

Abstract
Trinucleotide repeats can be highly unstable, mutating far more frequently than point mutations. Repeats typically mutate by addition or loss of units of the repeat. CAG repeat expansions in humans trigger neurological diseases that include myotonic dystrophy, Huntington disease, and several spinocerebellar ataxias. In human cells, diverse mechanisms promote CAG repeat instability, and in mice, the mechanisms of instability are varied and tissue-dependent. Dissection of mechanistic complexity and discovery of potential therapeutics necessitates quantitative and scalable screens for repeat mutation. We describe a GFP-based assay for screening modifiers of CAG repeat instability in human cells. The assay exploits an engineered intronic CAG repeat tract that interferes with expression of an inducible GFP minigene. Like the phenotypes of many trinucleotide repeat disorders, we find that GFP function is impaired by repeat expansion, in a length-dependent manner. The intensity of fluorescence varies inversely with repeat length, allowing estimates of repeat tract changes in live cells. We validate the assay using transcription through the repeat and engineered CAG-specific nucleases, which have previously been reported to induce CAG repeat instability. The assay is relatively fast and should be adaptable to large-scale screens of chemical and shRNA libraries.
Introduction
Expansions of CAG trinucleotide repeats (TNRs) cause several neurological diseases in humans, including Huntington disease, myotonic dystrophy type 1, and a number of spinocerebellar ataxias [1], [2]. Long CAG tracts are unstable during transmission between generations, giving rise to progeny with additional CAG units (expansions) or with fewer units (contractions), but usually with a bias toward expansions. Expansions in the germ line lead to earlier disease onset and increased severity in affected individuals [1], while expansions in specific neurons exacerbate disease symptoms [3], [4]. Reducing repeat expansions or promoting repeat contractions—even partial contractions—would significantly advance therapy for TNR disorders.
One obstacle to devising therapies for shrinking expanded CAG repeats is the diversity of pathways that destabilize repeat tracts. Studies in model organism have identified a broad spectrum of DNA transactions—replication, recombination, DNA repair, and transcription, to name a few—that can contribute to TNR instability [5], [6], [7], [8]. Virtually any protein or process that exposes single DNA strands in CAG repeat regions allows formation of hairpins and slipped duplexes, which trigger repeat instability [9], [10]. In addition, studies in mice have revealed that mechanisms of TNR instability differ from tissue to tissue [11], [12], [13], [14], [15].
Investigations into TNR instability depend on methods to assess repeat variation. Traditionally, small-pool PCR [16] and GeneScan [17], [18] have provided effective tools for assessing TNR instability, but these labor-intensive methods do not scale well. Emerging methods such as Illumina and PacBio sequencing are promising because they enable high-throughput and precise measurement of repeat length changes [19], [20], [21]; however, they are still cost prohibitive for large-scale screens. Selection assays in yeast [22], [23], [24], [25], [26] and mammalian cells [27], [28], [29], [30], [31] offer powerful methods for testing cellular processes and candidate genes for their effects on TNR instability. But the frequency of detected events in these assays is generally too low for high-throughput screens.
Here, we described a fast and scalable GFP-based fluorescence assay for assessment of CAG repeat instability. Like our selection assays in human cells, this fluorescence assay is based on the ability of long CAG tracts in an intron to interfere with gene expression. Importantly, the degree of fluorescence depends in an inverse manner on the length of the repeat tract, providing a noninvasive estimate of the length of a CAG repeat tract in living cells.
Materials and Methods
Plasmids
The GFP-Pem1 plasmid, a kind gift from Dr. Lei Li, was created by the insertion of portions of the large intron in the Rattus norvegicus Rhox5 gene into the eGFP gene on the pEGFP-N1 backbone (Clontech Laboratories, Mountain View, CA). The resulting GFP minigene contains an efficiently spliced intron approximately 1.5 kb in length. The plasmid was further modified to contain a polylinker in the intron [32]. We inserted a (CAG)89 repeat tract, along with 129 bp of flanking sequence, into the NotI site in the polylinker to generate plasmid pGFP-Pem1-CAG89. The CAG repeat tract, which was originally cloned from a myotonic dystrophy patient [33], retained 43 nucleotides 5′ and 19 nucleotides 3′ of human sequences flanking the repeat tract at the myotonic dystrophy locus.
To insert the modified GFP gene into the pcDNA5/FRT/TO vector (Invitrogen, Carlsbad, CA), which carries the inducible CMV/TetO2 hybrid promoter (cytomegalovirus immediate early—CMV—promoter plus two tetracycline operator 2—TetO2—sites) and an FRT site for insertion in T-REx HEK293 cells, we first introduced an XhoI linker at the MfeI restriction in pGFP-Pem1-CAG vector. We digested pGFP-Pem1-CAG-XhoI with PspOMI and XhoI, isolated the modified GFP gene, which was then inserted into compatible NotI and XhoI sites in the polylinker in pcDNA5/FRT/TO, generating plasmid pCAG89 (Figure 1). To create plasmid pCAG0, we digested pCAG89 with NotI, which removes the CAG repeat tract and 129 nucleotides of flanking DNA, and recircularized the backbone.
Figure 1. Construction of GFP(CAG)89 and GFP(CAG)0 cell lines.

The GFP(CAG)89 cell line was generated by FLP-mediated site-specific recombination between the FRT sites in plasmid pCAG89 and in the genome of T-REx 293 cells, creating a chromosomal integrant. The GFP(CAG)0 cell line was generated in a similar way using plasmid pCAG0, which was derived from pCAG89 by cleavage with NotI (N) and religation, a procedure that eliminates the CAG repeat along with the flanking human sequences. HygB is hygromycin B; Zeo is zeocin.
