et al, 2004), we demonstrated that expression of Rheb in PDK1(PHKI/PHKI), but not PDK1(155E/155E) knockin cells, induced activation of S6K (Figure 5C). Taken together, our findings indicate that it is the deficient phosphorylation of TSC2 by PKB resulting in a lack of activation of Rheb by IGF1 that explains why IGF1 fails to stimulate S6K in the PDK1(PHKI/PHKI) knockin ES cells (Figure 5E). To our knowledge this is the first genetic analysis in mammalian cells that has enabled the relative contributions of the PI 3-kinase-dependent and -independent pathways involved in the activation of S6K to be dissected independently.
In conclusion, our studies define the crucial role that binding of PDK1 to PtdIns(3,4,5)P3 plays in activation of PKB, and hence in enabling S6K to be activated through the PI 3-kinase pathway. We also believe that this is the first study to exploit knockin technology to probe the physiological roles of phosphoinositide binding to PH domains. Our results illustrate the power of knockin technology to explore the physiological roles of protein–lipid as well as protein–protein interactions in regulating the specificity of signal transduction pathways, without having to rely on overexpression approaches.
Materials and methods
A detailed Materials and methods section is provided in the Supplementary section.
Supplementary Material
Supplementary Figure 1
Acknowledgments
We thank John Lucocq for advice on cell size determination, Maria Deak for advice in generating the targeting construct and for cloning Rheb, Julia Carr and Leah Ronaldson for genotyping, Moustapha Aoubala and Shirley Rodgie for preparation of antibodies, Calum Thompson for tissue sectioning and the School of Life Sciences DNA Sequencing Service. EJM is the recipient of an MRC Predoctoral fellowship. DRA is supported by the Association for International Cancer Research, Diabetes UK, the Medical Research Council UK, the Moffat Charity and the pharmaceutical companies that support the Division of Signal Transduction Therapy (AstraZeneca, Boehringer-Ingelheim, GlaxoSmithKline, Merck & Co. Inc, Merck KgaA and Pfizer).
References
- Alessi DR, Deak M, Casamayor A, Caudwell FB, Morrice N, Norman DG, Gaffney P, Reese CB, MacDougall CN, Harbison D, Ashworth A, Bownes M (1997) 3-Phosphoinositide-dependent protein kinase-1 (PDK1): structural and functional homology with the Drosophila DSTPK61 kinase. Curr Biol 7: 776–789 [DOI] [PubMed] [Google Scholar]
- Anderson KE, Coadwell J, Stephens LR, Hawkins PT (1998) Translocation of PDK-1 to the plasma membrane is important in allowing PDK-1 to activate protein kinase B. Curr Biol 8: 684–691 [DOI] [PubMed] [Google Scholar]
- Balendran A, Biondi RM, Cheung PC, Casamayor A, Deak M, Alessi DR (2000a) A 3-phosphoinositide-dependent protein kinase-1 (PDK1) docking site is required for the phosphorylation of protein kinase Czeta (PKCzeta) and PKC-related kinase 2 by PDK1. J Biol Chem 275: 20806–20813 [DOI] [PubMed] [Google Scholar]
- Balendran A, Casamayor A, Deak M, Paterson A, Gaffney P, Currie R, Downes CP, Alessi DR (1999) PDK1 acquires PDK2 activity in the presence of a synthetic peptide derived from the carboxyl terminus of PRK2. Curr Biol 9: 393–404 [DOI] [PubMed] [Google Scholar]
- Balendran A, Hare GR, Kieloch A, Williams MR, Alessi DR (2000b) Further evidence that 3-phosphoinositide-dependent protein kinase-1 (PDK1) is required for the stability and phosphorylation of protein kinase C (PKC) isoforms. FEBS Lett 484: 217–223 [DOI] [PubMed] [Google Scholar]
- Biondi RM, Cheung PC, Casamayor A, Deak M, Currie RA, Alessi DR (2000) Identification of a pocket in the PDK1 kinase domain that interacts with PIF and the C-terminal residues of PKA. EMBO J 19: 979–988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biondi RM, Kieloch A, Currie RA, Deak M, Alessi DR (2001) The PIF-binding pocket in PDK1 is essential for activation of S6K and SGK, but not PKB. EMBO J 20: 4380–4390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brazil DP, Hemmings BA (2001) Ten years of protein kinase B signalling: a hard Akt to follow. Trends Biochem Sci 26: 657–664 [DOI] [PubMed] [Google Scholar]
- Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME (1999) Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96: 857–868 [DOI] [PubMed] [Google Scholar]
- Cho KS, Lee JH, Kim S, Kim D, Koh H, Lee J, Kim C, Kim J, Chung J (2001) Drosophila phosphoinositide-dependent kinase-1 regulates apoptosis and growth via the phosphoinositide 3-kinase-dependent signaling pathway. Proc Natl Acad Sci USA 98: 6144–6149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins BJ, Deak M, Arthur JS, Armit LJ, Alessi DR (2003) In vivo role of the PIF-binding docking site of PDK1 defined by knock-in mutation. EMBO J 22: 4202–4211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA (1995) Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 378: 785–789 [DOI] [PubMed] [Google Scholar]
- Currie RA, Walker KS, Gray A, Deak M, Casamayor A, Downes CP, Cohen P, Alessi DR, Lucocq J (1999) Role of phosphatidylinositol 3,4,5-trisphosphate in regulating the activity and localization of 3-phosphoinositide-dependent protein kinase-1. Biochem J 337: 575–583 [PMC free article] [PubMed] [Google Scholar]
- Frame S, Cohen P (2001) GSK3 takes centre stage more than 20 years after its discovery. Biochem J 359: 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frodin M, Antal TL, Dummler BA, Jensen CJ, Deak M, Gammeltoft S, Biondi RM (2002) A phosphoserine/threonine-binding pocket in AGC kinases and PDK1 mediates activation by hydrophobic motif phosphorylation. EMBO J 21: 5396–5407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frodin M, Gammeltoft S (1999) Role and regulation of 90 kDa ribosomal S6 kinase (RSK) in signal transduction. Mol Cell Endocrinol 151: 65–77 [DOI] [PubMed] [Google Scholar]
- Frodin M, Jensen CJ, Merienne K, Gammeltoft S (2000) A phosphoserine-regulated docking site in the protein kinase RSK2 that recruits and activates PDK1. EMBO J 19: 2924–2934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao T, Toker A, Newton AC (2001) The carboxyl terminus of protein kinase c provides a switch to regulate its interaction with the phosphoinositide-dependent kinase, PDK-1. J Biol Chem 276: 19588–19596 [DOI] [PubMed] [Google Scholar]
- Gundersen HJ (1986) Stereology of arbitrary particles. A review of unbiased number and size estimators and the presentation of some new ones, in memory of William R. Thompson. J Microsc 143: 3–45 [PubMed] [Google Scholar]
- Kovacina KS, Park GY, Bae SS, Guzzetta AW, Schaefer E, Birnbaum MJ, Roth RA (2003) Identification of a proline-rich Akt substrate as a 14-3-3 binding partner. J Biol Chem 278: 10189–10194 [DOI] [PubMed] [Google Scholar]
- Lawlor MA, Mora A, Ashby PR, Williams MR, Murray-Tait V, Malone L, Prescott AR, Lucocq JM, Alessi DR (2002) Essential role of PDK1 in regulating cell size and development in mice. EMBO J 21: 3728–3738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie NR, Biondi RM, Alessi DR (2001) Phosphoinositide-regulated kinases and phosphoinositide phosphatases. Chem Rev 101: 2365–2380 [DOI] [PubMed] [Google Scholar]
- Li Y, Corradetti MN, Inoki K, Guan KL (2004) TSC2: filling the GAP in the mTOR signaling pathway. Trends Biochem Sci 29: 32–38 [DOI] [PubMed] [Google Scholar]
- Manning BD, Cantley LC (2003) Rheb fills a GAP between TSC and TOR. Trends Biochem Sci 28: 573–576 [DOI] [PubMed] [Google Scholar]
- Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC (2002) Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol Cell 10: 151–162 [DOI] [PubMed] [Google Scholar]
- Milburn CC, Deak M, Kelly SM, Price NC, Alessi DR, Van Aalten DM (2003) Binding of phosphatidylinositol 3,4,5-trisphosphate to the pleckstrin homology domain of protein kinase B induces a conformational change. Biochem J 375: 531–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mora A, Davies AM, Bertrand L, Sharif I, Budas GR, Jovanovic S, Mouton V, Kahn CR, Lucocq JM, Gray GA, Jovanovic A, Alessi DR (2003) Deficiency of PDK1 in cardiac muscle results in heart failure and increased sensitivity to hypoxia. EMBO J 22: 4666–4676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mora A, Komander D, Van Aalten DM, Alessi DR (2004) PDK1, the master regulator of AGC kinase signal transduction. Semin Cell Dev Biol 15: 161–170 [DOI] [PubMed] [Google Scholar]
- Newton AC (2003) Regulation of the ABC kinases by phosphorylation: protein kinase C as a paradigm. Biochem J 370: 361–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rena G, Guo S, Cichy SC, Unterman TG, Cohen P (1999) Phosphorylation of the transcription factor forkhead family member FKHR by protein kinase B. J Biol Chem 274: 17179–17183 [DOI] [PubMed] [Google Scholar]
- Rintelen F, Stocker H, Thomas G, Hafen E (2001) PDK1 regulates growth through Akt and S6K in Drosophila. Proc Natl Acad Sci USA 98: 15020–15025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebolt-Leopold JS, Dudley DT, Herrera R, Van Becelaere K, Wiland A, Gowan RC, Tecle H, Barrett SD, Bridges A, Przybranowski S, Leopold WR, Saltiel AR (1999) Blockade of the MAP kinase pathway suppresses growth of colon tumors in vivo. Nat Med 5: 810–816 [DOI] [PubMed] [Google Scholar]
- Shamji AF, Nghiem P, Schreiber SL (2003) Integration of growth factor and nutrient signaling: implications for cancer biology. Mol Cell 12: 271–280 [DOI] [PubMed] [Google Scholar]
- Sterio DC (1984) The unbiased estimation of number and sizes of arbitrary particles using the disector. J Microsc 134: 127–136 [DOI] [PubMed] [Google Scholar]
- Stokoe D, Stephens LR, Copeland T, Gaffney PR, Reese CB, Painter GF, Holmes AB, McCormick F, Hawkins PT (1997) Dual role of phosphatidylinositol-3,4,5-trisphosphate in the activation of protein kinase B. Science 277: 567–570 [DOI] [PubMed] [Google Scholar]
- Vitari AC, Deak M, Collins BJ, Morrice N, Prescott AR, Phelan A, Humphreys S, Alessi DR (2004) WNK1, the kinase mutated in an inherited high blood pressure syndrome, is a novel PKB/Akt substrate. Biochem J 378: 257–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volarevic S, Thomas G (2001) Role of S6 phosphorylation and S6 kinase in cell growth. Prog Nucleic Acid Res Mol Biol 65: 101–127 [DOI] [PubMed] [Google Scholar]
- Williams MR, Arthur JS, Balendran A, van der Kaay J, Poli V, Cohen P, Alessi DR (2000) The role of 3-phosphoinositide-dependent protein kinase 1 in activating AGC kinases defined in embryonic stem cells. Curr Biol 10: 439–448 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1