

Abstract
The β-lactamase inhibitory proteins (BLIPs) are a model system for examining molecular recognition in protein-protein interactions. BLIP and BLIP-II are structurally unrelated proteins that bind and inhibit TEM-1 β-lactamase. Both BLIPs share a common binding interface on TEM-1 and make contacts with many of the same TEM-1 surface residues. BLIP-II, however, binds TEM-1 over 150-fold tighter than BLIP despite the fact that it has fewer contact residues and a smaller binding interface. The role of eleven TEM-1 amino acid residues that contact both BLIP and BLIP-II was examined by alanine mutagenesis and determination of the association (kon) and dissociation (koff) rate constants for binding each partner. The substitutions had little impact on association rates and resulted in a wide range of dissociation rates as previously observed for substitutions on the BLIP side of the interface. The substitutions also had less effect on binding affinity for BLIP than BLIP-II. This is consistent with the high affinity and small binding interface of the TEM-1-BLIP-II complex, which predicts per residue contributions should be higher for TEM-1 binding to BLIP-II versus BLIP. Two TEM-1 residues (E104 and M129) were found to be hotspots for binding BLIP while five (L102, Y105, P107, K111, and M129) are hotspots for binding BLIP-II with only M129 as a common hotspot for both. Thus, although the same TEM-1 surface binds to both BLIP and BLIP-II, the distribution of binding energy on the surface is different for the two target proteins, that is, different binding strategies are employed.
Keywords: protein–protein interactions, molecular recognition, beta-lactamase, binding kinetics, antibiotic resistance
Introduction
Nearly all biological processes are regulated by protein-protein interactions and understanding the principles guiding molecular recognition is fundamental to our ability to manipulate them.1,2 Many proteins participating in biological pathways have multiple binding partners, and understanding how a single binding site can accommodate a variety of binding partners with different structural elements is of significant interest.3 In this study, alanine-scanning mutagenesis was combined with kinetic analysis to examine how shared interface residues in TEM-1 β-lactamase contribute to affinity and specificity for different binding partner proteins.
The interactions of β-lactamase Inhibitory Proteins (BLIPs) with β-lactamases are an established model for studying protein–protein binding and molecular recognition.4–10 Many previous studies have described the presence of binding hotspots, which are clusters of residues responsible for a large portion of the binding energy for an interaction.4,5,8,11–13 Substitution of hotspot residues to alanine results in a greater than 10-fold increase of the dissociation constant (Kd) due to a reduction in binding free energy. In addition, the presence of specificity-determining residues in an interface plays a major role in the choice of binding partners for an interaction. By definition, substitution of a specificity-determining residue with alanine has different or even opposite effects on the affinity between different interaction partners.5,14 The existence of these residues allows rational engineering of protein interfaces with altered binding properties.
This study utilizes TEM-1 β-lactamase as a common binding partner for naturally occurring BLIPs.9,10 TEM-1 is an extensively studied representative of class A β-lactamases, which mediate bacterial resistance towards penicillin and cephalosporin antibiotics.15 Class A β-lactamases are serine hydrolases and the family includes a large number of enzymes from both Gram-positive and Gram-negative bacteria. Crystal structures of the TEM-1-BLIP and TEM-1-BLIP-II complexes reveal extensive interactions of each BLIP with the same interface at the center of the protruding loop-helix composed of residues 99–114 of TEM-1 β-lactamase.16,17
BLIP is a naturally occurring inhibitor of β-lactamases that is produced by the soil bacterium Streptomyces clavuligerus.18,19 BLIP inhibits class A enzymes with a wide range of affinities and its interaction with TEM-1 has been extensively studied using structural and biophysical methods. Crystal structures of BLIP, as well as the BLIP-TEM-1 β-lactamase complex, reveal a 17 kD protein that forms a concave surface which encapsulates the conserved loop-helix region of class A β-lactamases16,19 (Fig. 1). The BLIP-TEM-1 interface is relatively large with a buried surface area of 2636 Å2 that contains several aromatic residues.16 Aromatic amino acids are the most commonly found residues in binding hot spots and seem to be a common feature in protein–protein interfaces.20 The BLIP-TEM-1 interaction relies on a complex network of residues bridging the interface. This involves multiple hotspots or modules that act largely independent of one another but together contribute most of the binding free energy for the interaction.6 BLIP inhibits β-lactamases by inserting two loops into the enzyme active site that make contacts with catalytic residues and block antibiotic binding.16
Figure 1.
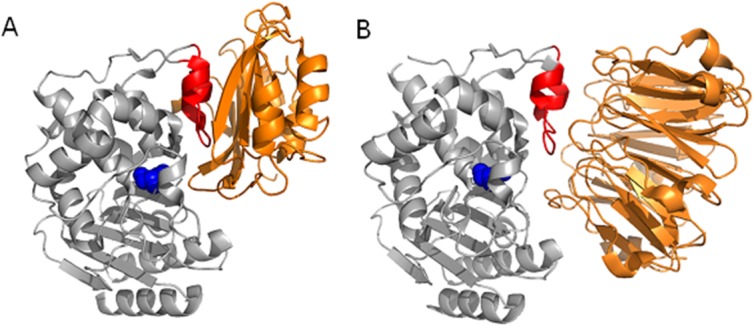
Structural comparison of BLIP-TEM-1 (PDB 1JTG) (A) and BLIP-II-TEM-1 (PDB 1JTD) (B). The protruding loop-helix region (red) and the catalytic serine 70 (blue space fill) of TEM-1 (gray) is shown as the primary binding region for both BLIP and BLIP-II (orange).
BLIP-II has a seven-bladed β-propeller structure and shares no structural or amino acid identity with BLIP (Fig. 1). The crystal structure of the BLIP-II-TEM-1 complex reveals that BLIP-II interacts with β-lactamase via a buried interaction surface area of 2,187 Å2, which is smaller than that observed with BLIP.17 Although BLIP and BLIP-II are unrelated proteins, they both interact with the same loop-helix region on TEM-1 β-lactamase16,17 (Fig. 1). Unlike BLIP, BLIP-II uses numerous turns to bind β-lactamase and sterically block access to the active site without direct interaction with any TEM-1 catalytic residues. BLIP-II is a tighter binding inhibitor of β-lactamases (low picomolar to femtomolar Ki values) than BLIP (micromolar to high picomolar Ki values) and has a much narrower range of binding affinities for β-lactamases (two orders of magnitude from weakest to tightest) when compared to those observed with BLIP (six orders of magnitude) despite binding to the same interface on the enzymes.5,19,21 Kinetic analysis of the binding reaction indicate the association rate constant for BLIP-II with TEM-1 (106 M−1s−1) is 10-fold faster than the on-rate for BLIP-TEM-1 and the off-rate of BLIP-II from TEM-1 (∼10−6 s−1) is ∼10-fold slower than that observed for BLIP-TEM-1.10,22 According to the cocrystal structures, 12 TEM-1 residues are involved in interactions with both BLIP and BLIP-II out of a total of 14 TEM-1 residues that interact with BLIP-II and 24 TEM-1 residues that interact with BLIP (Materials and Methods) (Table I).10,16,17 Therefore, BLIP-II uses significantly fewer contact residues than BLIP but binds TEM-1 with much higher affinity (∼150-fold). Understanding how BLIP-II uses fewer contact residues, has a smaller interface, and yet binds more tightly than BLIP would be a significant step towards understanding molecular recognition determinants of affinity and specificity. Alanine-scanning mutagenesis of residues on the TEM-1 β-lactamase side of the interface that are common between BLIP and BLIP-II was performed and the association and dissociation rate constants for each mutant were determined to assess the contributions of the individual TEM-1 positions for binding each BLIP.
Table I.