Cell Lines
To create human cells with chromosomally integrated targets, we co-transfected the Flp recombinase-encoding vector, pOG44, with the pCAG89 or the pCAG0 vector into the Flp-In T-REx 293 Cell Line (Invitrogen, Carlsbad, CA), using Lipofectamine 2000 Transfection Reagent (Invitrogen) (Figure 1). The cells were maintained in DMEM medium supplemented with 10% FBS at 37°C in 5% CO2 for two days. The cells were then trypsinized, diluted, and re-plated for colony formation. Individual colonies were screened for appropriate drug sensitivity. FLP-recombinase-mediated insertion of the pCAG89 and pCAG0 plasmids should confer resistance to 200 µg/mL hygromycin B (Invitrogen, Carlsbad, CA) and sensitivity to 100 µg/mL zeocin (Invitrogen). Resistance to blasticidin verified that the isolated clones retained the gene for the Tet Repressor, which controls the doxycycline-inducible TetO2 CMV promoter. Individual colonies were sequenced to determine the length of the CAG repeat tract in the chromosomal GFP minigene. One cell line with 89 CAG repeat units—designated GFP(CAG)89—and one cell line with 0 CAG repeats—designated GFP(CAG)0—were used in the experiments described here. The GFP(CAG)0 cell line serves as a control to identify the maximum level of GFP expression.
All cell lines were maintained in plates containing DMEM GIBCO 4.5 g/L D-Glucose; L-Glutamine (GIBCO, Grand Island, NY) supplemented with 10% HyClone Fetal Bovine Serum, Standard (Thermo Scientific, Logan, UT) in a humidified incubator at 37°C with 5% CO2.
Flow Cytometry
We performed flow cytometry analyses using the BD LSRFortessa Cell Analyzer (BD Biosciences, San Jose, CA). All data were analyzed with BD FACSDiva Software 6.1.2 (BD Biosciences). Prior to cytometry analysis, cells were trypsinized and diluted to 2×106 cells/mL in complete medium. We filtered cell solutions in 35 µm cell strainer cap tubes (BD Biosciences) and kept them on ice until analysis. For each sample, eGFP fluorescence was analyzed with a 488 nm wavelength blue laser. Because all cells with a GFP gene show some degree of fluorescence, we used gates to define the fluorescent population of interest. When we were interested in a subpopulation that was more fluorescent that the main population, we arbitrarily defined those cells that passed the appropriate gate as GFP+ cells. For transcription-induced repeat instability, we used the distribution of GFP(CAG)0 cells, fully induced with 2 µg/mL doxycycline for 24 hours, to define the gates for GFP+ cells. Frequencies of GFP+ cells were calculated as the number of GFP+ cells divided by the total number of cells counted.
Fluorescence-activated cell sorting was carried out on a BD FACSAria II Cell Sorter (BD Biosciences). We prepared cells identically for flow cytometry and cell sorting. The catch media was 50% serum and 50% complete medium, and the samples were kept at 4°C throughout sorting. Individual cells were sorted into individual wells of 96-well plates. The surviving cells—typically 10–20%—were grown into populations, and their repeat tracts were sequenced.
Analysis of CAG Tract Lengths
We determined CAG tract lengths by PCR amplification and sequence analysis. PCR mixtures consisted of 1 µL DNA, 0.3 mM dNTPs, 1.75 mM MgCl2, 1.2 mM betaine (Sigma-Aldrich, St. Louis, MO), 1 U ChromaTaq DNA Polymerase (Denville Scientific Inc., Denville, NJ), 4 µL 5× ChromaTaq Buffer, and dH2O to 20 µL. PCR mixtures contained 0.5 µM each forward (5′-AAGAGCTTCCCTTTACACAACG) and reverse (5′-TACCAGGACAGCAGTGGTCA) primer, which are located on either side of the repeat tract, about 250 nucleotides away. The PCR program consisted of 2 minutes at 94°C, followed by 39 cycles of 94°C for 15 seconds, 60°C for 30 seconds, and 72°C for 45 seconds, with a final extension cycle at 72°C for 10 minutes. PCR products were run on a 1.5% agarose gel and visualized with ethidium bromide. For sequence analysis, PCR products were isolated by either a QIAprep Spin Miniprep Kit (QIAGEN, Hilden, Germany) or a QIAquick Gel Extraction Kit (QIAGEN). Samples (100 ng) were sequenced at Lone Star Labs (Houston, Texas). To determine repeat length, AB Sequence Scanner Software v1.0 (Applied Biosystems, Foster City, CA) was used to analyze chromatographs of the sequencing reactions.
Quantitative Reverse Transcription PCR
We extracted RNA from trypsinized cell samples resuspended in PBS, using the RNeasy Mini Kit (QIAGEN, Hilden, Germany) supplemented with β-mercaptoethanol (Sigma-Aldrich), according to the manufacturer's protocol. First strand synthesis was performed on 1 µg RNA per sample in a final volume of 20 µL, using the iScript cDNA Synthesis Kit (Bio-Rad Laboratories, Hercules, CA), following the manufacturer's instructions. We prepared the quantitative PCR reaction using the QuantiFast SYBR Green PCR Kit (QIAGEN) and 2 µL cDNA reaction per sample in a final volume of 25 µL, according to the manufacturer's protocol. PCR reactions contained 0.5 µM of each primer. Primer set 1 (5′-CAGAAGAACGGCATCAAGGT and 5′-CTGGGTGCTCAGGTAGTGGT) and primer set 2 (5′-TATATCATGGCCGACAAGCA and 5′-GGGTGTTCTGCTGGTAGTGG) each amplified segments of GFP exon 2. As designed, each primer set should amplify equally well both the correct GFP spliced product and the aberrant spliced product that includes the CAG repeat tract. The amplifications were performed on a CFX96 Real-Time PCR Detection System (Bio-Rad Laboratories). The PCR program consisted of 5 minutes at 95°C, followed by 40 cycles of 95°C for 10 seconds and 60°C for 30 seconds. Following amplification, the temperature was ramped from 60°C to 95°C at a rate of 0.5°C every 5 seconds for a melt curve analysis. All samples were analyzed in duplicate and normalized to β-actin. Gene expression was determined by the ΔΔCt method [34].
Northern Blot
We generated a probe for EGFP mRNA by PCR-amplifying the complete EGFP gene in pEGFP-N1. The probe was radiolabeled using the DECAprime II Kit (Ambion, Austin, TX) and 32P-dCTP, according to the manufacturer's instructions. RNA was extracted from cultured cells with the RNeasy Mini Kit (QIAGEN) and subjected to electrophoresis on a 1% agarose denaturing formaldehyde gel (2.2 M formaldehyde; 200 mM MOPS, pH 7.0; 50 mM sodium acetate; 10 mM EDTA). The products were then transferred to a Amersham Hybond N+ nylon membrane (GE Healthcare, Piscataway, NJ) overnight and hybridized with the labeled probe, using ULTRAhyb Ultrasensitive Hybridization Buffer (Ambion).
