

Abstract
This review discusses main features of transmembrane (TM) proteins which distinguish them from water-soluble proteins and allow their adaptation to the anisotropic membrane environment. We overview the structural limitations on membrane protein architecture, spatial arrangement of proteins in membranes and their intrinsic hydrophobic thickness, co-translational and post-translational folding and insertion into lipid bilayers, topogenesis, high propensity to form oligomers, and large-scale conformational transitions during membrane insertion and transport function. Special attention is paid to the polarity of TM protein surfaces described by profiles of dipolarity/polarizability and hydrogen-bonding capacity parameters that match polarity of the lipid environment. Analysis of distributions of Trp resides on surfaces of TM proteins from different biological membranes indicates that interfacial membrane regions with preferential accumulation of Trp indole rings correspond to the outer part of the lipid acyl chain region—between double bonds and carbonyl groups of lipids. These “midpolar” regions are not always symmetric in proteins from natural membranes. We also examined the hydrophobic effect that drives insertion of proteins into lipid bilayer and different free energy contributions to TM protein stability, including attractive van der Waals forces and hydrogen bonds, side-chain conformational entropy, the hydrophobic mismatch, membrane deformations, and specific protein–lipid binding.
Keywords: protein–lipid interactions, hydrophobic thickness, polarity, database, protein stability, protein folding, membrane protein
Introduction
Biological membranes separate cells from the external environment by creating selective permeability barriers. In eukaryotic cells, membranes also divide cells into specialized compartments or organelles, such as nucleus, mitochondria, chloroplasts, endoplasmic reticulum, Golgi apparatus, lysosomes, peroxisomes, vacuoles, and transport vesicles. Different types of biological membranes grow, divide, evolve and function as complex macromolecular assemblies with specific lipid, protein, and carbohydrate compositions and distinct physical properties.
The lipid bilayer of cellular membranes is crowded with integral membrane proteins, with lipid:protein ratios ranging from 4:1 to 1:4 by mass.1 Membrane proteins may be classified as transmembrane (TM), which span the entire lipid bilayer, monotopic, which are permanently inserted into the membrane from one side, and peripheral proteins that bind to the membrane transiently. Membrane proteins participate in all vital cellular process, including protein and lipid biogenesis, cell shape regulation, transport, cell recognition and adhesion, energy production and homeostasis, signal transduction, and generation and propagation of electric impulse.
Because of the technical challenges, the pace of elucidation of three-dimensional (3D) structures of TM proteins is much slower than that of water-soluble proteins. Nevertheless, recent process in protein expression, crystallization, and the emerging crystallography on submicrometre-sized crystals has significantly accelerated membrane protein structure determination.2,3 In addition, improved NMR techniques have helped study structure and dynamics of TM β-barrels4,5 and multi-helical proteins,6,7 and allowed structural determination of dozens of oligomers formed by single-spanning TM proteins, for which crystallization usually fails to yield diffraction quality crystals.8–10
3D structures of TM proteins are provided by the Protein Data Bank (PDB).11 To date, more than 1000 distinct structures of more than 500 TM proteins and protein complexes have been determined. They are represented by ∼2000 PDB entries. Many of these structures are listed in Stephen White's database,12 the Membrane Proteins Data Bank13 and the transporter classification database.14 Three other specialized databases provide derivative PDB files with calculated locations of hydrophobic membrane boundaries: Protein Data Bank of Transmembrane Proteins,15 the Goarse-grained molecular dynamics simulation database,16,17 and the orientations of proteins in membranes (OPM) database.18 Collection of membrane proteins in specialized databases facilitates comparative analysis of membrane proteins. OPM is the most convenient resource for such analysis, because it simultaneously provides spatial positions, classification, biological origin, different conformations and oligomeric states, topology, and intracellular localization of TM, monotopic, and peripheral proteins. Spatial positions of proteins in membranes were determined in OPM by optimizing transfer energy of molecules from water to the anisotropic lipid environment.19 In addition, the underlying positioning of proteins in membranes (PPM 2.0) method provides methodology for analysis of polarity of protein surfaces.20
Adaptation of TM proteins to the lipid bilayer leads to a number of structural features which distinguish them from water-soluble proteins. Major features are briefly reviewed here, including the following: (a) limitations imposed by the lipid bilayer on protein architecture; (b) hydrophobic thickness and spatial arrangement of TM proteins in the lipid bilayer, (c) changes in TM protein surface polarity along the bilayer normal that can be described by polarity profiles; (d) the chaperone-assisted insertion and folding of proteins into membranes; (e) role of charged residues in topogenesis of TM proteins; (f) significant oligomerization propensity of proteins in the two-dimensional lipid matrix; (g) large-scale conformational transitions of proteins during their association with membranes and transport function. We also discuss physical forces that drive membrane-protein integration and specific interactions of TM proteins with each other and membrane lipids. The analysis uses 483 unique structures of TM proteins and other information provided by the OPM database (Supporting Information: Tables S1 and S2).
Structural Architectures of TM Proteins
The structural diversity of integral membrane proteins is limited by the restrictions imposed by their hydrophobic environment. TM α-helices, β-barrels, and β-helices are the only known protein architectures that fulfill the requirement to saturate the hydrogen bonding potential of their polar main-chain groups buried in membrane from water. Because of the limitations on protein structure, the probable number of membrane protein families was estimated as about 10 times fewer than that for soluble proteins.21 A recent estimate suggests that 80% of the sequence space for all polytopic membrane proteins may be covered by only ∼700 structures, while 70–80% of soluble protein domain sequences may be covered by ∼25,000 structures.22
TM α-helical proteins
The vast majority of TM proteins are α-helical (Fig. 1). These proteins are found in all types of biological membranes and are encoded by ∼25–30% genes in sequenced organisms.23 TM proteins are known as bitopic or polytopic if they cross the membrane only one or multiple times, respectively. Bitopic proteins represent the largest membrane protein class covering 15–18% and 30–47% of membrane proteomes in prokaryotes and eukaryotes, respectively.24 Genome-wide analysis of membrane proteins demonstrates that while proteins with small number of TM segments are prevalent in all organisms, prokaryotes also have a higher preference for proteins with 6 and 12 TM α-helices, and eukaryotes for proteins with 7 TM α-helices.25 It is noteworthy that proteins with even number of helices and with N- and C-termini located at the cytoplasmic membrane side are strongly overrepresented in eukaryotic and bacterial proteomes.26
Figure 1.
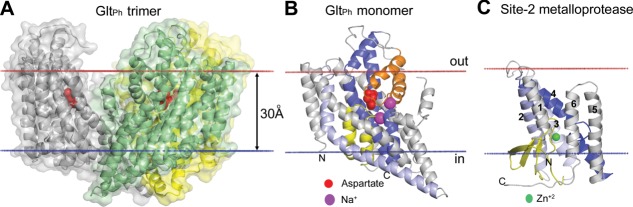
Complex structure of α-helical membrane proteins. (A) A trimer of sodium (Na+)-coupled aspartate transporter GltPh from Pyrococcus horikoshii in complex with aspartate (2NWL) has a bowl-shaped architecture with a crevice reaching halfway the lipid bilayer and open to the aqueous environment. Individual subunits in a trimer are colored grey, yellow, and green, aspartate molecules are shown by red spheres. (B) A monomer (2NWX, subunit A) has 8 TM helices and 2 reentrant loops from both membrane sides (yellow and orange). The highly tilted and bended 46 residue-long TM5 extends beyond the lipid bilayer on the cytoplasmic side (light blue). Reentrant loops and two marginally hydrophobic helices (discontinuous TM7 and TM8, blue) form the translocation pathway for substrate-aspartate (red spheres). The unwound region of TM4 defines the binding site for sodium ions (purple balls). (C) Site-2 Zn-metalloprotease from Methanocaldococcus jannaschii (3B4R) represents the TM six-helical α-bundle with three short helices (TM1, TM5, TM6) and a discontinuous TM4 helix. TMs 2–3 (light blue) and TM4 (blue) form protein core with a catalytic Zn2+ion (green sphere) in the active site, which is coordinated by two histidines and an aspartate. An unusual structural feature is a membrane-inserted four-stranded β-sheet (yellow) that occludes the active site. Calculated hydrophobic membrane boundaries from the OPM database are shown by lines: blue at the cytoplasmic side and red at the periplasmic/extracellular side.
To date more than 400 unique 3D structures of TM α-helical proteins and protein complexes are available for structural analysis (Table S1). However, the actual number of different structures is much greater due to the presence of PDB entries corresponding to different conformational, mutational, and oligomeric states, and complexes with distinct ligands or cofactors. Most structures represent prokaryotic proteins from archaebacteria (32 structures), Gram-negative bacteria (151 structures), and Gram-positive bacteria (33 structures). There are also 165 unique structures of eukaryotic TM α-helical proteins, most of which are from the plasma membrane and the endoplasmic reticulum, ER (103 and 21 structures, respectively), whereas only 7, 17, 14, and 3 structures represent α-helical proteins from the mitochondrial outer membrane (MOM), mitochondrial inner membranes (MIM), thylakoid, and vacuole/vesicule membranes, respectively.
Structural analysis27,28 of α-helical membrane proteins indicate the complexity of their architecture. TM α-helices are not necessarily straight, hydrophobic, and long enough to traverse the entire bilayer, rather they can be bent, distorted, or shorter than a half of the membrane thickness.29,30 Structural details of TM α-helical proteins have been extensively reviewed.27,31–36 Here we only briefly outline them.
TM α-helices of membrane proteins are generally hydrophobic. However, more than 30% of TM α-helices in polytopic proteins have low hydrophobicity.37–39 Such marginally hydrophobic TM segments are frequently functionally important and present in ion channels and transporters.40 Computational methods of TM helix prediction are based on search of long hydrophobic stretches and therefore often fail to recognize such helices.41,42 Though single-spanning helices of bitopic proteins are generally more hydrophobic than TM segments of polytopic proteins,43,44 around 20% of TM helices in bitopic proteins contain polar residues.24 The average number of polar residues per helix in bitopic proteins decreases from 4 in prokaryotic to 2 in eukaryotic proteins.24 The presence of polar and ionizable residues in single-spanning TM proteins was shown to promote helix–helix association.45–47
Lengths of TM α-helices range from 15 to 39 residues with an average length of ∼26 residues and preference for lengths greater than 20 residues. This differs from helices in water-soluble α-bundles that have an average length of ∼15–18 residues per helix and preference for shorter helices.32,33,48 Helices usually traverse the entire bilayer with tilt angle ranging from 5° to 35° relative to the membrane normal,31 and an average tilt 17° ± 11°.33 However, more tilted helices (tilt >45°) exist, for example, in structures of chloride-protein antiporter ClC (1KPL), sodium-hydantoin transporter Mhp1 (2JLN), sodium-dependent glutamate transporter GltPh (2NWL) [Fig. 1(A,B)], ammonia channel (1U7G), SecYEG translocon (subunit SecE, 3DIN), complex III (2FYU), ECF-transporter complex (subunit EcfS, 4HUQ, 4HZU). The very long and tilted helices usually form extensive contacts with 3–5 helices thus stabilizing protein structure.
TM α-helices may be straight or distorted due to the presence of kinks induced by Pro, tight turns of 310-helix, or wide turns of π-helix induced by insertions of a single residue into α-helix.34 Almost 40% of TM helices have irregularities, as compared to 19% in water-soluble proteins.32 TM α-bundle proteins may include a β-sheet, as it was found in structure of the site-2 meltalloproteinase (3B4R) [Fig. 1(C)]. Some α-helices are discontinuous (e.g., 3RKO), or penetrate only halfway into the membrane, thus forming reentrant loops.27,35,49 Re-entrant loops constitute a signature element in aquaporins (e.g., 3C02), voltage-gated potassium channels (e.g., 2R9R), rhomboid proteases (e.g., 4H1D), ClC H+/Cl− exchange transporters (e.g., 1kpl), H+-Glu− symporter (e.g., 3KBC), SecY translocases (e.g., 1RHZ). Exposed main-chain in discontinuous helices and non-helical regions of reentrant loops are often associated with binding sites of ions or substrates and regions with increased conformational flexibility.50
Many membrane proteins have repeated TM domains with similar tertiary structure.27 These tandem domains may have either parallel or antiparallel topology in the lipid bilayer. In the latter case, they are named inverted repeat domains. Proteins with duplicated TM domains in parallel orientation include ABC-transporters expressed as a single polypeptide (e.g., P-glycoprotein, 3G5U), and numerous secondary transporters: mitochondrial ATP-ADP carrier formed by three helical hairpins with a pseudo threefold symmetry (1OKC), different transporters from the major facilitator superfamily (1PW4, 2CFQ, 2XUT, 3WDO, 3O7Q, 3H5M, 4GC0, 4J29, 4M64) and from the resistance-nodulation-cell division51 family (2V50, 3NE5), where two halves of 12 TM α-bundle are related by a pseudo twofold axis. Among proteins with inverted repeat domains are aquaporins (1PW4), glycerol-conducting channel GlpF (1LDF), ammonia channels Amt1 (2B2F), SeqY translocase (1RHZ, 3DIN), transporters BtuCD (1L7V), ClC H+/Cl− exchange transporter (1KPL), Leu transporter LeuT (2A65), Na+/H+ antiporter NhaA (4BWZ).27,36 These proteins probably evolutionarily arise from duplication of the corresponding genes, which may lead to formation of covalently linked dual topology dimers.36,52
TM β-barrels
The second common type of TM proteins is a β-barrel (Fig. 2). TM β-barrels are abundant in outer membranes (OM) of Gram-negative bacteria, mycobacteria, mitochondria and plant plastids. In addition, various toxins create β-barrel pores in host cell membranes. A TM β-barrel can be formed either by a single polypeptide chain or by multiple chains. Currently there are 102 unique structures of TM β-barrels, among them 90 structures represent single-chain barrels and 12 structures are multi-chain barrels (Table S2). All known TM β-Barrels have the simplest sequential up-and-down topology of antiparallel β-strands. It was suggested that a β-hairpin serves as a principal evolutionary building block of such proteins.53 All TM β-barrels can be divided into three groups based on their topology and localization.
Figure 2.
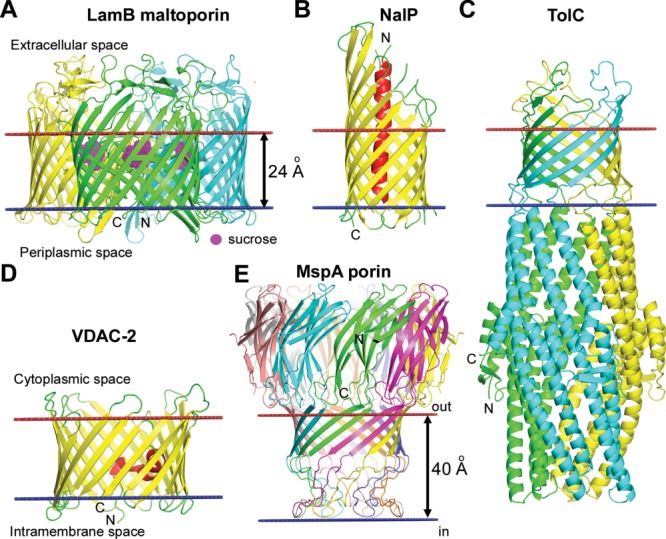
Different types of TM β-barrels. (A) Trimer of maltoporin LamB from the OM of E. coli with sucrose in the hydrophilic channel (1AF6). Each monomer of the sugar transporter represents a 18-stranded single-chain TM β-barrel with a channel that selects sugars through interaction with aromatic residues. (B) Monomer of NalP autotransporter from the OM of Neisseria meningitides (1UYO). NalP is a 12-stranded single-chain TM β-barrel with the N-terminal passenger domain (red helix) inside the pore that can be released after the self-cleavage. (C) Multichain β-barrel of TolC from the OM of E. coli (1EK9). 12-Stranded β-barrel of TolC is formed by homotrimer. (D) Single chain β-barrel of mitochondrial voltage dependent anion channel VDAC-2 from the MOM of Danio rerio (4BUM). VDAC-2 represents a 19-stranded TM β-barrel with N-terminal amphiphilic α-helix located inside the circular channel, which is involved in voltage sensing. (E) Multichain β-barrel of MspA porin from the OM of Mycobacterium smegmatis (1UUN). The stem domain of MspA represents a 16-stranded β-barrel formed by homo-octamer. Calculated membrane boundaries from the OPM database are shown by red and blue lines.
The first and the most numerous group includes single-chain β-barrels from the bacterial OM [Fig. 2(A,B)]. Around 2–3% proteins encoded by genomes of Escherichia coli and Pseudomonas aeruginosa are estimated as probable OM β-barrels.26,54,55 Structural features of OM β-barrels have been extensively reviewed56–60 and will be only mentioned here.
OM β-barrels are composed of an even number of antiparallel β-strands, and their N- and C-termini face the periplasm. β-Strands are tilted by ∼35–50° and connected via long loops on the extracellular side and short turns at the periplasmic side of the membrane. The internal pore has an oval shape with hydrophobic and aromatic residues being exposed to the lipid bilayer, while polar and charged residues face the interior of the pore. The most abundant OM proteins are porins that have 16 or 18 β-strands. Porins facilitate passive diffusion of small molecules across the membrane and can discriminate these molecules based on their size, charge, and lipohilicity. Some 8- to 12-stranded β-barrels act as enzymes, receptors, or adhesion molecules. Larger, 12- to 24-stranded β-barrels function as transporters of proteins involved in adhesion or biofilm formation. Because the OM lacks proton motive force, most β-barrels do not use energy. However, some of them can actively import molecules (e.g., iron complexes, nickel chelates, vitamin B12, and carbohydrates) against concentration gradients because they form complexes with active transporters from the bacterial inner membrane (IM). For example, 22-stranded TonB-dependent FhuA (1QFG) and BtuB (2GSK) transporters can use active transport mechanisms through interactions with TonB-ExbB-ExbD complex from the bacterial IM.61
The second group is represented by single-chain β-barrels formed by isoforms of the mitochondrial voltage-dependent anion channel (VDAC) [Fig. 2(D)], the most abundant proteins in the MOM.62 VDAC has an unusual topology and is not evolutionarily related to bacterial porins.53 Unlike bacterial OM β-barrels, VDAC-1 (3EMN) and VDAC-2 (4BUM) have an odd number of antiparallel β-strands that are closed through a pair of parallel β-strands (1st and 19th).5,63–65 Both isoforms have short loops at both membrane sides and N-terminal amphiphilic α-helix inserted into the round-shape channel.
The third group includes multi-chain β-barrels. Unlike single-chain β-barrels, they are obligatory homo- or hetero oligomers, where each subunit provides several β-strands to a β-barrel that encloses a channel with a relatively large inner diameter. The most extensively studied multi-chain β-barrels are TolC-like proteins (1EK9, 1YC9, 3PIK, 1WP1), the OM components of the bacterial Type I secretion system involved in extrusion of drugs and proteins66,67 [Fig. 2(C)]. Homotrimeric 12-stranded β-barrels were also found in bacterial autotransporter adhesins Hia (2GR7)68 and YadA (2LME).4 Another multi-chain β-barrel is the goblet-like mycobacterial MspA porin (1UUN), the most abundant protein from the OM of Micobacterium smegmatis that enable the transport of hydrophilic nutrients into mycobacteria69 [Fig. 2(E)].
Multi-chain TM β-barrels are also formed by diverse pore-forming proteins, such as mammalian perforins or certain toxins from bacteria, fungi, plants, and eukaryotic parasites.70,71 The known structures of TM β-barrels of bacterial toxins represent mushroom-shaped homo-heptamers (7AHL, 3O44, 4H56)72–74 or bicomponent octamers (3B07)75 [Fig. 3(C)].
Figure 3.
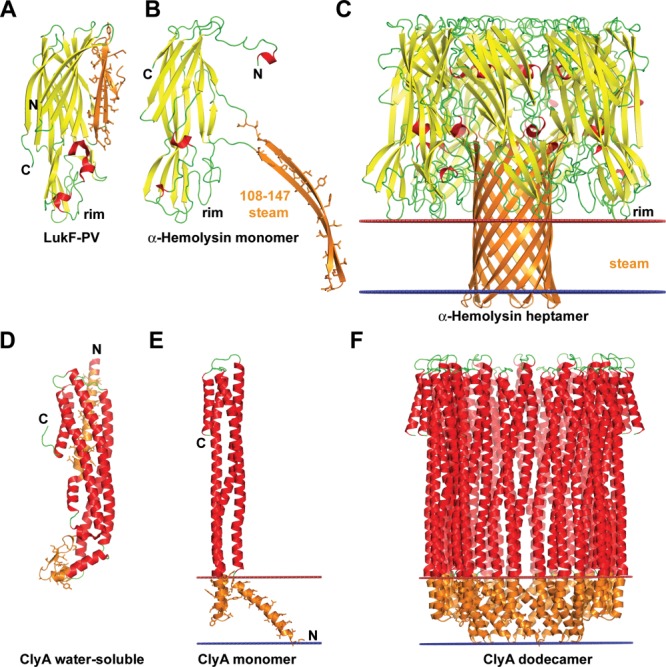
Conformational rearrangements of bacterial pore-forming toxins (PFT) upon binding to membranes. (A–C) Structures of β-PFT from Staphylococcus aureus: (A) soluble form of LukF monomer, a homologue of α-hemolysin (1PVL), (B) monomer of α-hemolysin from the oligomer (7AHL), (C) α-hemolysin heptamer (7AHL). Conformational changes in α-hemolysin include release of an amphiphilic β-hairpin (residues 108–147), oligomerization of seven molecules to create a stem domain with the central hydrophilic channel, and insertion of the stem domain into the membrane. (D–F) Structures of α-PFT from E. coli: (D) soluble form of ClyA monomer from E. coli (1QOY), (E) monomer of ClyA from the oligomer (2WCD), (F) ClyA dodecamer (2WCD). Conformational transitions of the ClyA toxin include detachment of β-tongue domain (residues 176–202) from the α-bundle, rearrangement of N-terminal α-helix, and formation of oligomers (octamers, dodecamers, or tridecamers) with circular pore inside the hydrophobic membrane core. The β-sheet is shown in yellow, the α-helices are shown in red, membrane-bound segments are shown in orange with side chains shown by sticks. Location of hydrophobic membrane boundaries are shown by lines: inner membrane leaflet is colored blue, outer leaflet is colored red.
TM β-helices
The β-Helix is another type of regular structure which can be stable in the lipid bilayer. It can be formed by peptides with alternating L- and D-amino acids, such as non-ribosomal antibiotics, gramicidin A, B, C.76,77 The cation-conducting structure of gramicidin A (1GRM) represents a TM head-to-head dimer of two right-handed single-stranded β6.3-helices (with 6.3 residues per helical turn)78–80 [Fig. 4(A)]. The size of the channel pore (diameter ∼4 Å)81 is large enough to enable the passage of monovalent cations.81,82 The gramicidin A channel is anchored to the membrane-water interface by four Trp near the C-terminus of each protomer. The hydrogen-bonding ability of Trp and dipole moment of the indole ring are essential for stabilization of the structure of the ion-conducting channel.83
Figure 4.