TEM-1 β-Lactamase Contact Residues with BLIP and BLIP-II
| TEM-1 residues | BLIP | BLIP-II |
|---|---|---|
| Gln99 | +a | + |
| Asn100 | + | + |
| Leu102 | + | + |
| Val103 | + | − |
| Glu104 | + | + |
| Tyr105 | + | + |
| Ser106 | + | − |
| Pro107 | + | + |
| Glu110 | + | + |
| Lys111 | + | + |
| His112 | + | − |
| Leu113 | − | + |
| Met129 | + | + |
| Ser130 | + | − |
| Pro167 | + | − |
| Glu168 | + | − |
| Asn170 | + | − |
| Lys215 | + | − |
| Val216 | + | + |
| Lys234 | + | − |
| Ser235 | + | − |
| Ala237 | + | + |
| Gly238 | + | − |
| Arg244 | + | − |
| Met272 | + | + |
| Asp273 | − | + |
Results
Kinetic parameters of TEM-1 β-lactamase alanine mutants
This study entails alanine scanning mutagenesis of the TEM-1 β-lactamase residues that contact both BLIP and BLIP-II according to the X-ray structures of the complexes.10,16,17 As stated above, 12 residues in TEM-1 contact both BLIP and BLIP-II, however, one of these (A237) is an alanine naturally and was not pursued further (Table I). As a first step, it was necessary to examine the impact of the alanine substitutions on the catalytic activity of the enzyme to ensure they retain structure and function. The β-lactamase mutants Q99A, N100A, L102A, E104A, E105A, P107A, E110A, K111A, M129A, V216A, and M272A were constructed by site-directed mutagenesis as described in Materials and Methods. Each mutant, as well as the wild type TEM-1 enzyme, was expressed and purified to homogeneity for enzyme kinetic analysis of nitrocefin hydrolysis. In general, the enzymes containing alanine substitutions exhibited kinetic parameters for nitrocefin hydrolysis similar to those of wild type (Table II). The L102A and P107A variants exhibited an ∼2-fold reduction in catalytic efficiency (kcat/Km) and the M272A variant displayed a 4-fold reduction in efficiency because of a decrease in kcat but nevertheless these mutants were still highly active with catalytic efficiency values in the 4 to 9 × 106 M−1 s−1 range (Table II).
Table II.
Kinetic Parameters for Nitrocefin Hydrolysis by TEM-1 β-Lactamase Wild Type and Contact Residue Alanine Mutants
| Mutant | Km (μM) | kcat (s−1) | kcat/Km (μM−1 s−1) |
|---|---|---|---|
| WT | 83 ± 16 | 1188 ± 133 | 14 |
| Q99A | 72 ± 17 | 1102 ± 89 | 15 |
| N100A | 71 ± 13 | 1057 ± 145 | 15 |
| L102A | 75 ± 14 | 450 ± 157 | 6 |
| E104A | 64 ± 19 | 992 ± 99 | 16 |
| Y105A | 75 ± 17 | 1126 ± 167 | 15 |
| P107A | 61 ± 16 | 573 ± 189 | 9 |
| E110A | 79 ± 13 | 1175 ± 190 | 15 |
| K111A | 58 ± 13 | 943 ± 178 | 16 |
| M129A | 67 ± 14 | 1004 ± 188 | 15 |
| V216A | 78 ± 19 | 952 ± 113 | 12 |
| M272A | 60 ± 8 | 237 ± 11 | 4 |
Determination of association rate constants
Having evaluated the enzyme activity of the alanine mutants, the next task was to determine the impact of the substitutions on TEM-1 binding to BLIP and BLIP-II. In order to gain information on what aspect of binding was affected by the substitutions, the association rate constant (kon) and the dissociation rate constant (koff) were determined for wild type TEM-1 and each mutant.
The association rate constants were determined for all 11 TEM-1 alanine mutants with both BLIPs by measuring the intrinsic tryptophan fluorescence using a stopped flow apparatus (Materials and Methods, Fig. 2). Overall, the association rate constant was not affected greatly by substitution of any of the TEM-1 contact residues. All of the association rates for TEM-1 alanine mutants were within 5-fold of each other for binding both BLIP and BLIP-II (Table III and Fig. 3). The association rates for TEM-1 binding BLIP were generally one order of magnitude slower than those observed for TEM-1 with BLIP-II. Wild type TEM-1 associated with BLIP at a rate of 1.1 × 105 M−1 s−1 while wild type TEM-1 and BLIP-II associated at a rate of 1.9 × 106 M−1 s−1. The M272A substitution enhanced the association rate constant 2-fold for TEM-1 binding BLIP while the L102A and E104A substitutions increased speed of association for binding BLIP-II by 2- and 3-fold, respectively (Table III). Nevertheless, taken together, the results indicate alanine substitutions of the TEM-1 β-lactamase contact residues do not greatly impact association rates.
Figure 2.
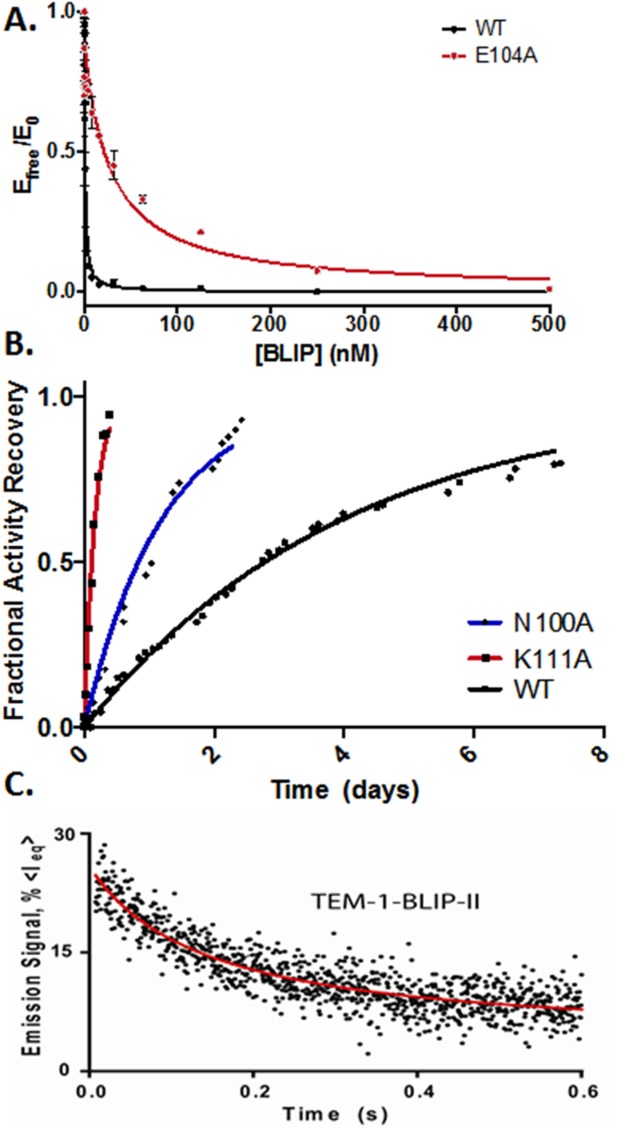
Kinetic characterization of TEM-1 alanine mutants association with BLIPs. A: Determination of BLIP Ki values for TEM-1 WT and E104A mutant β-lactamase for binding BLIP. B: Enzymatic activity-based measurements of dissociation between BLIP-II and TEM-1 (wild type, N100A, and K111A alanine variants). Data were fitted to first order kinetics to determine dissociation rate constants. C: Representative time-course of the stopped-flow tryptophan fluorescence to determine the association rate constants for BLIP-II/TEM-1 β-lactamase.
Table III.