Results
A GFP-Based Fluorescence Assay for CAG Repeat Instability
To develop a rapid assay for CAG repeat instability, one with a potential for rapid screening of drugs and candidate genes, we tested the effects of a long CAG repeat on expression of a chromosomal copy of a GFP minigene. In previous studies using the APRT gene and the HPRT minigene, we found that placing a long CAG repeat in an intron reduced expression below levels required for cell survival under selective conditions, giving rise to a selective assay for repeat contraction [28], [29]. To determine whether long CAG tracts also affected expression of the GFP minigene, we inserted a CAG89 repeat tract into the intron in the minigene, placed the minigene under control of a Tet-On promoter, and deposited it in a chromosome by FLP-mediated recombination into the FRT site in the genome of T-REx HEK293 cells (Figure 1). In parallel to the GFP(CAG)89 cell line, we also generated a GFP(CAG)0 cell line, which carries the same GFP minigene at the same chromosomal location, but without a CAG repeat tract (Figure 1).
To test inducibility of expression from the Tet-On promoter, we grew both cell lines in the presence or absence of doxycycline for two days and then observed them by microscopy (Figure 2A). As expected, neither cell line exhibited visible fluorescence in the absence of doxycycline. Upon addition of doxycycline, GFP(CAG)0 cells fluoresced intensely, indicating that the Tet-On system is functional and that the repeat-containing intron is correctly spliced. By contrast, GFP(CAG)89 cells did not reveal visible GFP fluorescence in the presence of doxycycline. Given that both cell lines contain the GFP gene in the same genomic location, we conclude that the lack of fluorescence in GFP(CAG)89 cells is due to the presence of the repeat tract.
Figure 2. GFP fluorescence in GFP(CAG)89 cells and in GFP(CAG)0 cells before and after addition of doxycycline.

A. Fluorescence microscopy. Brightfield and fluorescent images of the same cell populations are shown. B. Flow cytometry. In each cell line, with or without added doxycycline, 50,000 cells were analyzed by flow cytometry and plotted as a histogram. Although not shown, unmodified T-REx cells—in the presence or absence of doxycycline—give a distribution that is indistinguishable from uninduced GFP(CAG)89 cells.
To observe the distribution of GFP fluorescence in the cell populations, both cell lines were analyzed by flow cytometry. Without induction—with basal transcription only—GFP(CAG)0 cells fluoresced more intensely than the GFP(CAG)89 cells (Figure 2B). Addition of doxycycline increased the mean fluorescence intensities of both cell lines, with GFP(CAG)0 cells once again fluorescing more intensely than the GFP(CAG)89 cells, supporting the observations made by microscopy. In addition, given that the fluorescence intensities were higher in GFP(CAG)0 cells than in GFP(CAG)89 cells for both the induced and uninduced populations, these results further support the idea that the CAG89 repeat tract inhibits GFP expression. Importantly, the induced GFP(CAG)0 and GFP(CAG)89 cells formed discrete populations in histograms of fluorescence intensity.
In the GFP(CAG)0 cells, we noted the presence of a small population of highly fluorescent cells in the uninduced cell population, and also a small population of nonfluorescent cells in the induced population (Figure 2B). The highly fluorescent cells, which we will refer to as GFP+ cells, might arise by loss of the Tet repressor; the nonfluorescent cells could arise by loss of the GFP reporter gene. We tested those possibilities by preselecting cells with blasticidin, whose resistance gene is linked to the Tet repressor gene, and with hygromycin, whose resistance gene is linked to the GFP gene. Treatment with blasticidin reduced the background of GFP+ cells by 8-fold; treatment with hygromycin reduced the level of nonfluorescent cells by 4-fold. Thus, to increase the sensitivity of the assay, we routinely pre-treated cells with blasticidin and hygromicin.
To quantify GFP expression, we analyzed induced and uninduced cells by quantitative RT-PCR, using two independent primer sets. As shown in Figure 3A, doxycycline induced transcription of the GFP gene by approximately 100-fold in both cell lines. Although fold inductions were indistinguishable in the two cell lines, the presence of the CAG89 repeat tract was consistently associated with a 2- to 3-fold reduction in transcript levels relative to levels in cells with the CAG0 repeat tract. In GFP(CAG)89 cells, induction of transcription increased the frequency of highly fluorescent cells—GFP+ cells—in a time-dependent manner (Figure 3B), as we demonstrated previously for transcription-induced HPRT+ cells in our HPRT selection assay [28].
Figure 3. Doxycycline-induced transcription of the GFP gene.
