

Abstract
The β-amyloid (Aβ) peptide aggregates into a number of soluble and insoluble forms, with soluble oligomers thought to be the primary factor implicated in Alzheimer's disease pathology. As a result, a wide range of potential aggregation inhibitors have been developed. However, in addition to problems with solubility and protease susceptibility, many have inadvertently raised the concentration of these soluble neurotoxic species. Sandberg et al. previously reported a β-hairpin stabilized variant of Aβ42 that results from an intramolecular disulphide bridge (A21C/A31C; Aβ42cc), which generates highly toxic oligomeric species incapable of converting into mature fibrils. Using an intracellular protein-fragment complementation (PCA) approach, we have screened peptide libraries using E. coli that harbor an oxidizing environment to permit cytoplasmic disulphide bond formation. Peptides designed to target either the first or second β-strand have been demonstrated to bind to Aβ42cc, lower amyloid cytotoxicity, and confer bacterial cell survival. Peptides have consequently been tested using wild-type Aβ42 via ThT binding assays, circular dichroism, MTT cytotoxicity assays, fluorescence microscopy, and atomic force microscopy. Results demonstrate that amyloid-PCA selected peptides function by both removing amyloid oligomers as well as inhibiting their formation. These data further support the use of semirational design combined with intracellular PCA methodology to develop Aβ antagonists as candidates for modification into drugs capable of slowing or even preventing the onset of AD.
Keywords: amyloid, protein misfolding, protein–protein interactions, library screening, protein-fragment complementation assay, Alzheimer's disease
Introduction
It has been widely speculated that small soluble oligomers of Aβ play a major role in the pathology of Alzheimer's disease (AD).1–4 However, developing drugs that prevent the accumulation of these species has proven difficult. Initial studies that sought to breakdown large insoluble oligomers have in some instances led to an accumulation of small toxic species,5 while more recent work has shown that even dimers or trimers of Aβ are toxic and able to impair synaptic function.6 In screening libraries to identify peptides capable of modifying aggregation, an elegant approach would be to lock Aβ42 into a toxic conformation. This would ensure population of an oligomeric state that is pathogenically relevant to the disease. Sandberg et al. previously studied the effect of an intramolecular disulphide-bridged double mutant of Aβ42 (A21C/A31C; Aβ42cc).7 In this approach, conservative mutations were used to create a disulphide bridge and force an otherwise unstructured and monomeric Aβ42 into the β-hairpin conformation found in mature fibrils. The conformation, therefore, provides the structural basis for subsequent amyloidogenesis.8 Disulphide tethering was found to stabilize highly toxic Aβ oligomers that retained the conformational and biological properties of wild-type Aβ. Moreover, these molecules did not go on to convert into mature fibrils and were shown to contain SDS-stable dimeric and trimeric species. Experiments using SH-SY5Y human neuroblastoma cells demonstrated that Aβ42cc oligomers or protofibrillar species formed by these oligomers were 50 times more potent inducers of neuronal apoptosis than amyloid fibrils or samples of monomeric wild-type Aβ42, in which toxic aggregates were only transiently formed.
A wide variety of approaches have been used to create amyloid inhibitors, including small molecule, antibody, and peptide-based inhibitors. These have ranged from those that stabilize the native state, sequester monomers, inhibit amyloid growth, as well as those that initiate clearance via chaperones and proteases (see Ref.9 for an overview). In our own studies, we previously presented a novel variation of the intracellular protein-fragment complementation assay (PCA) selection system whereby bacteria were used to screen peptide libraries and generate Aβ aggregation antagonists10 and subsequently their retroinversed analogues.11 During PCA a “core recognition element” Aβ25–35, was used as the target since, along with Aβ15–21, it is known to form amyloid fibrils in isolation and instigate amyloid formation in the parental protein. Aβ25–35 was fused to one half of murine dihydrofolate reductase (mDHFR) with a peptide library fused to the second half. Library members that bound Aβ25–35 resulted in a colony under selective conditions. The most effective binders were then further enriched by growth competition to isolate from binders that led to lower growth rates. Reduced bacterial growth rates are due to the inherent propensity for Aβ to aggregate and lead to increased cytotoxic effects.12,13 Three outcomes are possible for any given library member:
Library members bind Aβ, reduce its toxicity and recombine mDHFR to confer cell survival.
Library members bind Aβ and recombine mDHFR but either populate or do not prevent population of a toxic species. These result in reduced cell growth relative to 1, or cell death.
Library members with no affinity for Aβ and, therefore, no effect on amyloid formation will not recombine mDHFR, resulting in cell death.
Intracellular selection means no assumptions are made regarding the mechanism of antagonist action or which amyloid state becomes populated during selection, aiding the removal of molecules with undesirable properties from the library such as those that are too hydrophobic hence insoluble, those tagged for degradation, those susceptible to protease action, and those that are nonspecific.
To further strengthen this approach, we report on the use of full length Aβ42cc as the target molecule rather than Aβ15–21 or the Aβ25–35 amyloidogenic regions of the molecule used previously.10 In this experiment, PCA is undertaken using Aβ42cc under oxidizing conditions to ensure that:
During selection the Aβ42cc target is constrained into the β-hairpin structure associated with the pathology of the disease
The “core recognition elements” of Aβ42cc associated with instigation of amyloid formation remain exposed, largely unmodified, and in the correct conformation.
Aβ42cc is able to form soluble oligomeric structures that are highly toxic and associated with AD pathology, thus ensuring that a larger number of structurally relevant epitopes are presented relative to larger and/or insoluble fibrils populated in the absence of the bridge.
Expressing the increased toxicity variant Aβ42cc raises the sensitivity of the PCA relative to Aβ42. This is because cells in scenario B above are predicted to die more readily or grow at reduced rates.
We find the peptides derived using this approach to be capable of binding to the disease relevant wild-type Aβ42 and reducing associated amyloid formation. To understand the mechanism of peptide action we have studied the effectiveness of peptides and found them to be capable of removing preformed fibrils as well as preventing amyloid from forming. These studies have successfully produced a number of lead peptide sequences that are expected to provide a scaffold for future drug candidates.
Results
We have combined semirational design with intracellular selection using amyloid-PCA to screen peptide libraries and select Aβ42 aggregation antagonists.10,14,15 A disulphide-bridged variant of Aβ42, known as Aβ42cc, has been used as the target for library screening since it constrains the protein into the β-hairpin structure found in amyloid structures that form oligomers associated with AD toxicity (Fig. 1). PCA libraries were initially screened and selected on M9 minimal agar plates. Following this initial single-step selection, colonies underwent competition selection where they were pooled and grown before dilution into liquid M9 minimal media. This process was repeated multiple times as passages to select for the most effective binding sequence. In this process, 1–2 high affinity binders that inhibit amyloid formation and reduce bacterial toxicity were found to display the fastest growth and dominate the bacterial pool. The selection process indicated that only a very limited subset of library members were able to bind Aβ42cc (<1%) and that these can be further separated during competition selection PCA to isolate the most effective binders. DNA sequencing continued throughout passaging until selection arrived at one discrete peptide. All subsequent aggregation studies were undertaken using Aβ42 as Aβ42cc is not found naturally, with ThT binding and TEM data demonstrating that fibrils do not form when the intramolecular disulphide bridge is intact.7 We demonstrate that the sequences identified using the Aβ42cc target can be transferred to the biologically relevant Aβ42.