Kinetic Constants for TEM-1 β-Lactamase Interaction with BLIP and BLIP-II
| BLIP-TEM-1 |
BLIP-II-TEM-1 |
|||||
|---|---|---|---|---|---|---|
| kon | Calculated koff | Ki | kon | koff | Calculated Kd | |
| Mutant | (105M−1 s−1)a | (10−7 s−1)b | (pM) | (105M−1 s−1)a | (10−7 s−1)c | (pM)d |
| WT | 1.08 ± 0.21 | 248 | 230 ± 15 | 19 ± 5.1 | 29 ± 2.0 | 1.5 |
| Q99A | 0.8 ± 0.17 | 498 | 622 ± 23 | 25±5.5 | 88 ± 18 | 3.6 |
| N100A | 0.67 ± 0.16 | 13 | 19 ± 3 | 17 ± 4.7 | 97 ± 19 | 5.8 |
| L102A | 0.45 ± 0.12 | 441 | 980 ± 49 | 46 ± 9.0 | 1269 ± 348 | 28 |
| E104A | 0.75 ± 0.16 | 18,865 | 25,153 ± 1700 | 63 ± 24 | 389 ± 68 | 6.2 |
| Y105A | 0.28 ± 0.09 | 55 | 196 ± 22 | 13 ± 2.3 | 866 ± 179 | 67 |
| P107A | 0.75 ± 0.16 | 257 | 343 ± 40 | 29 ± 6.8 | 801 ± 194 | 28 |
| E110A | 1.01 ± 0.20 | 25 | 25 ± 2 | 27 ± 5.6 | 42 ± 5 | 1.6 |
| K111A | 0.85 ± 0.17 | 46 | 54 ± 9 | 12 ± 2.5 | 697 ± 111 | 56 |
| M129A | 0.78 ± 0.15 | 2,292 | 2938 ± 230 | 18 ± 3.5 | 1894 ± 314 | 106 |
| V216A | 0.89 ± 0.17 | 77 | 86 ± 12 | 13 ± 2.6 | 21 ± 3 | 1.6 |
| M272A | 2.18 ± 0.20 | 460 | 211 ± 38 | 14 ± 0.4 | 34 ± 1 | 2.4 |
Stopped-flow tryptophan fluorescence spectrometry measurements at ambient temperature (23°C).
The dissociation rate (koff) was calculated from the inhibition constant (Ki) and the association rate constant at ambient temperature (23°C).
Activity-based dissociation experiments using inactive TEM-1 E166A-substituted enzyme in the competitive displacement assay. First-order reaction kinetics were used to determine the dissociation rate constants.
The dissociation constant (Kd) was calculated from the dissociation rate constant and association rate constant at ambient temperature (23°C).
Figure 3.
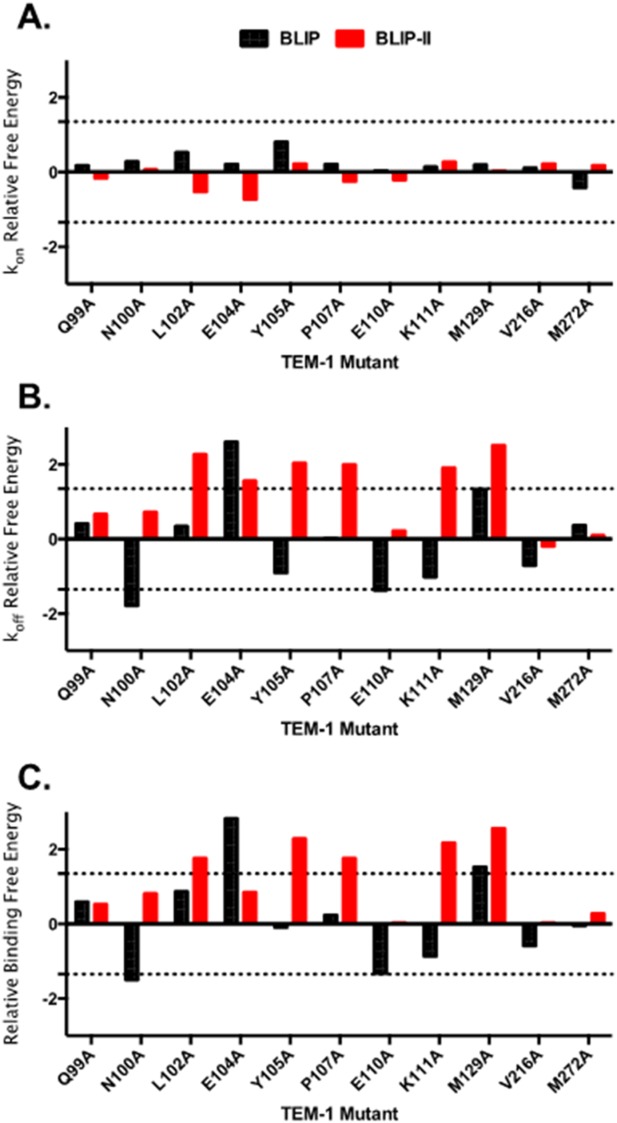
Comparison of the ΔΔG values of the TEM-1 alanine variants for binding BLIP (black) and BLIP-II (red) for association constants (A), dissociation rate constants (B) and relative binding energy change (C). Relative free energy changes are defined as the difference in energy between wild-type TEM-1 and the alanine mutant (ΔΔG = −RTln(Kd,wt/Kd,mut)). The top control line is set at 1.35 kcal/mol and the bottom control line is set at −1.35 kcal/mol, which indicates a 10-fold change.
Determination of dissociation rate constants
To measure the effects of the alanine substitutions on the rate of dissociation of TEM-1 and BLIP-II as well as the overall Kd of the interaction, an enzymatic recovery assay was used to measure the rate of BLIP-II-β-lactamase complex dissociation.10,22 The dissociation rate experiments were performed by forming a BLIP-II-β-lactamase complex and then adding 400-fold excess of the inactive TEM-1 β-lactamase mutant, E166A. This TEM variant exhibits very low levels of β-lactam hydrolysis activity but still binds to BLIP-II with similar affinity as wild type TEM-1.10 These characteristics allow for the hydrolysis activity of the wild type TEM-1 enzyme to be monitored as an indication of the off-rate because as BLIP-II dissociates from the active β-lactamase it binds and is sequestered by the large excess of inactive TEM E166A enzyme. This data was fitted to the first order rate equation and the dissociation rate constant was determined (Fig. 2). However, this assay was not an effective way to measure the BLIP-TEM-1 dissociation rate, which is too fast to determine koff accurately. Instead, an enzymatic inhibition assay was utilized to determine the Ki of the interaction between BLIP and the TEM-1 variants.21,23 The dissociation rate constant was then calculated using the following equation:
The alanine substituted TEM-1 β-lactamases showed dramatic changes in the dissociation rate constants for both binding partners. The weaker binding BLIP dissociates from wild type TEM-1 β-lactamase 9-fold faster (2.5 × 10−5 s−1) than does BLIP-II (2.9 × 10−6 s−1) (Table III). However, several TEM-1 alanine variants displayed large increases in dissociation rate for both BLIPs (Fig. 3). Therefore, the individual TEM-1 amino acid residues in the binding interface contribute largely to the rate of dissociation of the complexes with both BLIP and BLIP-II as was previously observed for BLIP-II alanine scanning experiments.22 Alanine substitutions in TEM-1 are surprisingly well tolerated in the BLIP interface (Table III). The TEM-1 E104A and M129A substitutions significantly increase the dissociation rate while the Q99A, L102A, Y105A, P107A, K111A, V216A, and M272A substitutions have only a modest effect and the N100A and E110A substitutions decrease the dissociation rate. In contrast, the interface with BLIP-II is more sensitive to substitutions (Table III). The TEM-1 Q99A, N100A, E110A, V216A, and M272A substitutions modestly affect dissociation rates while the L102A, E104A, Y105A, P107A, K111A, and M129A substitutions result in large increases in dissociation rate and no substitutions significantly decrease the dissociation rate (Table III). These results suggest BLIP-II is more highly optimized to bind to the TEM-1 surface in that the majority of TEM-1 residues contribute to the slow dissociation of BLIP-II. In contrast, BLIP is less optimized to bind the TEM-1 surface in that most TEM-1 positions can be converted to alanine without increasing the dissociation rate and, in fact, substitutions at several positions slow down dissociation. The increased optimization of BLIP-II versus BLIP for binding the TEM-1 surface is consistent with the fact that BLIP-II binds TEM-1 much tighter and with fewer contact residues than does BLIP.