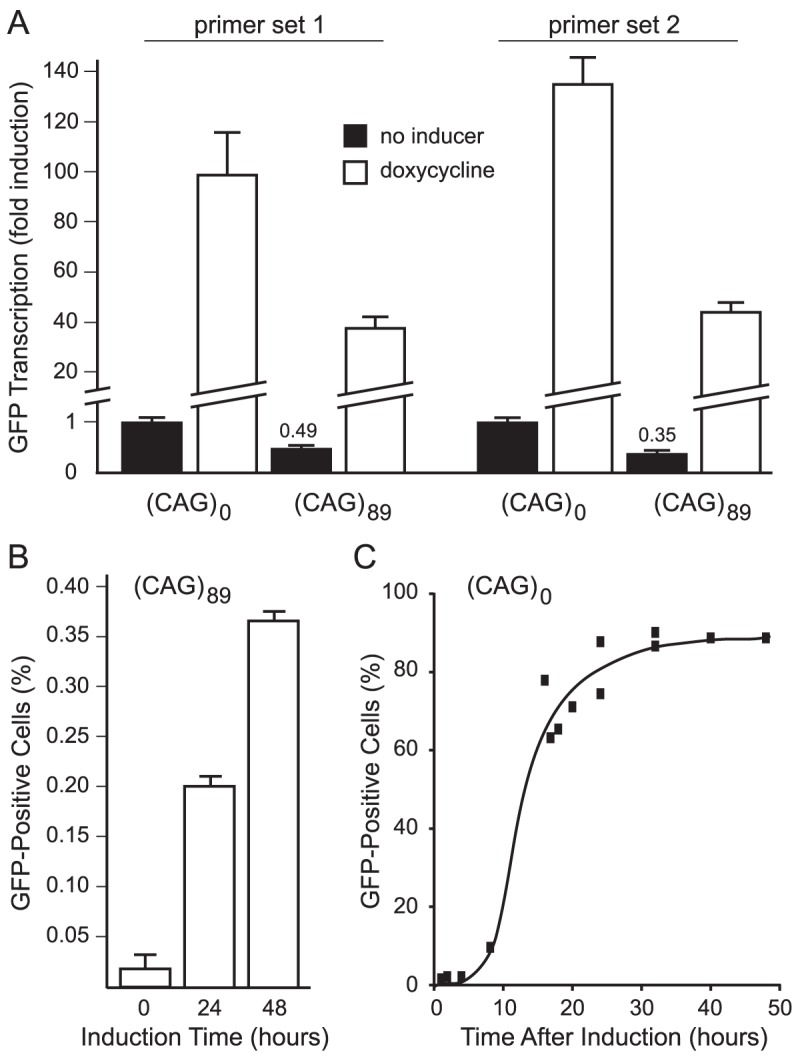
A. Quantification of doxycycline-induced GFP transcription in GFP(CAG)89 and GFP(CAG)0 cell lines. Cells were induced with doxycycline for three days prior to analysis. Two independent primer sets, both specific for GFP exon 2, were used to amplify GFP transcripts by quantitative RT-PCR. For each primer set, the individual results were internally normalized to β-actin and then to the levels of GFP transcripts in uninduced GFP(CAG)0 cells. The values for the uninduced levels of GFP transcripts in GFP(CAG)89 cells are indicated. The increase over uninduced levels in GFP(CAG)89 cells was 76-fold for primer set 1 and 98-fold for primer set 2. B. Transcription-induced changes in numbers of GFP+ cells in GFP(CAG)89 cells. Cells were treated with doxycycline for 0, 24, or 48 hours prior to analysis for GFP+ cells by flow cytometry, using the High gate indicated in Figure 4A to define the population of GFP+ cells. The frequencies of GFP+ cells at 0, 24, and 48 hours, respectively, were 0.02±0.013%, 0.20±0.02%, and 0.37±0.01%, as determined by counting three samples of 50,000 cells. C. Kinetics of induction of GFP expression in GFP(CAG)0 cells. Doxycycline (2 µg/mL) was added at time 0 to wells of 6-well plates containing 100,000 GFP(CAG)0 cells. Individual wells were harvested at the indicated times and analyzed by flow cytometry, using the High gate indicated in Figure 4A to define the population of GFP+ cells.
Fluorescence Intensity is Inversely Proportional to Repeat Length
Because transcription induces repeat instability, as we previously showed, we expected that addition of doxycycline here would do the same, allowing us to test the relationship between repeat length and fluorescence intensity. We induced GFP(CAG)89 cells with doxycycline for three days to stimulate repeat instability, and then sorted cells from this distribution based on GFP fluorescence intensity. The High intensity gates corresponded to the gates used to measure transcription-induced repeat instability, while the Medium and Low gates corresponded to regions of lesser fluorescence intensity (Figure 4A). Individual colonies were grown and analyzed by DNA sequencing and by FACS.
Figure 4. Relationship between GFP fluorescence intensity and CAG repeat tract length.

A. Isolation of cells from different parts of the distribution of fluorescence intensity. Induced GFP(CAG)89 cells were sorted according to the indicated gates, single cells were grown into colonies, and their CAG tracts were amplified by PCR and sequenced. B. Distribution of tract lengths in cells sorted by fluorescence intensity. C. Fluorescence intensity of cells with different CAG tract lengths.
Sequence analysis revealed that CAG tract length correlated inversely with the fluorescence intensity defined by the original gates (Figure 4B). Sorting through the High intensity gate yielded 15 colonies with repeats from 7 to 35 units in length; sorting through the Medium gate gave 15 colonies with repeats 49 to 58 units; and sorting through the Low gate yielded 17 colonies with repeats from 77 to 100 units. Notably, the Low gate yielded 4 clones with repeat tracts that were at least 5 units longer than the parental tract, including one that was 11 units longer. To verify the relationship between repeat length and fluorescence intensity, we grew several colonies into populations and analyzed them by flow cytometry. As shown in Figure 4C, there is a reasonable inverse correlation between CAG tract length and fluorescence intensity.
Long Repeat Tracts Cause Aberrant Splicing
In our previous studies, we had showed that long CAG tracts interfered with correct splicing of the APRT and HPRT genes [28], [29]. To determine the effects of CAG repeat tracts on splicing of the GFP minigene, we induced transcription in GFP(CAG)89 cells and subjected the RNA to RT-PCR, using primers in the GFP exons. As shown in Figure 5A, we observed two major products. Sequencing revealed that the lower band corresponded to the correctly spliced GFP transcript, encoding functional GFP (Figure 5B). The upper band corresponded to an aberrantly spliced transcript that contained approximately 300 extra nucleotides between the two GFP exons. The extra exon contained the CAG repeat tract plus 38 extra nucleotides immediately downstream of the repeat tract (Figure 5B), which matches exactly with the aberrant splicing we observed in the ARPT and HRPT selection assays [29]. The CAG repeat likely behaves the same in these different genes because the signals responsible for aberrant splicing—the donor and acceptor splice sites and the CAG repeat itself—are contained within the Not1 fragment used to move the repeat tract from gene to gene (Figure 5B). As expected, the GFP(CAG)0 cells, which lack the entire Not1 fragment, yield only the correctly spliced product (Figure 5C).
Figure 5. Aberrant splicing at the CAG repeat tract.