Figure 1.
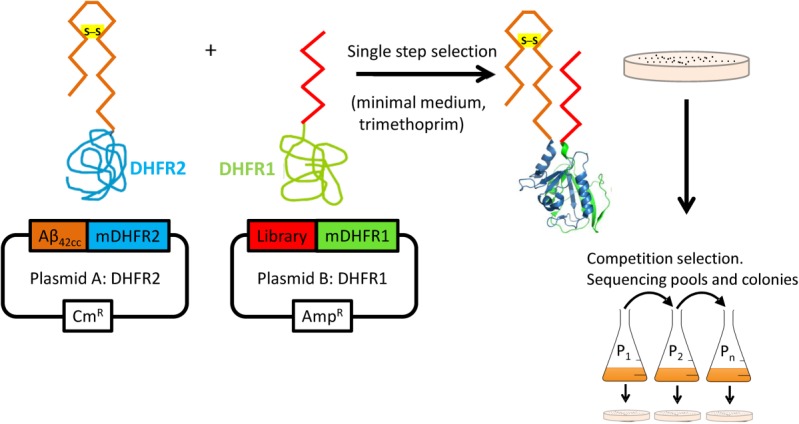
The PCA for amyloid systems. Selection is undertaken using SHuffle cells that harbor an oxidizing cytoplasmic environment to promote disulphide bond formation. Library members that bind to Aβ42cc lead to the recombination of murine DHFR and lead to a colony under selective conditions (bacterial DHFR is specifically inhibited using trimethoprim). Subsequent competition selection in liquid media isolates winners of highest efficacy. Those library members that bind the target and are able to confer faster growth rates by reducing the toxic effects of the amyloid protein most effectively will be selected. As the assay is performed in the cytoplasm of E.coli, any nonspecific, unstable, aggregation prone (insoluble), protease susceptible members are removed.
Library generation
PCA was undertaken using an Aβ42cc target with sequences corresponding to Aβ15–21 (QKLVFFA) Aβ29–35 (GAIIGLM), and Aβ36–42 (VGGVVIA) as library templates. The first of these two regions are known to aggregate into toxic fibrils in isolation16–18 and therefore provided a strong starting point for deriving peptides known to bind to Aβ42cc and potentially capable of inhibiting aggregation. The third sequence incorporates the two additional C-terminal amino acids (Ile-Ala) that renders Aβ42 much more toxic than Aβ40. The first library incorporated four fully randomized residues at positions 17–20 of Aβ15–21, generating a library size of 160,000 (15QKxxxxA21), and resulted in one winner (cys1521; Table I). Interestingly, this library had previously resulted in no colonies when using either Aβ42 or Aβ15–21 as target, suggesting an improved epitope for binding on introduction of the cysteine-bridge. Moreover, the selected sequence, QKVLLFA, bared remarkable similarity to the Aβ15–21 (QKLVFFA) parental sequence on which it was based, having been enriched during PCA from four fully randomized residues at positions 17–20. The Aβ15–21 peptide has been previously shown to form amyloid in isolation and found to be toxic in MTT experiments.19,20 The second library incorporated four fully randomized positions at 29–30 and 34–35 of Aβ29–35, with residues 31–33 fixed from a previous selection,10 generating a 160,000 member library (29xxKATxx35), and resulted in two amyloid-PCA winners (cys2935a and cys2935b; Table I) with no sequence homology to the amyloid template on which they were based. The last library fixed residues 38–40 of Aβ36–42 as “GVV” as is found in wild-type Aβ40 and Aβ42, but was fully randomized at residues 36–37 and importantly at residues 41–42 associated with the increased toxicity of Aβ42, generating a 160,000 member library (36xxGVVxx42), and resulting in one winner (cys3642; Table I).
Table I.
PCA Derived Sequences
| Name | Sequence |
|---|---|
| cys1521 | Ac-QKVLLFA-NH2 |
| cys2935a | Ac-AGKATGL-NH2 |
| cys2935b | Ac-GAKATAN-NH2 |
| cys3642 | Ac-RWGVVWG-NH2 |
| iAβ5 | Ac-LPFFD-NH2 |
Four libraries consisting of 160,000 members each and based around residues 15–21, 29–35, and 36–42 of the Aβ1–42 were fully randomised at positions shown underlined to allow options for all amino acids. A second library consisting of 160,000 members was fully randomised at positions 29–30 and 34–35 with the KAT sequence from the first selection fixed. Cys2935a/b is based on a previously selected sequence that targeted Aβ29–35.10 Acetylation and amidation of the protein was added to mimic the polypeptide backbone and provide additional potential hydrogen-bond acceptors and donors. The positive control sequence, iAβ5, is also shown.
Peptide characterization
PCA derived peptide sequences (Table I) were synthesized and characterized using a number of experiments that included thioflavin-T (ThT) dye binding, circular dichroism (CD), oblique angle fluorescence (OAF), and atomic force microscopy (AFM). The results demonstrate that the peptides do not aggregate in isolation, and are able to prevent Aβ42 aggregation and/or remove preformed fibrils. In addition, growth competition assays using E.coli under PCA conditions in M9 media and an MTT assay using PC12 cells, both using the Aβ42 parent peptide, were undertaken to establish cytotoxicity to bacterial and mammalian cells. The growth competition experiments simultaneously demonstrate that peptides bind to Aβ42 and reduce its associated toxicity during bacterial selection. MTT experiments were used to establish that the toxicity associated with extracellular Aβ42 to mammalian cells could be reduced when incubated in the presence of PCA selected peptides.
Cell growth experiments
The effect of inhibitors on the growth of E.coli harboring pES300d-Aβ42cc-DHFR2 target and pES230d-antagonist-DHFR1 fusion plasmids as present in the final PCA selection round were tested (Fig. 2). In this experiment, cells were grown in a shaking incubator from a starting OD600 of 0.02 under PCA conditions in M9 minimal media containing Cm, Amp, and Kan to retain target and antagonist expressing plasmids as well as pREP4 for expression of the lac repressor. In addition Tmp was included for inhibition of bacterial DHFR and IPTG to induce high levels of target and antagonist expression. This experiment monitors both mDHFR reassembly, and, therefore, binding of antagonist to the Aβ42cc target, as well as the toxicity of the oligomeric state that is populated. As expected, expression of the toxic Aβ42cc did not result in significant levels of growth even though the protein is well documented to self-associate (Fig. 2). In addition, western blots (Supporting Information Fig. S5) show that Aβ42cc is expressed in the soluble fraction, suggesting that the protein is both soluble and toxic and is, therefore, populating toxic protofibrillar structures. All four antagonists in this study, along with the positive control cJun-FosW, were clearly able to restore bacterial growth thereby providing strong evidence for direct binding and reduced toxicity in the context of this bacterial selection system.