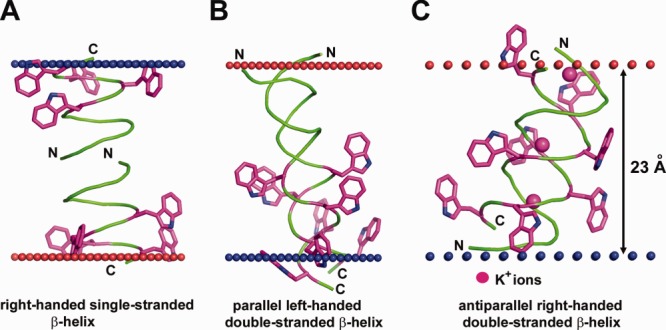
Different structures of antibiotic pentadecapeptide, gramicidin from Bacillus brevis. (A) Single-stranded right-handed β-helix of gramicidin A in the lipid bilayer (1MAG) forming a narrow ion channel with circular cross-section (diameter 4 Å). (B) Parallel left-handed double-stranded β-helix of gramicidin A in Ca+2–methanol solution (1MIC). (C) Antiparallel right-handed double-stranded β-helix of gramicidin D crystallized from methanol in complex with K+-ions (2IZQ). Insertion of gramicidin into the lipid bilayer decreases local hydrophobic thickness of the bilayer up to 20 Å and induces leakage of monovalent ions. Four Trp residues in each pentadecapeptide are shown by sticks colored purple (C-atoms) and blue (N-atoms). Calculated membrane boundaries from the OPM database are shown by red and blue spheres.
Changing the gramicidin A sequence (Trp substitutions) or environmental conditions (using organic solvents or lipid bilayers with acyl chains <10 or >20 carbons) promotes formation of very different double-stranded intertwined helices, which can be right- or left-handed with parallel- or antiparallel chains (2XDC, 2IZQ, 1MIC) [Fig. 4(B,C)]. Double-stranded helices can traverse the membrane but are unable to conduct ions.76,77,84
Folding and Targeting of TM Proteins
At appropriate conditions, a number of hydrophobic peptides,85,86 some α-helical proteins87–89 and β-barrels60 can fold and insert into the lipid bilayer independently of proteinaceous chaperones, assembly cofactors, and energy sources. However, such in vitro assembly is significantly slower and less efficient than similar processes occurring in vivo.
In vivo TM proteins fold and insert into membranes either co-translationally or post-translationally. Membrane protein insertion pathways are especially complex in eukaryotes, because the newly synthesized proteins must be targeted from ER or cytosol to the plasma membrane (PM) or different intracellular organelles. Biogenesis of membrane proteins in vivo is assisted by complex protein machineries aimed at recognition, translocation and integration of proteins to specific membranes in correct topology, conformation and oligomeric state. Here we briefly describe key TM chaperones with available 3D structures, which are involved in protein insertion into membranes.
Cotranslational protein folding and insertion into membranes
Most TM α-helical proteins insert into membranes through the co-translational pathway that involves the signal recognition particle (SRP) and Sec-translocon complex. This SRP/Sec-translocon system exists in ER of eukaryotic cells, cell membranes of Archaea and Eubacteria, thylakoid membranes, but is absent in mitochondria.90,91
SRP recognizes the cleavable N-terminal hydrophobic signal sequence or the first TM helix while it emerges from the ribosome. By interacting with the SRP receptor and the Sec-translocon, SRP targets the ribosome-nascent chain complex to the eukaryotic ER or the prokaryotic cell membrane. Structures of bacterial SecYEG (3DIN),92 archaebacterial SecYEβ (1RHZ),93 and canine Sec61αβγ (4CG5)94 demonstrate structural conservation of the SRP/Sec-translocon systems (Fig. 5). Ribosome binds directly to the Sec61α/SecY pore at the cytoplasmic site and pushes the nascent polypeptide chain through the protein translocation channel. GTP hydrolysis provides energy for chain elongation by the ribosome. A lateral gate located between TM2b and TM7 of Sec61α/SecY-translocon presumably provides exit of folded helical segments one by one or in pairs into the lipid environment.93,95–97 Insertion of chloroplast-encoded proteins (e.g., proteins from the reaction center of photosystem II) occurs from stroma into thylakoid membranes through similar cpSRP-pathway.98
Figure 5.
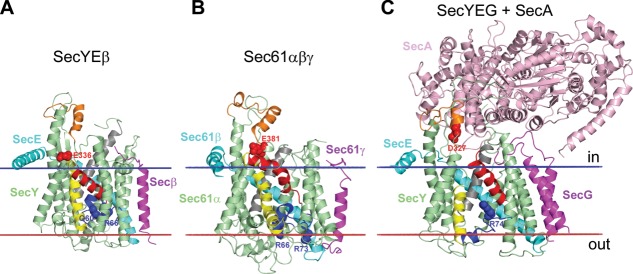
Protein translocation channels: (A) Archaebacterial SecYEβ from Methanococcus jannaschii (1RHZ); (B) canine Sec61αβγ (4CG5); (C) complex of SecYEG with SecA from E. coli (3DIN). The channel-forming Sec61α/SecY subunit has 10 TM α-helices arranged as two five-helix inverted repeats forming an hourglass-shaped channel between two repeats. The loop5–6 and loop8–9 (colored orange) contact with SecA or ribosome. The channel has a cytoplasmic funnel, the central hydrophobic ring, and external funnel filled by the TM2a helical plug (colored blue) with basic/polar residues (shown by blue spheres) that can electrostatically repel positive charges of translocated segments thus assisting in topology decision.322 Negatively charged residues from TM8 (shown by red spheres) also play topological role. In resting state (A, B), the lateral gate between TM2b (colored red) and TM7 (colored yellow) is closed, TM10 (colored gray) is moved outward, and the TM2a plug seals the pore ring, thus preventing ion leakage. In the SecYEG-SecA complex (C), insertion of two-helix finger of SecA into cytoplasmic funnel of SecY causes movement of the TM2a plug toward the periplasm and the displacement of the TM2b away from the TM7, thus creating a 5 Å-gap between these helices and providing partial opening of the translocation channel.92 In the open state, lateral gate between TM2b and TM7 is open and TM10 moves inward to the channel, which helps to release the translocated segment from channel into the lipid environment.94 Calculated membrane boundaries from the OPM database are shown by red and blue lines.
SecA ATPase assists SecYEG [Fig. 5(C)] in translocation of secretory proteins and membrane proteins with large periplasmic domains across the membrane.97 SecA-mediated translocation is stimulated by 12-helical SecDF dimer (3AQP), a member of the RND family of transporters that uses the proton-motive force99 [Fig. 6(B)].
Figure 6.
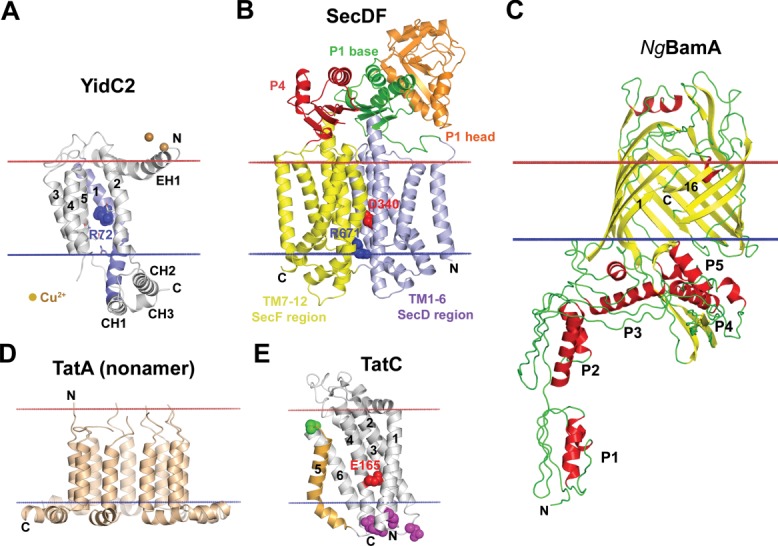
Structures of TM chaperones assisting membrane insertion of TM proteins. (A) YidC2 fragment (27–267) from Bacillus halodurans, (3WO7), an independent insertase and a membrane chaperone that cooperates with translocon SecYEG.196 YidC27–267 has five-helical TM core, N-terminal amphiphilic EH1 helix, and CH1, CH2, CH3 hydrophilic cytoplasmic helices. The long TM1 (colored blue) is bent in the middle at P77 and forms a continuous helix with the CH1 helix. TMs 1–5 form a positively charged hydrophilic grove with central R72 (blue sphere) and several solvent-exposed polar residues (shown by sticks). The grove is open to the lipids and cytoplasm and sealed toward the extracellular side. It was suggested that positively charged R72 in the grove (shown by blue spheres) transiently captures hydrophilic regions with acidic residues of translocated helices, thus facilitating their TM insertion.196 (B) Structure of SecDF (F-form) from Thermus thermophilus (3aqp), a chaperone powered by proton motive force that assists SecYEG. D340 (shown by red spheres) and R671 (shown by blue spheres) are residues involved in proton conduction.99 (C) BamA protein from Neisseria gonorrhoeae, the central component of BAM complex (4K3B). BamA, which has TM 16-stranded β-barrel and five periplasmic POTRA domains (P1-P5), coordinates recognition, folding and membrane insertion of TM β-barrels in the bacterial OM. BamA can undergo lateral opening between 1st and 16th β-strands, providing route from the interior channel to the lipid environment.323 (D) Nonamer of TatA from E. coli (2LZS), a protein-translocating element of Tat-system. TatA binds to TatBC/substrate complex and polymerizes to form substrate translocation channel. (E) TatC from Aquifex aeolicus (4B4A), a six-helical substrate-binding component of Tat-system. TatC forms complex with bitopic TatB protein via TM5 (colored orange with green spheres). Tween-arginine signal sequence of a substrate is recognized by TatC (contact region is shown by purple spheres) within TatBC complex. Bitopic TatA protein binds to negatively charged concave face of TatC with solvent-exposed E165 (shown by red spheres). Calculated membrane boundaries from the OPM database are shown by red (“out” side) and blue (“in” side) lines.
Bacterial SecYEG translocase co-operates with YidC chaperone [Fig. 6(A)] during folding and topogenesis of multi-spanning proteins, proteins complexes100–102 and proteins with large periplasmic domains.91,103 YidC can also function as an independent insertase of single and double membrane-spanning proteins (e.g., subunit C of F1F0-ATP syntase).
YidC homologues were also found in archaebacteria, mitochondria (Oxa1) and chloroplasts (Alb3). The YidC/Oxa1/Alb3 protein family mediates assembly of major energy-transducing protein complexes.102 Mitochondrial Oxa1, which functions as a voltage-gated and substrate-dependent translocase,104 is involved in co-translational insertion of polytopic TM proteins encoded by mitochondrial genome (13 core subunits of respiratory complexes I, III, IV and V) from the matrix to the MIM.105
In addition, the SecYEG-YidC machinery may co-operate with the TatABC-system [Fig. 6(D,E)] during integration of polytopic TM proteins with large globular domains bearing diverse cofactors.106 It was proposed that some membrane proteins, such as Rieske iron-sulfur protein, are initially inserted into membrane by the Sec-machinery and then use Tat-complex to translocate folded globular domain containing Fe-S cluster.107
Post-translational targeting, folding and membrane insertion
In contrast to the majority of TM α-helical proteins, which insert co-translationally, β-barrels fold and insert into membranes via post-translational pathways. The post-translational integration with membranes also occurs for C-tail anchored proteins (TA) and nuclear-encoded TM α-helical proteins targeted to mitochondria and chloroplasts.
In Gram-negative bacteria, the biogenesis of OM β-barrel proteins includes several steps108,109: (i) SecYEG-SecA-dependent translocation across the IM assisted by SecB chaperone; (ii) transit through the periplasm assisted by chaperones Scp, DEgP and SurA; and (iii) folding and insertion into the OM by the β-barrel assembly machinery (BAM) formed by BamA 16-stranded TM β-barrel and four accessory lipoproteins (BamBCDE). Comparison of available BamA structures (4K3B and 4K3C) suggests that the lateral gate from the protein interior to the lipid bilayer may be open due to transient separation of the 1st from the 16th β-strand [Fig. 6(C)].
C-tail anchored proteins have a large water-soluble N-terminal domain and a single C-terminal TM α-helix. Because the C-terminal membrane anchor emerges from the ribosome after termination of translation, it cannot interact with SRP and Sec-system and uses post-translational insertion pathways that involve cytosolic chaperones and specific membrane receptors.110,111 It was also suggested that TA proteins may require chaperone assistance only for delivery to the targeted membrane in an insertion-competent form (to prevent aggregation), but not for insertion into membranes.110
The nuclear-encoded mitochondrial preproteins are targeted to the organelle by either N-terminal cleavable presequences or internal targeting signals with medium hydrophobicity and positively charged flanking residues.112–115 Proteins targeted to the MOM usually have internal targeting signals, proteins targeted to the intermembrane space have “classical” amphipathic signals, while proteins directed to the MIM or matrix have either bipartite signals (amphipathic presequence followed by hydrophobic sorting sequence) or internal signals.114 Proteins in unfolded state are translocated across MOM by the Translocase of Outer Mitochondrial membrane (TOM)-complex composed of several receptors and TOM40 β-barrel channel.114,116,117 Almost all mitochondria-targeted proteins are recognized and transferred across the MOM by TOM complex, except TA and signal-anchored proteins, which insert into MOM spontaneously. Membrane insertion of these proteins depends on the lipid composition of target membranes, for example, on the presence of ergosterol and cardioplipin.118–120 TOM complex also assists insertion into MOM of some bitopic and polytopic proteins.117 β-barrels (VDAC, TOM40, and SAM50) are inserted into MOM from the intermembrane space by the Sorting and Assembly Machinery (SAM)-complex which is similar to bacterial BAM-complex.114,117
Protein translocation and insertion into MIM is assisted by translocases of the inner membrane (TIM) complexes. TIM22 complex inserts multi-spanning proteins containing internal signals, such as ADP/ATP and phosphate carriers.116 The twin-pore carrier TIM22 is an essential integral protein of the complex. It forms a hydrophilic high conductance channel activated by the inner membrane potential Δψ and regulated by internal targeting peptides.121 TIM23 complex interacts with presequence-translocase-associated import motor (PAM) complex. Together, they translocate proteins with cleavable presequences into mitochondrial matrix or insert them into MIM. TIM23 complex uses both the electric membrane potential Δψ and the ATP-dependent activity of PAM as driving forces for translocation of preproteins across MIM.120 Oxa1 may assist TIM23 complex in import of multi-spanning MIM proteins.114
Plant chloroplasts have three distinct membranes: the outer, the inner and the thylakoid ones. Targeting to chloroplasts is guided by cleavable N-terminal transit sequences or by internal sequences included into mature proteins.115,122 Proteins targeted to thylakoid lumen have bipartite presequences that contain a tandem of stromal and luminal sorting signals. Interior chloroplast proteins are translocated to stroma across both the outer and inner envelope membranes by the Translocons of the Outer and Inner envelopes of Chloroplast (TOC and TIC). They use GTP and ATP as energy sources.123,124 After translocation to the stroma and cleavage of the transit sequence, TM proteins can insert into thylakoid or inner envelope membranes. Proteins destined to thylakoid membranes can use one of three chaperone-assisted pathways (cpSec-, cpSRP-, or DpH-) or insert spontaneously.125,126 The first, cpSec-pathway requires SecYE, SecA proteins and ATP for translocation of proteins targeted to the lumen or TM proteins with large globular domains. The second, cpSRP-pathway works post-translationally and independently of cpSecY. It requires two SRPs with their receptors and Alb3.1 protein.127 This pathway is engaged in transport of the highly abundant nuclear-encoded light harvesting chlorophyll a/b-binding proteins (LHCPs) (1rtw).128 The third, DpH-dependent pathway uses the proton gradient (ΔpH and Δψ) as a driving force. It provides translocation of presequences with twin-arginine motif to the lumenal side of thylakoids (e.g., PsaN and PsbT from Photosystem II).129,130 DpH-system components are homologous to components of the bacterial Tat-system.129 Finally, the spontaneous insertion is restricted to a few single-spanning membrane proteins (CFoII of the ATPase, and PsbX, PsbW of the photosystem II) and some polytopic proteins (Elip2, PsbS).90
Spatial Arrangement in Membranes and Hydrophobic Thickness of TM Proteins
Each TM protein has large continuous non-polar surface that penetrates to the lipid acyl chain region. To provide burial of this non-polar surface from water, a TM protein acquires a certain spatial position in the lipid bilayer. This position can be investigated experimentally or calculated by minimizing transfer energy of the protein structure from water to membrane.131
Each TM protein has an additional important geometrical parameter: its intrinsic hydrophobic thickness, which can be defined as the width of a slice enclosed by two parallel planes separating the non-polar and polar regions. Localization and concentration of proteins with similar hydrophobic thicknesses in a certain membrane type may regulate the membrane width.132 Therefore, average hydrophobic thickness of TM proteins is an appropriate characteristic of a particular membrane type: proteins from different membrane types tend to have distinct intrinsic hydrophobic thicknesses (Fig. 7). For example, average hydrophobic thicknesses of α-helical proteins from eubacterial and archaebacterial cell membranes, eukaryotic ER, and thylakoid membranes are close to 31 Å. Average hydrophobic thicknesses are significantly larger for eukaryotic PM proteins (∼33 Å), but smaller for proteins from MIM (∼28 Å). This is in agreement with X-ray scattering data.132 The gradual decrease of hydrophobic thicknesses of proteins targeted to different membranes (PM>ER>MIM) may facilitate protein sorting during membrane biogenesis. Indeed, the post-translational targeting and unassisted insertion of TA proteins into MOM requires short and moderately hydrophobic helices with positively charged flanking residues, while ER-targeting is driven by longer and more hydrophobic helices.110,133
Figure 7.
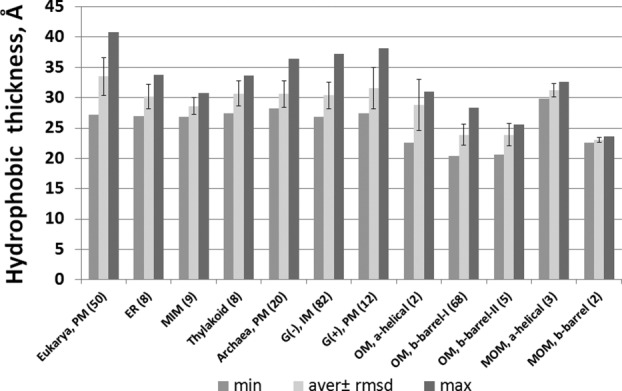
Intrinsic hydrophobic thicknesses of α-helical and β-barrel TM proteins from different membrane types: eukaryotic plasma membrane (PM), endoplasmic reticulum (ER), mitochondrial outer (MOM) and inner (MIM) membranes, PM of Archaebacteria and Gram-positive bacteria, outer (OM) and inner (IM) membranes of Gram-negative bacteria. Numbers of protein structures in each protein set are indicated in parenthesis (adopted from Ref.20).
All β-barrel proteins from the OM of Gram-negative bacteria and the MOM have significantly smaller hydrophobic thickness (∼24 Å) (Figs. 2 and 7), in agreement with NMR studies of detergent-embedded residues of OmpX, analysis of the thickness of detergent belt in crystals of OMPLA, and neutron scattering studies of LPS bilayers.134–138 In contrast, the stem domain of β-barrel porin MspA from the outer mycobacterial membrane has unusually large hydrophobic thickness of ∼40 Å [Fig. 2(E)], consistent with the expected thickness of this membrane, as it contains up to ∼60% of long-chain (C30 to C90) mycolic acids.139
Polarity Profiles, Membrane Interface, and Midpolar Regions
The lipid bilayer cannot be viewed merely as a hydrocarbon slab. This is a complex anisotropic environment that can be described by profiles of polarity and lateral pressure along the bilayer normal.140 While a protein in water is under isotropic pressure, a TM protein experience negative pressure at the level of lipid carbonyls, which is counterbalanced by positive pressure at the lipid head-group and acyl chain regions.140 The lateral pressure profiles in membranes cannot be directly measured; however, they can be estimated from MD simulation of artificial lipid bilayers.141–143
Polarity, elastic, and geometrical properties of artificial lipid bilayers can be experimentally obtained using scattering techniques.144–147 For example, the polarity of a fluid lipid bilayer can be quantified by a few parameters commonly used to describe solubility of molecules in organic solvents: hydrogen bonding donor and acceptor capacities (α and β) and dipolarity/polarizability parameter (π*).19 The transbilayer profiles of these parameters have been recently calculated for several single- and multi-component lipid bilayers using distribution of lipid fragments obtained from neutron diffraction and X-ray scattering data20 [Fig. 8(A,B)].
Figure 8.
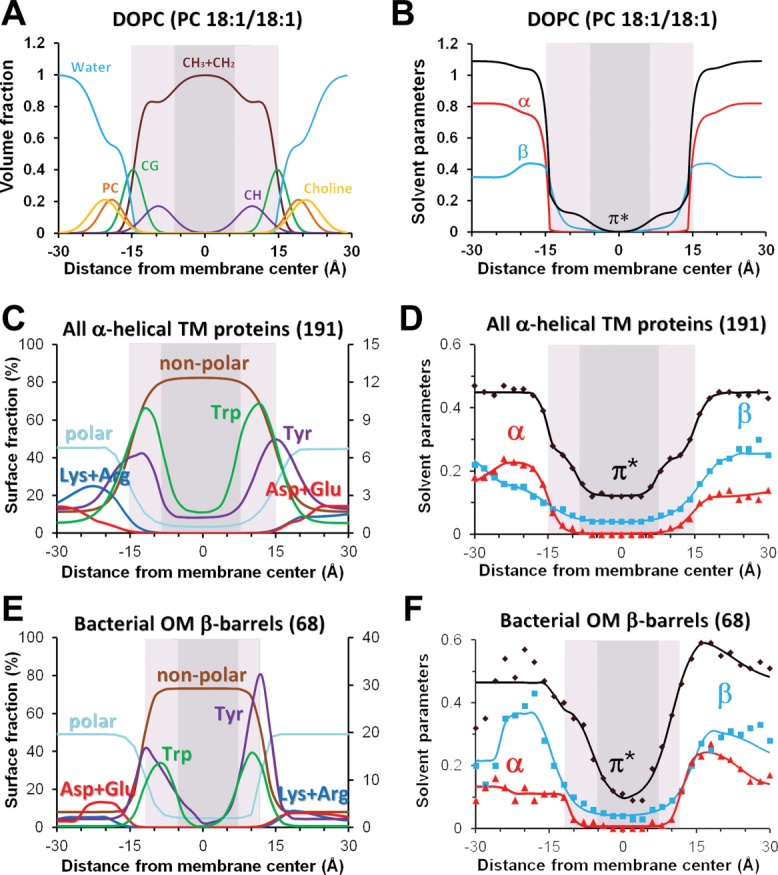
Distributions of chemical groups and polarity profiles in artificial lipid bilayers (A, B) and at the lipid-facing surfaces of TM proteins (C-F). (A) Volume fractions of lipid segments determined by X-ray and neutron scattering for fluid DOPC membrane.324 (B) Profiles of hydrogen-bonding donor (α) and acceptor (β) capacities and dipolarity/polarizability parameter (π*) calculated for DOPC bilayers.19 (C, E) Distribution of lipid-facing atoms in structures of 191 TM α-helical proteins from all membranes (C) and 68 OM β-barrels (E): polar atoms (N- and O-atoms) of all residues, non-polar atoms (C- and S- atoms of Val, Leu, Ile, Met, Cys, Phe, Tyr, and Trp), aromatic atoms and C-atoms from benzene ring of Tyr and indole ring of Trp, charged groups of Asp/Glu and Arg/Lys. (D, F). Transbilayer profiles of polarity parameters (α, β, and π*) were calculated for lipid-facing atoms on surface of TM α-helical (D) and OM β-barrel proteins (F) (see Ref.20 for details). Midpolar regions (colored light gray) were mapped as area of preferential accumulation of Trp indole rings in TM proteins (C–F), and based on locations lipid double bond in artificial bilayers (A,B). Central hydrophobic regions are colored dark gray.
Unfortunately, the polarity of natural biological membranes cannot be determined in the same way, because they have a highly diverse and yet unidentified lipid and protein composition. Instead, one can analyze hydrophobic thickness and polarity of surfaces in 3D structures of TM proteins, which is expected to match polarity of surrounding lipids. Indeed, all TM proteins have a central hydrophobic zone composed of aliphatic residues and two polar zones that contain numerous ionizable residues and crystallized water. Arg and Lys residues are embedded in the lipid head-group area, where they can form H-bonds and ion pairs with lipids. The intermediate region is marked by belts of Tyr and Trp residues that favorably interact with the membrane interface.148–153
Hence the polarity of protein surfaces may be described by distributions of different types of amino acid residues, such as polar, non-polar, aromatic, charged, and so forth. However, it can be described more quantitatively by calculating profiles of polarity parameters α, β, and π*. Unlike distributions of residues, these polarity profiles represent integral quantitative characteristics that account for hydrogen-bonding capacities and dipolarity/polarizability of atoms on the protein surface.
The corresponding polarity profiles were recently obtained for TM proteins from different biological membranes, including bacterial, archaebacterial and eukaryotic plasma membranes, inner mitochondrial, thylakoid, and bacterial outer membranes.20 As expected, the polarity of protein surfaces (described by α, β, and π*) changes gradually [Fig. 8(D,F)] and correlates with properties of the surrounding lipid bilayer. Polarity profiles can be approximated by sigmoidal curves whose midpoints coincide with midpoints in distributions of co-crystallized water molecules, with maxima in distributions of carbonyl groups from co-crystallized lipids, and match positions of calculated hydrophobic membrane boundaries. Interestingly, polar groups of Trp and Tyr residues of TM proteins often point to membrane boundaries where they may form hydrogen bonds with glycerol groups of co-crystallized lipids. However, distributions of aromatic rings of Tyr and Trp residues do not coincide: while maxima of distribution of Tyr benzene rings correspond to the lipid carbonyl region, maxima of distributions of Trp indole rings are shifted by 3–5 Å closer to the membrane center [Fig. 8(C,E)]. This tendency was observed for all proteins from different membrane types, including α-helical and β-barrel TM proteins.
Polarity profiles calculated for proteins from different membrane types appear to be quite similar at the center of membranes (Fig. 9). Thus, central regions of proteins derived from membranes with dissimilar composition demonstrate comparable hydrophobicity. This observation may explain the well-known tolerance of TM proteins to alteration of the lipid composition in native cells, in artificial bilayers, and during protein expression in different host organisms.154 Larger distinctions in polarity profiles are observed near hydrophobic boundaries and in the lipid head group areas. These dissimilarities may correlate with the large diversity of the lipid head-groups in different membranes though may also reflect biases associated with mechanisms of protein insertion into membranes.
Figure 9.
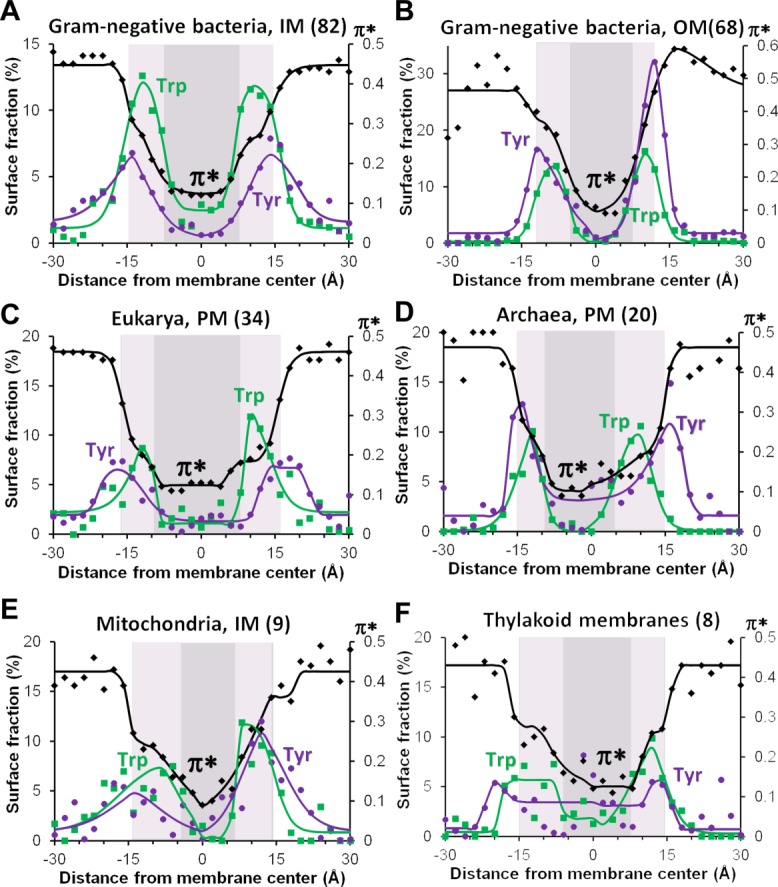
Comparison of surface polarity in TM proteins from different biological membranes: IM (A) and OM (B) of Gram-negative bacteria, PM of eukaryotic (C) and archaebacterial (D) cells, mitochondrial inner membranes (E) and thylakoid membranes (F). Transbilayers distributions of aromatic and C-atoms of benzene ring of Tyr (purple lines and dots) and indole ring of Trp (green lines and squares) are compared with profiles of dipolarity/polarizability parameter π* (black lines and diamonds) calculated from distributions of all solvent-exposed atoms in proteins from different membranes. Numbers of structures in each protein set are indicated in parenthesis. Midpolar regions mapped by the preferential accumulation of Trp indole rings are colored light gray, central hydrophobic regions are colored dark gray (see the set of proteins and details of calculations in Ref.20).
A closer examination of polarity profiles in TM proteins (Figs. 8 and 9) shows that the central, 30–31 Å-wide, non-polar region is not a uniform environment, but includes two peripheral, 5–8 Å-wide zones with increased dipolarity/polarizability parameter (π*) and hydrogen-bonding acceptor capacity (β). These peripheral zones with intermediate polarity can be named “midpolar” regions.155 In native biological membranes, location of “midpolar” regions can be deduced from the changes of polarity parameter π*, or mapped by distribution of Trp indole rings in TM proteins, which can be regarded as a reporter group. Preferential accumulation of the Trp indole rings in the area between lipid carbonyl groups and double bonds indicates a relatively small electrostatic penalty for the Trp dipole and a higher dielectric constant there, which may be partly attributed to the presence of residual water.
Depths of “midpolar” regions, as deduced from locations of Trp residues, depend on the type of membrane and may be different on the inner and outer sides. For example, the polarity profiles of proteins from the bacterial OM demonstrate significant asymmetry of their “midpolar” regions. This correlates with asymmetric lipid composition in OM, where the inner leaflet is composed of phospholipids, but the outer leaflet is formed by lipopolysaccharides (LPS).156 In contrast, proteins from the bacterial IM, which are composed entirely of phospholipids, show symmetric polarity profiles. Some degree of asymmetry in the “midpolar” regions was also observed for proteins from the eukaryotic and archaebacterial PM, MIM, and thylakoid membranes (Fig. 9). This observed asymmetry of polarity in eukaryotic PM proteins correlates with non-uniform distribution of different lipid species and cholesterol between leaflets of eukaryotic cell membranes.157,158
Topogenesis of TM α-Helical Proteins and Role of Charged Residues
In addition to the spatial arrangement in membranes and hydrophobic thickness, an important structural parameter of TM proteins is topology, or placement of their termini on the certain side of the membrane. This is another feature that distinguishes them from water-soluble proteins. TM proteins usually acquire unique topology, although there are rare examples of proteins with dual topology.37 For example, the bacterial multidrug transporter EmrE functions as an antiparallel homo-dimer of monomers with opposite topologies (3B5D).159,160 Based on their orientation in membranes, bitopic proteins were classified to topological classes I (with N-terminus facing to the outer space and N-terminal cleavable signal peptide), II (with N-terminus facing the inner space), and III (with N-terminus facing to the outer space), while all polytopic proteins are assigned to class IV regardless of their topology.1,161
The topology of TM α-helical proteins is usually defined during their co-translational insertion, folding, and assembly in membranes.37 Topogenesis of multi-spanning proteins may involve significant rearrangement and reorientation of helices during protein folding162–166 and post-translational insertion of reentrant loops.163 For example, marginally hydrophobic helices can be released from Sec-translocon peripherally in the lipid head group region and be refolded later in the course of helix assembly.163 Membrane insertion and spatial orientation of such helices may be promoted by adding charged or hydrophobic residues to the flanking regions, or guided by interactions with adjacent TM helices and their topological preferences.38,40,44,167,168 For example, insertion of highly charged S4 helix of voltage-sensing domain of the Shaker and KAT1 voltage-gated K-channels is assisted by electrostatic interactions with neighboring helices.169
The topology of proteins with co-translational membrane insertion depends on a number of factors. Among them are properties of TM segments (length, hydrophobicity, hydrophobic gradient of helical segment, position of aromatic residues near TM helix ends, charge difference at both ends of helix), folding and glycosylation of extramembrane domains, membrane electrochemical potential, and protein–lipid interactions.170–172
However, two major topological determinants are the hydrophobicity of TM segments and the asymmetric distribution of positively charged residues (Arg and Lys) in polar regions located at both membrane sides, the so-called positive-inside rule.173,174 This rule was suggested based on statistical analyses of amino acid residue distributions in TM α-helical proteins from Eubacteria, Archaea, and Eukarya (including proteins form mitochondria and thylakoids).161,174–181 Such analysis exposed the significant prevalence of positively charged residues in loops located on the side of the membrane from which proteins are inserted (the cis side or “inside”), which is a cytoplasmic side of bacterial IM, eukaryotic PM and ER, the stromal side of thylakoids, and the matrix side of MIM. The “inside” bias of positively charged residues appear to be stronger for prokaryotic proteins.174 Interestingly, the positive-inside rule is applicable to mitochondria-encoded MIM proteins, but not to nuclear-encoded MIM proteins which show more symmetric distribution of positively charged residues on both membrane sides, but significant “outside” bias for negatively charged Glu residues.180
Distributions of positive charges in 3D structures of TM proteins are consistent with the “positive inside” rule for α-helical proteins from all membranes, no matter whether one considers all protein residues182,183 or only solvent-accessible ones.20 Moreover, not only the distributions of cationic residues, but also distributions of net charge follow this pattern in protein structures from nearly all membranes: it is positive on the inner membrane side and close to zero or slightly negative on the outer side. Two exceptions are proteins from MIM and thylakoid membranes: MIM proteins have a positive net charge on both membrane sides, though it is larger at the inner leaflet, while thylakoid TM proteins have rather similar numbers of positively and negatively charged residues at each side, and therefore no pronounced maxima for net charge (Fig. 10).
Figure 10.
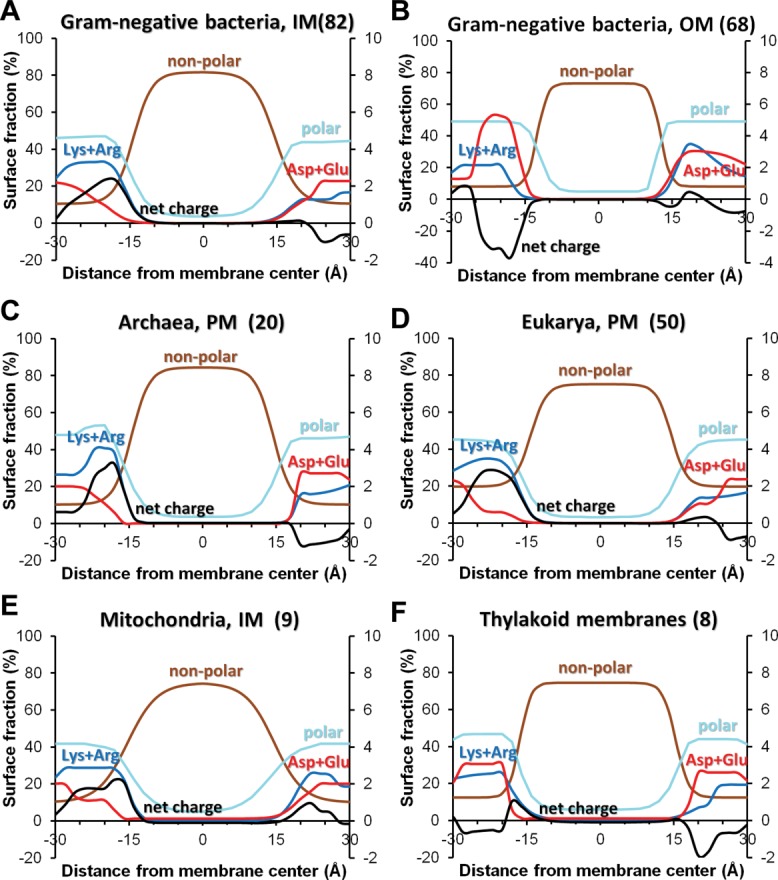
Distribution of lipid-facing atomic groups and charges in TM proteins from different biological membranes: IM (A) and OM (B) of Gram-negative bacteria, PM of archaebacterial (C) and eukaryotic (D) cells, mitochondrial inner membranes (E) and thylakoid membrane (F). Distribution of polar atoms (N- and O-atoms) of all residues are shown by light blue lines, non-polar atoms (C- and S- atoms of Val, Leu, Ile, Met, Cys, Phe, Tyr, and Trp) are shown by brown lines, positively charged groups of Arg and Lys are shown by blue lines, negatively charged groups of Asp and are shown by red lines, net charges are shown by black lines. Numbers of structures in each protein set are indicated in parenthesis (see the set of proteins and details of calculations in Ref.20).
Maxima of distributions of positively charged groups are located at approximately 20–22 Å-distance from the bilayer center in both membrane leaflets. This corresponds to locations of phosphodiester groups of phospholipids that may form ionic pairs with Arg and Lys residues. The unusual lack of maxima of net positive charges in proteins from thylakoid membranes may be explained by the unique membrane composition of these membranes. In thylakoid membranes, phosopholipids are mainly substituted by non-phosphorous glycolipids,184 which significantly decreases the amount of lipid groups involved in ionic interactions with basic protein residues.
The role of positive charges as topological determinants was experimentally supported for both signal peptides and integral membrane proteins. For example, it was found that adding or removing of a single basic residue at critical locations can invert the topology of individual TM segments,185 or entire proteins.164 Placing charged residues downstream of a signal sequence arrested protein chain movement through Sec-translocon186 and blocked protein secretion.187 This “charge block” effect was more pronounced in prokaryotic than eukaryotic systems, which can be attributed to the presence of an electrical potential (positive outside) across the bacterial IM.187
Acidic residues may also affect topology of proteins inserted by Sec-translocon, but to a lesser extent than basic residues, and only if present in high numbers.185 It was suggested that negative charges around hydrophobic segments play only a secondary role in fine-tuning the topology in Sec-dependent membrane insertion.185 In contrast, acidic residues in periplasmic regions of double-helical M13 and single-helical P3f coat proteins appear to be important for Sec-independent translocation/insertion that requires the proton motive force.188–190 Consistently with smaller topological role of acidic residues, it was found that distributions of these residues in sequences of membrane proteins is generally symmetric174 and show only moderate preference of Glu on the trans (“outside”) side of the MIM.161,180 Likewise, analysis of 3D structures of membrane proteins showed almost symmetric distribution of negatively charged residues at the both membrane sides.20,182,183
Physical mechanisms behind the positive-inside topological rule are not completely understood. It may be possible that the retention of positively charged residues on the cytoplasmic side partially results from their interactions with acidic phospholipids at the cytoplasmic leaflet191 or from the influence of the electrochemical potential on the bacterial IM (inside negative).190,192 However, the same positive-inside bias was observed for proteins that reside in membranes of acidophilic archaebacteria with reversed Δψ (inside positive).193 Hence the positive-inside rule is not strictly connected to the sign of the transmembrane potential. Instead, it is likely attributed to structural details of the Sec-translocation channel and YidC chaperone. Indeed, mutagenesis studies of the Sec61p-translocon demonstrated that orientation of signal sequence can be changed by reversing the positively charged residues in the TM2a plug (R67E, R74E) or by elimination of a negatively charged residue at the cytoplasmic end of the TM8 (E382Q)194,195 (Fig. 5). The insight on the topological role of acidic residues of single-spanning proteins was obtained from the inspection of the crystal structure of the bacterial YidC196: the translocation of N-terminal acidic residues flanking the single TM helix may be directed by conserved positive charge located in the hydrophilic grove of the YidC [Fig. 6(A)].
Unlike TM α-helical proteins, single-chain TM β-barrels from the bacterial OM demonstrate opposite “positive-outside”197 trend in distribution of solvent-exposed Lys and Arg residues [Fig. 10(B)]. Basic residues are highly abundant in outside-facing loops, where they interact with negatively charged LPS from the outer leaflet. In contrast, acidic residues occur much more frequently in periplasmic turns of β-barrels (“negative-inside” rule),20 where they interact with cationic periplasmic chaperones (e.g., Skp) that assist in folding and insertion of β-barrels into the OM.198
Oligomerization of TM Proteins
TM α-helical proteins frequently form dimers or higher order oligomers in membranes.199 The majority of membrane protein complexes (50–70%) are homo-oligomers,200,201 where homo-dimers represent the largest fraction (>65%).202 The prevalence of homomers is also observed in available structures of α-helical TM proteins, where homo-dimers and higher order homo-oligomers account for ∼20% and ∼30% of unique protein structures, respectively (Table S1), while hetero-oligomers account for ∼19% of structures. Large membrane protein complexes may also include peripheral proteins, for example in signaling cascades and electron-transfer chains.
Though oligomerization is a common property of both water-soluble and membrane proteins,203 the presence of a lipid bilayer reduces the area accessible to diffusion of proteins and therefore promotes non-specific aggregation due to several factors: increased concentration of proteins in two-dimensional space, microphase separation and lateral segregation of lipids, and formation of lipid rafts. The lipid bilayer also restrains positions of TM helix termini and forces helices to adopt nearly parallel or anti-parallel arrangements.
All functional protein–protein association in membrane is driven primarily by specific interactions, rather than these non-specific factors. Specific interactions may involve non-covalent binding of extramembrane and/or TM domains, domain swapping and even covalent linking.199,203–206
High affinity lateral association of TM α-helices critically depends on packing of complementary helical interfaces that may contain specific dimerization motifs or polar residues involved in interhelical salt bridges or hydrogen bonds.45,207–211 The extent and structure of TM protein aggregates may also depend on membrane curvature,212 be modulated by physico-chemical properties of the lipid bilayer213 and affected by the environmental conditions (pH, presence of ions, organic or macromolecular ligands, lipids, and detergents).214–216
While the physiological impact of experimentally observed transient oligomerization of some proteins, such as GPCRs, is not completely understood,217 in many cases, the functional significance of stable multi-protein complexes has been well-established. It is known that protein association increases protein stability, generates TM conductive pores (ion channels, bacterial secretion systems, toxins), forms ligand-binding sites at the oligomer interface, allows cooperativity between subunits, and provides an additional level of regulation.218
Some structural units are stable and functional in membranes only as oligomers.199,218 Among them are: α-helical dimers of potassium uptake proteins (3UM7), ABC transporters (2QI9, 3G5U, 3PUW, 4AYT), EmrE multidrug transporter (3B5D), MgtE transporter (2YVX), trimers of acid-sensing and ATP-gated channels (3H9V, 4NTX), tetramers of voltage-, ligand-, Ca2+-, or H+-gated (2R9R, 3EFF, 3LDC) potassium channels, pentamers of ligand-gated channels (3rhw), hexameric Gap junction-forming connexins (2ZW3) and CRAC-channels (4HKR), 4-, 5-, 7-meric mechanosensitive channels (2OAR, 2VV5, 3HZQ), F-, V-, and A-type proton and sodium-translocating ATPases with 10 to 15 subunits (1YCE, 2BL2, 2X2V, 2XQU, 3V3C, 4B2Q), octameric α-helical barrels of Wza (2J58), 14-meric α-helical core complex of type IV secretion system (3JQO), trimeric β-barrels of TolC-like bacterial proteins (1EK9), 12-meric α-pore-forming ClyA toxin (2WCD), and heptameric haemolysin (7AHL) or octameric (3B07) β-pore-forming toxins [Figs. 2(C,E) and 3(C,F)].
Large-Scale Conformational Transitions
TM proteins seem to undergo significant conformational transitions more frequently than water-soluble proteins due to several reasons. First, co-translational and post-translational integration of proteins in membranes frequently leads to significant conformational changes. These changes have been extensively studied for antimicrobial, cell-penetrating and fusogenic peptides, bitopic proteins, and pore-forming toxins.8,205,219–223 For example, membrane binding and insertion of many biologically-active peptides is accompanied by folding and stabilization of their regular secondary structure, the so-called partitioning-folding coupling.224,225 Pore-forming toxins also undergo large conformation rearrangements and oligomerization when converted from the water-soluble form to membrane-integrated pores.71 After binding to specific receptors (e.g., lipids, proteins, sugars, GPI anchors) in target cell membranes, these water-soluble toxins undergo activation, oligomerization, and folding into α-helical or β-barrel structures enclosing large (up to ∼26 Å inner diameter) water-filled channels.71 Bacterial, viral, and eukaryotic toxins can form different types of pores that either destroy membranes or facilitate toxin delivery into host cells. Though molecular details of toroidal pores that can be formed by actinoporin-like toxins are still poorly understood, structures of pores formed by α-helical dodecamer of E. coli ClyA toxin (2WCD) or by multi-chain β-barrels of bacterial α-, and γ-hemolysins (7AHL, 3O44, 4H56)72–74 and bicomponent leukoltoxin (Luk) (3B07)75 have been resolved. Comparison of structures of water-soluble and membrane-bound forms of pore-forming toxins uncovers the magnitude and nature of structural rearrangements during protein activation, oligomerization and insertion into the lipid bilayer (Fig. 3).
Furthermore, the large-scale conformational transitions between multiple states (“open,” “closed,” “inward-facing,” “outward-facing”) is a signature of proteins involved in transfer of various substances or signals across the membrane: receptors, channels, and transporters. Conformational transitions may include significant movements of “plug” and other structural elements and changes in positions of individual subunits, domains, or entire protein complexes with respect to the lipid bilayer. Structural transitions in secondary and primary transporters induced by binding of ligands, lipids, and nucleotides involve domain reorientation and movement of helical pairs.27,226–229 Rigid-body helix movements can be depicted as translation, piston, rotations parallel (pivot) and perpendicular to the membrane.230
For example, large protomer reorganization and reorientation in membranes occurs in energy-coupling factor (ECF), a member of ABC transporter superfamily. ECF transporters are composed of two TM domains (EcfT and EcfS) and two nucleotide-binding cytoplasmic domains (EcfA and EcfA').231,232 Substrate-binding domains EcfS are individually stable in membranes and may exist either independently (3RLB, 3P5N, 4DVE) or as part of a tetrameric complex (4HZU). Surprisingly, TM helices of EcfS are oriented along the membrane normal in isolated subunit, but almost perpendicular to membrane normal in tetramer (Fig. 11).
Figure 11.
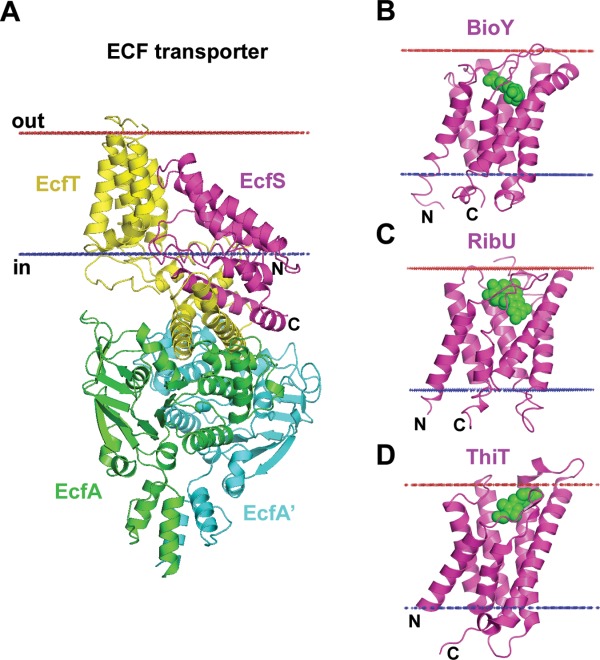
Reorientations of TM domains in energy coupling factor (ECF) transporters. (A) ECF transporter tetramer from Lactobacillus brevis (4HZU) is composed of TM energy-coupling component (EcfT) that links cytosolic ATPases, EcfA-EcfA', to the substrate-binding or S-protein (EcfS). In context of the ECF transporter tetramer, helices of the EcfS are highly tilted relative to the membrane plane (>45°) with the opening of the substrate-binding pocket approaching the cytoplasmic membrane interface. (B–D) Individual substrate-binding proteins are stable in membranes and are oriented along the membrane normal with substrate-binding pocket facing the extracellular space: (B) Biotin transporter S-protein, BioY, from Lactococcus lactis (4DVE); (C) Riboflavin transporter S-protein, RibU, from Staphylococcus aureus (3P5N); and (D) Thiamine transporter S-protein, ThiT, from Lactococcus lactis (3RLB). Substrates inside the binding pockets of S-proteins are shown by green spheres. Calculated hydrophobic membrane boundaries from the OPM database are shown by lines: blue at the cytoplasmic side and red at the extracellular side.
Conformational transitions may involve changes in subunit composition and quaternary structure of protein complexes. One notable example of substrate-dependent protomer rearrangement is the Tat-system found in bacterial, archaebacterial, and thylakoid membranes.233 The Tat-system is composed of three small proteins, single-spanning TatA (2LZS) and TatB (2MI2) and a hexa-helical TatC (4B4A). These subunits form large multimeric complexes for translocation across membranes of folded proteins with twin-arginine signal sequence. After binding of a translocated protein to TatBC complex, this complex recruits TatA protomers and oligomerize into a TM pore-like channels228,233,234 of variable diameter (30–70 Å) depending on the number of protomers in the complex (85–130 particles)235 [Fig. 6(D,E)].
Energetics of Protein–Membrane Interactions
3D structures of both water-soluble and membrane proteins correspond to a minimum of Gibbs free energy in their native environment.236 Physical forces and factors that contribute to structural stability of membrane proteins were discussed in the literature.237–243 A predictive and verifiable theory of membrane protein folding must account for all these forces and reproduce the experimentally determined membrane folding pathways and thermodynamic stabilities of TM α-helical and β-barrel proteins. However, development of such theory still remains a challenge.
Folding of α-helical peptides and proteins in membranes may be described by a thermodynamic cycle that includes several processes: (1) helix-coil transition in water, (2) transfer of folded α-helices into the lipid environment; (3) helix-helix association in membrane, (4) polypeptide chain binding and partial folding at membrane interface, (5) membrane insertion/folding of interface-bound polypeptide chain (Fig. 12). A similar cycle can be drawn for β-barrel proteins. Free energy changes during these processes were determined in experimental studies of protein folding and helix-coil transition in aqueous solution (ΔGfold,wat),244–246 peptide binding to membrane surface (ΔGbind),247–249 transmembrane insertion of peptides (ΔGfold,wat + ΔGtransf),250–252 membrane binding of folded peripheral proteins (ΔGtransf),253 and energies of α-helix association in membranes and micelles (ΔGassoc).254 In addition, equilibrium unfolding energies were measured for many TM α-helical and β-barrel proteins.255
Figure 12.
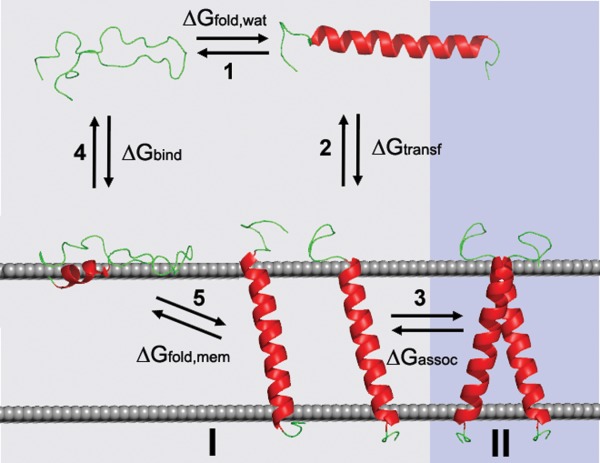
Folding, membrane insertion and oligomerization of an α-helical peptide. The five states of the peptide are: a coil, an α-helix in aqueous solution, a partially folded peptide at the surface of membrane, TM α-helix and TM α-helical dimer. Numbers in the thermodynamic cycle indicate different processes: (1) helix–coil transition in water, (2) transfer of folded α-helices into the lipid environment; (3) helix–helix association in membrane, (4) polypeptide chain binding and partial folding at membrane interface, (5) membrane insertion/folding of interface-bound polypeptide chain. Processes (4+5) and (3) correspond to steps (I) and (II) in two-stage model of membrane protein assembly in vitro:325 (I) α-helix folding and insertion; and (II) lateral association in the lipid bilayer.
These experimental free energy values can be used for development and verification of theoretical models that describe energetics of protein folding and association in membanes. For example, formation of a TM α-helical dimer can be represented as the sequence of steps (1), (2), and (3), that is, formation of an α-helix in water, helix transfer from water into the membrane, and association of helices in the membrane. Though these steps do not necessarily describe real events during protein folding and insertion in membranes, this cycle remains valid for energy calculations based on experimentally-derived parameters. Energy of helix-coil transition (step 1) can be calculated using experimental α-helix propensities, entropic and enthalpic contributions for polypeptide backbone, and energies of interactions between side-chains in water.244–246 Transfer energy of a protein from water to the fluid lipid bilayer (step 2) can be estimated using an implicit solvent model of the lipid bilayer.19,256 Energy of specific helix-helix association (step 3) can be evaluated based on empirical parameterization of hydrogen-bonds and van der Waals forces in non-polar media.257 The corresponding physical processes of transfer and association in membrane are described in more detail below.
Non-specific solvation of membrane proteins by the lipid bilayer
Insertion of membrane protein into the lipid bilayer is driven by the hydrophobic effect, as proteins tend to bury their large hydrophobic surfaces from water to non-polar environment. However, the hydrophobic interactions are unimportant for helix–helix association inside the lipid bilayer. This situation differs from folding of water-soluble proteins, where burial of non-polar residues from water drives the entire folding process.
The energy of insertion of TM proteins into membranes can be evaluated based on various hydrophobicity scales. Whole-residue hydrophobicity scales were derived from studies of partitioning of peptides or amino acid analogues between water and more hydrophobic organic solvents or lipid vesicles,258 from mutagenesis studies,259 from statistical analysis of residue distribution along the membrane normal,182,183,260–263 or MD simulations.264–267 All these scales correlate with each other, though absolute values vary by several fold.39,268,269 Both experimental and theoretical studies indicate the favorable insertion into the lipid bilayer of non-polar residues, but the high cost of insertion of polar and charged residues.
Recently, the so-called “biological” hydrophobicity scales have been derived based on of the efficiency of the insertion of hydrophobic segments into the ER membranes (mammalian and yeast), bacterial IM, and mitochondrial inner membranes, which were mediated by Sec61, SecYEG, YidC, and TIM23 translocons, respectively.43,270–273 It was found that “biological” scales developed for different systems are rather similar, though absolute difference between contribution for the most hydrophobic and most hydrophilic residues is larger in bacterial system and smaller in yeast than for the mammalian ER membranes. “Biological” scales appear to be rather similar to a “knowledge-based” scale derived from relative abundance of residues in membrane proteins182 and to the White-Wimley octanol scale.274
Although whole-residue hydrophobicity scales may be helpful for predicting TM α-helical segments and their topology,275,276 or positioning of proteins in membranes,183,262 they cannot properly account for the solvent exposure of protein residues. For example, the solvent-accessible surface area and the corresponding solvation energies are much greater for amino acid analogues than for less exposed residues included in an α-helix, a β-barrel, or the whole protein structure.19 Therefore, the correct estimation of solvation energy requires the full-atomic description of membrane proteins.
The estimation of transfer energies of proteins from water to membrane requires adequate representation of the lipid bilayer, where energy cost of residue insertion strongly depends on membrane penetration depth.43 It's noteworthy that even hydrocarbon slab representation of the lipid can produce very reasonable results for positioning of TM and peripheral proteins in membranes.253 Nevertheless, more complex description of anisotropic membrane properties and more advanced solvation models are desirable to account not only for the depth-dependent hydrophobic interactions, but also for electrostatic, hydrogen bonding, ionic, and other energy contributions that change along the membrane normal. For example, the recently proposed PPM method19,256 uses a new implicit solvent model and represents the anisotropic lipid bilayer by profiles of polarity parameters (α, β, ε, and π*) calculated from X-ray and neutron scattering data on lipid bilayers. This approach also allows characterization of polarity profiles for different biological membranes, including their hydrophobic thicknesses and polarity of interfacial regions.20
Folding and insertion of TM α-helical proteins in artificial membranes also depend on mechanical properties of the bilayer. These properties can be regulated by changing lipid composition and vesicle size. In particular, it was shown that curvature stress translated in increased lateral pressure at the center of the lipid bilayer enhanced folding of OmpA and galactose transporter GalP, but decreased folding of bacteriorhdopsin, diacylglycerol kinase, and EmrE transporter.277 Therefore, calculations of protein transfer energy into membranes, as well as the energy of protein association (see below), should account for some additional contributions from protein–lipid and lipid–lipid interactions, including curvature stress and mechanical deformation of the lipid bilayer.140 The energetic cost of bilayer deformations depends on material properties of the bilayer, such as thickness, intrinsic lipid curvature, and the elastic compression and bending moduli.278–280
Helix–helix association in the lipid bilayer
The association of TM α-helices within the non-polar lipid bilayer cannot be driven by hydrophobic forces, as for water-soluble proteins. Instead, it is primarily driven by van der Waals interactions of tightly packed helices and, to a smaller degree, by inter-helical hydrogen bonds and ionic interactions.204,205 To describe energetics of helix association in membranes, the following interactions should be considered: (1) attractive van der Waals forces and hydrogen bonds in the lipid environment, (2) side-chain conformational entropy changes which oppose helix–helix association; and (3) additional “solvation” energy contributions which come from protein–lipid and lipid–lipid interactions, including those responsible for mechanical deformation of membrane,281 and (4) electrostatic interactions of helix macrodipoes and rigid-body immobilization entropy of interacting helices.257,282
All intermolecular forces, including van der Waals interactions and H-bonds, are of electrostatic nature, and therefore depend on dielectric properties of the environment.283 The magnitudes of these interactions are similar in the non-polar interiors of water-soluble and TM proteins, ∼ −1 to −1.5 kcal mol−1 for hydrogen-bond and ∼0.02 kcal mol−1 Å2 for van der Waals interactions.257,284,285 It is noteworthy that helix-helix association is similar to crystallization or liquid-solid state transition.285,286 Therefore, these interactions are of the same nature as enthalpy of fusion of molecular crystals; they follow “like dissolves like” rule, and their energy is much weaker than “potential energy” in molecular mechanics and dynamics, which is parameterized using enthalpy of sublimation. This requires novel parameterization of interatomic potentials based on data for condensed media, such as ΔΔG values for protein mutants.257,285
The importance of van der Waals interactions for association of TM helices is supported by many observations. The mutual arrangements of TM α-helixes are limited to a few discrete packing modes, which provide knobs-into-holes intercalation of their side chains, as required for close packing of the quasi-parallel helices. These packing modes include parallel and antiparallel helix arrangement, with left-handed and right-handed crossing motifs.287 In parallel helices, the left-handed packing with positive crossing angles (∼+20°) is related to the presence of a heptad repeat, while the right-handed packing with negative crossing angles (∼−40°) is related to the presence of a tetrad repeat and GxxxG-like sequence motif.207,208 The structure of parallel TM dimers and symmetric oligomers are well superimposible (rmsd of 1–2 Å) with corresponding templates taken from water-soluble proteins (Fig. 13).
Figure 13.
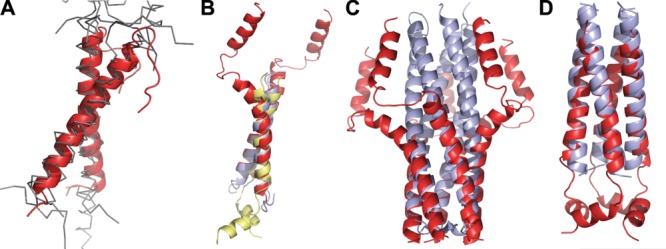
Structural similarity of TM and water-soluble α-helical oligomers. (A) Superposition (rmsd 1.1 to 2.2 Å) of right-handed (crossing angle ∼−40°) TM dimers of bitopic proteins with GxxxG motif: glycophorin A (1AFO, cartoon, colored red) and other unrelated TM homo- and hetero-dimers (2J5D, 2JWA, 2K1K, 2K9J, 2J7A, thin lines, colored grey). (B) Superposition (rmsd 1.1 to 1.2 Å) of left-handed (crossing angle ∼20°) TM helix pairs taken from bitopic protein complexes with heptad packing motif: zeta dimer (2HAC, colored blue), phospholamban pentamer (1ZLL, colored red), and M2 tetramer (2RLF, colored yellow). (C–D) Superposition (rmsd ∼1 Å) of TM oligomers with water-soluble coiled-coil proteins: (C) phospholamban (1ZLL, colored red) and water-soluble pentameric cartilage oligomeric matrix protein (1VDF, colored blue); (D) tetramer M2 channel (2RLF,red) and water-soluble tetrameric CGN4 leucine zipper mutant (1GCL, colored blue).
Furthermore, small and polar residues (Gly, Ala, Ser, Thr) occur more frequently at helix packing interface in TM proteins207 than in water-soluble α-bundle proteins.32,288 The presence of small residues (Gly>Ala>Va>Ile) is required for efficient helix association in membranes, because it allows close packing and significantly increases van-der Waals interactions of anti-parallel helices with heptad motifs.289 In contrast, efficient association of similar helices in water, which is driven by hydrophobic forces, required presence of larger side chains on the contact surface and show higher preference for the Ile side chain vs. Gly (Ile>Va>Ala>Gly).289
The important role of hydrogen-bonding and ionic interactions was demonstrated for self-assembly of hydrophobic TM α-helices bearing centrally located polar amino-acid residues (Asn, Asp, Gln, Glu, and His).290–292 It was shown that the affinity of oligo-Leu TM segments and membrane-soluble MS1 peptides in the lipid bilayer dramatically increased upon placement of an Asn residue within the TM segment,290,292 which was due to formation of intermolecular hydrogen bonds between Asn side chains.290 The role of ionic interactions between charged residues embedded into the lipid bilayer was demonstrated in a study of the T-cell receptor complex.293 Formation of α-helical complexes is also affected by electrostatic interactions of α-helix macrodipoles in membrane, which are stabilizing for antiparallel, but destabilizing for parallel α-helices.257,282
In addition to enthalpic contributions, the free energy of helix-helix interactions in membrane depends on entropic contributions that oppose the association. The most important of these represents the conformational entropy changes of flexible side-chains due to their immobilization during association.257 The loss of side-chain entropy is smaller for residues that are conformationally restricted in isolated α-helices (Gly, Ala, Val, Thr, Ile).
Another important factor for association of proteins in membranes is the hydrophobic mismatch, that is, the difference between hydrophobic thicknesses of TM proteins and surrounding lipid acyl chain region.294 A hydrophobic mismatch can be alleviated through changes of protein tilt angles, conformational transitions, oligomerization, or by thinning or stretching the lipid bilayer.295 Bilayer-mediated elastic forces caused by the hydrophobic mismatch can significantly affect specific and non-specific association of TM domains.209,211,212,296–299 For example, the hydrophobic mismatch between mitochondrial F1F0 ATP synthase (hydrophobic thickness 32.8 Å) and inner mithochondrial membrane (average hydrophobic thickness 28.6 ± 1.4 Å)20 may promote oligomerization of this protein. Indeed, oligomers of mitochondrial F1F0-ATP synthase were observed by EM as ribbon-like structures stacked in parallel along the crystae.300
Specific lipid–protein interactions
Insertion, positioning and 3D structure of proteins in membranes can be significantly affected by direct protein–lipid interactions.149,150,301–303 The strength of protein–lipid association can vary from high affinity specific binding to rotational immobilization of first-shell (or annular) lipids that undergo fast exchange with bulk lipids.279 Many TM proteins were co-crystallized with annular lipids or surrounding detergents, as well as with specifically bound bound lipids that usually originate from native membranes. Here we mention several such examples.
Acyl chains of annular lipids usually fill hydrophobic cavities on protein surfaces and form non-specific van der Waals interactions. Surprisingly, it was observed that hydrocarbon tails of different lipids or detergents in tetramers of mammalian aquaporin AQP0 (2b60, 3m9i, 1ymg) occupy similar cavities on protein surface, while their head-groups interact differently with polar residues of the protein.304,305
The locations of strongly bound lipids are also rather similar in structures of homologous proteins, such as mitochondrial (2DYR) or bacterial (1M56, 1QLE) cytochrome c oxidases.306 The structural conservation of lipid binding sites were reported for cholesterol in β2-adrenergic receptor (2RH1) and Na+-K+-ATPase (2ZXE), for cardiolipin (CL) in formate dehydrogenase–N (1KQF), and for phosphatidylglycerol (PG) in KcsA potassium channel (1K4C).303
The specific binding of lipids and other hydrophobic molecules, such as carotinoids, chlorophyll and other cofactors, can stabilize loosely packed TM α-bundles and large multimeric protein complexes.150,303 For example, folding, stability, and function of the plant light harvesting complex II, which is composed of loosely packed marginally hydrophobic TM helices (1RWT, 2BHW), requires presence the of lipids and numerous cofactors (chlorophylls and carotenoids).307 Lipids co-crystallized with cyanobacteria Photosystem II (3BZ1) include 11 monogalactosyl diacylglycerol, 7 digalactosyl diacylglycerol, 5 sulfoquinovosyl diacylglycerol, and 2 PG. This lipid composition of the preferential location of acidic lipids at the stroma leaflet reflects the relative contents (∼45 mol%, ∼25 mol%, ∼15–25 mol%, 5–15 mol%, respectively) and asymmetric distribution of these mostly non-phosphorus lipids in thylakoid membranes.308 Coordination of lipid molecules with photosynthetic proteins was shown to play an important structural role: lipids fill internal cavities near cofactors, stabilize oligomers, “lubricate” subunits that need to be structurally flexible, and, possibly, influence the biophysical properties of cofactors that affect the rate of electron transfer.309
Natural lipids and cofactors also stabilize trimeric complexes of archaebacterial retinal-containing bacteriorhodopsin (BR)-like ion pumps.310 Different cryo-electron microscopy and crystal structures of BR show the presence of up to 30 lipid-binding sites per trimer that are occupied by archaeols (sn-2,3-diphitanylglycerol) consisting of two C20 isoprenoid chains connected via an ether linkage to glycerol.311 Besides, in each trimeric assembly of all BR-like proteins, except BR itself, carotenoid bacterioruberin with C50 carbon atoms stabilizes the trimer by binding to crevices between adjacent protomers; while in the deltarhodopsin structure (4FBZ), three extended squalene molecules together with lipid molecules fill the central opening of the trimer [Fig. 14(A–C)].
Figure 14.

Specific protein–lipid interactions. (A) Bacteriorhodopsin trimer from Halobacterium salinarium (1IW6) with co-crystallized archaeol lipids (colored orange), including S-TGA-1 sulfoglycolipid (3-sulfate-Galpβ1–6Manpα1–2Glcpα1-archeol) colored cyan. (B) Deltarhodopsin trimer from Haloterrigena thermotolerans (4FBZ) with co-crystallized lipids/detergents (colored orange), 3 squalene molecules (colored cyan) occupying intertrimer space, and 3 bacterioruberin moleculs (colored purple) located at monomer interfaces. (C) Cruxrhodopsin-3 trimer from Haloarcula vallismortis (4JR8) with 3 co-crystallized bacterioruberin moleculs (colored purple) located at monomer interfaces. (D) Dimeric mitochondrial cytochrome c oxidase from bovine heart (2DYR) co-crystallysed with 26 lipids (colored orange) including four hemes A (colored red) and four CLs (colored cyan): two CLs (CL-1) at the intermembrane side that likely participate in dimer stabilization, two CLs (CL-2) molecules at the matrix side that may participate in the proton translocation along the protein surface.313 In the latter case CL in each monomer interacts with basic residues from subunits 3 and 7A1. (E) Ferrix hydroxamate uptake receptor FhuA from E. coli (colored by secondary structure: yellow β-strands, red helices, and green loops) in complex with LPS (colored orange) (1QFG). Lipid A of LPS forms specific interactions with a number of basic (shown as blue sticks), polar (white sticks) and aromatic residues (green sticks) of FhuA.
The crystal structure of dimeric mitochondrial cytochrome c oxidase (2DYR) includes 26 tightly bound phospholipids.312 Among these lipids, there are four CL molecules: two CLs facing the intermembrane side that supposedly participate in dimer stabilization, and two CLs facing matrix side that may function as proton traps that facilitate the proton translocation along the protein surface313 [Fig. 14(D)]. CL bearing four acyl chains and two negative charges is the signature phospholipid of mitochondrial membranes: it accounts up to 20 mol% of the inner membrane.314,315 CL is known to be critically required for biogenesis and stabilization of respiratory chain supercomplexes316,317 and for regulation of many other mitochondrial functions.318 Another highly abundant mitochondrial protein, the ADP/ATP carrier, represents a TM six-helical bundle formed by three repeated α-helical hairpins, each bound to a CL molecule.319 It was shown that CL is critical for carrier folding and translocation.320
Specific lipid-protein interactions may involve contacts of lipid hydrocarbon chains with aromatic residues and ionic and/or hydrogen bonding interactions of lipid phosphodiester groups with Tyr, Asn, Lys, His, Arg, Trp, Ser, and Gln located near lipid head groups.149,150 For example, a conserved structural motif for LPS recognition by the ferric hydroxamate uptake receptor FhuA, an OM β-barrel, [Fig. 14(E)] includes cluster of eight basic residues (K280, K306, K351, R382, R384, K439, K441, R474) and three polar residues (E304, Q353, D386) that form ionic and electrostatic interactions with the phosphate moieties. In addition, six aromatic residues of FhuA (F231, F235, Y284, F302, F355, F380) interact with six acyl chains of the lipid A.303,321
These examples demonstrate the essential role of annular and specifically bound lipids for structure, stability, and function of TM proteins and their complexes. Thus, both non-specific and specific protein–lipid interactions should be taken into account during computational modeling of membrane proteins.
Summary
This review summarizes our current understanding of membrane protein structures, different pathways of protein biogenesis, physical forces that define protein folding and integration into membranes, as well as common and distinctive properties of TM proteins targeted to different biological membranes. The available spatial architectures of TM proteins are limited to α-helical bundles, single-chain and multi-chain β-barrels and β-helices. However, their 3D structures are highly complex and include significant irregularities in secondary structure (310-helices, kinks, interruptions, β-bulges, turns of π-helix), reentrant loops, and repeated domains with identical or different topologies. TM proteins frequently form large complexes, which are crucial for their biological function. Structural examination demonstrates several helix-helix interaction motifs, suggests the role of ligands and cofactors, and highlights ability of these proteins to switch between different conformational states and oligomerization modes. The most significant conformational transitions are observed in pore-forming toxins that exist in water-soluble and membrane-bound forms and transporters that switch between multiple conformations. Analysis of polarity of TM protein surfaces uncovers the presence of a “midpolar” region inside the hydrocarbon region of the lipid bilayer. This membrane region is characterized by the increased dipolarity/polarizability and the hydrogen-bonding acceptor capacity and can be mapped by the preferential accumulation of indole rings of solvent-exposed Trp residues. The observed asymmetry of “midpolar” regions in proteins from different membranes correlates with asymmetric lipid composition of corresponding membranes. Comparison of TM proteins from different types of natural biological membranes shows protein adaptation to distinct lipid environments and correlation between properties of protein surfaces and thicknesses and polarity asymmetry of target membranes. Distinct hydrophobic thicknesses and polarity profiles obtained from analysis of TM proteins from different membranes can be used to describe anisotropic lipid environment in corresponding natural membranes. These models of biological membranes may be useful for development of advanced computational methods aimed at structural modeling and prediction of spatial localization, topology, and self-translocation ability of peptides, proteins and small drug-like molecules. Analysis of forces and factors that define folding and stability of TM proteins highlights the essential role of the hydrophobic effect that drives protein insertion into membrane and important contributions into protein stability of van der Waals attraction forces, hydrogen bonding and ionic interactions, side-chain conformational entropy, the hydrophobic mismatch, membrane deformations, and specific protein-lipid binding.
Glossary
- 3D
three-dimensional
- BAM
β-barrel assembly machinery
- BR
bacteriorhodopsin
- CL
cardiolipin
- ECF
energy-coupling factor
- ER
endoplasmic reticulum
- GPCR
G protein-coupled receptors
- IM
inner membrane
- LPS
lipopolysaccharide
- MD
molecular dynamics
- MIM
mitochondrial inner membrane
- MOM
mitochondrial outer membrane
- OM
outer membrane
- OPM
Orientations of Proteins in Membranes database
- PDB
Protein Data Bank
- PG
phosphatidylglycerol
- PM
plasma membrane
- PPM
Positioning of Proteins in Membranes method and web-server
- rmsd
root mean square deviation of Cα atom coordinates
- SRP
signal recognition particle
- TA
C-tail anchored proteins
- TM
transmembrane
- VDAC
voltage-dependent anion channel.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Supplementary Information
References
- 1.Tan S, Tan HT, Chung MC. Membrane proteins and membrane proteomics. Proteomics. 2008;8:3924–3932. doi: 10.1002/pmic.200800597. [DOI] [PubMed] [Google Scholar]
- 2.Bill RM, Henderson PJF, Iwata S, Kunji ERS, Michel H, Neutze R, Newstead S, Poolman B, Tate CG, Vogel H. Overcoming barriers to membrane protein structure determination. Nat Biotechnol. 2011;29:335–340. doi: 10.1038/nbt.1833. [DOI] [PubMed] [Google Scholar]
- 3.Johansson LC, Arnlund D, White TA, Katona G, Deponte DP, Weierstall U, Doak RB, Shoeman RL, Lomb L, Malmerberg E, Davidsson J, Nass K, Liang M, Andreasson J, Aquila A, Bajt S, Barthelmess M, Barty A, Bogan MJ, Bostedt C, Bozek JD, Caleman C, Coffee R, Coppola N, Ekeberg T, Epp SW, Erk B, Fleckenstein H, Foucar L, Graafsma H, Gumprecht L, Hajdu J, Hampton CY, Hartmann R, Hartmann A, Hauser G, Hirsemann H, Holl P, Hunter MS, Kassemeyer S, Kimmel N, Kirian RA, Maia FR, Marchesini S, Martin AV, Reich C, Rolles D, Rudek B, Rudenko A, Schlichting I, Schulz J, Seibert MM, Sierra RG, Soltau H, Starodub D, Stellato F, Stern S, Struder L, Timneanu N, Ullrich J, Wahlgren WY, Wang X, Weidenspointner G, Wunderer C, Fromme P, Chapman HN, Spence JC, Neutze R. Lipidic phase membrane protein serial femtosecond crystallography. Nat Methods. 2012;9:263–265. doi: 10.1038/nmeth.1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shahid SA, Bardiaux B, Franks WT, Krabben L, Habeck M, van Rossum BJ, Linke D. Membrane-protein structure determination by solid-state NMR spectroscopy of microcrystals. Nat Methods. 2012;9:1212–1217. doi: 10.1038/nmeth.2248. [DOI] [PubMed] [Google Scholar]
- 5.Hiller S, Garces RG, Malia TJ, Orekhov VY, Colombini M, Wagner G. Solution structure of the integral human membrane protein VDAC-1 in detergent micelles. Science. 2008;321:1206–1210. doi: 10.1126/science.1161302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gautier A. Structure determination of alpha-helical membrane proteins by solution-state NMR: emphasis on retinal proteins. Biochim Biophys Acta. 2014;1837:578–588. doi: 10.1016/j.bbabio.2013.06.009. [DOI] [PubMed] [Google Scholar]
- 7.Van Horn WD, Kim HJ, Ellis CD, Hadziselimovic A, Sulistijo ES, Karra MD, Tian C, Sonnichsen FD, Sanders CR. Solution nuclear magnetic resonance structure of membrane-integral diacylglycerol kinase. Science. 2009;324:1726–1729. doi: 10.1126/science.1171716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Call ME, Chou JJ. A view into the blind spot: solution NMR provides new insights into signal transduction across the lipid bilayer. Structure. 2010;18:1559–1569. doi: 10.1016/j.str.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gayen S, Li Q, Kang C. Solution NMR study of the transmembrane domain of single-span membrane proteins: opportunities and strategies. Curr Protein Pept Sci. 2012;13:585–600. doi: 10.2174/138920312803582979. [DOI] [PubMed] [Google Scholar]
- 10.Bocharov EV, Volynsky PE, Pavlov KV, Efremov RG, Arseniev AS. Structure elucidation of dimeric transmembrane domains of bitopic proteins. Cell Adh Migr. 2010;4:284–298. doi: 10.4161/cam.4.2.11930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Berman HM, Battistuz T, Bhat TN, Bluhm WF, Bourne PE, Burkhardt K, Iype L, Jain S, Fagan P, Marvin J, Padolla D, Ravichandran V, Schneider B, Thanki N, Weissig H, Westbrook JD, Zardecki C. The Protein Data Bank. Acta Crystallogr D Biol Crystallogr. 2002;58:899–907. doi: 10.1107/s0907444902003451. [DOI] [PubMed] [Google Scholar]
- 12.White SH, Snaider C. 2014. Database. Available at: http://blanco.biomol.uci.edu/mpstruc White laboratory at UC Irvine.
- 13.Raman P, Cherezov V, Caffrey M. The membrane Protein Data Bank. Cell Mol Life Sci. 2006;63:36–51. doi: 10.1007/s00018-005-5350-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saier MH, Yen MR, Noto K, Tamang DG, Elkan C. The transporter classification database: recent advances. Nucleic Acids Res. 2009;37:D274–D278. doi: 10.1093/nar/gkn862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tusnady GE, Dosztanyi Z, Simon I. PDB_TM: selection and membrane localization of transmembrane proteins in the protein data bank. Nucleic Acids Res. 2005;33:D275–D278. doi: 10.1093/nar/gki002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sansom MSP, Scott KA, Bond PJ. Coarse-grained simulation: a high-throughput computational approach to membrane proteins. Biochem Soc Trans. 2008;36:27–32. doi: 10.1042/BST0360027. [DOI] [PubMed] [Google Scholar]
- 17.Chetwynd AP, Scott KA, Mokrab Y, Sansom MSP. CGDB: a database of membrane protein/lipid interactions by coarse-grained molecular dynamics simulations. Mol Membr Biol. 2008;25:662–669. doi: 10.1080/09687680802446534. [DOI] [PubMed] [Google Scholar]
- 18.Lomize MA, Pogozheva ID, Joo H, Mosberg HI, Lomize AL. OPM database and PPM web server: resources for positioning of proteins in membranes. Nucleic Acids Res. 2012;40:370–376. doi: 10.1093/nar/gkr703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lomize AL, Pogozheva ID, Mosberg HI. Anisotropic solvent model of the lipid bilayer. 2. Energetics of insertion of small molecules, peptides, and proteins in membranes. J Chem Inf Model. 2011;51:930–946. doi: 10.1021/ci200020k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pogozheva ID, Tristram-Nagle S, Mosberg HI, Lomize AL. Structural adaptations of proteins to different biological membranes. Biochim Biophys Acta. 2013;1828:2592–2608. doi: 10.1016/j.bbamem.2013.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu W, Eilers M, Patel AB, Smith SO. Helix packing moments reveal diversity and conservation in membrane protein structure. J Mol Biol. 2004;337:713–729. doi: 10.1016/j.jmb.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 22.Oberai A, Ihm Y, Kim S, Bowie JU. A limited universe of membrane protein families and folds. Protein Sci. 2006;15:1723–1734. doi: 10.1110/ps.062109706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Almen MS, Nordstrom KJ, Fredriksson R, Schioth HB. Mapping the human membrane proteome: a majority of the human membrane proteins can be classified according to function and evolutionary origin. BMC Biol. 2009;7:50. doi: 10.1186/1741-7007-7-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Worch R, Bokel C, Hofinger S, Schwille P, Weidemann T. Focus on composition and interaction potential of single-pass transmembrane domains. Proteomics. 2010;10:4196–4208. doi: 10.1002/pmic.201000208. [DOI] [PubMed] [Google Scholar]
- 25.Wallin E, von Heijne G. Genome-wide analysis of integral membrane proteins from eubacterial, archaean, and eukaryotic organisms. Protein Sci. 1998;7:1029–1038. doi: 10.1002/pro.5560070420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Elofsson A, von Heijne G. Membrane protein structure: prediction versus reality. Annu Rev Biochem. 2007;76:125–140. doi: 10.1146/annurev.biochem.76.052705.163539. [DOI] [PubMed] [Google Scholar]
- 27.Vinothkumar KR, Henderson R. Structures of membrane proteins. Q Rev Biophys. 2010;43:65–158. doi: 10.1017/S0033583510000041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McLuskey K, Roszak AW, Zhu Y, Isaacs NW. Crystal structures of all-alpha type membrane proteins. Eur Biophys J. 2010;39:723–755. doi: 10.1007/s00249-009-0546-6. [DOI] [PubMed] [Google Scholar]
- 29.Arce J, Sturgis JN, Duneau J-P. Dissecting membrane protein architecture: an annotation of structural complexity. Biopolymers. 2009;91:815–829. doi: 10.1002/bip.21255. [DOI] [PubMed] [Google Scholar]
- 30.Hedin LE, Illergard K, Elofsson A. An introduction to membrane proteins. J Proteome Res. 2011;10:3324–3331. doi: 10.1021/pr200145a. [DOI] [PubMed] [Google Scholar]
- 31.Bowie JU. Helix packing in membrane proteins. J Mol Biol. 1997;272:780–789. doi: 10.1006/jmbi.1997.1279. [DOI] [PubMed] [Google Scholar]
- 32.Gimpelev M, Forrest LR, Murray D, Honig B. Helical packing patterns in membrane and soluble proteins. Biophys J. 2004;87:4075–4086. doi: 10.1529/biophysj.104.049288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eyre TA, Partridge L, Thornton JM. Computational analysis of alpha-helical membrane protein structure: implications for the prediction of 3D structural models. Protein Eng Des Sel. 2004;17:613–624. doi: 10.1093/protein/gzh072. [DOI] [PubMed] [Google Scholar]
- 34.Popot JL, Engelman DM. Helical membrane protein folding, stability, and evolution. Annu Rev Biochem. 2000;69:881–922. doi: 10.1146/annurev.biochem.69.1.881. [DOI] [PubMed] [Google Scholar]
- 35.Yan C, Luo J. An analysis of reentrant loops. Protein J. 2010;29:350–354. doi: 10.1007/s10930-010-9259-z. [DOI] [PubMed] [Google Scholar]
- 36.Pornillos O, Chang G. Inverted repeat domains in membrane proteins. FEBS Lett. 2006;580:358–362. doi: 10.1016/j.febslet.2005.12.054. [DOI] [PubMed] [Google Scholar]
- 37.von Heijne G. Membrane-protein topology. Nat Rev Mol Cell Biol. 2006;7:909–918. doi: 10.1038/nrm2063. [DOI] [PubMed] [Google Scholar]
- 38.Ojemalm K, Halling KK, Nilsson I, von Heijne G. Orientational preferences of neighboring helices can drive er insertion of a marginally hydrophobic transmembrane helix. Mol Cell. 2012;45:529–540. doi: 10.1016/j.molcel.2011.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhao G, London E. An amino acid “transmembrane tendency” scale that approaches the theoretical limit to accuracy for prediction of transmembrane helices: relationship to biological hydrophobicity. Protein Sci. 2006;15:1987–2001. doi: 10.1110/ps.062286306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hedin LE, Ojemalm K, Bernsel A, Hennerdal A, Illergard K, Enquist K, Kauko A, Cristobal S, von Heijne G, Lerch-Bader M, Nilsson I, Elofsson A. Membrane insertion of marginally hydrophobic transmembrane helices depends on sequence context. J Mol Biol. 2010;396:221–229. doi: 10.1016/j.jmb.2009.11.036. [DOI] [PubMed] [Google Scholar]
- 41.Rath EM, Tessier D, Campbell AA, Lee HC, Werner T, Salam NK, Lee LK, Church WB. A benchmark server using high resolution protein structure data, and benchmark results for membrane helix predictions. BMC Bioinform. 2013;14:111. doi: 10.1186/1471-2105-14-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rath A, Deber CM. Protein structure in membrane domains. Annu Rev Biophys. 2012;41:135–155. doi: 10.1146/annurev-biophys-050511-102310. [DOI] [PubMed] [Google Scholar]
- 43.Hessa T, Meindl-Beinker NM, Bernsel A, Kim H, Sato Y, Lerch-Bader M, Nilsson I, White SH, von Heijne G. Molecular code for transmembrane-helix recognition by the Sec61 translocon. Nature. 2007;450:1026–1030. doi: 10.1038/nature06387. [DOI] [PubMed] [Google Scholar]
- 44.von Heijne G. Membrane proteins: from bench to bits. Biochem Soc Trans. 2011;39:747–750. doi: 10.1042/BST0390747. [DOI] [PubMed] [Google Scholar]
- 45.Bano-Polo M, Baeza-Delgado C, Orzaez M, Marti-Renom MA, Abad C, Mingarro I. Polar/ionizable residues in transmembrane segments: effects on helix–helix packing. PLoS One. 2012;7:e44263. doi: 10.1371/journal.pone.0044263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Herrmann JR, Fuchs A, Panitz JC, Eckert T, Unterreitmeier S, Frishman D, Langosch D. Ionic interactions promote transmembrane helix-helix association depending on sequence context. J Mol Biol. 2010;396:452–461. doi: 10.1016/j.jmb.2009.11.054. [DOI] [PubMed] [Google Scholar]
- 47.Meindl-Beinker NM, Lundin C, Nilsson I, White SH, von Heijne G. Asn- and Asp-mediated interactions between transmembrane helices during translocon-mediated membrane protein assembly. EMBO Rep. 2006;7:1111–1116. doi: 10.1038/sj.embor.7400818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Baeza-Delgado C, Marti-Renom MA, Mingarro I. Structure-based statistical analysis of transmembrane helices. Eur Biophys J. 2013;42:199–207. doi: 10.1007/s00249-012-0813-9. [DOI] [PubMed] [Google Scholar]
- 49.Viklund H, Granseth E, Elofsson A. Structural classification and prediction of reentrant regions in alpha-helical transmembrane proteins: application to complete genomes. J Mol Biol. 2006;361:591–603. doi: 10.1016/j.jmb.2006.06.037. [DOI] [PubMed] [Google Scholar]
- 50.Screpanti E, Hunte C. Discontinuous membrane helices in transport proteins and their correlation with function. J Struct Biol. 2007;159:261–267. doi: 10.1016/j.jsb.2007.01.011. [DOI] [PubMed] [Google Scholar]
- 51.Kazi A, Sun JZ, Doi K, Sung SS, Takahashi Y, Yin H, Rodriguez JM, Becerril J, Berndt N, Hamilton AD, Wang HG, Sebti SM. The BH3 alpha-helical mimic BH3-M6 disrupts Bcl-X(L), Bcl-2, and MCL-1 protein-protein interactions with Bax, Bbak, Bad, or Bim and induces apoptosis in a Bax- and Bim-dependent manner. J Biol Chem. 2011;286:9382–9392. doi: 10.1074/jbc.M110.203638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rapp M, Seppala S, Granseth E, von Heijne G. Emulating membrane protein evolution by rational design. Science. 2007;315:1282–1284. doi: 10.1126/science.1135406. [DOI] [PubMed] [Google Scholar]
- 53.Zeth K, Thein M. Porins in prokaryotes and eukaryotes: common themes and variations. Biochem J. 2010;431:13–22. doi: 10.1042/BJ20100371. [DOI] [PubMed] [Google Scholar]
- 54.Wimley WC. Toward genomic identification of beta-barrel membrane proteins: composition and architecture of known structures. Protein Sci. 2002;11:301–312. doi: 10.1110/ps.29402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhai YF, Saier MH. The beta-barrel finder (BBF) program, allowing identification of outer membrane beta-barrel proteins encoded within prokaryotic genomes. Protein Sci. 2002;11:2196–2207. doi: 10.1110/ps.0209002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Buchanan SK. Beta-barrel proteins from bacterial outer membranes: structure, function and refolding. Curr Opin Struct Biol. 1999;9:455–461. doi: 10.1016/S0959-440X(99)80064-5. [DOI] [PubMed] [Google Scholar]
- 57.Schulz GE. Beta-barrel membrane proteins. Curr Opin Struct Biol. 2000;10:443–447. doi: 10.1016/s0959-440x(00)00120-2. [DOI] [PubMed] [Google Scholar]
- 58.Schulz GE. The structure of bacterial outer membrane proteins. Biochim Biophys Acta. 2002;1565:308–317. doi: 10.1016/s0005-2736(02)00577-1. [DOI] [PubMed] [Google Scholar]
- 59.Fairman JW, Noinaj N, Buchanan SK. The structural biology of beta-barrel membrane proteins: a summary of recent reports. Curr Opin Struct Biol. 2011;21:523–531. doi: 10.1016/j.sbi.2011.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Buchanan SK, Yamashita Y, Fleming KG. Structure and folding of outer membrane proteins. In: Engelman EH, Tamm LK, editors. Comprehensive biophysics. Oxford: Academic Press; 2012. pp. 139–163. [Google Scholar]
- 61.Noinaj N, Guillier M, Barnard TJ, Buchanan SK. TonB-dependent transporters: regulation, structure, and function. Annu Rev Microbiol. 2010;64:43–60. doi: 10.1146/annurev.micro.112408.134247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bay DC, Hafez M, Young MJ, Court DA. Phylogenetic and coevolutionary analysis of the beta-barrel protein family comprised of mitochondrial porin (VDAC) and Tom40. Biochim Biophys Acta. 2012;1818:1502–1519. doi: 10.1016/j.bbamem.2011.11.027. [DOI] [PubMed] [Google Scholar]
- 63.Bayrhuber M, Meins T, Habeck M, Becker S, Giller K, Villinger S, Vonrhein C, Griesinger C, Zweckstetter M, Zeth K. Structure of the human voltage-dependent anion channel. Proc Natl Acad Sci USA. 2008;105:15370–15375. doi: 10.1073/pnas.0808115105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ujwal R, Cascio D, Colletier JP, Faham S, Zhang J, Toro L, Ping PP, Abramson J. The crystal structure of mouse VDAC1 at 2.3 angstrom resolution reveals mechanistic insights into metabolite gating. Proc Natl Acad Sci USA. 2008;105:17742–17747. doi: 10.1073/pnas.0809634105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schredelseker J, Paz A, Lopez CJ, Altenbach C, Leung CS, Drexler MK, Chen JN, Hubbell WL, Abramson J. High-resolution structure and double electron-electron resonance of the zebrafish voltage dependent anion channel 2 reveal an oligomeric population. J Biol Chem. 2014;289:12566–12577. doi: 10.1074/jbc.M113.497438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Andersen C. Channel-tunnels: outer membrane components of type I secretion systems and multidrug efflux pumps of Gram-negative bacteria. Rev Physiol Biochem Pharmacol. 2003;147:122–165. doi: 10.1007/s10254-003-0008-y. [DOI] [PubMed] [Google Scholar]
- 67.Andersen C, Hughes C, Koronakis V. Protein export and drug efflux through bacterial channel-tunnels. Curr Opin Cell Biol. 2001;13:412–416. doi: 10.1016/s0955-0674(00)00229-5. [DOI] [PubMed] [Google Scholar]
- 68.Meng G, Surana NK, St Geme JW, III, Waksman G. Structure of the outer membrane translocator domain of the haemophilus influenzae Hia trimeric autotransporter. EMBO J. 2006;25:2297–2304. doi: 10.1038/sj.emboj.7601132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Faller M, Niederweis M, Schulz GE. The structure of a mycobacterial outer-membrane channel. Science. 2004;303:1189–1192. doi: 10.1126/science.1094114. [DOI] [PubMed] [Google Scholar]
- 70.Bischofberger M, Iacovache I, van der Goot FG. Pathogenic pore-forming proteins: function and host response. Cell Host Microbe. 2012;12:266–275. doi: 10.1016/j.chom.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 71.Iacovache I, Degiacomi MT, van der Goot FG. Pore-forming toxins. In: Egelman EH, editor. Comprehensive biophysics. Academic Press, New York: Elsevier B.V; 2012. pp. 164–188. v.5. [Google Scholar]
- 72.De S, Olson R. Crystal structure of the vibrio cholerae cytolysin heptamer reveals common features among disparate pore-forming toxins. Proc Natl Acad Sci USA. 2011;108:7385–7390. doi: 10.1073/pnas.1017442108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Song L, Hobaugh MR, Shustak C, Cheley S, Bayley H, Gouaux JE. Structure of staphylococcal alpha-hemolysin, a heptameric transmembrane pore. Science. 1996;274:1859–1866. doi: 10.1126/science.274.5294.1859. [DOI] [PubMed] [Google Scholar]
- 74.Savva CG, Fernandes da Costa SP, Bokori-Brown M, Naylor CE, Cole AR, Moss DS, Titball RW, Basak AK. Molecular architecture and functional analysis of NetB, a pore-forming toxin from clostridium perfringens. J Biol Chem. 2013;288:3512–3522. doi: 10.1074/jbc.M112.430223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yamashita K, Kawai Y, Tanaka Y, Hirano N, Kaneko J, Tomita N, Ohta M, Kamio Y, Yao M, Tanaka I. Crystal structure of the octameric pore of staphylococcal gamma-hemolysin reveals the beta-barrel pore formation mechanism by two components. Proc Natl Acad Sci USA. 2011;108:17314–17319. doi: 10.1073/pnas.1110402108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kelkar DA, Chattopadhyay A. The gramicidin ion channel: a model membrane protein. Biochim Biophys Acta. 2007;1768:2011–2025. doi: 10.1016/j.bbamem.2007.05.011. [DOI] [PubMed] [Google Scholar]
- 77.Wallace BA. Gramicidin channels and pores. Annu Rev Biophys Biophy Chem. 1990;19:127–157. doi: 10.1146/annurev.bb.19.060190.001015. [DOI] [PubMed] [Google Scholar]
- 78.Arseniev AS, Barsukov IL, Bystrov VF, Lomize AL, Ovchinnikov Yu A. 1H NMR study of gramicidin a transmembrane ion channel. Head-to-head right-handed, single-stranded helices. FEBS Lett. 1985;186:168–174. doi: 10.1016/0014-5793(85)80702-x. [DOI] [PubMed] [Google Scholar]
- 79.Lomize AL, Orekhov V, Arseniev AS. Refinement of the spatial structure of the gramicidin a ion channel. Bioorg Khim. 1992;18:182–200. [PubMed] [Google Scholar]
- 80.Ketchem RR, Lee KC, Huo S, Cross TA. Macromolecular structural elucidation with solid-state NMR-derived orientational constraints. J Biomol NMR. 1996;8:1–14. doi: 10.1007/BF00198135. [DOI] [PubMed] [Google Scholar]
- 81.Smart OS, Goodfellow JM, Wallace BA. The pore dimensions of gramicidin a. Biophys J. 1993;65:2455–2460. doi: 10.1016/S0006-3495(93)81293-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Urry DW, Goodall MC, Glickson JD, Mayers DF. The gramicidin a transmembrane channel: characteristics of head-to-head dimerized (l,d) helices. Proc Natl Acad Sci USA. 1971;68:1907–1911. doi: 10.1073/pnas.68.8.1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sun H, Greathouse DV, Andersen OS, Koeppe RE. The preference of tryptophan for membrane interfaces—insights from N-methylation of tryptophans in gramicidin channels. J Biol Chem. 2008;283:22233–22243. doi: 10.1074/jbc.M802074200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Koeppe RE, Schmutzer SE, Andersen OS. Gramicidin channels as cation nanotubes. In: Hayden O, Nielsch K, editors. Molecular- and nano-tubes. Berlin: Springer-Verlag Berlin; 2011. pp. 11–30. [Google Scholar]
- 85.Killian JA. Synthetic peptides as models for intrinsic membrane proteins. FEBS Lett. 2003;555:134–138. doi: 10.1016/s0014-5793(03)01154-2. [DOI] [PubMed] [Google Scholar]
- 86.Deber CM, Liu LP, Wang C. Perspective: peptides as mimics of transmembrane segments in proteins. J Pept Res. 1999;54:200–205. doi: 10.1034/j.1399-3011.1999.00118.x. [DOI] [PubMed] [Google Scholar]
- 87.Booth PJ, Curnow P. Membrane proteins shape up: understanding in vitro folding. Curr Opin Struct Biol. 2006;16:480–488. doi: 10.1016/j.sbi.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 88.Harris NJ, Booth PJ. Folding and stability of membrane transport proteins in vitro. Biochim Biophys Acta. 2012;1818:1055–1066. doi: 10.1016/j.bbamem.2011.11.006. [DOI] [PubMed] [Google Scholar]
- 89.DiBartolo ND, Booth PJ. The membrane factor: biophysical studies of alpha helical transmembrane protein folding. In: Egelman EH, editor. Comprehensive biophysics. Academic Press, New York: Elsevier B.V; 2012. pp. 290–316. v.3. [Google Scholar]
- 90.Dalbey RE, Kuhn A. Evolutionarily related insertion pathways of bacterial, mitochondrial, and thylakoid membrane proteins. Annu Rev Cell Dev Biol. 2000;16:51–87. doi: 10.1146/annurev.cellbio.16.1.51. [DOI] [PubMed] [Google Scholar]
- 91.Dalbey RE, Wang P, Kuhn A. Assembly of bacterial inner membrane proteins. Annu Rev Biochem. 2011;80:161–187. doi: 10.1146/annurev-biochem-060409-092524. [DOI] [PubMed] [Google Scholar]
- 92.Zimmer J, Nam Y, Rapoport TA. Structure of a complex of the ATPase Seca and the protein-translocation channel. Nature. 2008;455:936–943. doi: 10.1038/nature07335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Van den Berg B, Clemons WM, Jr, Collinson I, Modis Y, Hartmann E, Harrison SC, Rapoport TA. X-ray structure of a protein-conducting channel. Nature. 2004;427:36–44. doi: 10.1038/nature02218. [DOI] [PubMed] [Google Scholar]
- 94.Gogala M, Becker T, Beatrix B, Armache JP, Barrio-Garcia C, Berninghausen O, Beckmann R. Structures of the Sec61 complex engaged in nascent peptide translocation or membrane insertion. Nature. 2014;506:107–110. doi: 10.1038/nature12950. [DOI] [PubMed] [Google Scholar]
- 95.White SH, von Heijne G. The machinery of membrane protein assembly. Curr Opin Struct Biol. 2004;14:397–404. doi: 10.1016/j.sbi.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 96.Bowie JU. Solving the membrane protein folding problem. Nature. 2005;438:581–589. doi: 10.1038/nature04395. [DOI] [PubMed] [Google Scholar]
- 97.Park E, Rapoport TA. Mechanisms of Sec61/SecY-mediated protein translocation across membranes. Annu Rev Biophys. 2012;41:21–40. doi: 10.1146/annurev-biophys-050511-102312. [DOI] [PubMed] [Google Scholar]
- 98.Zhang L, Paakkarinen V, Suorsa M, Aro EM. A SecY homologue is involved in chloroplast-encoded D1 protein biogenesis. J Biol Chem. 2001;276:37809–37814. doi: 10.1074/jbc.M105522200. [DOI] [PubMed] [Google Scholar]
- 99.Tsukazaki T, Mori H, Echizen Y, Ishitani R, Fukai S, Tanaka T, Perederina A, Vassylyev DG, Kohno T, Maturana AD, Ito K, Nureki O. Structure and function of a membrane component SecDF that enhances protein export. Nature. 2011;474:235–238. doi: 10.1038/nature09980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kol S, Nouwen N, Driessen AJ. Mechanisms of YidC-mediated insertion and assembly of multimeric membrane protein complexes. J Biol Chem. 2008;283:31269–31273. doi: 10.1074/jbc.R800029200. [DOI] [PubMed] [Google Scholar]
- 101.van der Laan M, Bechtluft P, Kol S, Nouwen N, Driessen AJ. F1F0 ATP synthase subunit c is a substrate of the novel YidC pathway for membrane protein biogenesis. J Cell Biol. 2004;165:213–222. doi: 10.1083/jcb.200402100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.van der Laan M, Nouwen NP, Driessen AJ. YidC—an evolutionary conserved device for the assembly of energy-transducing membrane protein complexes. Curr Opin Microbiol. 2005;8:182–187. doi: 10.1016/j.mib.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 103.Kiefer D, Kuhn A. YidC as an essential and multifunctional component in membrane protein assembly. Int Rev Cytol. 2007;259:113–138. doi: 10.1016/S0074-7696(06)59003-5. [DOI] [PubMed] [Google Scholar]
- 104.Kruger V, Deckers M, Hildenbeutel M, van der Laan M, Hellmers M, Dreker C, Preuss M, Herrmann JM, Rehling P, Wagner R, Meinecke M. The mitochondrial oxidase assembly protein1 (Oxa1) insertase forms a membrane pore in lipid bilayers. J Biol Chem. 2012;287:33314–33326. doi: 10.1074/jbc.M112.387563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Herrmann JM, Neupert W, Stuart RA. Insertion into the mitochondrial inner membrane of a polytopic protein, the nuclear-encoded Oxa1p. EMBO J. 1997;16:2217–2226. doi: 10.1093/emboj/16.9.2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Jermy A. Bacterial secretion: Sec and Tat collaborate in a Rieske business. Nat Rev Microbiol. 2012;10:801. doi: 10.1038/nrmicro2912. [DOI] [PubMed] [Google Scholar]
- 107.Keller R, de Keyzer J, Driessen AJ, Palmer T. Co-operation between different targeting pathways during integration of a membrane protein. J Cell Biol. 2012;199:303–315. doi: 10.1083/jcb.201204149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hagan CL, Silhavy TJ, Kahne D. Beta-barrel membrane protein assembly by the Bam complex. Annu Rev Biochem. 2011;80:189–210. doi: 10.1146/annurev-biochem-061408-144611. [DOI] [PubMed] [Google Scholar]
- 109.Rigel NW, Silhavy TJ. Making a beta-barrel: assembly of outer membrane proteins in gram-negative bacteria. Curr Opin Microbiol. 2012;15:189–193. doi: 10.1016/j.mib.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Borgese N, Fasana E. Targeting pathways of c-tail-anchored proteins. Biochim Biophys Acta. 2011;1808:937–946. doi: 10.1016/j.bbamem.2010.07.010. [DOI] [PubMed] [Google Scholar]
- 111.Hegde RS, Keenan RJ. Tail-anchored membrane protein insertion into the endoplasmic reticulum. Nat Rev Mol Cell Biol. 2011;12:787–798. doi: 10.1038/nrm3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Waizenegger T, Stan T, Neupert W, Rapaport D. Signal-anchor domains of proteins of the outer membrane of mitochondria: structural and functional characteristics. J Biol Chem. 2003;278:42064–42071. doi: 10.1074/jbc.M305736200. [DOI] [PubMed] [Google Scholar]
- 113.Kanaji S, Iwahashi J, Kida Y, Sakaguchi M, Mihara K. Characterization of the signal that directs Tom20 to the mitochondrial outer membrane. J Cell Biol. 2000;151:277–288. doi: 10.1083/jcb.151.2.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Fox TD. Mitochondrial protein synthesis, import, and assembly. Genetics. 2012;192:1203–1234. doi: 10.1534/genetics.112.141267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.von Heijne G, Steppuhn J, Herrmann RG. Domain structure of mitochondrial and chloroplast targeting peptides. Eur J Biochem. 1989;180:535–545. doi: 10.1111/j.1432-1033.1989.tb14679.x. [DOI] [PubMed] [Google Scholar]
- 116.Rehling P, Brandner K, Pfanner N. Mitochondrial import and the twin-pore translocase. Nat Rev Mol Cell Biol. 2004;5:519–530. doi: 10.1038/nrm1426. [DOI] [PubMed] [Google Scholar]
- 117.Dukanovic J, Rapaport D. Multiple pathways in the integration of proteins into the mitochondrial outer membrane. Biochim Biophys Acta. 2011;1808:971–980. doi: 10.1016/j.bbamem.2010.06.021. [DOI] [PubMed] [Google Scholar]
- 118.Merklinger E, Gofman Y, Kedrov A, Driessen AJ, Ben-Tal N, Shai Y, Rapaport D. Membrane integration of a mitochondrial signal-anchored protein does not require additional proteinaceous factors. Biochem J. 2012;442:381–389. doi: 10.1042/BJ20111363. [DOI] [PubMed] [Google Scholar]
- 119.Kemper C, Habib SJ, Engl G, Heckmeyer P, Dimmer KS, Rapaport D. Integration of tail-anchored proteins into the mitochondrial outer membrane does not require any known import components. J Cell Sci. 2008;121:1990–1998. doi: 10.1242/jcs.024034. [DOI] [PubMed] [Google Scholar]
- 120.Dimmer KS, Rapaport D. Unresolved mysteries in the biogenesis of mitochondrial membrane proteins. Biochim Biophys Acta. 2012;1818:1085–1090. doi: 10.1016/j.bbamem.2011.08.027. [DOI] [PubMed] [Google Scholar]
- 121.Kovermann P, Truscott KN, Guiard B, Rehling P, Sepuri NB, Muller H, Jensen RE, Wagner R, Pfanner N. Tim22, the essential core of the mitochondrial protein insertion complex, forms a voltage-activated and signal-gated channel. Mol Cell. 2002;9:363–373. doi: 10.1016/s1097-2765(02)00446-x. [DOI] [PubMed] [Google Scholar]
- 122.Lee DW, Kim JK, Lee S, Choi S, Kim S, Hwang I. Arabidopsis nuclear-encoded plastid transit peptides contain multiple sequence subgroups with distinctive chloroplast-targeting sequence motifs. Plant Cell. 2008;20:1603–1622. doi: 10.1105/tpc.108.060541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wickner W, Schekman R. Protein translocation across biological membranes. Science. 2005;310:1452–1456. doi: 10.1126/science.1113752. [DOI] [PubMed] [Google Scholar]
- 124.Soll J, Schleiff E. Protein import into chloroplasts. Nat Rev Mol Cell Biol. 2004;5:198–208. doi: 10.1038/nrm1333. [DOI] [PubMed] [Google Scholar]
- 125.Robinson C, Thompson SJ, Woolhead C. Multiple pathways used for the targeting of thylakoid proteins in chloroplasts. Traffic. 2001;2:245–251. doi: 10.1034/j.1600-0854.2001.1r010.x. [DOI] [PubMed] [Google Scholar]
- 126.Skalitzky CA, Martin JR, Harwood JH, Beirne JJ, Adamczyk BJ, Heck GR, Cline K, Fernandez DE. Plastids contain a second Sec translocase system with essential functions. Plant Physiol. 2011;155:354–369. doi: 10.1104/pp.110.166546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Jaru-Ampornpan P, Shen K, Lam VQ, Ali M, Doniach S, Jia TZ, Shan SO. ATP-independent reversal of a membrane protein aggregate by a chloroplast Srp subunit. Nat Struct Mol Biol. 2010;17:696–702. doi: 10.1038/nsmb.1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Tu CJ, Schuenemann D, Hoffman NE. Chloroplast ftsy, chloroplast signal recognition particle, and GTP are required to reconstitute the soluble phase of light-harvesting chlorophyll protein transport into thylakoid membranes. J Biol Chem. 1999;274:27219–27224. doi: 10.1074/jbc.274.38.27219. [DOI] [PubMed] [Google Scholar]
- 129.Cline K, Dabney-Smith C. Plastid protein import and sorting: different paths to the same compartments. Curr Opin Plant Biol. 2008;11:585–592. doi: 10.1016/j.pbi.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kapazoglou A, Sagliocco F, Dure L., III PSII-T, a new nuclear encoded lumenal protein from photosystem II. Targeting and processing in isolated chloroplasts. J Biol Chem. 1995;270:12197–12202. doi: 10.1074/jbc.270.20.12197. [DOI] [PubMed] [Google Scholar]
- 131.Lomize AL, Pogozheva ID, Lomize MA, Mosberg HI. Positioning of proteins in membranes: a computational approach. Protein Sci. 2006;15:1318–1333. doi: 10.1110/ps.062126106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Mitra K, Ubarretxena-Belandia T, Taguchi T, Warren G, Engelman DM. Modulation of the bilayer thickness of exocytic pathway membranes by membrane proteins rather than cholesterol. Proc Natl Acad Sci USA. 2004;101:4083–4088. doi: 10.1073/pnas.0307332101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Borgese N, Brambillasca S, Soffientini P, Yabal M, Makarow M. Biogenesis of tail-anchored proteins. Biochem Soc Trans. 2003;31:1238–1242. doi: 10.1042/bst0311238. [DOI] [PubMed] [Google Scholar]
- 134.Fernandez C, Hilty C, Wider G, Wuthrich K. Lipid-protein interactions in DHPC micelles containing the integral membrane protein OmpX investigated by NMR spectroscopy. Proc Natl Acad Sci USA. 2002;99:13533–13537. doi: 10.1073/pnas.212515099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Snijder HJ, Timmins PA, Kalk KH, Dijkstra BW. Detergent organisation in crystals of monomeric outer membrane phospholipase A. J Struct Biol. 2003;141:122–131. doi: 10.1016/s1047-8477(02)00579-8. [DOI] [PubMed] [Google Scholar]
- 136.Abraham T, Schooling SR, Nieh MP, Kučerka N, Beveridge TJ, Katsaras J. Neutron diffraction study of pseudomonas aeruginosa lipopolysaccharide bilayers. J Phys Chem B. 2007;111:2477–2483. doi: 10.1021/jp066012+. [DOI] [PubMed] [Google Scholar]
- 137.Kucerka N, Papp-Szabo E, Nieh MP, Harroun TA, Schooling SR, Pencer J, Nicholson EA, Beveridge TJ, Katsaras J. Effect of cations on the structure of bilayers formed by lipopolysaccharides isolated from pseudomonas aeruginosa PAO1. J Physl Chem B. 2008;112:8057–8062. doi: 10.1021/jp8027963. [DOI] [PubMed] [Google Scholar]
- 138.Tamm LK, Hong H, Liang BY. Folding and assembly of beta-barrel membrane proteins. Biochim Biophys Acta. 2004;1666:250–263. doi: 10.1016/j.bbamem.2004.06.011. [DOI] [PubMed] [Google Scholar]
- 139.Niederweis M, Danilchanka O, Huff J, Hoffmann C, Engelhardt H. Mycobacterial outer membranes: in search of proteins. Trends in Microbiol. 2009;18:109–116. doi: 10.1016/j.tim.2009.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Fiedler S, Broecker J, Keller S. Protein folding in membranes. Cell Mol Life Sci. 2010;67:1779–1798. doi: 10.1007/s00018-010-0259-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Marsh D. Lateral pressure profile, spontaneous curvature frustration, and the incorporation and conformation of proteins in membranes. Biophys J. 2007;93:3884–3899. doi: 10.1529/biophysj.107.107938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Cantor RS. Lateral pressures in cell membranes: a mechanism for modulation of protein function. J Phys Chem B. 1997;101:1723–1725. [Google Scholar]
- 143.Cantor RS. Lipid composition and the lateral pressure profile in bilayers. Biophys J. 1999;76:2625–2639. doi: 10.1016/S0006-3495(99)77415-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Pabst G, Kucerka N, Nieh MP, Rheinstadter MC, Katsaras J. Applications of neutron and X-ray scattering to the study of biologically relevant model membranes. Chem Phys Lipids. 2010;163:460–479. doi: 10.1016/j.chemphyslip.2010.03.010. [DOI] [PubMed] [Google Scholar]
- 145.Raghunathan M, Zubovski Y, Venable RM, Pastor RW, Nagle JF, Tristram-Nagle S. Structure and elasticity of lipid membranes with genistein and daidzein bioflavinoids using X-ray scattering and MD simulations. J Phys Chem B. 2012;116:3918–3927. doi: 10.1021/jp211904j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Boscia AL, Treece BW, Mohammadyani D, Klein-Seetharaman J, Braun AR, Wassenaar TA, Klosgen B, Tristram-Nagle S. X-ray structure, thermodynamics, elastic properties and MD simulations of cardiolipin/dimyristoylphosphatidylcholine mixed membranes. Chem Phys Lipids. 2014;178:1–10. doi: 10.1016/j.chemphyslip.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Boscia AL, Akabori K, Benamram Z, Michel JA, Jablin MS, Steckbeck JD, Montelaro RC, Nagle JF, Tristram-Nagle S. Membrane structure correlates to function of LLP2 on the cytoplasmic tail of HIV-1 gp41 protein. Biophys J. 2013;105:657–666. doi: 10.1016/j.bpj.2013.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Killian JA, von Heijne G. How proteins adapt to a membrane-water interface. Trends Biochem Sci. 2000;25:429–434. doi: 10.1016/s0968-0004(00)01626-1. [DOI] [PubMed] [Google Scholar]
- 149.Lee AG. Lipid–protein interactions in biological membranes: a structural perspective. Biochim Biophys Acta. 2003;1612:1–40. doi: 10.1016/s0005-2736(03)00056-7. [DOI] [PubMed] [Google Scholar]
- 150.Palsdottir H, Hunte C. Lipids in membrane protein structures. Biochim Biophys Acta. 2004;1666:2–18. doi: 10.1016/j.bbamem.2004.06.012. [DOI] [PubMed] [Google Scholar]
- 151.Yau WM, Wimley WC, Gawrisch K, White SH. The preference of tryptophan for membrane interfaces. Biochemistry. 1998;37:14713–14718. doi: 10.1021/bi980809c. [DOI] [PubMed] [Google Scholar]
- 152.Hong HD, Park S, Jimenez RHF, Rinehart D, Tamm LK. Role of aromatic side chains in the folding and thermodynamic stability of integral membrane proteins. J Am Chem Soc. 2007;129:8320–8327. doi: 10.1021/ja068849o. [DOI] [PubMed] [Google Scholar]
- 153.Liu W, Caffrey M. Interactions of tryptophan, tryptophan peptides, and tryptophan alkyl esters at curved membrane interfaces. Biochemistry. 2006;45:11713–11726. doi: 10.1021/bi0608414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Sanders CR, Mittendorf KF. Tolerance to changes in membrane lipid composition as a selected trait of membrane proteins. Biochemistry. 2011;50:7858–7867. doi: 10.1021/bi2011527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Hanshaw RG, Stahelin RV, Smith BD. Noncovalent keystone interactions controlling biomembrane structure. Chemistry. 2008;14:1690–1697. doi: 10.1002/chem.200701589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Nikaido H. Outer membranes, Gram-negative bacteria. In: Schaechter M, editor. Encyclopedia of microbiology. Oxford, UK: Academic Press; 2009. pp. 439–452. [Google Scholar]
- 157.van Meer G, Voelker DR, Feigenson GW. Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol. 2008;9:112–124. doi: 10.1038/nrm2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Maxfield FR, van Meer G. Cholesterol, the central lipid of mammalian cells. Curr Opin Cell Biol. 2010;22:422–429. doi: 10.1016/j.ceb.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Chen YJ, Pornillos O, Lieu S, Ma C, Chen AP, Chang G. X-ray structure of EmrE supports dual topology model. Proc Natl Acad Sci USA. 2007;104:18999–19004. doi: 10.1073/pnas.0709387104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Korkhov VM, Tate CG. An emerging consensus for the structure of EmrE. Acta Crystallogr D. 2009;65:186–192. doi: 10.1107/S0907444908036640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.von Heijne G, Gavel Y. Topogenic signals in integral membrane proteins. Eur J Biochem. 1988;174:671–678. doi: 10.1111/j.1432-1033.1988.tb14150.x. [DOI] [PubMed] [Google Scholar]
- 162.Kauko A, Hedin LE, Thebaud E, Cristobal S, Elofsson A, von Heijne G. Repositioning of transmembrane alpha-helices during membrane protein folding. J Mol Biol. 2010;397:190–201. doi: 10.1016/j.jmb.2010.01.042. [DOI] [PubMed] [Google Scholar]
- 163.Pitonzo D, Skach WR. Molecular mechanisms of aquaporin biogenesis by the endoplasmic reticulum Sec61 translocon. Biochim Biophys Acta. 2006;1758:976–988. doi: 10.1016/j.bbamem.2006.04.021. [DOI] [PubMed] [Google Scholar]
- 164.Seppala S, Slusky JS, Lloris-Garcera P, Rapp M, von Heijne G. Control of membrane protein topology by a single C-terminal residue. Science. 2010;328:1698–1700. doi: 10.1126/science.1188950. [DOI] [PubMed] [Google Scholar]
- 165.Ojemalm K, Watson HR, Roboti P, Cross BC, Warwicker J, von Heijne G, High S. Positional editing of transmembrane domains during ion channel assembly. J Cell Sci. 2013;26:464–472. doi: 10.1242/jcs.111773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Lu Y, Turnbull IR, Bragin A, Carveth K, Verkman AS, Skach WR. Reorientation of aquaporin-1 topology during maturation in the endoplasmic reticulum. Mol Biol Cell. 2000;11:2973–2985. doi: 10.1091/mbc.11.9.2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Norholm MH, Cunningham F, Deber CM, von Heijne G. Converting a marginally hydrophobic soluble protein into a membrane protein. J Mol Biol. 2011;407:171–179. doi: 10.1016/j.jmb.2011.01.035. [DOI] [PubMed] [Google Scholar]
- 168.Bano-Polo M, Martinez-Gil L, Wallner B, Nieva JL, Elofsson A, Mingarro I. Charge pair interactions in transmembrane helices and turn propensity of the connecting sequence promote helical hairpin insertion. J Mol Biol. 2013;425:830–840. doi: 10.1016/j.jmb.2012.12.001. [DOI] [PubMed] [Google Scholar]
- 169.Zhang L, Sato Y, Hessa T, von Heijne G, Lee JK, Kodama I, Sakaguchi M, Uozumi N. Contribution of hydrophobic and electrostatic interactions to the membrane integration of the shaker K+ channel voltage sensor domain. Proc Natl Acad Sci USA. 2007;104:8263–8268. doi: 10.1073/pnas.0611007104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Dowhan W, Bogdanov M. Lipid-dependent membrane protein topogenesis. Ann Rev Biochem. 2009;78:515–540. doi: 10.1146/annurev.biochem.77.060806.091251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.von Heijne G. Formation of transmembrane helices in vivo—is hydrophobicity all that matters? J Gen Physiol. 2007;129:353–356. doi: 10.1085/jgp.200709740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.van Dalen A, de Kruijff B. The role of lipids in membrane insertion and translocation of bacterial proteins. Biochim Biophys Acta. 2004;1694:97–109. doi: 10.1016/j.bbamcr.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 173.von Heijne G. Membrane protein structure prediction. Hydrophobicity analysis and the positive-inside rule. J Mol Biol. 1992;225:487–494. doi: 10.1016/0022-2836(92)90934-c. [DOI] [PubMed] [Google Scholar]
- 174.Nilsson J, Persson B, von Heijne G. Comparative analysis of amino acid distributions in integral membrane proteins from 107 genomes. Proteins Struct Funct Bioinform. 2005;60:606–616. doi: 10.1002/prot.20583. [DOI] [PubMed] [Google Scholar]
- 175.von Heijne G. Control of topology and mode of assembly of a polytopic membrane protein by positively charged residues. Nature. 1989;341:456–458. doi: 10.1038/341456a0. [DOI] [PubMed] [Google Scholar]
- 176.von Heijne G. The distribution of positively charged residues in bacterial inner membrane-proteins correlates with the trans-membrane topology. EMBO J. 1986;5:3021–3027. doi: 10.1002/j.1460-2075.1986.tb04601.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Dalbey RE. Positively charged residues are important determinants of membrane protein topology. Trends Biochem Sci. 1990;15:253–257. doi: 10.1016/0968-0004(90)90047-f. [DOI] [PubMed] [Google Scholar]
- 178.Sipos L, von Heijne G. Predicting the topology of eukaryotic membrane proteins. Eur J Biochem. 1993;213:1333–1340. doi: 10.1111/j.1432-1033.1993.tb17885.x. [DOI] [PubMed] [Google Scholar]
- 179.Gavel Y, Steppuhn J, Herrmann R, von Heijne G. The “positive-inside rule” applies to thylakoid membrane proteins. FEBS Lett. 1991;282:41–46. doi: 10.1016/0014-5793(91)80440-e. [DOI] [PubMed] [Google Scholar]
- 180.Gavel Y, von Heijne G. The distribution of charged amino acids in mitochondrial inner-membrane proteins suggests different modes of membrane integration for nuclearly and mitochondrially encoded proteins. Eur J Biochem. 1992;205:1207–1215. doi: 10.1111/j.1432-1033.1992.tb16892.x. [DOI] [PubMed] [Google Scholar]
- 181.Nakashima H, Nishikawa K. The amino acid composition is different between the cytoplasmic and extracellular sides in membrane proteins. FEBS Lett. 1992;303:141–146. doi: 10.1016/0014-5793(92)80506-c. [DOI] [PubMed] [Google Scholar]
- 182.Ulmschneider MB, Sansom MSP, Di Nola A. Properties of integral membrane protein structures: derivation of an implicit membrane potential. Proteins Struct Funct Bioinform. 2005;59:252–265. doi: 10.1002/prot.20334. [DOI] [PubMed] [Google Scholar]
- 183.Schramm CA, Hannigan BT, Donald JE, Keasar C, Saven JG, Degrado WF, Samish I. Knowledge-based potential for positioning membrane-associated structures and assessing residue-specific energetic contributions. Structure. 2012;20:924–935. doi: 10.1016/j.str.2012.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Benning C. Mechanisms of lipid transport involved in organelle biogenesis in plant cells. Annu Rev Cell Develop Biol. 2009;25:71–91. doi: 10.1146/annurev.cellbio.042308.113414. [DOI] [PubMed] [Google Scholar]
- 185.Nilsson I, von Heijne G. Fine-tuning the topology of a polytopic membrane protein: role of positively and negatively charged amino acids. Cell. 1990;62:1135–1141. doi: 10.1016/0092-8674(90)90390-z. [DOI] [PubMed] [Google Scholar]
- 186.Fujita H, Yamagishi M, Kida Y, Sakaguchi M. Positive charges on the translocating polypeptide chain arrest movement through the translocon. J Cell Sci. 2011;124:4184–4193. doi: 10.1242/jcs.086850. [DOI] [PubMed] [Google Scholar]
- 187.Johansson M, Nilsson I, von Heijne G. Positively charged amino acids placed next to a signal sequence block protein translocation more efficiently in Escherichia coli than in mammalian microsomes. Mol Gen Genet. 1993;239:251–256. doi: 10.1007/BF00281625. [DOI] [PubMed] [Google Scholar]
- 188.Kiefer D, Hu X, Dalbey R, Kuhn A. Negatively charged amino acid residues play an active role in orienting the Sec-independent Pf3 coat protein in the Escherichia coli inner membrane. EMBO J. 1997;16:2197–2204. doi: 10.1093/emboj/16.9.2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Delgado-Partin VM, Dalbey RE. The proton motive force, acting on acidic residues, promotes translocation of amino-terminal domains of membrane proteins when the hydrophobicity of the translocation signal is low. J Biol Chem. 1998;273:9927–9934. doi: 10.1074/jbc.273.16.9927. [DOI] [PubMed] [Google Scholar]
- 190.Schuenemann TA, Delgado-Nixon VM, Dalbey RE. Direct evidence that the proton motive force inhibits membrane translocation of positively charged residues within membrane proteins. J Biol Chem. 1999;274:6855–6864. doi: 10.1074/jbc.274.11.6855. [DOI] [PubMed] [Google Scholar]
- 191.van Klompenburg W, Nilsson I, von Heijne G, de Kruijff B. Anionic phospholipids are determinants of membrane protein topology. EMBO J. 1997;16:4261–4266. doi: 10.1093/emboj/16.14.4261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Andersson H, von Heijne G. Membrane-protein topology—effects of delta-mu(h)+ on the translocation of charged residues explain the positive inside rule. EMBO J. 1994;13:2267–2272. doi: 10.1002/j.1460-2075.1994.tb06508.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.van de Vossenberg JL, Albers SV, van der Does C, Driessen AJ, van Klompenburg W. The positive inside rule is not determined by the polarity of the delta psi (transmembrane electrical potential) Mol Microbiol. 1998;29:1125–1127. doi: 10.1046/j.1365-2958.1998.01001.x. [DOI] [PubMed] [Google Scholar]
- 194.Goder V, Junne T, Spiess M. Sec61p contributes to signal sequence orientation according to the positive-inside rule. Mol Biol Cell. 2004;15:1470–1478. doi: 10.1091/mbc.E03-08-0599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Junne T, Schwede T, Goder V, Spiess M. Mutations in the Sec61p channel affecting signal sequence recognition and membrane protein topology. J Biol Chem. 2007;282:3201–33209. doi: 10.1074/jbc.M707219200. [DOI] [PubMed] [Google Scholar]
- 196.Kumazaki K, Chiba S, Takemoto M, Furukawa A, Nishiyama K-i, Sugano Y, Mori T, Dohmae N, Hirata K, Nakada-Nakura Y, Maturana AD, Tanaka Y, Mori H, Sugita Y, Arisaka F, Ito K, Ishitani R, Tsukazaki T, Nureki O. Structural basis of Sec-independent membrane protein insertion by YidC. Nature. 2014;509:516–520. doi: 10.1038/nature13167. [DOI] [PubMed] [Google Scholar]
- 197.Jackups R, Liang J. Interstrand pairing patterns in beta-barrel membrane proteins: the positive-outside rule, aromatic rescue, and strand registration prediction. J Mol Biol. 2005;354:979–993. doi: 10.1016/j.jmb.2005.09.094. [DOI] [PubMed] [Google Scholar]
- 198.Qu J, Behrens-Kneip S, Holst O, Kleinschmidt JH. Binding regions of outer membrane protein a in complexes with the periplasmic chaperone Skp. A site-directed fluorescence study. Biochemistry. 2009;48:4926–4936. doi: 10.1021/bi9004039. [DOI] [PubMed] [Google Scholar]
- 199.Cymer F, Schneider D. Oligomerization of polytopic alpha-helical membrane proteins: causes and consequences. Biol Chem. 2012;393:1215–1230. doi: 10.1515/hsz-2012-0231. [DOI] [PubMed] [Google Scholar]
- 200.Pereira-Leal JB, Levy ED, Teichmann SA. The origins and evolution of functional modules: lessons from protein complexes. Philos Trans R Soc Lond B Biol Sci. 2006;361:507–517. doi: 10.1098/rstb.2005.1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Venkatakrishnan AJ, Levy ED, Teichmann SA. Homomeric protein complexes: evolution and assembly. Biochem Soc Trans. 2010;38:879–882. doi: 10.1042/BST0380879. [DOI] [PubMed] [Google Scholar]
- 202.Nishi H, Hashimoto K, Madej T, Panchenko AR. Evolutionary, physicochemical, and functional mechanisms of protein homooligomerization. Prog Mol Biol Transl Sci. 2013;117:3–24. doi: 10.1016/B978-0-12-386931-9.00001-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Marianayagam NJ, Sunde M, Matthews JM. The power of two: protein dimerization in biology. Trends Biochem Sci. 2004;29:618–625. doi: 10.1016/j.tibs.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 204.Ridder A, Langosch D. In: Transmembrane domains in membrane protein folding, oligomerization, and function. Buchner J, Kiefhaber T, editors. Weinheim, Germany: Wiley-VCH Verlag GmbH; 2008. pp. 876–918. Protein Folding Handbook. [Google Scholar]
- 205.Langosch D, Arkin IT. Interaction and conformational dynamics of membrane-spanning protein helices. Protein Sci. 2009;18:1343–1358. doi: 10.1002/pro.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Schneider D, Finger C, Prodohl A, Volkmer T. From interactions of single transmembrane helices to folding of alpha-helical membrane proteins: analyzing transmembrane helix–helix interactions in bacteria. Curr Protein Pept Sci. 2007;8:45–61. doi: 10.2174/138920307779941578. [DOI] [PubMed] [Google Scholar]
- 207.Senes A, Engel DE, DeGrado WF. Folding of helical membrane proteins: the role of polar, gxxxg-like and proline motifs. Curr Opin Struct Biol. 2004;14:465–479. doi: 10.1016/j.sbi.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 208.Curran AR, Engelman DM. Sequence motifs, polar interactions and conformational changes in helical membrane proteins. Curr Opin Struct Biol. 2003;13:412–417. doi: 10.1016/s0959-440x(03)00102-7. [DOI] [PubMed] [Google Scholar]
- 209.Cymer F, Veerappan A, Schneider D. Transmembrane helix–helix interactions are modulated by the sequence context and by lipid bilayer properties. Biochim Biophys Acta. 2012;1818:963–973. doi: 10.1016/j.bbamem.2011.07.035. [DOI] [PubMed] [Google Scholar]
- 210.Melnyk RA, Kim S, Curran AR, Engelman DM, Bowie JU, Deber CM. The affinity of GxxxG motifs in transmembrane helix–helix interactions is modulated by long-range communication. J Biol Chem. 2004;279:16591–16597. doi: 10.1074/jbc.M313936200. [DOI] [PubMed] [Google Scholar]
- 211.Li E, Wimley WC, Hristova K. Transmembrane helix dimerization: beyond the search for sequence motifs. Biochim Biophys Acta. 2012;1818:183–193. doi: 10.1016/j.bbamem.2011.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Parton DL, Klingelhoefer JW, Sansom MS. Aggregation of model membrane proteins, modulated by hydrophobic mismatch, membrane curvature, and protein class. Biophys J. 2011;101:691–699. doi: 10.1016/j.bpj.2011.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Hong H, Bowie JU. Dramatic destabilization of transmembrane helix interactions by features of natural membrane environments. J Am Chem Soc. 2011;133:11389–11398. doi: 10.1021/ja204524c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Cross TA, Sharma M, Yi M, Zhou HX. Influence of solubilizing environments on membrane protein structures. Trends Biochem Sci. 2011;36:117–125. doi: 10.1016/j.tibs.2010.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Grigoryan G, Moore DT, DeGrado WF. Transmembrane communication: general principles and lessons from the structure and function of the M2 proton channel, K(+) channels, and integrin receptors. Annu Rev Biochem. 2011;80:211–237. doi: 10.1146/annurev-biochem-091008-152423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Ubarretxena-Belandia I, Stokes DL. Membrane protein structure determination by electron crystallography. Curr Opin Struct Biol. 2012;22:520–528. doi: 10.1016/j.sbi.2012.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Gurevich VV, Gurevich EV. How and why do GPCRs dimerize? Trends Pharmacol Sci. 2008;29:234–240. doi: 10.1016/j.tips.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Meng GY, Fronzes R, Chandran V, Remaut H, Waksman G. Protein oligomerization in the bacterial outer membrane. Mol Membr Biol. 2009;26:136–145. doi: 10.1080/09687680802712422. [DOI] [PubMed] [Google Scholar]
- 219.Bechinger B. A dynamic view of peptides and proteins in membranes. Cell Mol Life Sci. 2008;65:3028–3039. doi: 10.1007/s00018-008-8125-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Song C, Weichbrodt C, Salnikov ES, Dynowski M, Forsberg BO, Bechinger B, Steinem C, de Groot BL, Zachariae U, Zeth K. Crystal structure and functional mechanism of a human antimicrobial membrane channel. Proc Natl Acad Sci USA. 2013;110:4586–4591. doi: 10.1073/pnas.1214739110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Bechinger B, Resende JM, Aisenbrey C. The structural and topological analysis of membrane-associated polypeptides by oriented solid-state nmr spectroscopy: established concepts and novel developments. Biophys Chem. 2011;153:115–125. doi: 10.1016/j.bpc.2010.11.002. [DOI] [PubMed] [Google Scholar]
- 222.Poschner BC, Fischer K, Herrmann JR, Hofmann MW, Langosch D. Structural features of fusogenic model transmembrane domains that differentially regulate inner and outer leaflet mixing in membrane fusion. Mol Membr Biol. 2011;27:1–10. doi: 10.3109/09687680903362044. [DOI] [PubMed] [Google Scholar]
- 223.Iacovache I, van der Goot FG, Pernot L. Pore formation: an ancient yet complex form of attack. Biochim Biophys Acta. 2008;1778:1611–1623. doi: 10.1016/j.bbamem.2008.01.026. [DOI] [PubMed] [Google Scholar]
- 224.White SH, Wimley WC, Ladokhin AS, Hristova K. Protein folding in membranes: determining energetics of peptide-bilayer interactions. Energ Biol Macromol B. 1998;295:62–87. doi: 10.1016/s0076-6879(98)95035-2. [DOI] [PubMed] [Google Scholar]
- 225.Wimley WC, White SH. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat Struct Biol. 1996;3:842–848. doi: 10.1038/nsb1096-842. [DOI] [PubMed] [Google Scholar]
- 226.Marcoux J, Wang SC, Politis A, Reading E, Ma J, Biggin PC, Zhou M, Tao H, Zhang Q, Chang G, Morgner N, Robinson CV. Mass spectrometry reveals synergistic effects of nucleotides, lipids, and drugs binding to a multidrug resistance efflux pump. Proc Natl Acad Sci USA. 2013;110:9704–9709. doi: 10.1073/pnas.1303888110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Thogersen L, Nissen P. Flexible P-type ATPases interacting with the membrane. Curr Opin Struct Biol. 2012;22:491–499. doi: 10.1016/j.sbi.2012.05.009. [DOI] [PubMed] [Google Scholar]
- 228.Rollauer SE, Tarry MJ, Graham JE, Jaaskelainen M, Jager F, Johnson S, Krehenbrink M, Liu SM, Lukey MJ, Marcoux J, McDowell MA, Rodriguez F, Roversi P, Stansfeld PJ, Robinson CV, Sansom MS, Palmer T, Högbom M, Berks BC, Lea SM. Structure of the TatC core of the twin-arginine protein transport system. Nature. 2012;492:210–214. doi: 10.1038/nature11683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Shi Y. Common folds and transport mechanisms of secondary active transporters. Annu Rev Biophys. 2013;42:51–72. doi: 10.1146/annurev-biophys-083012-130429. [DOI] [PubMed] [Google Scholar]
- 230.Matthews EE, Zoonens M, Engelman DM. Dynamic helix interactions in transmembrane signaling. Cell. 2006;127:447–450. doi: 10.1016/j.cell.2006.10.016. [DOI] [PubMed] [Google Scholar]
- 231.Wang T, Fu G, Pan X, Wu J, Gong X, Wang J, Shi Y. Structure of a bacterial energy-coupling factor transporter. Nature. 2013;497:272–276. doi: 10.1038/nature12045. [DOI] [PubMed] [Google Scholar]
- 232.Xu K, Zhang M, Zhao Q, Yu F, Guo H, Wang C, He F, Ding J, Zhang P. Crystal structure of a folate energy-coupling factor transporter from Lactobacillus brevis. Nature. 2013;497:268–271. doi: 10.1038/nature12046. [DOI] [PubMed] [Google Scholar]
- 233.Palmer T, Berks BC. The twin-arginine translocation (Tat) protein export pathway. Nat Rev Microbiol. 2012;10:483–496. doi: 10.1038/nrmicro2814. [DOI] [PubMed] [Google Scholar]
- 234.Frobel J, Rose P, Muller M. Twin-arginine-dependent translocation of folded proteins. Philos Trans R Soc Lond B Biol Sci. 2012;367:1029–1046. doi: 10.1098/rstb.2011.0202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Gohlke U, Pullan L, McDevitt CA, Porcelli I, de Leeuw E, Palmer T, Saibil HR, Berks BC. The tata component of the twin-arginine protein transport system forms channel complexes of variable diameter. Proc Natl Acad Sci USA. 2005;102:10482–10486. doi: 10.1073/pnas.0503558102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.White SH. Translocons, thermodynamics, and the folding of membrane proteins. FEBS Lett. 2003;555:116–121. doi: 10.1016/s0014-5793(03)01153-0. [DOI] [PubMed] [Google Scholar]
- 237.Haltia T, Freire E. Forces and factors that contribute to the structural stability of membrane proteins. Biochim Biophys Acta. 1995;1241:295–322. doi: 10.1016/0304-4157(94)00161-6. [DOI] [PubMed] [Google Scholar]
- 238.White SH, Wimley WC. Membrane protein folding and stability: physical principles. Annu Rev Biophys Biomol Struct. 1999;28:319–365. doi: 10.1146/annurev.biophys.28.1.319. [DOI] [PubMed] [Google Scholar]
- 239.Booth PJ, Templer RH, Meijberg W, Allen SJ, Curran AR, Lorch M. In vitro studies of membrane protein folding. Crit Rev Biochem Mol Biol. 2001;36:501–603. doi: 10.1080/20014091074246. [DOI] [PubMed] [Google Scholar]
- 240.Minetti CA, Remeta DP. Energetics of membrane protein folding and stability. Arch Biochem Biophys. 2006;453:32–53. doi: 10.1016/j.abb.2006.03.023. [DOI] [PubMed] [Google Scholar]
- 241.MacKenzie KR. Folding and stability of alpha-helical integral membrane proteins. Chem Rev. 2006;106:1931–1977. doi: 10.1021/cr0404388. [DOI] [PubMed] [Google Scholar]
- 242.Stanley AM, Fleming KG. The process of folding proteins into membranes: challenges and progress. Arch Biochem Biophys. 2008;469:46–66. doi: 10.1016/j.abb.2007.09.024. [DOI] [PubMed] [Google Scholar]
- 243.Bowie JU. Membrane protein folding: how important are hydrogen bonds? Curr Opin Struct Biol. 2011;21:42–49. doi: 10.1016/j.sbi.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Chakrabartty A, Baldwin RL. Stability of alpha-helices. Adv Protein Chem. 1995;46:141–176. [PubMed] [Google Scholar]
- 245.Lomize AL, Mosberg HI. Thermodynamic model of secondary structure for alpha-helical peptides and proteins. Biopolymers. 1997;42:239–269. doi: 10.1002/(SICI)1097-0282(199708)42:2<239::AID-BIP12>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 246.Makhatadze GI. Thermodynamics of alpha-helix formation. Adv Protein Chem. 2005;72:199–226. doi: 10.1016/S0065-3233(05)72008-8. [DOI] [PubMed] [Google Scholar]
- 247.Ladokhin AS, White SH. Protein chemistry at membrane interfaces: non-additivity of electrostatic and hydrophobic interactions. J Mol Biol. 2001;309:543–552. doi: 10.1006/jmbi.2001.4684. [DOI] [PubMed] [Google Scholar]
- 248.Seelig J. Thermodynamics of lipid-peptide interactions. Biochim Biophys Acta. 2004;1666:40–50. doi: 10.1016/j.bbamem.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 249.Ziegler A. Thermodynamic studies and binding mechanisms of cell-penetrating peptides with lipids and glycosaminoglycans. Adv Drug Deliv Rev. 2008;60:580–597. doi: 10.1016/j.addr.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 250.Reshetnyak YK, Andreev OA, Segala M, Markin VS, Engelman DM. Energetics of peptide (PHLIP) binding to and folding across a lipid bilayer membrane. Proc Natl Acad Sci USA. 2008;105:15340–15345. doi: 10.1073/pnas.0804746105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Wimley WC, White SH. Designing transmembrane alpha-helices that insert spontaneously. Biochemistry. 2000;39:4432–4442. doi: 10.1021/bi992746j. [DOI] [PubMed] [Google Scholar]
- 252.Kyrychenko A, Rodnin MV, Posokhov YO, Holt A, Pucci B, Killian JA, Ladokhin AS. Thermodynamic measurements of bilayer insertion of a single transmembrane helix chaperoned by fluorinated surfactants. J Mol Biol. 2012;416:328–334. doi: 10.1016/j.jmb.2011.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Lomize AL, Pogozheva ID, Lomize MA, Mosberg HI. The role of hydrophobic interactions in positioning of peripheral proteins in membranes. BMC Struct Biol. 2007;7:44. doi: 10.1186/1472-6807-7-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.MacKenzie KR, Fleming KG. Association energetics of membrane spanning alpha-helices. Curr Opin Struct Biol. 2008;18:412–419. doi: 10.1016/j.sbi.2008.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Booth PJ, Curnow P. Folding scene investigation: membrane proteins. Curr Opin Struct Biol. 2009;19:8–13. doi: 10.1016/j.sbi.2008.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Lomize AL, Pogozheva ID, Mosberg HI. Anisotropic solvent model of the lipid bilayer. 1. Parameterization of long-range electrostatics and first solvation shell effects. J Chem Inf Model. 2011;51:918–929. doi: 10.1021/ci2000192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Lomize AL, Pogozheva ID, Mosberg HI. Quantification of helix-helix binding affinities in micelles and lipid bilayers. Protein Sci. 2004;13:2600–2612. doi: 10.1110/ps.04850804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.White SH, Wimley WC. Hydrophobic interactions of peptides with membrane interfaces. Biochim Biophys Acta. 1998;1376:339–352. doi: 10.1016/s0304-4157(98)00021-5. [DOI] [PubMed] [Google Scholar]
- 259.Moon CP, Fleming KG. Side-chain hydrophobicity scale derived from transmembrane protein folding into lipid bilayers. Proc Natl Acad Sci USA. 2011;108:10174–10177. doi: 10.1073/pnas.1103979108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Adamian L, Nanda V, DeGrado WF, Liang J. Empirical lipid propensities of amino acid residues in multispan alpha helical membrane proteins. Proteins Struct Funct Bioinform. 2005;59:496–509. doi: 10.1002/prot.20456. [DOI] [PubMed] [Google Scholar]
- 261.Koehler J, Woetzel N, Staritzbichler R, Sanders CR, Meiler J. A unified hydrophobicity scale for multispan membrane proteins. Proteins Struct Funct Bioinform. 2009;76:13–29. doi: 10.1002/prot.22315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Senes A, Chadi DC, Law PB, Walters RFS, Nanda V, DeGrado WF. E-z, a depth-dependent potential for assessing the energies of insertion of amino acid side-chains into membranes: derivation and applications to determining the orientation of transmembrane and interfacial helices. J Mol Biol. 2007;366:436–448. doi: 10.1016/j.jmb.2006.09.020. [DOI] [PubMed] [Google Scholar]
- 263.Hsieh D, Davis A, Nanda V. A knowledge-based potential highlights unique features of membrane alpha-helical and beta-barrel protein insertion and folding. Protein Sci. 2012;21:50–62. doi: 10.1002/pro.758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Schow EV, Freites JA, Cheng P, Bernsel A, von Heijne G, White SH, Tobias DJ. Arginine in membranes: the connection between molecular dynamics simulations and translocon-mediated insertion experiments. J Membr Biol. 2011;239:35–48. doi: 10.1007/s00232-010-9330-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Johansson ACV, Lindahl E. Position-resolved free energy of solvation for amino acids in lipid membranes from molecular dynamics simulations. Proteins Struct Funct Bioinform. 2008;70:1332–1344. doi: 10.1002/prot.21629. [DOI] [PubMed] [Google Scholar]
- 266.MacCallum JL, Bennett WFD, Tieleman DP. Partitioning of amino acid side chains into lipid bilayers: results from computer simulations and comparison to experiment. J Gen Physiol. 2007;129:371–377. doi: 10.1085/jgp.200709745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.MacCallum JL, Bennett WF, Tieleman DP. Distribution of amino acids in a lipid bilayer from computer simulations. Biophys J. 2008;94:3393–3404. doi: 10.1529/biophysj.107.112805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Zhao G, London E. Strong correlation between statistical transmembrane tendency and experimental hydrophobicity scales for identification of transmembrane helices. J Membr Biol. 2009;229:165–168. doi: 10.1007/s00232-009-9178-0. [DOI] [PubMed] [Google Scholar]
- 269.MacCallum JL, Tieleman DP. Hydrophobicity scales: a thermodynamic looking glass into lipid–protein interactions. Trends Biochem Sci. 2011;36:653–662. doi: 10.1016/j.tibs.2011.08.003. [DOI] [PubMed] [Google Scholar]
- 270.Hessa T, Kim H, Bihlmaier K, Lundin C, Boekel J, Andersson H, Nilsson I, White SH, von Heijne G. Recognition of transmembrane helices by the endoplasmic reticulum translocon. Nature. 2005;433:377–381. doi: 10.1038/nature03216. [DOI] [PubMed] [Google Scholar]
- 271.Hessa T, Reithinger JH, von Heijne G, Kim H. Analysis of transmembrane helix integration in the endoplasmic reticulum in Scerevisiae. J Mol Biol. 2009;386:1222–1228. doi: 10.1016/j.jmb.2009.01.027. [DOI] [PubMed] [Google Scholar]
- 272.Xie K, Hessa T, Seppala S, Rapp M, von Heijne G, Dalbey RE. Features of transmembrane segments that promote the lateral release from the translocase into the lipid phase. Biochemistry. 2007;46:15153–15161. doi: 10.1021/bi701398y. [DOI] [PubMed] [Google Scholar]
- 273.Botelho SC, Osterberg M, Reichert AS, Yamano K, Bjorkholm P, Endo T, von Heijne G, Kim H. Tim23-mediated insertion of transmembrane alpha-helices into the mitochondrial inner membrane. EMBO J. 2011;30:1003–1011. doi: 10.1038/emboj.2011.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.White SH, von Heijne G. How translocons select transmembrane helices. Annu Rev Biophys. 2008;37:23–42. doi: 10.1146/annurev.biophys.37.032807.125904. [DOI] [PubMed] [Google Scholar]
- 275.Fagerberg L, Jonasson K, von Heijne G, Uhlen M, Berglund L. Prediction of the human membrane proteome. Proteomics. 2010;10:1141–1149. doi: 10.1002/pmic.200900258. [DOI] [PubMed] [Google Scholar]
- 276.Bernsel A, Viklund H, Falk J, Lindahl E, von Heijne G, Elofsson A. Prediction of membrane-protein topology from first principles. Proc Natl Acad Sci USA. 2008;105:7177–7181. doi: 10.1073/pnas.0711151105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Mitchell DC. Progress in understanding the role of lipids in membrane protein folding. Biochim Biophys Acta. 2012;1818:951–956. doi: 10.1016/j.bbamem.2011.12.029. [DOI] [PubMed] [Google Scholar]
- 278.Andersen OS, Koeppe RE. Bilayer thickness and membrane protein function: an energetic perspective. Annu Rev Biophys Biomol Struct. 2007;36:107–130. doi: 10.1146/annurev.biophys.36.040306.132643. [DOI] [PubMed] [Google Scholar]
- 279.Marsh D. Protein modulation of lipids, and vice-versa, in membranes. Biochim Biophys Acta. 2008;1778:1545–1575. doi: 10.1016/j.bbamem.2008.01.015. [DOI] [PubMed] [Google Scholar]
- 280.Phillips R, Ursell T, Wiggins P, Sens P. Emerging roles for lipids in shaping membrane-protein function. Nature. 2009;459:379–385. doi: 10.1038/nature08147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Yano Y, Yamamoto A, Ogura M, Matsuzaki K. Thermodynamics of insertion and self-association of a transmembrane helix: a lipophobic interaction by phosphatidylethanolamine. Biochemistry. 2011;50:6806–6814. doi: 10.1021/bi200560c. [DOI] [PubMed] [Google Scholar]
- 282.Ben-Tal N, Ben-Shaul A, Nicholls A, Honig B. Free-energy determinants of alpha-helix insertion into lipid bilayers. Biophys J. 1996;70:1803–1812. doi: 10.1016/S0006-3495(96)79744-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Israelachvili JN. Intermolecular and surface forces. London: Academic Press; 1992. [Google Scholar]
- 284.Faham S, Yang D, Bare E, Yohannan S, Whitelegge JP, Bowie JU. Side-chain contributions to membrane protein structure and stability. J Mol Biol. 2004;335:297–305. doi: 10.1016/j.jmb.2003.10.041. [DOI] [PubMed] [Google Scholar]
- 285.Lomize AL, Reibarkh MY, Pogozheva ID. Interatomic potentials and solvation parameters from protein engineering data for buried residues. Protein Sci. 2002;11:1984–2000. doi: 10.1110/ps.0307002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Shakhnovich EI, Finkelstein AV. Theory of cooperative transitions in protein molecules. I. Why denaturation of globular protein is a first-order phase transition. Biopolymers. 1989;28:1667–1680. doi: 10.1002/bip.360281003. [DOI] [PubMed] [Google Scholar]
- 287.Grigoryan G, DeGrado WF. Probing designability via a generalized model of helical bundle geometry. J Mol Biol. 2011;405:1079–1100. doi: 10.1016/j.jmb.2010.08.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Eilers M, Patel AB, Liu W, Smith SO. Comparison of helix interactions in membrane and soluble alpha-bundle proteins. Biophys J. 2002;82:2720–2736. doi: 10.1016/S0006-3495(02)75613-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Zhang Y, Kulp DW, Lear JD, DeGrado WF. Experimental and computational evaluation of forces directing the association of transmembrane helices. J Am Chem Soc. 2009;131:11341–11343. doi: 10.1021/ja904625b. [DOI] [PubMed] [Google Scholar]
- 290.Zhou FX, Cocco MJ, Russ WP, Brunger AT, Engelman DM. Interhelical hydrogen bonding drives strong interactions in membrane proteins. Nat Struct Biol. 2000;7:154–160. doi: 10.1038/72430. [DOI] [PubMed] [Google Scholar]
- 291.Zhou FX, Merianos HJ, Brunger AT, Engelman DM. Polar residues drive association of polyleucine transmembrane helices. Proc Natl Acad Sci USA. 2001;98:2250–2255. doi: 10.1073/pnas.041593698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Choma C, Gratkowski H, Lear JD, DeGrado WF. Asparagine-mediated self-association of a model transmembrane helix. Nat Struct Biol. 2000;7:161–166. doi: 10.1038/72440. [DOI] [PubMed] [Google Scholar]
- 293.Call ME, Pyrdol J, Wiedmann M, Wucherpfennig KW. The organizing principle in the formation of the T cell receptor-CD3 complex. Cell. 2002;111:967–979. doi: 10.1016/s0092-8674(02)01194-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Marsh D. Energetics of hydrophobic matching in lipid–protein interactions. Biophys J. 2008;94:3996–4013. doi: 10.1529/biophysj.107.121475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Holt A, Killian JA. Orientation and dynamics of transmembrane peptides: the power of simple models. Eur Biophys J. 2010;39:609–621. doi: 10.1007/s00249-009-0567-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Ursell T, Huang KC, Peterson E, Phillips R. Cooperative gating and spatial organization of membrane proteins through elastic interactions. PLoS Comput Biol. 2007;3:e81. doi: 10.1371/journal.pcbi.0030081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.de Planque MR, Killian JA. Protein–lipid interactions studied with designed transmembrane peptides: role of hydrophobic matching and interfacial anchoring. Mol Membr Biol. 2003;20:271–284. doi: 10.1080/09687680310001605352. [DOI] [PubMed] [Google Scholar]
- 298.Benjamini A, Smit B. Robust driving forces for transmembrane helix packing. Biophys J. 2012;103:1227–1235. doi: 10.1016/j.bpj.2012.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Sparr E, Ash WL, Nazarov PV, Rijkers DT, Hemminga MA, Tieleman DP, Killian JA. Self-association of transmembrane alpha-helices in model membranes: importance of helix orientation and role of hydrophobic mismatch. J Biol Chem. 2005;280:39324–39331. doi: 10.1074/jbc.M502810200. [DOI] [PubMed] [Google Scholar]
- 300.Thomas D, Bron P, Weimann T, Dautant A, Giraud MF, Paumard P, Salin B, Cavalier A, Velours J, Brethes D. Supramolecular organization of the yeast F1Fo-ATP synthase. Biol Cell. 2008;100:591–601. doi: 10.1042/BC20080022. [DOI] [PubMed] [Google Scholar]
- 301.Lee AG. Lipid–protein interactions. Biochem Soc Trans. 2011;39:761–766. doi: 10.1042/BST0390761. [DOI] [PubMed] [Google Scholar]
- 302.Findlay HE, Booth PJ. The biological significance of lipid–protein interactions. J Phys Condens Matter. 2006;18:S1281–S1291. doi: 10.1088/0953-8984/18/28/S11. [DOI] [PubMed] [Google Scholar]
- 303.Adamian L, Naveed H, Liang J. Lipid-binding surfaces of membrane proteins: evidence from evolutionary and structural analysis. Biochim Biophys Acta. 2011;1808:1092–1102. doi: 10.1016/j.bbamem.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Hite RK, Gonen T, Harrison SC, Walz T. Interactions of lipids with aquaporin-0 and other membrane proteins. Pflugers Arch. 2008;456:651–661. doi: 10.1007/s00424-007-0353-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Hite RK, Li Z, Walz T. Principles of membrane protein interactions with annular lipids deduced from aquaporin-0 2D crystals. EMBO J. 2010;29:1652–1658. doi: 10.1038/emboj.2010.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Qin L, Hiser C, Mulichak A, Garavito RM, Ferguson-Miller S. Identification of conserved lipid/detergent-binding sites in a high-resolution structure of the membrane protein cytochrome c oxidase. Proc Natl Acad Sci USA. 2006;103:16117–16122. doi: 10.1073/pnas.0606149103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Horn R, Paulsen H. Folding in vitro of light-harvesting chlorophyll a/b protein is coupled with pigment binding. J Mol Biol. 2002;318:547–556. doi: 10.1016/S0022-2836(02)00115-8. [DOI] [PubMed] [Google Scholar]
- 308.Kern J, Guskov A. Lipids in photosystem II: multifunctional cofactors. J Photochem Photobiol B Biol. 2011;104:19–34. doi: 10.1016/j.jphotobiol.2011.02.025. [DOI] [PubMed] [Google Scholar]
- 309.Jones MR. Lipids in photosynthetic reaction centres: structural roles and functional holes. Prog Lipid Res. 2007;46:56–87. doi: 10.1016/j.plipres.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 310.Krebs MP, Isenbarger TA. Structural determinants of purple membrane assembly. Biochim Biophys Acta. 2000;1460:15–26. doi: 10.1016/s0005-2728(00)00126-2. [DOI] [PubMed] [Google Scholar]
- 311.Pebay-Peyroula E, Rosenbusch JP. High-resolution structures and dynamics of membrane protein–lipid complexes: a critique. Curr Opin Struct Biol. 2001;11:427–432. doi: 10.1016/s0959-440x(00)00228-1. [DOI] [PubMed] [Google Scholar]
- 312.Shinzawa-Itoh K, Aoyama H, Muramoto K, Terada H, Kurauchi T, Tadehara Y, Yamasaki A, Sugimura T, Kurono S, Tsujimoto K, Mizushima T, Yamashita E, Tsukihara T, Yoshikawa S. Structures and physiological roles of 13 integral lipids of bovine heart cytochrome c oxidase. EMBO J. 2007;26:1713–1725. doi: 10.1038/sj.emboj.7601618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Arnarez C, Marrink SJ, Periole X. Identification of cardiolipin binding sites on cytochrome c oxidase at the entrance of proton channels. Sci Rep. 2013;3:1263. doi: 10.1038/srep01263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.de Kroon AI, Dolis D, Mayer A, Lill R, de Kruijff B. Phospholipid composition of highly purified mitochondrial outer membranes of rat liver and neurospora crassa. Is cardiolipin present in the mitochondrial outer membrane? Biochim Biophys Acta. 1997;1325:108–116. doi: 10.1016/s0005-2736(96)00240-4. [DOI] [PubMed] [Google Scholar]
- 315.Cosentino K, Garcia-Saez AJ. Mitochondrial alterations in apoptosis. Chem Phys Lipids. 2014;181:62–75. doi: 10.1016/j.chemphyslip.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 316.Zhang M, Mileykovskaya E, Dowhan W. Gluing the respiratory chain together. Cardiolipin is required for supercomplex formation in the inner mitochondrial membrane. J Biol Chem. 2002;277:43553–43556. doi: 10.1074/jbc.C200551200. [DOI] [PubMed] [Google Scholar]
- 317.Schagger H. Respiratory chain supercomplexes of mitochondria and bacteria. Biochim Biophys Acta. 2002;1555:154–159. doi: 10.1016/s0005-2728(02)00271-2. [DOI] [PubMed] [Google Scholar]
- 318.Schlame M, Rua D, Greenberg ML. The biosynthesis and functional role of cardiolipin. Prog Lipid Res. 2000;39:257–288. doi: 10.1016/s0163-7827(00)00005-9. [DOI] [PubMed] [Google Scholar]
- 319.Pebay-Peyroula E, Dahout-Gonzalez C, Kahn R, Trezeguet V, Lauquin GJ, Brandolin G. Structure of mitochondrial ADP/ATP carrier in complex with carboxyatractyloside. Nature. 2003;426:39–44. doi: 10.1038/nature02056. [DOI] [PubMed] [Google Scholar]
- 320.Hoffmann B, Stockl A, Schlame M, Beyer K, Klingenberg M. The reconstituted ADP/ATP carrier activity has an absolute requirement for cardiolipin as shown in cysteine mutants. J Biol Chem. 1994;269:1940–1944. [PubMed] [Google Scholar]
- 321.Ferguson AD, Welte W, Hofmann E, Lindner B, Holst O, Coulton JW, Diederichs K. A conserved structural motif for lipopolysaccharide recognition by procaryotic and eucaryotic proteins. Structure. 2000;8:585–592. doi: 10.1016/s0969-2126(00)00143-x. [DOI] [PubMed] [Google Scholar]
- 322.Rapoport TA, Goder V, Heinrich SU, Matlack KE. Membrane-protein integration and the role of the translocation channel. Trends Cell Biol. 2004;14:568–575. doi: 10.1016/j.tcb.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 323.Noinaj N, Kuszak AJ, Gumbart JC, Lukacik P, Chang H, Easley NC, Lithgow T, Buchanan SK. Structural insight into the biogenesis of beta-barrel membrane proteins. Nature. 2013;501:385–390. doi: 10.1038/nature12521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Kucerka N, Nagle JF, Sachs JN, Feller SE, Pencer J, Jackson A, Katsaras J. Lipid bilayer structure determined by the simultaneous analysis of neutron and X-ray scattering data. Biophys J. 2008;95:2356–2367. doi: 10.1529/biophysj.108.132662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Popot JL, Engelman DM. Membrane-protein folding and oligomerization—the 2-stage model. Biochemistry. 1990;29:4031–4037. doi: 10.1021/bi00469a001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information