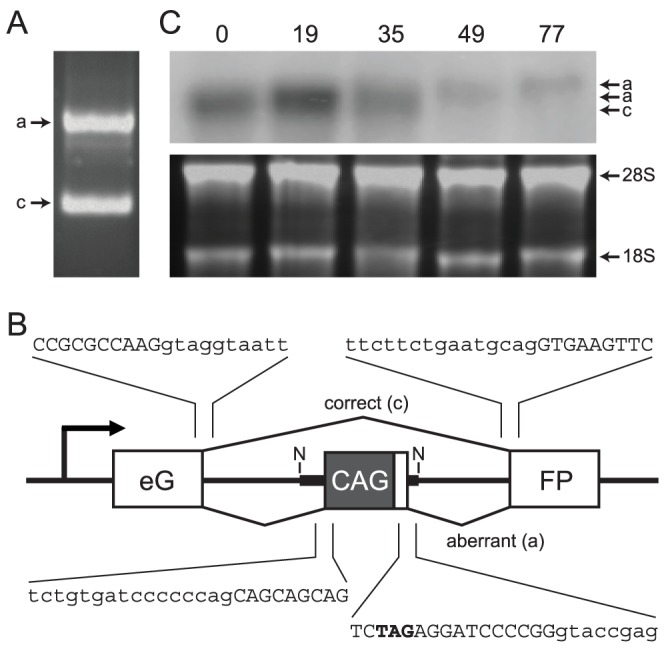
A. Amplification of GFP transcripts from induced GFP(CAG)89 cells by RT-PCR. B. Splice junctions in normal and aberrantly spliced GFP mRNA. The lower band in A corresponds to correctly spliced GFP mRNA, with sequences around the splice sites indicated above the gene. The upper band in A corresponds to aberrantly spliced mRNA that included the CAG repeat as an extra exon. Splice sites at the ends of the CAG exon are indicated below the gene. In all cases, upper case letters were retained in the spliced product and lower case letter were eliminated. An in-frame stop codon is highlighted in bold. The Not1 site that was used to clone the CAG repeat tract, along with its 129 bp of flanking sequences, is indicated as N. C. Northern blot analysis of GFP transcripts in induced cell lines with different lengths of CAG repeat tracts. Numbers above the lanes indicate the length of the CAG repeat tract. The lane labeled “0” is from GFP(CAG)0 cells. The upper panel shows the Northern blot, using a hybridization probe made from the full-length EGFP gene. The lower panel shows rRNA bands present in the ethidium bromide-stained gel prior to transfer for blot analysis.
The extra exon included in the aberrant splice product in GFP(CAG)89 cells renders the transcript incapable of making functional GFP because it contains an in-frame termination codon, highlighted in Figure 5B, and because it alters the reading frame of the downstream GFP exon. As shown in the Northern blot analysis in Figure 5C, the correctly spliced (short) transcript was not visible with longer-length repeat tracts. In addition, the amount of aberrantly spliced transcript tended to decrease with increasing length of the CAG tract. Both of these observations are consistent with our previous results [29]. Thus, long CAG repeat tracts decrease production of correct GFP mRNA, in part by promoting inclusion of the CAG tract in aberrantly spliced GFP mRNA and in part by decreasing overall transcription.
Transcription Versus Other Treatments
Visualizing GFP fluorescence requires transcription of the GFP gene, yet transcription itself causes repeat instability (Figure 3B). For GFP fluorescence to be broadly useful as an assay for CAG repeat instability, it must be possible to detect the effects of other treatments above those induced by transcription. Because transcription-induced CAG contractions accumulate with time of transcription [28], one way to limit the background due to transcription is to reduce the time of transcription to the minimum required for robust GFP expression. To determine the kinetics of GFP expression, we added doxycycline to GFP(CAG)0 cells and measured the percentage of the population that expressed GFP over time. As shown in Figure 3C, expression of GFP reached a plateau at about 24 hours.
To determine whether we could see a stimulation of contractions over the background level of GFP+ cells induced by transcription, we treated GFP(CAG)89 cells with a zinc-finger nuclease (ZFN50/ZFN51) that cleaves CAG repeat tracts [35], [36]. We transfected ZFNs into cells and then after 72 hours added doxycycline for 24 hours to induce GFP expression. Gates were selected in the control population transfected with cleavage dead (cd) versions of the ZFNs to distinguish the highest 10%, 1%, 0.1%, and 0.01% of cells, according to their fluorescence intensity (Figure 6). Populations of cells transfected with ZFN50/ZFN51 showed a significant increase in GFP+ cells in every gate relative to control cells (Figure 6). In the two most stringent gates (0.1% and 0.01%), the active ZFN showed a 10-fold increase in GFP+ cells, a result that matches our previous experiments with our selective APRT and HRPT systems [35]. Characterization of the repeat tracts in isolated GFP+ cells showed that all carried modifications of the CAG repeat tract: 13 contained contractions and 1 contained a deletion that removed the repeat tract. These results confirm that it is possible to see effects on CAG repeat instability above the background of events due to transcription.
Figure 6. Stimulation of GFP+ cells by transfection with ZFN50/ZFN51.
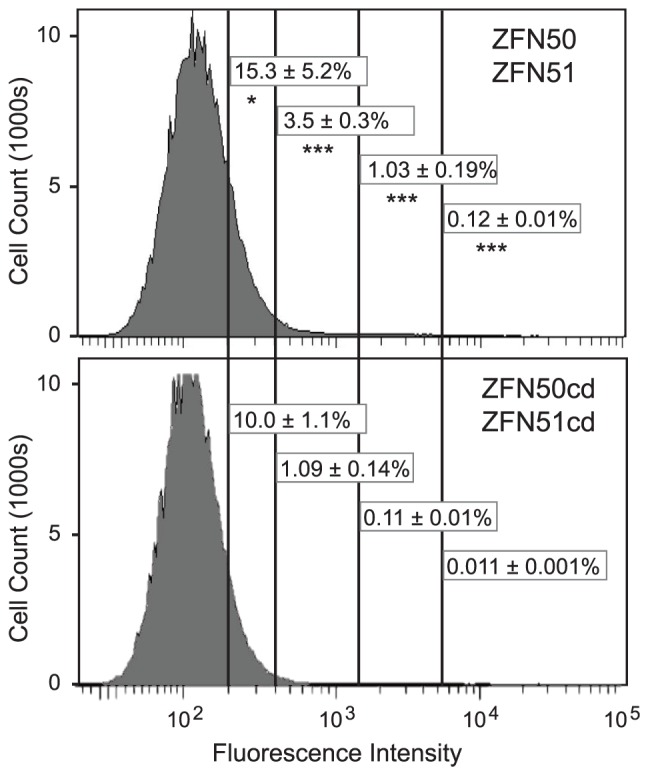
GFP(CAG)89 cells were transfected with 1.25 µg of each ZFN plasmid to a total of 2.5 µg per well of a 6-well plate. Cleavage-dead (cd) versions of ZFN50 and ZFN51, which carry the inactivating D450A mutation in the FokI cleavage domain and the R487D or the D483R obligate-heterodimer mutations in their dimerization domains, were used as transfection controls; they do not cleave CAG repeat tracts in vitro, nor do they stimulate CAG repeat contractions in GFP(CAG)89 cells [36]. The numbers presented in the figure show the mean and standard deviation from two experiments, each with three independent samples, for a total of six measurements for each ZFN. The numbers associated with each gate include all cells to the right of the gate. The numbers and gates used to derive them are displayed on a pair of histograms from parallel transfection samples for each ZFN. A total of 2.0×106 cells were analyzed for ZFN50/ZFN51 transfections, and 2.2×106 cells were analyzed for the cleavage-dead control ZFNs. Statistical significance was determined by a two tailed t-test of means of the corresponding gates of the active and inactive ZFNs (* = P<.05; *** = P<0.0001).