Figure 2.
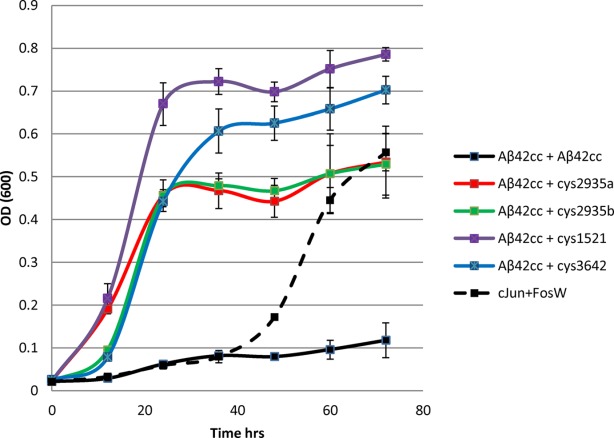
To confirm that expression of Aβ42cc-DHFR1 /Aβ42cc-DHFR2 fusions impedes the growth rate of E. coli, and to ascertain substitution with antagonist-DHFR1 fusions are able to reverse this effect, growth competition experiments were undertaken in M9 liquid media in an identical manner to that during the PCA selection process. In these experiments, cells expressed either i) Aβ42cc + Aβ42cc, ii) Aβ42cc + antagonist, or iii) nontoxic cJun+FosW. In this experiment, only scenarios ii) and iii) led to significant growth rates in E. coli, providing evidence that a high affinity interaction is formed and that toxicity associated with expression of Aβ42cc has been lowered, leading to significant growth rates relative to i).
ThT experiments demonstrate a reduction in amyloid content
To determine the ability of PCA derived peptides to prevent fibril assembly (inhibition) or to breakdown preformed fibrils (reversal), ThT fluorescence was used as an indicator of the degree to which Aβ42 aggregates into amyloid fibrils. In these assays, Aβ42 was rendered monomeric21 and freeze-dried before being resuspended and incubated at 37°C, leading to formation of amyloid aggregates. For the inhibition assay, PCA-selected peptides were added on Day 0 and their ability to prevent amyloid formation was assessed after 3 days. For the reversal assay Aβ42 was incubated in isolation for three days to allow amyloid formation before addition of peptide, followed by incubation for a further 3 days to monitor the ability of peptides to breakdown amyloid aggregates. In all cases, a positive control peptide from the literature (iAβ522,23) known to perform well in ThT assays was used. Figure 3 shows the results of inhibition and reversal experiments for the four single PCA-selected peptides and two examples of peptides administered in combination. Both inhibition and reversal experiments have been undertaken at five different Aβ42:peptide ratios ranging from 10-fold substoichiometric to 10-fold superstoichiometric. For every peptide, and at the majority of stoichiometries, the ThT-bound signal was reduced for both inhibition and reversal experiments, indicating that peptides are able to bind Aβ42 and lower the amount of ThT-bound amyloid in solution. Consistent with previous studies, changes in Aβ42:inhibitor stoichiometry altered binding although it was not clear if this was dependent on dose.10,24 However, on average the effects observed at ratios of 1:0.1 are small while ratios of 1:1 are lower than 1:2 or 1:4 (see Supporting Information Fig. S4). These ratios were required to consistently elicit an effect in both inhibition and reversal experiments. Increasing the ratio to 1:10 led to poorer levels of inhibition for several peptides (and consequently increased ThT fluorescence). What is also clear from these experiments is that ThT-bound amyloid can be frequently reduced to less than 50% of the signal found in the Aβ42 positive control, with peptides performing well in both inhibition and reversal experiments. Reassuringly, the positive control iAβ5 peptide was also able to reduce the ThT signal by >40%.
Figure 3.

ThT Inhibition and Reversal data for a) individual peptides and b) peptides used in combination. The data show the effects of different stoichiometries of the inhibitors cys1521, cys2935a, cys2935b, cys3642, cys1521 + cys2935a, cys1521 + cys3642, and iAβ5 on the aggregation of 50 μM Aβ42. The assay was undertaken at 3 days for the inhibition assay. For the reversal assay, Aβ42 was incubated in isolation before addition of peptides on Day 3 with the assay undertaken on Day 6. The assay was performed at 10 μM Aβ42 concentrations. All errors are expressed as the standard error of the mean.
In addition, we have conducted experiments where amyloid-PCA derived peptides predicted to target different regions of the disulphide tethered Aβ42cc have been combined [Supporting Information Fig. 3(b)]. These combinations were (a) cys1521/cys2935a and (b) cys1521/cys3642. In both instances, these peptides were derived from libraries based on (and, therefore, predicted to independently target) the first and second strands within the tethered β-hairpin structure of Aβ42cc. Indeed, cys2935a is based on a sequence that was selected using the Aβ25–35 target,10 with the same library unable to yield any binding sequences when using the wild-type Aβ42 or Aβ15–21 as a target. In addition, the library used to generate cys1521 was also unable generate any hits against wild-type Aβ42. This is significant since it suggests that Aβ42cc represents a far more accessible target for intracellular library screening and antagonist selection. Despite this, ThT experiments in which these peptides were combined generated only one instance where two peptides assayed together were, within error, consistently more effective across the stoichiometries than the average of the individual component peptides [Fig. 3(b)]. This was observed for inhibition experiments that combined cys1521 and cys2935a. In this case, an additional 25–35% reduction in ThT fluorescence was observed over the average of the individual peptides at 1:0.1, 1:2, 1:4, and 1:10 stoichiometries, indicating a benefit in combining them [Fig. 3(b), green vs. purple]. At 1:1, large cumulative errors precluded any interpretation. In contrast, combinations of cys1521 and cys3642 gave no consistent benefit in inhibition experiments. For peptides tested in combination during reversal experiments, no consistent benefit was observed over either peptide assayed alone. Rather, combinations of cys1521 and cys2935a displayed signs of synergy at 1:1 to 1:2 molar ratios with the trend reversing for 1:4 and 1:10, indicating that higher concentrations of peptide can lead to increased ThT fluorescence. As increasing the molar ratio did not lead to improvements in the reversal of amyloid in most cases, this indicated that a twofold excess of peptide:Aβ may be sufficient to reverse fibril formation. A similar trend was true of combining cys1521 and cys3642 in reversal experiments. However, in both cases the trend was not observed in inhibition assays, perhaps reflecting the fact that more binding sites are available in monomerized Aβ42 to which inhibitor can bind and, therefore, remain soluble relative to aged and aggregated samples. Lastly, it cannot be ruled out that the combined peptides do not interact with each other; however, mixing peptides does not result in any gain in amyloid formation as increased ThT signals are not observed.