Functional epitopes for BLIP and BLIP-II
Residues that result in >10-fold decrease in binding affinity are considered in this study as hotspot residues belonging to the functional epitope.4,5 Therefore, the functional epitope for TEM-1 binding to BLIP consists of only two residues including E104 and M129 (Fig. 4). Because the TEM-1-BLIP-II binding interface is smaller than that of TEM-1-BLIP and the interaction is 150-fold tighter, one might expect the proportion of residues contributing strongly to the TEM-1-BLIP-II interface to be correspondingly larger. This was found to be the case in that only 2/11 TEM-1 residues are hotspots for BLIP binding while 5/11 are hotspots for binding BLIP-II (Table IV and Fig. 4). This finding reflects the increased dissociation rates of the TEM-1 mutants for binding BLIP-II versus BLIP as discussed above. A comparison of the impact of the alanine substitutions for the two targets reveals M129A results in a large decrease in binding affinity for both BLIP and BLIP-II due to increased dissociation rates. In contrast, residue E104 contributes to binding affinity for BLIP but not BLIP-II and residues L102, Y105, P107 and K111 contribute to binding BLIP-II but not BLIP. Finally, TEM-1 residues Q99, N100, E110, V216, and M272A do not strongly contribute to binding either BLIP or BLIP-II. The general picture that emerges from these results is that, although BLIP and BLIP-II bind to the same region of TEM-1 β-lactamase, they do so by utilizing remarkably different binding energy contributions from the individual TEM-1 residues.
Figure 4.
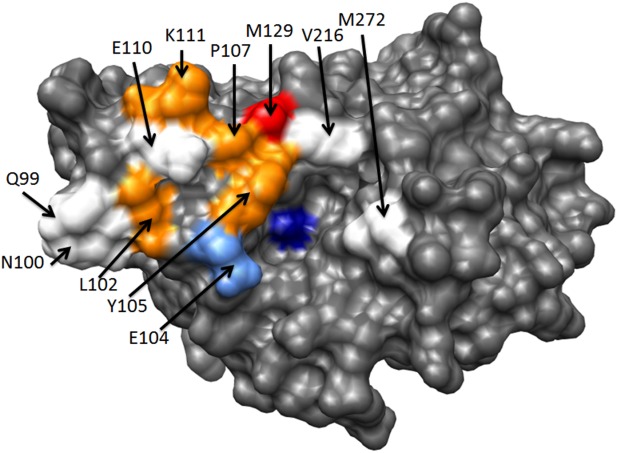
TEM-1 β-lactamase binding hotpots. The TEM-1 β-lactamase structure is shown in gray (PDB ID 1XPB). The catalytic serine 70 residues is colored dark blue. A residue is defined as a hotspot if the alanine substitution reduces binding by >10-fold (Table IV). TEM-1 positions Q99, N100, E110, V216, and M272 are not hotpots for binding either BLIP or BLIP-II and are colored white. TEM-1 residue E104 is a hotspot for binding BLIP but not BLIP-II and is colored light blue. Positions L102, Y105, P107, and K111 are hotspots for binding BLIP-II but not BLIP and are colored orange. TEM-1 residue M129 is a hotspot for binding both BLIP and BLIP-II and is colored red.
Table IV.
TEM-1 β-Lactamase Contact Residues with BLIP and BLIP-II
| BLIP-TEM-1 |
BLIP-II-TEM-1 |
|||||
|---|---|---|---|---|---|---|
| Mutant | TEM-1 mut/wt Kia | BLIP contact/distance (Å)b | Contact mut/wt Kic | TEM-1 mut/wt Kdd | BLIP-II contact/distance (Å)e | Contact mut/wt Kdf |
| Q99 | 2.7 | W150/3.7Å | 368 | 2.4 | D206/2.9Å | 3.6 |
| H148/3.9* | 42 | Y208/4.0 | 30.6 | |||
| N100 | 0.1 | R160/2.8* | 22 | 3.9 | Y208/3.6 | 30.6 |
| W150/3.5 | 368 | R247/3.8 | ND | |||
| L102 | 4.3 | W112/3.9 | 26 | 18.7 | Y191/3.1 | 261.5 |
| F230/3.8 | 801.9 | |||||
| E104 | 109 | K74/2.7 | 92 | 4.1 | R286/4.8 | 4.5 |
| Y105 | 0.9 | K74/3.6 | 92 | 44.7 | R286/3.5 | 4.5 |
| G49/3.9 | ND | |||||
| P107 | 1.5 | Y53/3.9 | 42 | 18.7 | Y73/3.6 | 204.4 |
| H41/3.9 | 68 | F74/3.8 | 74.2 | |||
| E110 | 0.1 | S71/2.6 | 0.4 | 1.1 | E268/2.5 | 22.7 |
| S113/2.9 | 0.2 | N112/2.9 | 7.7 | |||
| K111 | 0.2 | S39/3.2 | 0.6 | 37.3 | D131/2.8 | 27.7 |
| W152/3.3 | 243.4 | |||||
| M129 | 12.8 | F36/3.3 | 68 | 70.7 | F74/3.6 | 74.2 |
| Y50/3.4 | 0.02 | W53/4.1 | 10.5 | |||
| V216 | 0.4 | Y50/3.4 | 0.02 | 1.1 | W53/3.6 | 10.5 |
| D52/4.1 | 12.8 | |||||
| M272A | 0.9 | G48/3.4 | 1.4 | 1.7 | N50/3.6 | 9.0 |
| N51/4.5 | ND | |||||
Ratio of the TEM-1 alanine mutant versus wild type Ki for binding BLIP as determined in this study. For example, for the TEM-1 Q99A interaction with BLIP in the top line, left panel, the ratio of the Ki for TEM-1 Q99A/TEM-1 wt is 2.7.
BLIP contact residue and distance from the TEM-1 residue. For example, for TEM-1 Q99 in the top line, left panel residue TEM-1 Q99 is 3.7 Å from BLIP residue W150. Asterisk indicates a contact with a main chain atom in TEM-1.
Ratio of the BLIP alanine mutant versus wild type for binding TEM-1 as determined by Zhang.4 For example, for TEM-1 Q99 in the top line, left panel, the ratio of BLIP W150A/BLIP wt is 368 based on previously published values.
Ratio of the TEM-1 alanine mutant versus wild type Kd for binding BLIP-II as determined in this study.
BLIP-II contact residue and distance from the TEM-1 residue.
Ratio of the BLIP-II alanine mutant versus wild type for binding TEM-1 as determined by Brown et al.22
ND, not determined.
Discussion
While the individual contributions of BLIP and BLIP-II interface residues for binding to Class A enzymes have been described, the functional significance of the TEM-1 contact residues shared by these two proteins remains largely unknown.5,8,22,24 The alanine scanning approach taken here identified key hotspot residues for the interaction of TEM-1 β-lactamase with BLIP and BLIP-II. Extremely tight binding interactions between BLIP-II and TEM-1 have been characterized by determining both the individual association and dissociation rate constants to derive the Kd. In general, the association rate constants (kon) showed little variation for TEM-1 alanine mutants, while the dissociation rate constants (koff) exhibited large variations. These results are consistent with previous BLIP and BLIP-II mutagenesis studies indicating that dissociation constants, which are highly dependent on short range contacts, are most easily disrupted by alanine substitutions.22,25 On the other hand, association rates, which are strongly influenced by long-range electrostatic attraction forces that depend on electrostatic complementarity, are less sensitive to disruption by single alanine substitutions.26,27
The BLIP-TEM-1 binding interface is large with 2636 Å2 of surface buried upon complex formation.16 Despite the large binding interface, the majority of the binding energy is contained in two patches of BLIP hotspot residues (F36, H41, D49, Y53, K74, W112, F142, H148, W150, R160, W162) which show a bias towards large aromatic amino acids.5,8 The hotspot residues in TEM-1 for binding BLIP are in contact with BLIP hotspot residues. For example, TEM-1 E104 forms a salt bridge with BLIP K74 that is buried upon complex formation.16 In a previous study it was found that the BLIP K74A substitution results in a 92-fold decrease in binding affinity for TEM-1 and therefore the TEM-1 E104 and BLIP K74 hotspots match across the interface (Table IV). Also, TEM-1 residue M129 contacts BLIP residue F36 and the F36A substitution was previously shown to result in a 68-fold loss in affinity for TEM-1. In addition, several TEM-1 residues that are not hotspots, including E110, K111, and V216, contact BLIP residues that are not hotspots (Table IV). Several exceptions exist, however, in that TEM-1 residues Q99, N100, L102, Y105, and P107 make contacts with BLIP residues that are hotspots (Table IV). In addition, the M129 TEM-1 hotspot residue for BLIP also contacts BLIP residue Y50 and the Y50A substitution increases binding affinity for TEM-1.4 A possible explanation for these findings is that the BLIP substitutions may result in changes in the interface that alter positioning of other key residues, such as those in contact with TEM-1 hotspots, thereby altering binding energy. Support for the idea that amino acid substitutions of interface residues could impact the positioning of other amino acids beyond the site of mutation comes from the X-ray structures of complexes of BLIP and TEM-1 mutants. For example, Reichmann et al. showed that a E104Y:Y105N double mutant of TEM-1 results in a major structural rearrangement of the residue 46–51 loop of BLIP in the complex.20 In addition, the structure of the BLIP W150A mutant in complex with TEM-1 revealed a 4 Å change in the position of BLIP D49, which is 25 Å away from W150.25
A similar analysis of hotspots in the BLIP-II-TEM-1 interface reveals good matching between the TEM-1 hotspot residues and those residues in BLIP-II previously determined to be hotpots for binding TEM-122 (Table IV). For example, TEM-1 hotspot residues L102, P107, K111, and M129 all make direct contact with residues previously determined to be hotspots in BLIP-II (Table IV). Among the TEM-1 hotspot residues for binding BLIP-II, only Y105 does not contact a hotspot residue in BLIP-II and one of its contact residues in BLIP-II is G49, which was not examined (Table IV). Similarly, the majority of the contacts from BLIP-II hotspot residues are to TEM-1 hotspot residues, with exceptions being contacts between BLIP-II hotspots and the nonhotspot TEM-1 residues N100, E110, and V216 (Table IV).22
In general, alanine substitutions in TEM-1 β-lactamase have less of an impact on binding affinity for BLIP than BLIP-II (Table IV and Fig. 3). The average increase in Ki(d) for TEM-1 alanine mutants versus wild type is 12.1-fold for BLIP as compared to 18.6-fold for BLIP-II. This reflects the fact that only two of the eleven TEM-1 residues tested are hotspots for BLIP binding. In addition, the low average increase in Ki for TEM-1 binding to BLIP is influenced by residues N100 and E110, K111 and V216 where the alanine substitution results in tighter binding to BLIP (Table IV). The lower average increase in Kd for TEM-1 mutants binding BLIP versus BLIP-II can be rationalized if one considers that the TEM-1-BLIP interaction is over 150-fold weaker than that for TEM-1-BLIP-II while also utilizing more area in the binding interface compared to TEM-1-BLIP-II, which predicts the per residue contributions to binding affinity should be less for TEM-1 binding BLIP versus BLIP-II.
The TEM-1 N100A, E110A, K111A, and V216A substitutions are interesting in that they significantly increase binding affinity for BLIP suggesting the wild type TEM-1 residue at these positions is detrimental for binding (Table III). The majority of the improvement in binding affinity for each of these substitutions is due to a decrease in the dissociation rate (Table III). These findings suggest that removal of these TEM-1 side chains by alanine substitutions optimize short range contacts in the interface either directly by removal of an unfavorable interaction or by a cooperative effect on multiple interactions in the interface. TEM-1 residue N100 is at the edge of the interface of the complex and makes a main chain hydrogen bond to the side chain of BLIP R160 and a more distant side chain contact with BLIP W150 (Table IV). It is unclear why this alanine substitution reduces the dissociation rate. The mutant would retain the main chain bond to BLIP R160 but lose the interaction with W150. TEM-1 E110 is buried in the interface and forms hydrogen bonds with the hydroxyl groups of BLIP S71 and S113. The BLIP S71A and S113A substitutions do not alter binding affinity and so this region does not contribute to affinity of the complex and removal of the E110 side chain may facilitate new interactions in the interface.4 Similarly, TEM-1 K111 forms a hydrogen bond with BLIP S39 and the S39A substitution does not affect binding affinity.4 Interestingly, TEM-1 V216 has its closest interaction with BLIP Y50 and previous results have shown that the BLIP Y50A substitution increases binding affinity for TEM-1 by 50-fold.4 Therefore, the creation of extra space in this region allows for a tighter interaction between BLIP and TEM-1.
Computational programs that can predict changes in binding affinity upon mutation are a valuable asset for advancement of protein engineering and drug design. It was therefore of interest to compare the results of computational alanine scanning of the TEM-1 interface with BLIP and BLIP-II to the experimental results presented here. Alanine mutations of the TEM-1 residues tested in this study were performed computationally using Robetta and BeAtMuSiC online servers.28,29 The ΔΔG values generated by the online servers and the experimental values are listed in Table V and plotted in Figure 5. The average of the standard deviations of the prediction values for each position versus the experimental results for each position for TEM-1 binding BLIP was 1.2 kcal mol−1 for both programs. For the interface residues examined between TEM-1 and BLIP-II, the average of the standard deviations was 0.7 kcal mol−1 for Robetta and 0.6 kcal mol−1 for BeAtMuSiC. Overall, both programs were more successful at predicting free energy changes upon mutation in the TEM-1/BLIP-II interface than the TEM-1/BLIP interface. This could be due to the fact that BLIP-II is a rigid protein while mutations at the interface between BLIP and TEM-1 have been shown to cause significant movement in the BLIP structure to accommodate these changes at the interface as described above.10,20,25
Table V.
ΔΔG Values of the TEM-1 Alanine Variants for Binding BLIP and BLIP-II
| BLIP |
BLIP-II |
|||||
|---|---|---|---|---|---|---|
| Mutant | Robetta | BeAtMuSiC | Experimental | Robetta | BeAtMuSiC | Experimental |
| Q99A | 1.40 | 1.05 | 0.60 | 2.12 | 1.20 | 0.53 |
| N100A | 0.30 | 0.7 | −1.50 | 0.23 | 0.55 | 0.81 |
| L102A | 0.50 | 1.89 | 0.87 | 0.60 | 1.97 | 1.76 |
| E104A | 2.10 | 1.32 | 2.83 | −0.24 | 0.51 | 0.85 |
| Y105A | 4.49 | 4.22 | −0.10 | 2.03 | 3.63 | 2.29 |
| P107A | n/a | 1.04 | 0.24 | n/a | 1.08 | 1.76 |
| E110A | 1.71 | 1.16 | −1.34 | 1.82 | 1.61 | 0.04 |
| K111A | 1.58 | 0.78 | −0.87 | 1.03 | 1.47 | 2.18 |
| M129A | 0.52 | 1.8 | 1.53 | 0.57 | 1.53 | 2.56 |
| V216A | 0.53 | 1.77 | −0.59 | 0.44 | 1.99 | 0.04 |
| M272A | 0.50 | 1.23 | −0.05 | 0.67 | 1.10 | 0.28 |
n/a, data for position not generated in alanine scanning of interface.
Figure 5.
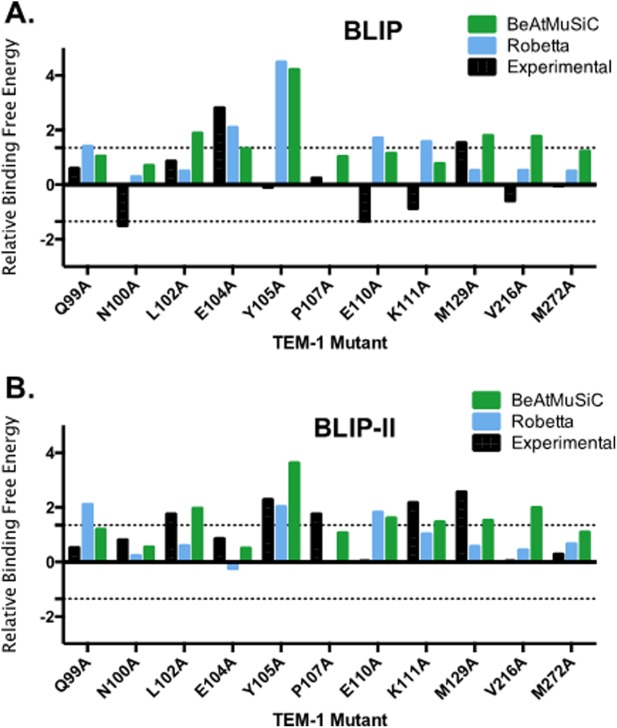
Comparison of the ΔΔG values of the TEM-1 alanine variants for binding BLIP (A) and BLIP-II (B) as determined experimentally, and predicted by BeAtMuSiC and Robetta. For experimental data, relative free energy changes are defined as the difference in energy between wild-type TEM-1 and the alanine mutant (ΔΔG = −RTln(Kd,wt/Kd,mut)). The top control line is set at 1.35 kcal/mol and the bottom control line is set at −1.35 kcal/mol, which indicates a 10-fold change or hotspot for binding. BeAtMuSiC results are shown in green, Robetta results are shown in blue and the experimental results are shown in black.
The β-lactamase-BLIP system is an example of a common surface on a protein (TEM-1) that interacts with proteins of disparate structure (BLIP or BLIP-II). There are many examples in biology of a protein using a single surface to bind different target proteins including protein kinase A regulatory domain, Ran GTPase, calmodulin, Fc receptor, and BirA, among others.30–35 In the TEM-1 β-lactamase-BLIP system studied here it was found that largely different residues on TEM-1 serve as hotspots for binding BLIP versus BLIP-II, that is, different binding strategies are used to bind disparate partners. This is similar to the situation with BirA, which uses a single surface to either homodimerize or to form a heterodimer with the biotin carboxyl carrier protein of acetyl coenzyme A carboxylase.35 Amino acid substitutions in the BirA binding surface revealed some overlap in residues required for both interactions but hotspots specific for one but not the other were also found.35,36 In addition, it has been shown by comparison of a database of homologous proteins binding to heterogeneous partner proteins that promiscuous binding, that is, the binding of a single protein surface to multiple, unrelated partner proteins, is largely achieved by alternate binding strategies for the different partners rather than convergent evolution to use the same binding interactions for different partners.37 The experiments reported here extend these observations to the TEM-1 β-lactamase-BLIP system.
Class A β-lactamase enzymes are the most prevalent β-lactamases worldwide and are diverse in sequence.15 Since the introduction of newer β-lactam derivatives, class A enzymes capable of hydrolyzing extended-spectrum cephalosporins, monobactams, and carbapenems have evolved or emerged.38,39 There is a pressing need for new inhibitors and improved detection methods for β-lactamases. Better understanding of the forces driving the interaction of β-lactamases and BLIPs can aid in design of better diagnostic tools for detection of those enzymes.
Materials and Methods
Construction of alanine substitutions
The TEM-1 residues chosen for mutation were based on the previously performed cluster analysis of the BLIP–TEM-1 and BLIP-II-TEM-1 interfaces8,10 (Table I). The TEM-1 residues of the TEM-1 BLIP interfaces were mutated to alanine by site-directed mutagenesis using the Quikchange method (Stratagene) using the Pfu turbo polymerase (Stratagene) and the pET-TEM-1 plasmid as template DNA.40 DpnI restriction enzyme was then added to the solution to remove the parental strands. The Quikchange product was introduced into E. coli XL-1 Blue cells (Stratagene). DNA sequencing confirmed the mutations and that no extraneous mutations occurred in the TEM-1 gene (Lonestar Labs).
Protein purification
The TEM-1 wild type and alanine-substituted enzymes were purified as previously described.40,41 BLIP and BLIP-II were purified using the TALON Metal Affinity Resin (Clontech) using an N-terminal His-tag on BLIP and a C-terminal his-tag on BLIP-II as previously described.10,23 The protein concentrations were determined by a Bradford assay, and compared with a β-lactamase standard curve calibrated by quantitative amino acid analysis.
β-Lactamase inhibition assay
The inhibition assay for evaluating the binding constant of tight-binding inhibitors has been described previously.21,23 Various concentrations of BLIP were incubated with 0.2 nM of TEM-1 β-lactamase variants for 1 h at room temperature in 50 mM phosphate buffer pH 7.0. The percentage of β-lactamase bound by BLIP was determined by monitoring the initial velocity of TEM-1-β-lactamase-mediated nitrocefin (chromogenic cephalosporin analog) hydrolysis at room temperature (23°C) in a spectrophotometer at OD485nm. Nitrocefin was used at a concentration of 40 µM for the binding assays and the experiments were repeated a minimum of two times. The Kiapp values for BLIP inhibition of each TEM-1 mutant were determined by fitting the initial velocities to the Morrison tight-binding equation [Eq. (1)]:42
| (1) |
where Efree is the concentration of free enzyme determined by residual activity of TEM-1 β-lactamase by comparison with the initial velocity of nitrocefin hydrolysis by the uninhibited β-lactamase, [E0] is the total enzyme concentration and [I0] is the total inhibitor concentration. The Ki values were calculated from the Kiapp values using Eq. (2) below as described previously.21 The Km value for nitrocefin hydrolysis for wild type and each TEM-1 β-lactamase mutant from Table II was used for calculation of the Ki values.
| (2) |
Stopped-flow tryptophan fluorescence spectrometric measurements of association rate
The association rate constants for binding of BLIP-II and BLIP to TEM-1 β-lactamase were determined as previously described using a Kintek stopped-flow spectrofluorometer with binary injection mode to monitor the tryptophan intrinsic fluorescence over a time course.10 The proteins were dialyzed into 10 mM Tris, 150 mM NaCl at pH 7.0. Two injection syringes containing equal protein concentrations of 10 µM for BLIP and TEM-1, and 5 µM for BLIP-II and TEM-1, were engaged on the instrument and used for mixing. The excitation and emission wavelengths were 286 and 340 nm with bandwidths of 2 and 8 nm, respectively. The data was collected at 10-ms intervals with a 10-ms mixing dead time. Multiple traces (>10) were collected at room temperature (23°C) and averaged together. The averaged trace of the change in intrinsic fluorescence was fitted with a second order kinetic time course [Eq. (3)] to determine the association constant using the program Graphpad Prism 5,
| (3) |
where Ft is the intrinsic fluorescence at time t, ΔF0 is the amplitude of the total change in the intrinsic fluorescence signal, F∞ is the background intrinsic fluorescence after the association is complete, and TC is the time parameter determined by fitting the data to the equation above. The association rate constant, kon, is then calculated with Eq. (4),
| (4) |
where [M0] is the molar concentration of the unbound proteins.
Enzymatic determination of the dissociation rate constants
The dissociation rate constants for BLIP-II and TEM-1 were determined as previously described.10,22 The slow dissociation rate constants (koff) were determined by measuring the recovery of the wild-type β-lactamase activity by competitive displacement with an inactive TEM-1 variant (E166A).10,22 The E166A mutation prevents the deacylation step of the catalytic mechanism rendering the enzyme inactive after one round of acylation and allows for the mutant enzyme to remain in large excess but still allows the measurement of the wild-type β-lactamase activity. The TEM-1 E166 residue is not at the binding interface with BLIP-II and the E166A substitution does not affect binding to BLIP-II.10 TEM-1 was incubated in a 2-fold excess of BLIP-II for one hour. The BLIP-II-β-lactamase complexes were then diluted into a ≥400 molar excess TEM-1 E166A competitor solution. The final TEM-1 enzyme concentrations were 5 nM for Q99A, N100A, E104A, E105A, E110A, K111A, M129A, V216A, M272A, and 10 nM for mutants L102A and P107A (Table II). The buffer used in these experiments was 50 mM sodium phosphate, pH 7.0, supplemented with 1 mg/mL BSA and reactions were performed at room temperature (23°C). A 400 µM concentration of nitrocefin was used for all dissociation kinetics experiments. The experiments were repeated at least twice. The amount of active β-lactamase over the time course was fitted with first order kinetics to determine the kinetic parameters [Eq. (5)],
| (5) |
where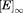 . is the amount of free β-lactamase when the dissociation reaction had reached completion as estimated by the enzymatic activity when uninhibited by BLIP-II.
. is the amount of free β-lactamase when the dissociation reaction had reached completion as estimated by the enzymatic activity when uninhibited by BLIP-II.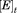 . is the amount of free β-lactamase estimated by enzymatic activity at time (t) and t is the time after mixing the BLIP-II-β-lactamase complex with the inactive TEM-1 E166A enzyme. C is the curve fitting constant representing the background rate of nitrocefin hydrolysis (including the activity of the TEM-1 E166A enzyme) and koff is the dissociation rate constant extrapolated from fitting the data. Because of the long time course of the experiment, positive and negative controls of the β-lactamase and inactive TEM-1 E166A alone, respectively, were used to assess the stability of the β-lactamases during the experiment. Due to fast dissociation rates (koff) of BLIP from TEM-1, this method was not suitable for their accurate determination and they were calculated from the inhibition constant (Ki) and the association rate constant at ambient temperature (23°C).
. is the amount of free β-lactamase estimated by enzymatic activity at time (t) and t is the time after mixing the BLIP-II-β-lactamase complex with the inactive TEM-1 E166A enzyme. C is the curve fitting constant representing the background rate of nitrocefin hydrolysis (including the activity of the TEM-1 E166A enzyme) and koff is the dissociation rate constant extrapolated from fitting the data. Because of the long time course of the experiment, positive and negative controls of the β-lactamase and inactive TEM-1 E166A alone, respectively, were used to assess the stability of the β-lactamases during the experiment. Due to fast dissociation rates (koff) of BLIP from TEM-1, this method was not suitable for their accurate determination and they were calculated from the inhibition constant (Ki) and the association rate constant at ambient temperature (23°C).
Computational prediction of the effect of TEM-1 mutations on binding affinity
Two programs were used to predict changes in binding affinity of the TEM-1 mutants on complex formation with BLIP or BLIP-II. Computational alanine scanning of TEM-1 interface residues was performed using Robetta, an online structure prediction and analysis server that makes use of protein–protein docking methodologies.28 Robetta uses a simple free energy function to perform the alanine scanning of the interface residues that the program defines. Pro107 was not included in the alanine scanning performed by Robetta because the program-defined interface did not include this residue. BeAtMuSiC was also used and this server relies on a set of statistical potentials derived from known protein structures and predicts the changes in binding affinity by combining the effect of the mutation on the overall stability of the complex and the interface.29 PDB IDs 1JTG (BLIP and TEM-1) and 1JTD (BLIP-II and TEM-1) chains A and B were submitted to the online servers for analysis.16,17
Acknowledgments
The authors thank Dar-Chone Chow and Hiram Gilbert for comments on the manuscript.
REFERENCES
- 1.Perkins JR, Diboun I, Dessailly BH, Lees JG, Orengo C. Transient protein-protein interactions: structural, functional, and network properties. Structure. 2010;18:1233–1243. doi: 10.1016/j.str.2010.08.007. [DOI] [PubMed] [Google Scholar]
- 2.Khan SH, Ahmad F, Ahmad N, Flynn DC, Kumar R. Protein-protein interactions: principles, techniques, and their potential role in new drug development. J Biomol Struct Dyn. 2011;28:929–938. doi: 10.1080/07391102.2011.10508619. [DOI] [PubMed] [Google Scholar]
- 3.Schreiber G, Keating AE. Protein binding specificity versus promiscuity. Curr Opin Struc Biol. 2011;21:50–61. doi: 10.1016/j.sbi.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang Z, Palzkill T. Determinants of binding affinity and specificity for the interaction of TEM-1 and SME-1 β-lactamase with β-lactamase inhibitory protein. J Biol Chem. 2003;278:45706–45712. doi: 10.1074/jbc.M308572200. [DOI] [PubMed] [Google Scholar]
- 5.Zhang Z, Palzkill T. Dissecting the protein-protein interface between beta-lactamase inhibitory protein and class A beta-lactamases. J Biol Chem. 2004;279:42860–42866. doi: 10.1074/jbc.M406157200. [DOI] [PubMed] [Google Scholar]
- 6.Reichmann D, Rahat O, Albeck S, Meged R, Dym O, Schreiber G. The modular architecture of protein-protein binding interfaces. Proc Natl Acad Sci USA. 2005;102:57–62. doi: 10.1073/pnas.0407280102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reynolds KA, Thomson JM, Corbett KD, Bethel CR, Berger JM, Kirsch JF, Bonomo RA, Handel TM. Structural and computational characterization of the SHV-1 beta-lactamase-beta-lactamase inhibitor protein interface. J Biol Chem. 2006;281:26745–26753. doi: 10.1074/jbc.M603878200. [DOI] [PubMed] [Google Scholar]
- 8.Reichmann D, Cohen M, Abramovich R, Dym O, Lim D, Strynadka NCJ, Schreiber G. Binding hot spots in the TEM1-BLIP interface in light of its modular architecture. J Mol Biol. 2007;365:663–679. doi: 10.1016/j.jmb.2006.09.076. [DOI] [PubMed] [Google Scholar]
- 9.Gretes M, Lim DC, de Castro L, Jensen S, Kang SG, Lee KJ, Strynadka NCJ. Insights into positive and negative requirements for protein-protein interactions by crystallographic analysis of the β-lactamase inhibitory proteins BLIP, BLIP-I, and BLP. J Mol Biol. 2009;389:289–305. doi: 10.1016/j.jmb.2009.03.058. [DOI] [PubMed] [Google Scholar]
- 10.Brown NG, Chow D-C, Sankaran B, Zwart P, Prasad BV, Palzkill T. Analysis of the binding forces driving the tight binding between β-lactamase inhibitory protein II (BLIP-II) and class A β-lactamases. J Biol Chem. 2011;286:32723–32725. doi: 10.1074/jbc.M111.265058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Keskin O, Ma B, Nussinov R. Hot regions in protein–protein interactions: the organization and contribution of structurally conserved hot spot residues. J Mol Biol. 2005;345:1281–1294. doi: 10.1016/j.jmb.2004.10.077. [DOI] [PubMed] [Google Scholar]
- 12.Moreira IS, Fernandes PA, Ramos MJ. Hot spots-A review of the protein-protein interface determinant amino acid residues. Proteins. 2007;68:803–812. doi: 10.1002/prot.21396. [DOI] [PubMed] [Google Scholar]
- 13.Kozakov D, Hall DR, Chuang GY, Cencic R, Brenke R, Grove LE, Beglov D, Pelletier J, Whitty A, Vajda S. Structural conservation of druggable hot spots in protein-protein interfaces. Proc Natl Acad Sci U.S.A. 2011;108:13528–13533. doi: 10.1073/pnas.1101835108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.De Lano WL. Unraveling hot spots in binding interfaces: progress and challenges. Curr Opin Struct Biol. 2002;12:14–20. doi: 10.1016/s0959-440x(02)00283-x. [DOI] [PubMed] [Google Scholar]
- 15.Bush K, Fisher JF. Epidemiological expansion, structural studies, and clinical challenges of new β-lactamases from Gram-negative bacteria. Ann Rev Microbiol. 2011;65:455–478. doi: 10.1146/annurev-micro-090110-102911. [DOI] [PubMed] [Google Scholar]
- 16.Strynadka NCJ, Jensen SE, Alzari PM, James MNG. A potent new mode of β-lactamase inhibition revealed by the 1.7Å X-ray crystallographic structure of the TEM-1-BLIP complex. Nature Struct Biol. 1996;3:290–297. doi: 10.1038/nsb0396-290. [DOI] [PubMed] [Google Scholar]
- 17.Lim D, Park HU, De Castro L, Kang SG, Lee HS, Jensen S, Lee KJ, Strynadka NCJ. Crystal structure and kinetic analysis of β-lactamase inhibitor protein-II in complex with TEM-1 β-lactamase. Nat Struct Biol. 2001;8:848–852. doi: 10.1038/nsb1001-848. [DOI] [PubMed] [Google Scholar]
- 18.Doran JL, Leskiw BK, Aippersbach S, Jensen SE. Isolation and characterization of a β-lactamase-inhibitory protein from Streptomyces clavuligerus and cloning and analysis of the corresponding gene. J Bacteriol. 1990;172:4909–4918. doi: 10.1128/jb.172.9.4909-4918.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Strynadka NCJ, Jensen SE, Johns K, Blanchard H, Page M, Matagne A, Frere J-M, James MNG. Structural and kinetic characterization of a β-lactamase-inhibitor protein. Nature. 1994;368:657–660. doi: 10.1038/368657a0. [DOI] [PubMed] [Google Scholar]
- 20.Reichmann D, Rahat O, Cohen M, Neuvirth H, Schreiber G. The molecular architecture of protein-protein binding sites. Curr Opin Struct Biol. 2007;17:67–76. doi: 10.1016/j.sbi.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 21.Brown NG, Palzkill T. Identification and characterization of beta-lactamase inhibitor protein-II (BLIP-II) interactions with beta-lactamases using phage display. Protein Eng Des Sel. 2010;23:469–478. doi: 10.1093/protein/gzq017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brown NG, Chow D-C, Ruprecht KE, Palzkill T. Identification of the β-lactamase inhibitor protein-II (BLIP-II) interface residues essential for binding affinity and specificity for class A β-lactamases. J Biol Chem. 2013;288:17156–17166. doi: 10.1074/jbc.M113.463521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Petrosino J, Rudgers G, Gilbert H, Palzkill T. Contributions of aspartate 49 and phenylalanine 142 residues of a tight binding inhibitory protein of β-lactamases. J Biol Chem. 1999;274:2394–2400. doi: 10.1074/jbc.274.4.2394. [DOI] [PubMed] [Google Scholar]
- 24.Hanes MS, Jude KM, Berger JM, Bonomo RA, Handel TM. Structural and biochemical characterization of the interaction between KPC-2 beta-lactamase and beta-lactamase inhibitor protein. Biochemistry. 2009;48:9185–9193. doi: 10.1021/bi9007963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang J, Palzkill T, Chow DC. Structural insight into the kinetics and DeltaCp of interactions between TEM-1 beta-lactamase and beta-lactamase inhibitory protein (BLIP) J Biol Chem. 2009;284:595–609. doi: 10.1074/jbc.M804089200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Selzer T, Schreiber G. Predicting the rate enhancement of protein complex formation from the electrostatic energy of interaction. J Mol Biol. 1999;287:409–419. doi: 10.1006/jmbi.1999.2615. [DOI] [PubMed] [Google Scholar]
- 27.Kozer N, Schreiber G. Effect of crowding on protein-protein association rates: fundamental differences between low and high mass crowding agents. J Mol Biol. 2004;336:763–774. doi: 10.1016/j.jmb.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 28.Kim DE, Chivian D, Baker D. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 2004;32:526–531. doi: 10.1093/nar/gkh468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dehouck Y, Kwasigroch JM, Rooman M, Gilis D. BeAtMuSiC: prediction of changes in the protein-protein binding affinity on mutations. Nucleic Acids Res. 2013;41:333–339. doi: 10.1093/nar/gkt450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Colledge M, Scott JD. AKAPs: from structure to function. Trends Cell Biol. 1999;9:916–921. doi: 10.1016/s0962-8924(99)01558-5. [DOI] [PubMed] [Google Scholar]
- 31.DeLano WL, Ultsch MH, de Vos AM, Wells JA. Convergent solution to binding at a protein-protein interface. Science. 2000;287:1279–1283. doi: 10.1126/science.287.5456.1279. [DOI] [PubMed] [Google Scholar]
- 32.Stewart M. Molecular mechanism of the nuclear protein import cycle. Nat Rev Mol Cell Biol. 2007;8:195–208. doi: 10.1038/nrm2114. [DOI] [PubMed] [Google Scholar]
- 33.Chang CA, McLaughlin WA, Baron R, Wang W, McCammon JA. Entropic contributions and the influence of the hydrophobic environment in promiscuous protein-protein association. Proc Natl Acad Sci USA. 2008;105:7456–7461. doi: 10.1073/pnas.0800452105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fromer M, Shifman JM. Tradeoff between stability and multispecificity in the design of promiscuous proteins. PLOS Comp Biol. 2009;5:e1000627. doi: 10.1371/journal.pcbi.1000627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao H, Naganathan S, Beckett D. Thermodynamic and structural investigation of bispecificity in protein-protein interactions. J Mol Biol. 2009;389:336–348. doi: 10.1016/j.jmb.2009.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Adikaram PR, Beckett D. Functional versatility of a single protein surface in two protein:protein interactions. J Mol Biol. 2012;419:223–233. doi: 10.1016/j.jmb.2012.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Martin J-F. Beauty is in the eye of the beholder: proteins can recognize binding sites of homologous proteins in more than one way. PLOS Comp Biol. 2010;6:e1000821. doi: 10.1371/journal.pcbi.1000821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Perez F, Endimiani A, Hujer KM, Bonomo RA. The continuing challenge of ESBLs. Curr Opin Pharmacol. 2007;7:459–469. doi: 10.1016/j.coph.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Papp-Wallace KM, Endimiani A, Taracila MA, Bonomo RA. Carbapenems: past, present, and future. Antimicrob Agents Chemother. 2011;55:4943–4960. doi: 10.1128/AAC.00296-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marciano DC, Brown NG, Palzkill T. Analysis of the plasticity of location of positive charge within the active site of the TEM-1 β-lactamase. Protein Sci. 2009;18:2080–2089. doi: 10.1002/pro.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Marciano DC, Pennington JM, Wang X, Wang J, Chen Y, Thomas VL, Shoichet BK, Palzkill T. Genetic and structural characterization of an L201P global suppressor substitution in TEM-1 beta-lactamase. J Mol Biol. 2008;384:151–164. doi: 10.1016/j.jmb.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Murphy DJ. Determination of accurate Ki values for tight-binding enzyme inhibitors: an in silico study of experimental error and assay design. Anal Biochem. 2004;327:61–67. doi: 10.1016/j.ab.2003.12.018. [DOI] [PubMed] [Google Scholar]