Discussion
In this report, we describe a GFP-based fluorescence assay for analysis of the instability of CAG repeat tracts. The assay is based on the ability of long, intronic CAG repeat tracts to interfere with expression of the gene in which they reside. Importantly—and uniquely—this assay provides the first method for estimating the length of CAG repeat tracts in living cells.
The GFP-based fluorescence assay has several advantages over the selection assays we have used previously [28], [29], [31], [37], [38], [39], [40], [41]. First, the fluorescence assay is faster, taking just a few days instead of two to three weeks. Second, the fluorescence assay permits detection and analysis of a wider range of tract lengths, whereas our selection assays only detect cells that have fewer than 38 repeats. For example, Changes in the spectrum of events potentially can provide insights into the mechanism of instability induced by a treatment. Third, the absolute values for the frequencies of contractions—induced by transcription and ZFNs—are higher than for similar treatments in our selection assays, for reasons that are not entirely clear. For example, ZFN treatments in our selection assay gave frequencies of 0.01%, whereas those same treatments in the fluorescence assay gave frequencies above 1%. Similarly, transcription in our selective system gave frequencies around 0.001%, whereas in the fluorescence assay it gave frequencies of 0.2%. The higher frequencies make the fluorescence assay amenable to high-throughput protocols for screening chemical and shRNA libraries for effects of CAG repeat instability. Thus, the GFP-based fluorescence assay represents a significant advance over our previous selection assays for investigating CAG repeat instability, combining the speed and flexibility of cell culture with the screening power of flow cytometry.
One surprising and useful feature of this fluorescence assay is that GFP expression is inversely dependent on the length of the CAG repeat tract. The relationship between tract length and GFP expression means that the assay can be used to test a broad spectrum of changes to the repeat tract, potentially including expansions as well as contractions. This property of the system is likely a combination of factors. As we discussed previously [29], the CAG tract may behave as an exonic splicing enhancer (ESE), mimicking the CA-rich ESE motifs that function to promote splicing in vivo and in vitro [42], [43], [44], [45], [46], [47]. Because the 5′ splice site immediately downstream of the CAG tract is fairly weak (ESEfinder score of 4.4 [42]), its function may depend on the presence of the ESE, with longer CAG tracts promoting more efficient use of the splicing signal [29]. A second contributing factor may be that CAG repeat tracts interfere with transcription in a length-dependent manner. We have shown that a CAG89 tract decreases transcript levels by 2- to 3-fold (Figure 3). Supporting this idea, several in vitro studies have shown that transcription stalls at DNA sequences such as CAG tracts that can form secondary structures [48], [49], [50], [51], [52], [53], [54]. Third, the presence of a premature termination codon in the “CAG exon” may induce nonsense-mediated decay, although its position near the splice junction may limit its effectiveness [55]. Finally, it is possible that CAG repeats may induce heterochromatin-mediated silencing of the GFP gene in a length-dependent manner, as has been observed in other systems [56]. Regardless of the mechanism, fluorescence intensity, by serving as a reporter for the length of the repeat, provides a powerful tool for elucidating mechanisms of repeat instability.
This fluorescence-based assay has a potential limitation. As described here, the activity of the GFP gene must pass a threshold of expression for GFP+ cells to be detected by flow cytometry. As shown here, cells can become GFP+ by contraction of the CAG repeat tract (Figure 4B). In principle, however, treatments that increased expression of the GFP reporter gene—by increasing transcription or by stabilizing either the mRNA or protein—could increase the number of GFP+ cells, without increasing the number of cells carrying contractions of the CAG repeat tract. This eventuality would show up during analysis of the repeat tracts in GFP+ cells.
In this report, we have focused on the use of the fluorescence assay to detect repeat contractions. As we have discussed previously in relation to our selection systems, contractions in human cells are good predictors of processes that influence repeat instability in model organisms and, by extension, in human patients [39]. The fluorescence assay, however, offers the possibility of directly detecting CAG repeat expansions. Repeat lengths in the range of 50 to 60 CAG repeat units should provide sufficient fluorescence intensity that decreases in intensity caused by expansions of the repeat can be detected. In addition, because both contractions and expansions will be visible in the same assay, treatments that cause differential effects on contractions and expansions will be immediately apparent. Processes that cause a bias toward expansions may be more relevant to the disease processes in patients, which display a strong bias toward expansions in germ line and somatic tissues. By contrast, treatments that cause a strong bias toward contractions may provide clues to treatments designed to shrink repeat tracts in patients.
Acknowledgments
We thank the members of the Wilson laboratory for helpful comments and discussion. We appreciate the expert assistance of Joel M. Sederstrom for flow cytometry and cell sorting.
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper.