CD experiments demonstrate a reduction in β-sheet content
As amyloid fibrils are predominately β-sheet, we used CD spectroscopy to provide an end-point characterization of the aggregates formed once ThT experiments were complete. Consistent with a β-sheet structure, spectra show a negative peak at 218 nm. At higher molar ratios (1:2 or greater for inhibition and 1:4 or greater for reversal) an additional large minima of up to −11 millidegrees was observed at 200 nm for the majority of peptides, indicating the adoption of a mixture of β-sheet (Aβ42) and random coil-like structures (peptides). This minima was noticeably less pronounced for the cys3642 peptide and was not observed for iAβ5, which adopted a spectrum consistent with a β-sheet structure at all concentrations. During CD experiments, we observed a decrease in β-sheet content in the majority of samples, supporting the ThT data by demonstrating that peptides reduce the global β-sheet content relative to the Aβ42 sample and, therefore, the amyloid content. These experiments therefore suggest that peptides can bind Aβ42 and exert their effect by reducing the amount of protein in the amyloid form. Lastly, ThT and CD spectroscopy experiments undertaken on peptides in isolation that have been incubated at 50 μM for 3 days under conditions identical to those used in aggregation assays using Aβ42 demonstrate that peptides do not bind significant amounts of ThT, and that the CD signal for all peptides (at 0:1) is consistent with that of a random coil or weakly helical conformation.
OAF microscopy
Samples used in ThT and CD experiments were also imaged using OAF microscopy for both inhibition and reversal experiments. To prevent bias toward any one sample the experiment was carried out blind.25 This technique allows for surface associated and stacked aggregates of amyloid fibers to be directly imaged. It was possible to assess the amount of protein deposited as amyloid and its morphology. OAF experiments were undertaken for both inhibition and reversal at all of the molar ratios assayed in ThT experiments (Fig. 4). In almost every case, for both inhibition and reversal experiments, a reduction in the amount of amyloid was observed relative to Aβ42 incubated in isolation under identical conditions. In a number of cases amyloid deposits much smaller in size were observed.
Figure 4.

OAF Microscopy data for all inhibitors at all stoichiometries. During inhibition experiments, Aβ42 was grown with inhibitor for 3 days and assayed for peptide induced inhibition of amyloid formation. During reversal experiments, Aβ42 was grown without inhibitor for 3 days, after which inhibitor was added at a stoichiometry of 1:1 and followed by a further 3 days incubation to assay for peptide induced reversal of amyloid deposition. Each sample was then imaged by fluorescence microscopy and panels showing representative images obtained. To quantify amyloid deposition the mean grey value over a 160 px × 160 px (13 μm × 13 μm) area randomly chosen for five separate images is plotted as fluorescence intensity. Each data point is scaled to overcome the “background noise” by taking Aβ (1:0) as the maximum and iAβ5 as the minimum [i.e., (signal-iAβ5)/(Aβ42-iAβ5)]. This defines the range over the positive and negative controls.
Atomic force microscopy
Again samples used in ThT and CD experiments were imaged using AFM as a second direct qualitative measure of fibril formation (Fig. 5). A stoichiometry of 1:2 (50 µM:100 µM) was chosen for AFM experiments as this was the concentration at which peptide inhibitors were on average deemed to be most effective in ThT experiments. AFM was undertaken for both inhibition and reversal experiments on cys1521, cys2935a, and cys1521+cys2935a as an additional measure of the extent to which these peptides are effective when mixed. A major reduction in the amount of amyloid was observed for all samples relative to the Aβ42 control. No fibrils were observed for peptides in the absence of Aβ42, either alone or when mixed. For inhibition experiments in the case of cys1521 + cys2935a no fibrils were observed; with mixing appearing to remove fibrils present for the cys1521 sample. For reversal experiments, the residual fibrils observed in the cys2935a were also largely removed on mixing with cys1521.
Figure 5.

AFM data. To demonstrate that inhibitors cys1521, cys2935a, and cys2935a + cys1521 do not aggregate in isolation, they were grown alone for 3 days at a concentration of 100 µM (i.e., 0:2 molar ratio) under the same conditions as inhibition experiments. In all cases, no fibril formation was observed. Inhibition experiments were undertaken at an Aβ42:inhibitor stoichiometry of 1:2 (row two). In this experiment, the same sample was used as for ThT experiments, in which Aβ42 was grown with inhibitor for 3 days at 37°C. All of the peptides are inhibitory, in particular the combination of cys2935 + cys1521. For reversal experiments, Aβ42 was grown alone for 3 days, after which inhibitor was added at a stoichiometry of 1:2 and followed by a further 3 days incubation to assay for peptide induced reversal of amyloid deposition. Each of the samples were imaged by AFM and panels showing representative images obtained. Also in reversal experiments, it can be clearly observed that for all of inhibitors there is a reduction of fibril, although this is less pronounced than for inhibition experiments. Scale bars are shown.
Cell-toxicity experiments
MTT ((3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide)) cell toxicity experiments were performed using Rat phaeochromocytoma (PC12) neuronal-like cells to reflect the nature of toxicity in the disease via the reduction of a redox dye (Fig. 6). We assessed the toxicity of extracellular Aβ42 deposits on PC12 cell integrity and its amelioration by preincubation of Aβ42 with peptides. Cells incubated with buffer alone resulted in a reduction of the redox dye MTT, leading to a color change from yellow to purple that can be measured by a change at A540. In contrast, incubation of cells with Aβ42 resulted in a large decrease in cell viability and consequently the ability to reduce MTT, leading to a smaller signal change. To study, the ability of inhibitors to reverse the effect on cell viability MTT assays were performed with a range of molar ratios relative to Aβ42 and normalized relative to cells in isolation (normalized as 0% death) and cells incubated with Aβ42 alone (normalized as 100% death). Although the changes are small and the cumulative errors resulting from comparatively small changes can be large, we were nonetheless able to draw some general conclusions from this assay. In particular, we observed that effective peptides were able to generate modest reductions in toxicity of 5–20% in the majority of cases. Neither cys2935a or cys2935b appeared effective in this assay, displaying almost no improvement across all stoichiometries. Cys1521 was able to elicit a small decrease of 5–15% in a nondose dependent manner. Cys3642 demonstrated a decrease across all molar ratios of 10–20%. A combination of cys1521 + cys2935a or cys1521 + cys3642 did not conclusively lead to additional lowering of toxicity [Fig. 6(b)]. Although reductions in toxicity greater than the average of the component peptides were observed, these were small; 5% for cys1521/cys2935a at 1:10 and 10% for cys1521/cys3642 at 1:10. At other molar ratios the errors were either too large or no additional benefits over Aβ42 alone were observed. It should be noted that as PCA is undertaken inside bacteria it is possible that intracellular selection of peptides that are nontoxic to E.coli may not be as effective when transferred into a mammalian cell system. There, exposure to different off-target interactions and a different protease pool will occur, in addition to the necessity for the peptide to function in the extracellular space, where Aβ is known to aggregate in the brain. This may explain the discrepancy between the effects on growth rates in E.coli and efficacy in MTT experiments on peptides incubated with PC12 cells.
Figure 6.

Shown are a) the results from the MTT toxicity assay using Aβ42 and selected inhibitors using different molar ratios after 24 h of incubation with PC12 cells. The assay was performed with 10 μM Aβ42 and different concentrations of inhibitor, for example, 1:0.1 (1 μM), 1:1 (10 μM), 1:4 (40 μM), 1:10 (100 μM). b) a comparison of the MTT results for individual peptides against those used in combination. All errors are expressed as the standard error of the mean.
Discussion
We have used an intracellular PCA combined with semirational library design to identify peptides that can reverse and inhibit Aβ42 aggregation by up to 80% as well as lead to a modest reduction (5–20%) in its toxicity. As a target during bacterial selection, we utilized a double cysteine mutant Aβ42cc (A21C/A30C7) that has been shown to lead to the population of pathogenically relevant soluble oligomers in the cytoplasm of an E.coli strain that harbors an oxidizing cytoplasm environment.10,26 An advantage of using PCA with Aβ42cc is that the disulphide bridge forces Aβ into the β-hairpin conformation that is associated with toxicity in AD. The Aβ42cc mutant has been shown to lead to the population of highly toxic oligomers, suggesting that this mutant can be used to improve the stringency of the amyloid-PCA assay. In addition, as oligomers are adopted, we envisage that this conformationally restricted mutant generates an increase in the number of exposed structural epitopes relative to fibrils formed by wild-type Aβ42, meaning that more mDHFR activity can be attained within cells harboring binders of this disulphide mutant. To target, the two β-strands either side of the hairpin, we used Aβ15–21, Aβ29–35, and Aβ36–42 as design scaffolds and fully randomized residues at four positions to create three libraries of 160,000 members. All three libraries resulted in the identification of sequences able to bind the target sequence. In the case of wild-type Aβ42, it should be noted that it is possible that the peptide inhibitors may be able to function by stabilizing monomeric Aβ42 before it misfolds into the β-hairpin structure, thus preventing conversion into a pathogenic structure. By using the restrained Aβ42cc protein, and because we were unable to identify full length Aβ42 binders using many of the same libraries, we infer that the wild-type structure is a poor target for antagonist recognition, possibly due to a lack of well-defined structural epitopes in the nonamyloid or early oligomeric conformations of the molecule. Thus, the generalized approach of using Aβ42cc to screen libraries and generate peptide hits before testing selected peptides on wild-type Aβ42 is potentially very exciting.
Combination experiments
As previously noted, we have identified hits from the same 15–21 library that was unable to yield binders for either wild-type Aβ42 or Aβ25–35 target proteins (unpublished data). The failure to identify binders in these previous PCA screens suggests that the epitope presented was insufficient for effective binding. Thus, we believe that the restricted conformation of Aβ42 in the disulphide bridged version represents an improved target for our “recognition-element” based libraries. As the 15–21 library was unable to provide colonies under PCA conditions with Aβ42, we instead focused our efforts on the 25–35 region of the molecule. We therefore envisage that the cys1521 winner is able to target the first β-strand in the constrained amyloid conformation of Aβ42cc, with the cys2935 and cys3642 libraries targeting the second β-strand. To look for synergy when used in combination, we therefore incubated the cys1521 winner with either cys2935a or cys3642. In the case of cys1521 and cys2935a, we found a reduction in bound-ThT signal of 20% over the average of the two individual peptides, indicating that there is an added benefit in combining peptides, and that they may occupy different regions within Aβ. MTT data produced large errors, however, improvements in cell viability of 5–10% at some molar ratios were observed, indicating that the combined effect of the inhibitors is greater than the average of the two. In agreement with this, OAF and AFM experiments also suggest that there is an additional benefit in mixing the cys1521 and cys2935a peptides.
This study has led to a number of peptides that are capable of preventing assembly of amyloid aggregates as well as breaking down preformed amyloid and further investigation is warranted in the search for molecules that can ultimately slow or even prevent the onset of AD.
Materials and Methods
Mutagenesis and protein engineering
pES300d-Aβ42cc-DHFR2 was created using overlap extension PCR with codon changes corresponding to A21C and A30C. These sites have been previously chosen to constrain the peptide into a β-hairpin conformation7 and are located on nonhydrogen bonded sites between β-strands either side of the hairpin and in close proximity to each other.27
Choice of E.coli strain
PCA were undertaken using E. coli SHuffle Express cells (New England Biolabs). SHuffle cells have been engineered to possess an oxidative cytoplasmic environment that favors disulphide bond formation. In particular, SHuffle cells have been engineered to lack thioredoxin reductase (trxB) and glutathione reductase (gor) with an additional suppressor mutation (ahpC) which is required to restore viability, allowing the formation of stable disulphide bonds in the cytoplasm. Under these conditions, thioredoxins are in their oxidized state, converting them from reductases to oxidases. Proteins that require disulphide bonds for their folding can, therefore, be oxidized and form stable disulphide bonds within the cytoplasm. This cell line also constitutively expresses a chromosomal copy of the disulphide bond isomerase DsbC. DsbC promotes the correction of misoxidized proteins into their correct form.10,26
Single step selection PCA
pES300d-Aβ42cc-DHFR2 and pREP4 (Qiagen; for expression of the lac repressor protein) were cotransformed into SHuffle Express cells (New England Biolabs) and plated onto LB agar with the appropriate antibiotics (Kan and Cm). These cells were next made electrocompetent before transformation with the appropriate pES230d-library-DHFR1 library plasmid (E.coli XL-1 cells were used for construction and cloning of libraries, as described previously10,14). Transformed cells were next plated on three different media; 1/20th of the cells were plated onto LB agar with three antibiotics (Kan, Amp, and Cm) as a positive control of transformation efficiency. A further 1/20th of the solution was plated onto M9 minimal medium agar with 1 μg/ml trimethoprim and the same three antibiotics as a negative control. Finally, the remaining 90% of transformed cells were plated onto M9 minimal agar in the presence of the three antibiotics, 1 μg/ml trimethoprim, and 1 mM IPTG (isopropyl-β-D-thiogalactopyranoside), to induce expression of the two DHFR fragment fused peptides). This single-step PCA selection typically led to approximately 50–100 colonies from libraries of 160,000 members, meaning that at least 99% of all library members are removed at this stage owing to their inability to bind Aβ42cc and reduce toxicity.