Funding Statement
This work was supported by: Baylor Research Advocates for Student Scientists to BAS (https://www.bcm.edu/giving/get-involved/brass); The Cytometry and Cell Sorting Core at Baylor College of Medicine with funding from the National Institutes of Health (AI036211, CA125123, and RR024574) (http://www.nih.gov/); A National Institutes of General Medical Sciences training grant (T32 GM08307) to BAS (http://www.nih.gov/); A National Eye Institute training grant (T32 EY07001) to CM (http://www.nih.gov/); grants from the National Institutes of Health (NS079926) to DM (http://www.nih.gov/); and a grant from the National Institutes of Health (GM38219 and EY11731) to JHW (http://www.nih.gov/). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Pearson CE, Edamura KN, Cleary JD (2005) Repeat instability: mechanisms of dynamic mutations. Nat Rev Genet 6:729–742. [DOI] [PubMed] [Google Scholar]
- 2. Orr HT, Zoghbi HY (2007) Trinucleotide repeat disorders. Annu Rev Neurosci 30:575–621. [DOI] [PubMed] [Google Scholar]
- 3. Shelbourne PF, Keller-McGandy C, Bi WL, Yoon SR, Dubeau L, et al. (2007) Triplet repeat mutation length gains correlate with cell-type specific vulnerability in Huntington disease brain. Hum Mol Genet 16:1133–1142. [DOI] [PubMed] [Google Scholar]
- 4. Gomes-Pereira M, Monckton DG (2006) Chemical modifiers of unstable expanded simple sequence repeats: what goes up, could come down. Mutat Res 598:15–34. [DOI] [PubMed] [Google Scholar]
- 5. Mirkin SM (2007) Expandable DNA repeats and human disease. Nature 447:932–940. [DOI] [PubMed] [Google Scholar]
- 6. Lin Y, Hubert L Jr, Wilson JH (2009) Transcription destabilizes triplet repeats. Mol Carcinog 48:350–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gemayel R, Vinces MD, Legendre M, Verstrepen KJ (2010) Variable tandem repeats accelerate evolution of coding and regulatory sequences. Annu Rev Genet 44:445–477. [DOI] [PubMed] [Google Scholar]
- 8. Richard GF, Kerrest A, Dujon B (2008) Comparative genomics and molecular dynamics of DNA repeats in eukaryotes. Microbiol Mol Biol Rev 72:686–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pearson CE, Sinden RR (1996) Alternative structures in duplex DNA formed within the trinucleotide repeats of the myotonic dystrophy and fragile X loci. Biochemistry 35:5041–5053. [DOI] [PubMed] [Google Scholar]
- 10. Gacy AM, Goellner G, Juranic N, Macura S, McMurray CT (1995) Trinucleotide repeats that expand in human disease form hairpin structures in vitro. Cell 81:533–540. [DOI] [PubMed] [Google Scholar]
- 11. Hubert L Jr, Lin Y, Dion V, Wilson JH (2011) Xpa deficiency reduces CAG trinucleotide repeat instability in neuronal tissues in a mouse model of SCA1. Hum Mol Genet 20:4822–4830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kovtun IV, Johnson KO, McMurray CT (2011) Cockayne Syndrome B protein antagonizes OGG1 in modulating CAG repeat length in vivo. Aging (Albany NY) 3:509–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kovtun IV, Liu Y, Bjoras M, Klungland A, Wilson SH, et al. (2007) OGG1 initiates age-dependent CAG trinucleotide expansion in somatic cells. Nature 447:447–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tome S, Panigrahi GB, Lopez Castel A, Foiry L, Melton DW, et al. (2011) Maternal germline-specific effect of DNA ligase I on CTG/CAG instability. Hum Mol Genet 20:2131–2143. [DOI] [PubMed] [Google Scholar]
- 15. Dion V, Lin Y, Hubert L Jr, Waterland RA, Wilson JH (2008) Dnmt1 deficiency promotes CAG repeat expansion in the mouse germline. Hum Mol Genet 17:1306–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang L, Leeflang EP, Yu J, Arnheim N (1994) Studying human mutations by sperm typing: instability of CAG trinucleotide repeats in the human androgen receptor gene. Nat Genet 7:531–535. [DOI] [PubMed] [Google Scholar]
- 17. Kovtun IV, McMurray CT (2001) Trinucleotide expansion in haploid germ cells by gap repair. Nat Genet 27:407–411. [DOI] [PubMed] [Google Scholar]
- 18. Lee JM, Zhang J, Su AI, Walker JR, Wiltshire T, et al. (2010) A novel approach to investigate tissue-specific trinucleotide repeat instability. BMC Syst Biol 4:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Highnam G, Franck C, Martin A, Stephens C, Puthige A, et al. (2013) Accurate human microsatellite genotypes from high-throughput resequencing data using informed error profiles. Nucleic Acids Res 41:e32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fondon JW 3rd, Martin A, Richards S, Gibbs RA, Mittelman D (2012) Analysis of microsatellite variation in Drosophila melanogaster with population-scale genome sequencing. PLoS One 7:e33036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Doi K, Monjo T, Hoang PH, Yoshimura J, Yurino H, et al. (2013) Rapid detection of expanded short tandem repeats in personal genomics using hybrid sequencing. Bioinformatics. [DOI] [PMC free article] [PubMed]
- 22. Richard GF, Viterbo D, Khanna V, Mosbach V, Castelain L, et al. (2014) Highly specific contractions of a single CAG/CTG trinucleotide repeat by TALEN in yeast. PLoS One 9:e95611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shah KA, Shishkin AA, Voineagu I, Pavlov YI, Shcherbakova PV, et al. (2012) Role of DNA polymerases in repeat-mediated genome instability. Cell Rep 2:1088–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shishkin AA, Voineagu I, Matera R, Cherng N, Chernet BT, et al. (2009) Large-scale expansions of Friedreich's ataxia GAA repeats in yeast. Mol Cell 35:82–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Debacker K, Frizzell A, Gleeson O, Kirkham-McCarthy L, Mertz T, et al. (2012) Histone deacetylase complexes promote trinucleotide repeat expansions. PLoS Biol 10:e1001257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rolfsmeier ML, Dixon MJ, Pessoa-Brandao L, Pelletier R, Miret JJ, et al. (2001) Cis-elements governing trinucleotide repeat instability in Saccharomyces cerevisiae. Genetics 157:1569–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lin Y, Dion V, Wilson JH (2005) A novel selectable system for detecting expansion of CAG.CTG repeats in mammalian cells. Mutat Res 572:123–131. [DOI] [PubMed] [Google Scholar]
- 28. Lin Y, Dion V, Wilson JH (2006) Transcription promotes contraction of CAG repeat tracts in human cells. Nat Struct Mol Biol 13:179–180. [DOI] [PubMed] [Google Scholar]
- 29. Gorbunova V, Seluanov A, Dion V, Sandor Z, Meservy JL, et al. (2003) Selectable system for monitoring the instability of CTG/CAG triplet repeats in mammalian cells. Mol Cell Biol 23:4485–4493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Concannon C, Lahue RS (2013) The 26S proteasome drives trinucleotide repeat expansions. Nucleic Acids Res 41:6098–6108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hubert L Jr, Lin Y, Dion V, Wilson JH (2011) Topoisomerase 1 and Single-Strand Break Repair Modulate Transcription-Induced CAG Repeat Contraction in Human Cells. Mol Cell Biol 31:3105–3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Seluanov A, Mittelman D, Pereira-Smith OM, Wilson JH, Gorbunova V (2004) DNA end joining becomes less efficient and more error-prone during cellular senescence. Proc Natl Acad Sci U S A 101:7624–7629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Iyer RR, Pluciennik A, Rosche WA, Sinden RR, Wells RD (2000) DNA polymerase III proofreading mutants enhance the expansion and deletion of triplet repeat sequences in Escherichia coli. J Biol Chem 275:2174–2184. [DOI] [PubMed] [Google Scholar]
- 34. Pfaffl MW (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29:e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mittelman D, Moye C, Morton J, Sykoudis K, Lin Y, et al. (2009) Zinc-finger directed double-strand breaks within CAG repeat tracts promote repeat instability in human cells. Proc Natl Acad Sci U S A 106:9607–9612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moye C (2013) Generation of Instability within Trinucleotide Repeats. PhD Thesis, Baylor College of Medicine.