Competition selection PCA
To increase the selection stringency, growth competition experiments were undertaken. Using this approach, selected colonies were pooled from the plate and grown in M9 minimal media under selective conditions (containing Kan, Amp, Cm, trimethoprim, and IPTG) and serially diluted over 5–10 passages. Using these sequential rounds of competition selection, subtle differences in growth rate are amplified, increasing the stringency of selection relative to the single-step method. Competition selection, therefore, allows the most effective 1–2 sequences to be isolated from the 50–100 Aβ42cc binders that are initially identified during single step selection. At each passage glycerol stocks were prepared and sequencing results were obtained (GATC biotech, London) for DNA pools as well as individual colonies. For each passage, 50 μL of liquid culture was added to 50 ml of fresh M9 minimal media, resulting in an approximate OD600 of 0.01. Cells were incubated at 37°C until an OD600 of ∼0.4 was reached (typically 2–3 days), before moving to the next passage.
Aβ peptide preparation
Aβ42 was purchased as a pure recombinant peptide from rPeptide (Stratech) and was used for all of the experiments described. Prior to use, a quantity of peptide was weighed using an analytical balance before being treated to three rounds of dissolution in hexofluoro-2-propanol (HFIP), sonication, drying, dissolution in trifluoroacetic acid (TFA), sonication and drying, according to the Zagorski protocol,21 and then aliquoted into appropriately sized batches for subsequent assays and dried via lyophilisation before being dissolved in 10 mM potassium phosphate buffer (pH 7.4) to generate a final concentration of 50 μM. TFA/HFIP treatment was used to ensure that amyloid growth always proceeds from the same monomeric state, thus reducing errors in amyloid formation measurements.
Peptide inhibitors
Peptides identified using amyloid-PCA (cys1521, cys2935a, cys2935b, cys3642, and a positive control from the literature, iAβ522), were obtained by Peptide Protein Research (Fareham, UK) as pure lyophilized peptides. Peptides were weighed using an analytical balance and stock solutions of 1 mM concentration were subsequently dissolved in ultrapure water. At this concentration (2–200x excess of that used in inhibition and reversal experiments) no aggregation or precipitate was observed. In addition, bioinformatics tools (e.g., Waltz,28 Amylpred,29 Pasta,30 Zyggregator,31 and Tango32) do not predict that any of the peptides contain amyloidogenic sequences or aggregate in isolation. Lastly, CD and ThT experiments on peptide antagonists in isolation demonstrate that these sequences do not bind ThT and form random coil-like species in isolation (see Supporting Information Fig. S1).
Growth competition experiments
To confirm that expression of Aβ42cc-DHFR2 fragment fusions impedes the growth rate of E. coli, and to ascertain that amyloid-PCA derived peptides fused to the DHFR1 fragment are able to reverse this effect, growth competition experiments were undertaken. These experiments were performed in M9 minimal media in an identical manner to that during the PCA competition selection process. In these experiments (Fig. 2), cells expressed either (i) Aβ42cc control, (ii) Aβ42cc + PCA derived peptide, or (iii) the nontoxic control cJun+FosW. The latter PCA-derived pair is known to form a high affinity interaction, leading to significant growth rates relative to (i).
Circular Dichroism
Far-UV CD spectra were recorded on an Applied Photophysics Chirascan at 20°C using the same samples as for ThT experiments. Spectra were recorded over the 200–300 nm range at a scan rate of 10 nm/min with step size of 1 nm. Spectra were recorded as the average of two scans. Peptide (10 μM in 10 mM Potassium Phosphate buffer pH 7.4) was added to a 0.1 cm cuvette (Hellma). Spectra were recorded as raw ellipticity.
Thioflavin T assays
ThT inhibition assays were performed with 50 μM Zagorski treated21 monomeric Aβ42 in 200 μL of 10 mM potassium phosphate, pH 7.4, with or without each peptide inhibitor at a concentration of 5 μM (for 1:0.1 molar ratio), 50 μM (for 1:1 molar ratio), 100 μM (for 1:2 molar ratio), 200 μM (for 1:4 molar ratio), and 0.5 mM (for 1:10 molar ratio). For the inhibition assays, the target-inhibitor mixtures were incubated together on day zero at 37°C. Single ThT readings were taken on day three at which maximal ThT binding was found. For the reversal assays, 200 μL of 50 μM target was incubated alone at 37°C for 3 days before adding to the required amount of lyophilized peptide. The vortexed target-inhibitor solutions were then incubated at 37°C for a further 3 days, during which time single ThT readings were taken on postmix Day 3. Experiments were undertaken in duplicate and errors expressed as the standard error of the mean. The ThT assay solution was prepared from a 25× stock containing 500 μM ThT in 250 mM Tris, pH 7.4. The stock was aliquoted and kept frozen until required. It was then allowed to thaw at room temperature for 10 min before dilution into the appropriate Tris buffer, giving the required freshly prepared ThT assay solution containing 20 μM ThT in 10 mM Tris and buffer at pH 7.4. A total of 2960 μL of the ThT assay solution was then added into 40 μL of each inhibition/reversal assay mixture, thoroughly vortexed and transferred into an appropriate cuvette. The fluorescence of amyloid-bound ThT was measured by fluorescence spectroscopy using a Cary Eclipse fluorescence spectrophotometer; bound ThT exhibits a new excitation maxima at 450 nm and an enhanced emission maxima at 482 nm.33
OAF microscopy experiments
Samples were imaged on a custom built OAF25 system. The OAF system permits high signal to noise imaging with greater sample depth penetration. Fluorescence imaging was achieved through a 500LP dichroic and clean up filter (Chroma, VT) before entering an Optosplit III triple color image splitter (Cairn, UK), and was detected using an EMCCD camera (Andor DU897, Andor, UK). The emission wavelength range used was 500–565 nm, therefore, both the excitation and emission wavelengths were off peak for ThT, however, the image quality was excellent. All samples were prestained with 10 μM ThT, pipetted onto a clean glass slide, air dried and then imaged in 10 mM potassium phosphate buffer, pH 7.4, supplemented with 100 mM DTT to minimize photobleaching. For consistency and cross-correlation, the same samples were used for inhibition/reversal imaging as those in ThT and CD experiments. In addition, all OAF experiments were performed blind to prevent bias toward any one sample.
AFM experiments
Samples were imaged in noncontact mode using a XE-120 Atomic Force Microscope (Park Systems, South Korea). NSC 15 silicon nitride cantilevers with a spring constant of 40 N/m were used for imaging at a scan rate of 1.0 Hz and a resolution of 256 × 256 pixels. All images were taken at room temperature. The AFM data were taken from inhibitors alone as well as inhibition and reversal experiments, using cys1521, cys2935a, and the cys1521 + cys2935a mixture. 5 µL sample was placed on freshly cleaved mica (thickness 0.3 mm). Following adsorption of the protein aggregates (2 min), the mica was washed with 5 mL of double distilled water. Excess water was removed and the samples were dried using a stream of nitrogen gas. Samples were immediately analyzed by AFM.
3-(4,5 Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide cell-toxicity assay
MTT experiments were undertaken using Rat phaeochromocytoma (PC12) cells to assess the effect of the toxicity of Aβ42. PC12 cells are known to be particularly sensitive and their use in this assay is well established.34 The MTT Vybrant® MTT Cell Proliferation Assay Kit (Invitrogen) was used to measure the conversion of the water soluble MTT dye to formazan, which is then solubilized, and the concentration determined by a color change monitored via absorbance measurement at 570 nm. The change in absorbance can then be converted to a percentage MTT reduction which can be used as an indicator of the PC12 cell health in the assay. The assay was performed with 10 μM Aβ42 and varying molar ratios of peptide corresponding to 1:0.1 (0.1 μM), 1:1 (10 μM), 1:4 (40 μM), 1:10 (100 μM). PC12 cells were maintained in RPMI 1640 + 2 mM Glutamine medium mixed with 10% Horse Serum, 5% Fetal Bovine Serum, supplemented with a 20 mg/mL Gentamicin. Cells were transferred to a sterile 96-well plate with 30,000 cells per well and experiments performed in triplicate. Briefly, different concentrations of peptides were screened in the presence of 10 μM Aβ42. The required volume from peptide and target stock solutions was freeze-dried overnight. The freeze-dried peptide and Aβ42 target were resuspended in 100% dimethyl sulfoxide (DMSO), each at 100× stock concentration (i.e., 1, 2, 4, or 10 mM). For example, for the molar ratio 1:1 a total of 5 μL from each of the resuspended inhibitor/DMSO and target/DMSO was mixed in a well of a 96-well preparation plate, thus giving 10 μL of 1:1 mM inhibitor/target concentration ratio in 100% DMSO. A total of 90 μL of RPMI media was added to the 10 μL inhibitor/target mixture (100:100 μM inhibitor/target ratio in 10% DMSO). A total of 10 μL of the 50:50 μM inhibitor/target mixture in 10% DMSO was then dispensed into 90 μL of media/PC12 cells, at final peptide inhibitor and target concentrations of 10 μM. These were incubated for 24 h at 37°C, 5% CO2, prior to the addition of the MTT dye. A total of 10 μL of the dye was added to each well and incubated for a further 4 h at 37°C, 5% CO2. A total of 100 μL of the DMSO (stop/solubilization solution) was then added to each well and was allowed to stand for 10 min. The absorbance was measured at 570 nm using a 96-well Versamax tunable microplate reader. Assay values for incubation with buffer were taken as 100% and inhibition of cell function by incubation with buffer containing Aβ42 alone taken as 0%. Raw data were then scaled as follows: (raw data point − Aβ42 mean)/(buffer mean − Aβ42 mean) × 100. The scaled mean for each data set was then plotted with the error given by the standard deviation.
Acknowledgments
The authors acknowledge the use made of the Park Systems XE-120 Atomic Force Microscope which was on loan from the EPSRC (Engineering and Physical Sciences Research Council) Engineering Instrument Pool. The authors wish to thank Dr Miao Yu and Dr Victoria Allen-Baume for excellent technical support throughout the project.
Glossary
- Aβ42
β-amyloid 1–42 variant
- CD
circular dichroism
- HFIP
hexafluoroisopropanol
- MTT
(3-(4,5-Dimethylthiazol-2-yl)−2,5-diphenyltetrazolium bromide
- PCA
protein-fragment complementation assay
- PPI
protein–protein interaction
- TEM
transmission electron microscopy
- TFA
trifluoroacetic acid
- ThT
Thioflavin-T
- OAF
oblique angle fluorescence
- AFM
atomic force microscopy
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Supplementary Information
References
- 1.Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Diffusible, nonfibrillar ligands derived from Abeta1–42 are potent central nervous system neurotoxins. Proc Natl Acad Sci USA. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Klein WL, Krafft GA, Finch CE. Targeting small Abeta oligomers: the solution to an Alzheimer's disease conundrum? Trends Neurosci. 2001;24:219–224. doi: 10.1016/s0166-2236(00)01749-5. [DOI] [PubMed] [Google Scholar]
- 3.Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- 4.Walsh DM, Selkoe DJ. A beta oligomers—a decade of discovery. J Neurochem. 2007;101:1172–1184. doi: 10.1111/j.1471-4159.2006.04426.x. [DOI] [PubMed] [Google Scholar]
- 5.Comery TA, Martone RL, Aschmies S, Atchison KP, Diamantidis G, Gong X, Zhou H, Kreft AF, Pangalos MN, Sonnenberg-Reines J, Jacobsen JS, Marquis KL. Acute gamma-secretase inhibition improves contextual fear conditioning in the Tg2576 mouse model of Alzheimer's disease. J Neurosci. 2005;25:8898–8902. doi: 10.1523/JNEUROSCI.2693-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.O'Nuallain B, Freir DB, Nicoll AJ, Risse E, Ferguson N, Herron CE, Collinge J, Walsh DM. Amyloid beta-protein dimers rapidly form stable synaptotoxic protofibrils. J Neurosci. 2010;30:14411–14419. doi: 10.1523/JNEUROSCI.3537-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sandberg A, Luheshi LM, Sollvander S, Pereira de Barros T, Macao B, Knowles TP, Biverstal H, Lendel C, Ekholm-Petterson F, Dubnovitsky A, Lannfelt L, Dobson CM, Hard T. Stabilization of neurotoxic Alzheimer amyloid-beta oligomers by protein engineering. Proc Natl Acad Sci USA. 2010;107:15595–15600. doi: 10.1073/pnas.1001740107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Petkova AT, Ishii Y, Balbach JJ, Antzutkin ON, Leapman RD, Delaglio F, Tycko R. A structural model for Alzheimer's beta-amyloid fibrils based on experimental constraints from solid state NMR. Proc Natl Acad Sci USA. 2002;99:16742–16747. doi: 10.1073/pnas.262663499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hard T, Lendel C. Inhibition of amyloid formation. J Mol Biol. 2012;421:441–465. doi: 10.1016/j.jmb.2011.12.062. [DOI] [PubMed] [Google Scholar]
- 10.Acerra N, Kad NM, Mason JM. Combining Intracellular selection with protein-fragment complementation to derive Aβ interacting peptides. Protein Eng Des Sel. 2013;26:463–470. doi: 10.1093/protein/gzt021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Acerra N, Kad NM, Griffith DA, Ott S, Crowther DC, Mason JM. Retro-inversal of intracellular selected ABeta interacting peptides: implications for a novel Alzheimers disease treatment. Biochemistry. 2014;53:2101–2111. doi: 10.1021/bi5001257. [DOI] [PubMed] [Google Scholar]
- 12.Sharpe S, Yau WM, Tycko R. Expression and purification of a recombinant peptide from the Alzheimer's beta-amyloid protein for solid-state NMR. Protein Expr Purif. 2005;42:200–210. doi: 10.1016/j.pep.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 13.Soscia SJ, Kirby JE, Washicosky KJ, Tucker SM, Ingelsson M, Hyman B, Burton MA, Goldstein LE, Duong S, Tanzi RE, Moir RD. The Alzheimer's disease-associated amyloid beta-protein is an antimicrobial peptide. PLoS One. 2010;5:e9505. doi: 10.1371/journal.pone.0009505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mason JM, Schmitz MA, Muller KM, Arndt KM. Semirational design of Jun-Fos coiled coils with increased affinity: Universal implications for leucine zipper prediction and design. Proc Natl Acad Sci USA. 2006;103:8989–8994. doi: 10.1073/pnas.0509880103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Remy I, Campbell-Valois FX, Michnick SW. Detection of protein-protein interactions using a simple survival protein-fragment complementation assay based on the enzyme dihydrofolate reductase. Nat Protoc. 2007;2:2120–2125. doi: 10.1038/nprot.2007.266. [DOI] [PubMed] [Google Scholar]
- 16.Pike CJ, Walencewicz-Wasserman AJ, Kosmoski J, Cribbs DH, Glabe CG, Cotman CW. Structure-activity analyses of beta-amyloid peptides: contributions of the beta 25–35 region to aggregation and neurotoxicity. J Neurochem. 1995;64:253–265. doi: 10.1046/j.1471-4159.1995.64010253.x. [DOI] [PubMed] [Google Scholar]
- 17.Hughes E, Burke RM, Doig AJ. Inhibition of toxicity in the beta-amyloid peptide fragment beta -(25–35) using N-methylated derivatives: a general strategy to prevent amyloid formation. J Biol Chem. 2000;275:25109–25115. doi: 10.1074/jbc.M003554200. [DOI] [PubMed] [Google Scholar]
- 18.Hung LW, Ciccotosto GD, Giannakis E, Tew DJ, Perez K, Masters CL, Cappai R, Wade JD, Barnham KJ. Amyloid-beta peptide (Abeta) neurotoxicity is modulated by the rate of peptide aggregation: Abeta dimers and trimers correlate with neurotoxicity. J Neurosci. 2008;28:11950–11958. doi: 10.1523/JNEUROSCI.3916-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tjernberg LO, Naslund J, Lindqvist F, Johansson J, Karlstrom AR, Thyberg J, Terenius L, Nordstedt C. Arrest of beta-amyloid fibril formation by a pentapeptide ligand. J Biol Chem. 1996;271:8545–8548. doi: 10.1074/jbc.271.15.8545. [DOI] [PubMed] [Google Scholar]
- 20.Lowe TL, Strzelec A, Kiessling LL, Murphy RM. Structure-function relationships for inhibitors of beta-amyloid toxicity containing the recognition sequence KLVFF. Biochemistry. 2001;40:7882–7889. doi: 10.1021/bi002734u. [DOI] [PubMed] [Google Scholar]
- 21.Zagorski MG, Yang J, Shao H, Ma K, Zeng H, Hong A. Methodological and chemical factors affecting amyloid beta peptide amyloidogenicity. Methods Enzymol. 1999;309:189–204. doi: 10.1016/s0076-6879(99)09015-1. [DOI] [PubMed] [Google Scholar]
- 22.Soto C, Kindy MS, Baumann M, Frangione B. Inhibition of Alzheimer's amyloidosis by peptides that prevent beta-sheet conformation. Biochem Biophys Res Commun. 1996;226:672–680. doi: 10.1006/bbrc.1996.1413. [DOI] [PubMed] [Google Scholar]
- 23.Soto C, Sigurdsson EM, Morelli L, Kumar RA, Castano EM, Frangione B. Beta-sheet breaker peptides inhibit fibrillogenesis in a rat brain model of amyloidosis: implications for Alzheimer's therapy. Nat Med. 1998;4:822–826. doi: 10.1038/nm0798-822. [DOI] [PubMed] [Google Scholar]
- 24.Kokkoni N, Stott K, Amijee H, Mason JM, Doig AJ. N-Methylated peptide inhibitors of beta-amyloid aggregation and toxicity. Optimization of the inhibitor structure. Biochemistry. 2006;45:9906–9918. doi: 10.1021/bi060837s. [DOI] [PubMed] [Google Scholar]
- 25.Kad NM, Wang H, Kennedy GG, Warshaw DM, Van Houten B. Collaborative dynamic DNA scanning by nucleotide excision repair proteins investigated by single-molecule imaging of quantum-dot-labeled proteins. Mol Cell. 2010;37:702–713. doi: 10.1016/j.molcel.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Levy R, Weiss R, Chen G, Iverson BL, Georgiou G. Production of correctly folded Fab antibody fragment in the cytoplasm of Escherichia coli trxB gor mutants via the coexpression of molecular chaperones. Protein Expr Purif. 2001;23:338–347. doi: 10.1006/prep.2001.1520. [DOI] [PubMed] [Google Scholar]
- 27.Mason JM, Gibbs N, Sessions RB, Clarke AR. The influence of intramolecular bridges on the dynamics of a protein folding reaction. Biochemistry. 2002;41:12093–12099. doi: 10.1021/bi026398o. [DOI] [PubMed] [Google Scholar]
- 28.Maurer-Stroh S, Debulpaep M, Kuemmerer N, Lopez de la Paz M, Martins IC, Reumers J, Morris KL, Copland A, Serpell L, Serrano L, Schymkowitz JW, Rousseau F. Exploring the sequence determinants of amyloid structure using position-specific scoring matrices. Nat Methods. 2010;7:237–242. doi: 10.1038/nmeth.1432. [DOI] [PubMed] [Google Scholar]
- 29.Frousios KK, Iconomidou VA, Karletidi CM, Hamodrakas SJ. Amyloidogenic determinants are usually not buried. BMC Struct Biol. 2009;9:44. doi: 10.1186/1472-6807-9-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Trovato A, Seno F, Tosatto SC. The PASTA server for protein aggregation prediction. Protein Eng Des Sel. 2007;20:521–523. doi: 10.1093/protein/gzm042. [DOI] [PubMed] [Google Scholar]
- 31.Tartaglia GG, Vendruscolo M. The Zyggregator method for predicting protein aggregation propensities. Chem Soc Rev. 2008;37:1395–1401. doi: 10.1039/b706784b. [DOI] [PubMed] [Google Scholar]
- 32.Fernandez-Escamilla AM, Rousseau F, Schymkowitz J, Serrano L. Prediction of sequence-dependent and mutational effects on the aggregation of peptides and proteins. Nat Biotechnol. 2004;22:1302–1306. doi: 10.1038/nbt1012. [DOI] [PubMed] [Google Scholar]
- 33.LeVine H., III Thioflavine T interaction with synthetic Alzheimer's disease beta-amyloid peptides: detection of amyloid aggregation in solution. Protein Sci. 1993;2:404–410. doi: 10.1002/pro.5560020312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shearman MS, Ragan CI, Iversen LL. Inhibition of PC12 cell redox activity is a specific, early indicator of the mechanism of beta-amyloid-mediated cell death. Proc Natl Acad Sci USA. 1994;91:1470–1474. doi: 10.1073/pnas.91.4.1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information