- 37. Lin Y, Wilson JH (2007) Transcription-induced CAG repeat contraction in human cells is mediated in part by transcription-coupled nucleotide excision repair. Mol Cell Biol 27:6209–6217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mittelman D, Sykoudis K, Hersh M, Lin Y, Wilson JH (2010) Hsp90 modulates CAG repeat instability in human cells. Cell Stress Chaperones 15:753–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lin Y, Leng M, Wan M, Wilson JH (2010) Convergent transcription through a long CAG tract destabilizes repeats and induces apoptosis. Mol Cell Biol 30:4435–4451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lin Y, Dent SY, Wilson JH, Wells RD, Napierala M (2010) R loops stimulate genetic instability of CTG.CAG repeats. Proc Natl Acad Sci U S A 107:692–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gorbunova V, Seluanov A, Mittelman D, Wilson JH (2004) Genome-wide demethylation destabilizes CTG.CAG trinucleotide repeats in mammalian cells. Hum Mol Genet 13:2979–2989. [DOI] [PubMed] [Google Scholar]
- 42. Cartegni L, Wang J, Zhu Z, Zhang MQ, Krainer AR (2003) ESEfinder: A web resource to identify exonic splicing enhancers. Nucleic Acids Res 31:3568–3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang Z, Hoffmann HM, Grabowski PJ (1995) Intrinsic U2AF binding is modulated by exon enhancer signals in parallel with changes in splicing activity. RNA 1:21–35. [PMC free article] [PubMed] [Google Scholar]
- 44. Tian M, Maniatis T (1992) Positive control of pre-mRNA splicing in vitro. Science 256:237–240. [DOI] [PubMed] [Google Scholar]
- 45. Lynch KW, Maniatis T (1995) Synergistic interactions between two distinct elements of a regulated splicing enhancer. Genes Dev 9:284–293. [DOI] [PubMed] [Google Scholar]
- 46. Gersappe A, Pintel DJ (1999) CA- and purine-rich elements form a novel bipartite exon enhancer which governs inclusion of the minute virus of mice NS2-specific exon in both singly and doubly spliced mRNAs. Mol Cell Biol 19:364–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Coulter LR, Landree MA, Cooper TA (1997) Identification of a new class of exonic splicing enhancers by in vivo selection. Mol Cell Biol 17:2143–2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Belotserkovskii BP, Mirkin SM, Hanawalt PC (2013) DNA sequences that interfere with transcription: implications for genome function and stability. Chem Rev 113:8620–8637. [DOI] [PubMed] [Google Scholar]
- 49. Tornaletti S, Park-Snyder S, Hanawalt PC (2008) G4-forming sequences in the non-transcribed DNA strand pose blocks to T7 RNA polymerase and mammalian RNA polymerase II. J Biol Chem 283:12756–12762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Parsons MA, Sinden RR, Izban MG (1998) Transcriptional properties of RNA polymerase II within triplet repeat-containing DNA from the human myotonic dystrophy and fragile X loci. J Biol Chem 273:26998–27008. [DOI] [PubMed] [Google Scholar]
- 51. Grabczyk E, Usdin K (2000) The GAA*TTC triplet repeat expanded in Friedreich's ataxia impedes transcription elongation by T7 RNA polymerase in a length and supercoil dependent manner. Nucleic Acids Res 28:2815–2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ditlevson JV, Tornaletti S, Belotserkovskii BP, Teijeiro V, Wang G, et al. (2008) Inhibitory effect of a short Z-DNA forming sequence on transcription elongation by T7 RNA polymerase. Nucleic Acids Res 36:3163–3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Belotserkovskii BP, De Silva E, Tornaletti S, Wang G, Vasquez KM, et al. (2007) A triplex-forming sequence from the human c-MYC promoter interferes with DNA transcription. J Biol Chem 282:32433–32441. [DOI] [PubMed] [Google Scholar]
- 54. Liu CR, Chang CR, Chern Y, Wang TH, Hsieh WC, et al. Spt4 is selectively required for transcription of extended trinucleotide repeats. Cell 148:690–701. [DOI] [PubMed] [Google Scholar]
- 55. Maquat LE (2004) Nonsense-mediated mRNA decay: splicing, translation and mRNP dynamics. Nat Rev Mol Cell Biol 5:89–99. [DOI] [PubMed] [Google Scholar]
- 56. Saveliev A, Everett C, Sharpe T, Webster Z, Festenstein R (2003) DNA triplet repeats mediate heterochromatin-protein-1-sensitive variegated gene silencing. Nature 422:909–913. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper.