

The molecular machinery that catalyzes S-acylation reactions at the Golgi comprises zDHHC enzymes with major differences in substrate affinity and S-acylation activity. It is proposed that the coexistence of these two groups of enzymes in cells is important to allow the Golgi S-acylation machinery to modify a wide and diverse set of substrates.
Abstract
S-acylation, the attachment of fatty acids onto cysteine residues, regulates protein trafficking and function and is mediated by a family of zDHHC enzymes. The S-acylation of peripheral membrane proteins has been proposed to occur at the Golgi, catalyzed by an S-acylation machinery that displays little substrate specificity. To advance understanding of how S-acylation of peripheral membrane proteins is handled by Golgi zDHHC enzymes, we investigated interactions between a subset of four Golgi zDHHC enzymes and two S-acylated proteins—synaptosomal-associated protein 25 (SNAP25) and cysteine-string protein (CSP). Our results uncover major differences in substrate recognition and S-acylation by these zDHHC enzymes. The ankyrin-repeat domains of zDHHC17 and zDHHC13 mediated strong and selective interactions with SNAP25/CSP, whereas binding of zDHHC3 and zDHHC7 to these proteins was barely detectable. Despite this, zDHHC3/zDHHC7 could S-acylate SNAP25/CSP more efficiently than zDHHC17, whereas zDHHC13 lacked S-acylation activity toward these proteins. Overall the results of this study support a model in which dynamic intracellular localization of peripheral membrane proteins is achieved by highly selective recruitment by a subset of zDHHC enzymes at the Golgi, combined with highly efficient S-acylation by other Golgi zDHHC enzymes.
INTRODUCTION
S-acylation, a reversible posttranslational modification (PTM) involving the attachment of fatty acids onto cysteine residues, plays a major role in regulating the trafficking and function of modified proteins (Linder and Deschenes, 2007; Fukata and Fukata, 2010; Salaun et al., 2010). This PTM is commonly referred to as palmitoylation, reflecting the fact that palmitic acid is the predominant fatty acid added to proteins in this way (Muszbek et al., 1999). Proteomic analyses have identified upward of 300 S-acylated proteins (Kang et al., 2008; Martin and Cravatt, 2009; Yount et al., 2010), which include both transmembrane and peripheral membrane proteins. For peripheral membrane proteins, a major function of S-acylation is to provide a stable membrane anchor; hence this PTM is essential for membrane association of signaling molecules such as H-Ras, N-Ras, TC10, and Gα subunits and for membrane fusion proteins such as synaptosomal-associated protein 25 (SNAP25) and cysteine-string protein (CSP; Salaun et al., 2010).
S-acylation reactions in mammals are catalyzed by a family of 24 zDHHC S-acyl-transferase enzymes (Fukata et al., 2004). These enzymes are predicted to be polytopic membrane proteins, with four to six transmembrane domains and a conserved 51–amino acid zDHHC-CR domain (aspartate-histidine-histidine-cysteine motif in a cysteine-rich, zinc finger–like domain). At the heart of this zDHHC-CR domain is a highly conserved DHHC tetrapeptide that is critical for S-acylation activity. Although the exact mechanism of S-acylation is not known, it has been long assumed, and recently demonstrated for purified zDHHC2, zDHHC3 and yeast Erf2, that it involves a two-step mechanism in which the zDHHCs form an acyl–enzyme intermediate (autoacylation), with the acyl group later transferred to the target protein (Mitchell et al., 2010; Jennings and Linder, 2012). Furthermore, the kinetics of autoacylation suggest a stoichiometry of one acyl group per enzyme molecule (Mitchell et al., 2010).
zDHHC enzymes are associated with the endoplasmic reticulum, Golgi, plasma membrane, and endosomes (Ohno et al., 2006; Greaves et al., 2011), with the catalytic zDHHC-CR domain facing the cytoplasm (Mitchell et al., 2006). Recent work has indicated that the Golgi apparatus is a hotspot for the S-acylation of peripheral membrane proteins, with the corresponding zDHHC enzymes being incapable of recognizing structural features of substrate proteins, surrounding the target cysteine(s); therefore, it has been suggested that zDHHC enzymes may lack substrate specificity for certain S-acylated proteins (Rocks et al., 2010). On the contrary, another study revealed specific substrate interactions and/or S-acylation activity for a subset of neuronal zDHHC enzymes (Huang et al., 2009).
To advance our understanding of how S-acylation reactions are handled by Golgi zDHHC enzymes, we examined the interactions between a subset of these enzymes and two S-acylated peripheral membrane proteins (SNAP25 and CSP). The specific aims of the study were to 1) determine whether individual Golgi zDHHC enzymes exhibit selective interactions with either of these two substrate proteins, 2) identify potential mechanisms involved in substrate recognition, and 3) determine the importance of zDHHC–substrate binding for subsequent S-acylation activity. The results presented suggest that there are marked differences in substrate specificity and S-acylation efficiency of Golgi zDHHC enzymes that modify peripheral membrane proteins. These enzymes displayed either strong and selective interactions with their substrates or nearly undetectable binding. Surprisingly, a complex nonlinear relationship was uncovered between the strength of zDHHC–substrate interactions and subsequent S-acylation, implying that Golgi zDHHC enzymes have major differences in their intrinsic S-acyl-transferase activity. The simultaneous expression of these two groups of zDHHC enzymes might therefore be important to allow the Golgi S-acylation machinery to modify a wide and diverse set of substrate proteins.
RESULTS
zDHHC17 displays strong and specific interaction with CSP and SNAP25
To assess binding of peripheral membrane proteins to Golgi zDHHC enzymes, we used the very well characterized S-acylated proteins cysteine-string protein (CSP) and SNAP25, and the Golgi-localized S-acyl-transferases zDHHC3, zDHHC7, and zDHHC17, since all these three enzymes can S-acylate both CSP (Greaves et al., 2008) and all members of the SNAP25 family (Greaves et al., 2010). By using hexahistidine (His6)-tagged forms of SNAP25/CSP to capture hemagglutinin (HA)-tagged versions of the foregoing enzymes from HEK293T cell lysates, we found that only zDHHC17 displayed detectable interaction with SNAP25 (Figure 1A) and CSP (Figure 1B).
FIGURE 1:
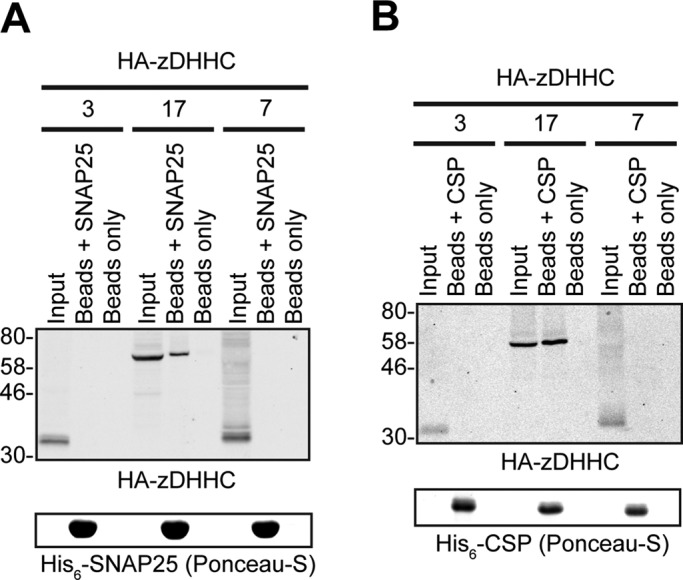
SNAP25 and CSP bind preferentially to zDHHC17 compared to zDHHC3 or zDHHC7. (A) HA-tagged zDHHCs were transfected into HEK293T cells, and corresponding cell lysates were incubated with Ni2+-NTA beads and either 75 μg of His6-SNAP25 or equivalent volume of PBS; 1/16 of input and 1/3 of bound fractions were subjected to SDS–PAGE, transfer to nitrocellulose, Ponceau-S staining, and Western blot analysis using HA antibody. (B) HA-tagged zDHHCs were transfected into HEK293T cells, and corresponding lysates were incubated with Ni2+-NTA beads that had been preincubated with either 75 μg of His6-CSP or an equivalent volume of PBS; 1/16 of input and 1/3 of bound fractions were subjected to SDS–PAGE, transfer to nitrocellulose, Ponceau-S staining, and Western blot analysis using HA antibody. Positions of molecular weight standards are shown on the left of Western blots.
To confirm the CSP/SNAP25 binding preference toward zDHHC17 over other Golgi zDHHC enzymes, we also used the mating-based split-ubiquitin system (SUS) in yeast; in this system, interactions of a bait-transmembrane protein (zDHHC enzyme) with another protein prey (CSP/SNAP25) can be assessed in vivo due to interaction-dependent reporter activation of genes required for yeast growth (Figure 2A; more detailed description of the SUS is given in Materials and Methods). SUS analysis also revealed preferential binding of SNAP25 and CSP to zDHHC17, than to zDHHC3, and this was even more prominent when zDHHC3/zDHHC17 expression was restricted by addition of methionine in the growth medium (Figure 2B). zDHHC17–CSP/SNAP25 interactions were found to be very strong and specific, since growth of CSP/SNAP25–zDHHC17 yeast matings was barely affected by methionine-induced reduction of zDHHC17 expression; furthermore, their growth was more prominent than the positive control (NubI–zDHHC17) matings (Figure 2, B and C). Specificity of the reported interaction was assessed by comparison with two other S-acylated proteins that are not known to be substrates of zDHHC17 (N-RAS, TC10). In contrast to SNAP25 and CSP, there was very little interaction observed between zDHHC17 and either N-RAS or TC10; indeed, at the highest methionine concentration, yeast growth was barely detectable (Figure 2C). To eliminate the possibility that the strong zDHHC17 interaction with CSP and SNAP25, observed by SUS, may be due to their increased membrane association caused by zDHHC-catalyzed S-acylation, we also assessed the foregoing interactions when the catalytic cysteine of zDHHC17 was mutated (C467A). This mutation had no effect on either the strength or the specificity of zDHHC17 interaction toward CSP and SNAP25 (Figure 2C).
FIGURE 2:

Assessment of zDHHC17 specificity toward SNAP25 and CSP, using the mating-based SUS. (A) Schematic illustration of the SUS. Ubiquitin is split into halves, with the Nub half fused to the N-terminus of a prey protein and the Cub half fused between a bait membrane protein and a PLV composed of protein A, a DNA-binding domain (LexA), and a transcriptional activating domain (VP16). Coexpression of the bait (e.g., zDHHC enzyme) with the prey (e.g., S-acylated substrate) could lead, upon interaction, to the reassembly of ubiquitin, which can now be recognized by ubiquitin-specific proteases. Spontaneous reassembly of ubiquitin cannot occur, due to a single amino acid substitution in Nub (I13G). The cleavage of ubiquitin will release the transcriptional reporter complex PLV, leading to the activation of auxotrophy reporter genes, allowing detection of interaction via growth analysis. Expression of bait and prey proteins can be assessed by Western blotting, using anti-VP16 and anti-HA antibodies, respectively. (B) SUS assessment of CSP and SNAP25 interactions with zDHHC3 and zDHHC17. Diploid yeast at corresponding OD600 was dropped on SD medium supplemented with adenine and histidine to verify equal dropping densities, and on SD medium (supplemented or not with methionine) to verify interactions. Plates were scanned after incubation at 30°C for the number of days indicated. Spontaneous reassembly of the C-terminal ubiquitin of zDHHC17-Cub-PLV with wild-type (NubI) or mutated (NubG) N-terminal ubiquitin was assessed in parallel. Expression of Cub-PLV, C-terminally tagged bait (zDHHC17) and NubG-2HA–tagged preys from haploid-yeast lysates was assessed by Western blotting using anti-VP16 and anti-HA antibodies, respectively. Positions of molecular weight markers are shown on the left. (C) SUS assessment of zDHHC17 (wild type and C467A catalytically inactive mutant) interaction with CSP, SNAP25, N-RAS, and TC10 and corresponding Western blots.
The N-terminal ankyrin-repeat domain of zDHHC17 is necessary and sufficient for interaction with SNAP25 and CSP
To identify the region of zDHHC17 involved in interaction with CSP and SNAP25, we constructed a series of zDHHC17 truncation mutants lacking the extreme N-terminus before the ankyrin repeat (ANK) domain (17-ΔN), the N-terminal region containing the ANK domain (17-ΔNAnk), the whole region after the fourth transmembrane domain and including the zDHHC domain (17-ΔCDHHC), or the C-terminal cytosolic region (17-ΔC; Figure 3A); all of these mutants were expressed at similar levels to wild-type (17-WT) zDHHC17-Cub-PLV (Figure 3B, right). The yeast growth patterns showed that all zDHHC17 truncation mutants, with the exception of 17-ΔNAnk, interacted with SNAP25 and CSP with efficiency similar to wild-type zDHHC17 (17-WT). The 17-ΔNAnk mutant showed a marked loss of interaction with both substrate proteins, which was clearly apparent when zDHHC17 expression was restricted by addition of 50 μM methionine (Figure 3B, left). Thus the ANK domain of zDHHC17 is essential for interaction with both SNAP25 and CSP.
FIGURE 3:

The ANK domain of zDHHC17 is both necessary and sufficient for binding to CSP and SNAP25. (A) Schematic diagram of zDHHC17 truncation mutants used for SUS analysis. (B) Detection of CSP and SNAP25 interactions with zDHHC17 wild type (17-WT) and 17-ΔN, 17-ΔNAnk, 17-ΔCDHHC, and 17-ΔC truncation mutants using SUS (growth assay). zDHHC17– CSP/SNAP25 matings at corresponding OD600 were dropped on SD medium supplemented with adenine and histidine to verify equal dropping densities, and on SD medium (supplemented or not with methionine) to verify interactions. Plates were scanned after incubation at 30°C for 2 d. Spontaneous reassembly of the C-terminal ubiquitin of zDHHC17-Cub-PLV with wild-type (NubI) or mutated (NubG) N-terminal ubiquitin was assessed in parallel. Top left, Western blot detection of zDHHC17-Cub-PLV truncation mutants (anti-VP16 antibody) and NubG-2HA-CSP/SNAP25 (anti-HA antibody) from haploid-yeast lysates, along with positions of molecular weight markers. (C) Interaction of GST-tagged, N-terminal cytosolic domain (amino acids11–305) of zDHHC17 (GST-17NAnk) with SNAP25 and CSP from rat brain. GST-precleared rat brain lysate was incubated with glutathione beads and either GST or GST-17NAnk; bound proteins were eluted after boiling in sample buffer and centrifugation. Input (I; 0.2%), unbound (U; 0.2%), and bound fractions (B; 1%) were subjected to SDS–PAGE, transfer to nitrocellulose, Ponceau-S staining, and Western blot analysis using the antibodies indicated. Positions of molecular weight markers are shown on the left. (D) Top, interaction of GST-17NAnk with palmitoylated (p) and nonpalmitoylated (np) Myc-tagged CSP. HEK293T cells were either transfected with Myc-CSP alone or cotransfected with Myc-CSP and HA-zDHHC3 plasmids; precleared HEK293T lysate was incubated with glutathione beads and purified GST or GST-17NAnk; bound proteins were eluted after boiling in sample buffer and centrifugation. Input (I; 6%), unbound (U; 5%), and bound fractions (B; 20%) were subjected to SDS–PAGE, transfer to nitrocellulose, Ponceau-S staining, and Western blot analysis using anti-Myc antibody. Positions of molecular weight markers are shown on the left. Bottom, percentage of Myc-CSP recovered was quantified for both p and np (n = 4; error bars, SEM), and differences were statistically analyzed by unpaired Student's t test (***p < 0.001).
The finding that the zDHHC17 construct lacking both the zDHHC-CR domain and C-terminus (17-ΔCDHHC) interacted with SNAP25 and CSP suggests that the ANK domain is sufficient for interaction with these substrates. To test this, and confirm the results of the SUS, we prepared a glutathione S-transferase (GST)–tagged version (GST-17NAnk) of the cytosolic N-terminus of zDHHC17 (residues 11–305), and incubated it with a rat brain lysate. Immunoblotting revealed that the GST-17NAnk construct (but not GST alone) captured SNAP25, and to a lesser degree CSP, but not other non–zDHHC17-interacting proteins (like synuclein) from the brain lysate (Figure 3C). We assumed that the very weak binding of CSP to GST-17NAnk may be due to the extensive S-acylation of this protein in brain tissue (Greaves et al., 2012), and this modification may hinder its interaction with zDHHC17's ANK domain. Indeed, when assessing binding of palmitoylated and nonpalmitoylated myc-CSP to GST-17NAnk, we observed sixfold-lower binding of palmitoylated CSP, irrespective of starting amount of palmitoylated and nonpalmitoylated CSP (Figure 3D). Given that removal of the extreme N-terminus of zDHHC17 (17-ΔN) did not alter zDHHC17 interaction with either CSP or SNAP25 (Figure 3B), these results indicate that the ANK domain of zDHHC17 is not only essential, but is also sufficient for substrate binding. Furthermore, the decrease in the in vitro binding of GST-17NAnk to S-acylated CSP might indicate a possible mechanism of in vivo zDHHC17 function, by which substrate can be disassociated from enzyme upon modification.
The ANK domain and C-terminus of zDHHC17 are required for S-acylation of SNAP25 and CSP
Because the ANK domain of zDHHC17 is both essential and adequate for substrate binding, it would be expected that zDHHC17 lacking this would not be able to S-acylate its substrates. To test this, we assessed the ability of each of the zDHHC17 truncation mutants (apart from ΔCDHHC, which lacks the catalytic domain) to induce S-acylation of enhanced green fluorescent protein (EGFP)–tagged SNAP25 and CSP, upon coexpression in HEK293T cells. Deletion of zDHHC17's N-terminal region containing the ANK domain (17-ΔNAnk) resulted in a significant loss of both SNAP25 and CSP S-acylation; surprisingly, however, deletion of the C-terminus (17-ΔC) resulted in significant loss of S-acylation of these substrates too (Figure 4, A and B). Hence, although the ANK domain of zDHHC17 is required for both substrate binding and subsequent S-acylation of SNAP25 and CSP, the C-terminus of zDHHC17 is involved only in S-acyl-transferase activity of zDHHC17, at least toward these substrates. None of these zDHHC17 truncations affected the Golgi localization of exogenous zDHHC17 in HEK293T cells (Figure 4C).
FIGURE 4:

The ANK domain and cytosolic C-terminal domain of zDHHC17 are required for S-acylation of SNAP25 and CSP. HEK293T cells were cotransfected with EGFP-SNAP25 (A) or EGFP-CSP (B), and an appropriate amount of the indicated HA-tagged zDHHC17 constructs (per well) or control (empty HA-pEF-BOS vector) to achieve similar zDHHC17 expression levels (17-WT and 17-ΔN, 2.4 μg;17-ΔNAnk and control, 3.2 μg; 17-ΔC, 1.6 μg). EGFP-SNAP25 and HA-zDHHC enzymes were detected by immunoblotting with GFP and HA antibodies, respectively, and incorporation of radiolabel was detected with the aid of a Kodak Biomax Transcreen LE. Positions of molecular weight markers are shown on the left. SNAP25 palmitoylation was assessed by the amount of 3H-palmitic acid incorporated (after metabolic labeling) relative to protein levels, and CSP palmitoylation was assessed, after separation of its palmitoylated (p) and nonpalmitoylated (np) forms by SDS–PAGE of cell lysates and immunoblotting with GFP, by calculating the ratio of palmitoylated EGFP-CSP (p) to the total protein (p + np). Percentage increase in palmitoylation (vs. control) was quantified (n = 4; error bars, SEM) and differences from 17-WT were analyzed by Tukey posttest, following a one-way analysis of variance (ANOVA; n.s., p ≥ 0.05, *p < 0.05, **p < 0.01, ***p < 0.001). Subcellular distribution of zDHHC17 constructs was assessed by cotransfection with Golgi marker GRASP65 (mCherry construct) and immunofluorescence using an HA antibody (C). Scale bars, 5 μm.
zDHHC13 also interacts with SNAP25 and CSP via its N-terminal region containing an ANK domain
We and others previously reported that coexpression of zDHHC13 with SNAP25 or CSP in mammalian cells does not lead to a significant increase in S-acylation of these substrates (Greaves et al., 2008, 2010; Huang et al., 2009). However, more recent work reported a small but significant decrease in SNAP25 S-acylation in brain samples from zDHHC13-knockout mice (Sutton et al., 2013), which indicates that zDHHC13 may contribute to SNAP25 S-acylation in vivo, possibly via interaction to zDHHC13's ANK domain. To determine whether zDHHC13 interacts with SNAP25 and CSP, we also assessed with SUS interactions of this enzyme with these substrates. zDHHC13 was expressed quite poorly in yeast but at comparable levels to zDHHC7 (Figure 5B, right), which allowed comparison between these two enzymes with respect to CSP/SNAP25 binding. SUS analysis revealed that zDHHC13 interacts with CSP and SNAP25 and, furthermore, that these interactions are much stronger than the ones observed for zDHHC7 (Figure 5B, left). In addition, the much increased growth of corresponding zDHHC13-CSP/SNAP25 matings compared with positive control ones (NubI-zDHHC13) suggests a very strong and specific zDHHC13 interaction with these two proteins (Figure 5B). We further assessed with SUS, using zDHHC13 truncation mutants (Figure 5A), whether the ANK-containing cytosolic region or the zDHHC–CR and C-terminal regions are important for these interactions. The zDHHC13 truncation mutants were expressed at higher than wild-type levels (Figure 5C, right), and thus required different growth periods; however, despite these expression-caused limitations, it was clear that zDHHC13 interaction was lost after removal of its N-terminus and ANK domains (13-ΔNAnk) but not after removal of its zDHHC-CR and C-terminal domains (13-ΔCDHHC; Figure 5C, left). The foregoing data suggest that the N-terminal region containing the ANK domain of zDHHC13 is also essential and sufficient for CSP and SNAP25 interaction.
FIGURE 5:

zDHHC13 interacts with CSP and SNAP25 via its NAnk domain. (A). Schematic diagram of zDHHC13 transmembrane constructs (13-WT, 13-ΔNAnk, and 13-ΔCDHHC) used for SUS analysis and of zDHHC17/13 cytosolic constructs (17-NAnk and 13-NAnk) used for pull-down assays. Comparison of zDHHC–CSP/SNAP25 interactions between zDHHC7 and zDHHC13 (B) or among different transmembrane zDHHC13 constructs (C) was assayed using the mating-based SUS (growth assay). zDHHC–CSP/SNAP25 matings at corresponding OD600 were dropped on SD medium supplemented with adenine and histidine to verify equal dropping densities, and on SD medium (supplemented or not with 50 μM methionine) to verify interactions. For comparison of interactions between zDHHC7 and zDHHC13, plates were scanned after incubation at 30°C for 4 d, whereas for comparison of interactions between zDHHC13 truncation mutants and wild type, plates were scanned after incubation at 30°C for the appropriate number of days (indicated) to achieve similar NubI-mating growth among different zDHHC13 constructs. Spontaneous reassembly of the C-terminal ubiquitin of zDHHC13-Cub-PLV with wild-type (NubI) or mutated (NubG) N-terminal ubiquitin was assessed in parallel. Protein levels of zDHHC-Cub-PLV truncation mutants were assessed by Western blotting, using an anti-VP16 antibody, with positions of molecular weight markers shown on the left of the blots. Assessment of His6-SNAP25 (D) and His6-CSP (E) binding to HA-tagged 17-NAnk, 13-NAnk, and full-length zDHHC13 (13-WT) was assayed by pull-down assays, as in Figure 1.
To further confirm the interactions of zDHHC13's ANK domain with CSP and SNAP25, and compare the interaction strength in a more direct way to zDHHC17's ANK domain, we used pull-down assays with purified His6-tagged CSP/SNAP25 to capture N-terminal HA-tagged zDHHC17/13 fragments (Figure 5A) from HEK293T lysates. These assays revealed that the zDHHC13 and zDHHC17 NAnk regions bind with similar strength to either SNAP25 (Figure 5D) or CSP (Figure 5E); binding of full-length zDHHC13 to SNAP25 and CSP was also confirmed using the same approach (Figure 5, D and E).
zDHHC3 and zDHHC7 can S-acylate SNAP25 and CSP more efficiently than zDHHC17
Surprisingly, when zDHHC3, 7, 13, and 17 S-acylation efficiencies toward SNAP25 and CSP were compared in mammalian cells, it was found that zDHHC3 and zDHHC7 were significantly more active against these substrates than zDHHC17 (Figure 6, A and B); zDHHC13, on the other hand, was found to be inactive toward these substrates (Figure 6, A and B), as previously reported (Greaves et al., 2008, 2010). Levels of zDHHC3/7 autoacylation were also higher, with zDHHC17/13 autoacylation being below detection level (Figure 6A). Thus differences in S-acylation efficiencies of zDHHC3, zDHHC7, zDHHC13, and zDHHC17 toward CSP and SNAP25 seem to be disproportional to their differences in substrate binding. Furthermore, the higher S-acylation activity of zDHHC3 and zDHHC7, despite their reduced substrate interactions, implies that these enzymes have a much greater S-acyl-transferase activity than zDHHC17 and zDHHC13.
FIGURE 6:
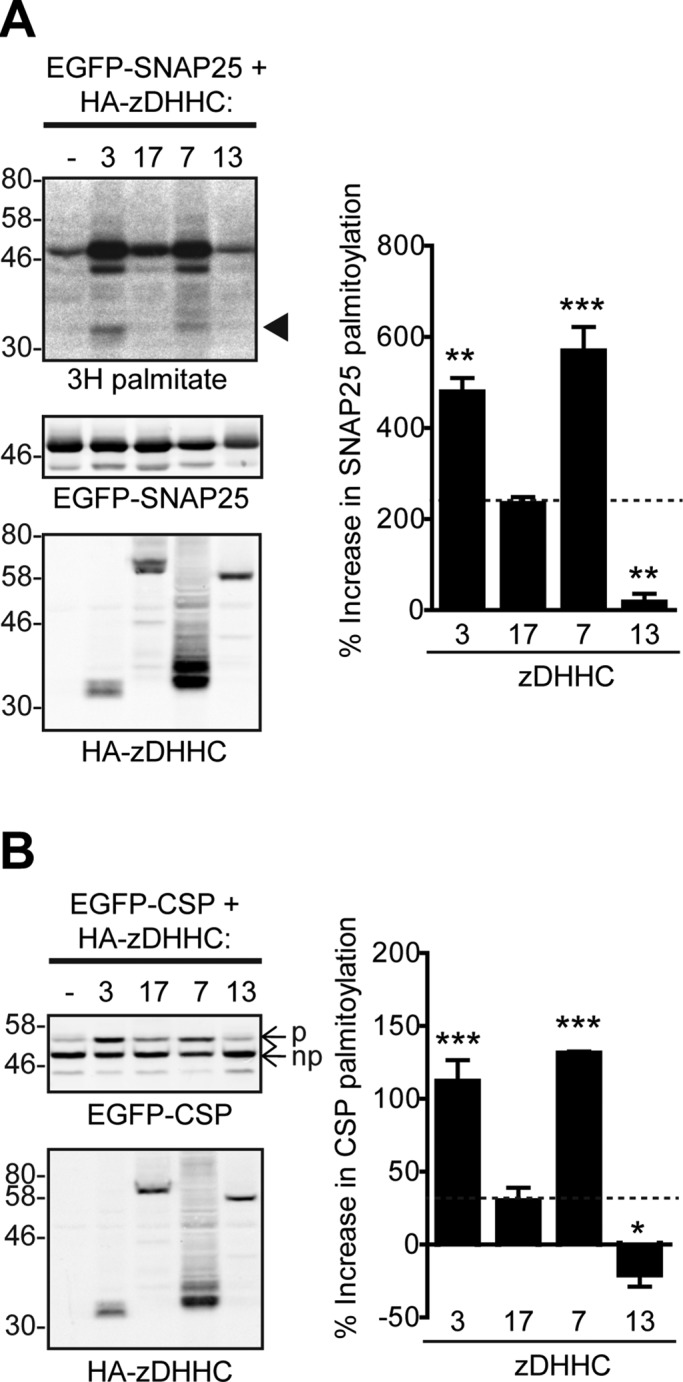
zDHHC3 and zDHHC7 can S-acylate CSP and SNAP25 more efficiently than zDHHC17, whereas zDHHC13 is inactive toward these substrates. (A) S-acylation of SNAP25 by zDHHC3, 17, 7, and 13. HEK293T cells were cotransfected with EGFP-SNAP25 and the indicated HA-tagged zDHHCs (or empty vector as control) and labeled with 3H-palmitate as described in Materials and Methods. EGFP-SNAP25 and HA-zDHHC enzymes were detected by immunoblotting with GFP and HA antibodies, respectively, and incorporation of radiolabel was detected with the aid of a Kodak Biomax Transcreen LE. Positions of molecular weight markers are shown on the left. Arrowhead indicates autoacylated zDHHC3/zDHHC7. SNAP25 palmitoylation was assessed by the amount of 3H-palmitic acid incorporated relative to protein level. Percentage increase in palmitoylation (vs. control) was quantified (n = 4; error bars, SEM), and differences from zDHHC17-induced palmitoylation were analyzed by Tukey posttest, following a one-way ANOVA (*p < 0.05, **p < 0.01, ***p < 0.001). (B) S-acylation of CSP by zDHHC3, 17, 7, and 13. HEK293T cells were cotransfected with EGFP-CSP and the indicated HA-tagged zDHHC. EGFP-CSP and HA-zDHHC enzymes were detected by immunoblotting with GFP and HA antibodies, respectively. CSP palmitoylation was assessed, after separation of its palmitoylated (p) and nonpalmitoylated (np) forms by SDS–PAGE of cell lysates, by calculating the ratio of palmitoylated EGFP-CSP (p) to the total protein (p + np). Percentage increase in palmitoylation (vs. control) was quantified (n = 4; error bars, SEM), and differences from zDHHC17-induced palmitoylation were analyzed by Tukey posttest, following a one-way ANOVA (*p < 0.05, **p < 0.01, ***p < 0.001).
The N-terminal ANK-containing domains of zDHHC17 and zDHHC13 are not functionally interchangeable
Because both zDHHC13 and zDHHC17 can interact strongly with SNAP25 and CSP via their ANK domains, we examined whether their N-terminal cytosolic regions (which contain their ANK domain) are functionally interchangeable. Although zDHHC13 having the NAnk domain of zDHHC17 (13-NAnk17; Figure 7A) remained inactive toward either SNAP25 or CSP (Figure 7, B and C), zDHHC17 containing the NAnk domain of zDHHC13 (17-NAnk13; Figure 7A) became largely inactive too, losing completely its ability to S-acylate CSP (Figure 7C) and retaining only partial activity against SNAP25 (Figure 7B). Swapping of the NAnk domains did not result in altered Golgi localization of either of the two enzymes (Figure 7D). Thus, although the NAnk domain of zDHHC13 interacts with both SNAP25 and CSP, it cannot efficiently support the S-acylation of these substrates by an adjacent zDHHC13 or zDHHC17 core.
FIGURE 7:

The ankyrin-repeat-containing cytosolic domains of zDHHC13 and zDHHC17 are not interchangeable. (A) Schematic diagram of zDHHC17/13 chimeras used for S-acylation assays. HEK293T cells were cotransfected with EGFP-SNAP25 or EGFP-CSP and the indicated HA-tagged zDHHC. EGFP-SNAP25, EGFP-CSP, and HA-zDHHC enzymes were detected by immunoblotting with GFP and HA antibodies, respectively, and incorporation of radiolabel was detected with the aid of a Kodak Biomax Transcreen LE. Positions of molecular weight markers are shown on the left. SNAP25 palmitoylation was assessed by the amount of 3H-palmitic acid incorporated (following metabolic labeling) relative to protein level (B), and CSP palmitoylation was assessed, after separation of its palmitoylated (p) and nonpalmitoylated (np) forms by SDS–PAGE of cell lysates, by calculating the ratio of palmitoylated EGFP-CSP (p) to the total protein (p + np). (C) Percentage increase in palmitoylation (vs. control) was quantified (SNAP25, n = 10; CSP, n = 4; error bars, SEM). Statistical analysis was performed using a one-way ANOVA and Tukey posttest (n.s., p ≥ 0.05, *p < 0.05, **p < 0.01, ***p < 0.001). (D) Subcellular distribution of zDHHC17/13 constructs was assessed by cotransfection with Golgi marker GRASP65 (mCherry construct) and immunofluorescence using an HA antibody. Scale bars, 5 μm.
Introducing a DQHC motif into zDHHC17 blocks S-acylation of SNAP25 and CSP
The fact that coexpression of zDHHC13 does not promote S-acylation of either SNAP25 or CSP (Greaves et al., 2008) is somewhat surprising, given that zDHHC13 interacts efficiently with these substrates, as observed with both SUS analysis and pull-down experiments. However, one conspicuous feature of zDHHC13 distinguishing it from all other mammalian DHHC enzymes is its unique DQHC motif, which could limit S-acyl-transferase activity and thus restrict the substrates to be S-acylated. A histidine-to-alanine mutation in the equivalent position of yeast Erf2 was previously shown to inactivate the enzyme, by reducing the rate of palmitoyl-Erf2 intermediate formation, and abolishing the palmitate transfer to Ras2 substrate (Mitchell et al., 2010). To investigate how the DQHC motif might affect zDHHC13 S-acyl-transferase activity, we introduced a DHHC motif into zDHHC13 and a DQHC motif into zDHHC17. Although the introduction of a DQHC motif (H465Q mutation; Figure 8A) into zDHHC17 completely abolished the ability of this enzyme to S-acylate either SNAP25 or CSP, zDHHC13 containing a DHHC motif (Q454H mutation; Figure 8A) remained inactive toward both SNAP25 and CSP (Figure 8, B and C). None of these mutations altered zDHHC13/17 localization (Figure 8D). Hence, although H465 is necessary for zDHHC17 activity, Q454 at the equivalent position of zDHHC13 cannot account on its own for zDHHC13's inactivity toward CSP and SNAP25.
FIGURE 8:

Effect of the DQHC/DHHC motif in zDHHC13 and zDHHC17 on S-acylation of SNAP25 and CSP. (A) Schematic diagram of zDHHC17/13 mutants used for S-acylation assays. HEK293T cells were cotransfected with either EGFP-SNAP25 or EGFP-CSP and the indicated HA-tagged zDHHC construct. EGFP-SNAP25, EGFP-CSP, and HA-zDHHC enzymes were detected by immunoblotting with GFP and HA antibodies, respectively, and incorporation of radiolabel was detected with the aid of a Kodak Biomax Transcreen LE. Positions of molecular weight markers are shown on the left. SNAP25 palmitoylation was assessed by the amount of 3H-palmitic acid incorporated (after metabolic labeling) relative to protein level (B), and CSP palmitoylation was assessed after separation of its palmitoylated (p) and nonpalmitoylated (np) forms by SDS–PAGE of cell lysates by calculating the ratio of palmitoylated EGFP-CSP (p) to the total protein (p + np). (C) Percentage increase in palmitoylation (vs. control) was quantified (SNAP25, n = 4; CSP, n = 6; error bars, SEM). Statistical analysis was performed using a one-way ANOVA and Tukey posttest (n.s., p ≥ 0.05, *p < 0.05, **p < 0.01, ***p < 0.001). (D) Subcellular distribution of zDHHC17/13 mutants was assessed by cotransfection with Golgi marker GRASP65 (mCherry construct) and immunofluorescence using an HA antibody. Scale bars, 5 μm.
DISCUSSION
The results presented in this study identify vast differences in substrate affinity and S-acyl-transferase activity among Golgi zDHHC enzymes, with zDHHC17 and zDHHC13 being equipped for protein binding, and zDHHC3 and zDHHC7 being highly efficient in protein S-acylation.
The zDHHC17 and zDHHC13 S-acyl-transferases were found to display strong and specific interactions with CSP and SNAP25; these interactions were mediated by a seven-ankyrin repeat domain (absent in other S-acyl-transferases) located in the cytosolic N-termini of these enzymes. Ankyrin repeats exist in numerous proteins, and they all serve a sole function: mediating protein–protein interactions (Li et al., 2006). Hence the ANK domain of zDHHC17 and 13 may be well fitted to be the recruiting module, at least for zDHHC17, for S-acylation of zDHHC17 substrates. Indeed, complete deletion of this module resulted in impaired CSP/SNAP25 S-acylation (this study), whereas partial deletion was shown to affect huntingtin (HTT) S-acylation too, a protein that appears to be both a substrate and regulator of this enzyme (Huang et al., 2009, 2011).
In addition to the ANK domain and catalytic DHHC-CR domain, the cytoplasmic C-terminus of zDHHC17 was also found to be essential for substrate S-acylation. Although it is possible that the C-terminus may couple to substrate proteins to facilitate S-acyl transfer, it is very unlikely that binding per se, and thus initial substrate recruitment, requires this region, since we did not observe any loss of binding to SNAP25 or CSP when this domain was deleted. Thus, this region is probably required for other steps of S-acylation, linked to the ability of zDHHC17 to form acyl intermediates and/or transfer them to the recruited proteins.
Binding of zDHHC3/zDHHC7 to SNAP25 and CSP was either very weak (SUS) or undetectable (pull downs). Nevertheless, zDHHC3 and zDHHC7 appeared to catalyze S-acylation of SNAP25/CSP more efficiently than zDHHC17, whereas zDHHC13 was inactive toward these proteins. Hence the much-increased S-acylation efficiency of zDHHC3/7 to SNAP25/CSP relative to zDHHC17/13 must be a result of the intrinsic ability of these enzymes to transfer acyl groups to target proteins. Indeed, autoacylation, which is often used as a measure of S-acylation activity, was previously found to be higher for zDHHC3 and zDHHC7 than for zDHHC17 and zDHHC13, with the latter exhibiting the lowest autoacylation activity (Ohno et al., 2012; Ren et al., 2013); this agrees with what we observed in our S-acylation assays (this study) and previous membrane targeting assays (Greaves et al., 2008, 2009, 2010) with these sets of enzymes and substrates. Similarly, we also observed prominent autoacylation of zDHHC3 and zDHHC7, but no detectable S-acylation for zDHHC17 and zDHHC13, in our S-acylation assays. Although some zDHHC enzymes have been shown to be S-acylated outside their DHHC domains (Yang et al., 2010), there is no evidence that acyl chains are present outside the DHHC domain of zDHHC3/7, and so this is unlikely to explain the increased autoacylation of these proteins relative to zDHHC17/13. Lack of substrate specificity and increased rate of S-acyl-transfer in zDHHC3- and zDHHC7-catalyzed reactions could explain not only why these enzymes are so versatile in the number of substrates they can S-acylate in various in-cell S-acylation assays, but also why most of the substrates are shared for these two enzymes (Greaves and Chamberlain, 2011).
Although the data presented clearly show that zDHHC3 and zDHHC7 are much more capable of S-acylating SNAP25 and CSP than zDHHC17 (or zDHHC13), additional factors may influence the in vivo S-acylation of these substrates by endogenously expressed enzymes, including 1) the cell-specific expression levels of various zDHHC-enzymes, 2) their possible compartmentalization within different subdomains of the Golgi, which might influence substrate accessibility, and 3) their affinity to (and/or local availability of) acyl-CoA as a substrate in the S-acylation reaction.
The lack of positive correlation between the strength of zDHHC–substrate interactions and S-acylation efficiency was most pronounced for zDHHC13, which bound tightly to SNAP25 and CSP but failed to S-acylate these proteins. To investigate why zDHHC13 has no detectable S-acylation activity toward CSP and SNAP25, we used two approaches: 1) swapping zDHHC13's and zDHHC17's cytosolic NAnk domains, and 2) swapping the DQHC/DHHC motifs within their zDHHC-CR region. Both approaches failed to activate zDHHC13 to S-acylate either SNAP25 or CSP. However, in both cases, the ability of zDHHC17 to modify these substrates was perturbed. Thus, there may be a difference in the way the NAnk domains of these enzymes present substrates to the catalytic domain. In addition, the DQHC motif in zDHHC13, although detrimental for zDHHC17 (and potentially for zDHHC13) S-acylation activity, cannot account on its own for zDHHC13's inability to modify these substrates. zDHHC13 has been shown to be the only other S-acyl-transferase apart from zDHHC17 to efficiently interact with, and S-acylate, HTT (Huang et al., 2011) and ClipR59 (Ren et al., 2013), but has not been found to have any activity toward other zDHHC17's substrates. We propose that the DQHC motif negatively affects zDHHC13's acyl-transfer ability, and thus its S-acylation ability is restricted to strongly bound substrates with appropriate orientation, limiting the substrates that can be S-acylated by this enzyme. Hence CSP and SNAP25, unlike HTT and ClipR59, may lack the orientation required upon binding for efficient S-acylation by zDHHC13. Of interest, however, a small but significant reduction of SNAP25 S-acylation was observed in zDHHC13-deficient mice (Sutton et al., 2013). Because in-cell S-acylation assays do not indicate S-acyl-transferase activity of zDHHC13 toward SNAP25, it is possible that this enzyme contributes indirectly to SNAP25 in vivo S-acylation, perhaps by recruiting it to the Golgi for S-acylation by other zDHHCs. Indeed, it will be interesting in future studies to confirm that the ability of zDHHC13 to increase S-acylation of HTT and ClipR59 is lost after mutation of its catalytic cysteine. This will rule out any indirect effect of zDHHC13 on the S-acylation of these substrates.
Although we focused on the role of zDHHC17 and zDHHC13 as S-acyl-transferases, the participation of the ANK domain of these enzymes in wider protein interaction networks may contribute to its cellular functions, independently of S-acyl-transferase function. Indeed, the unusually high proportion of interacting proteins shared by zDHHC17 and HTT, including the (non–S-acylated by zDHHC17) optineurin (Butland et al., 2014), indicates the existence of such complexes inside the cell. Furthermore, an S-acylation–independent scaffolding function for the ANK domain of zDHHC17 resulting in JNK3 phosphorylation has been documented (Yang and Cynader, 2011).
Our work also highlights the strength of the yeast split-ubiquitin system for the study of protein interactions between full-length, membrane-inserted zDHHC enzymes and their substrates. Although various factors that could influence zDHHC–substrate interactions, including other interacting partners or posttranslational modifications, may not be faithfully recapitulated in yeast cells, this system offers a valuable mechanism with which to interrogate the underlying mechanisms that govern specificity of zDHHC–substrate interactions. Indeed, we verified the findings made using this system with pull-down experiments from mammalian cell extracts. In future work, it will be interesting to implement techniques that allow zDHHC–substrate interactions to be monitored in real time in mammalian cells; this could be achieved through the use of Forster resonance energy transfer experiments using fluorescently tagged proteins. In addition, in vitro kinetic analysis with recombinant proteins is likely to shed more light on the differences in autoacylation and substrate S-acylation among various zDHHC enzymes (Jennings and Linder, 2012) observed in the present study.
What role might selective and nonselective S-acylation of substrate proteins by different zDHHC enzymes serve in the cell? For peripheral membrane proteins, an important requirement for S-acylation is that substrate proteins have a mechanism to associate transiently with cell membranes to allow their recognition by membrane-bound zDHHC enzymes. Proteins such as Ras use farnesylation as this initial membrane targeting signal, whereas SNAP25 and CSP appear to have a weak intrinsic membrane affinity (Greaves et al., 2008, 2009). The strength of this initial membrane interaction of nonacylated proteins would be the major factor that dictates how efficiently they become modified by an S-acylation machinery that lacks any specificity or mechanism of substrate recruitment. However the presence of enzymes such as zDHHC17, which interact more strongly with their substrates, may be important to ensure the efficient S-acylation of substrates that have weaker membrane affinities. Indeed, previous work in our lab suggests that disruption of the interaction of SNAP25 with zDHHC17 leads to a loss of stable membrane binding of this protein in neuroendocrine cells (Greaves et al., 2009), showing the importance of zDHHC–substrate specificity for S-acylation and intracellular targeting of this protein. Specificity in zDHHC–substrate interactions is not limited to zDHHC17/zDHHC13, as the PDZ ligands of zDHHC5 and zDHHC8 are involved in recognition of at least some of their substrates (Thomas et al., 2012). The differences in substrate binding and S-acylation efficiencies among zDHHCs, which are demonstrated in this study, imply vast differences in their S-acyl-transferase activities. Such diversity in S-acyl-transfer ability among zDHHC enzymes, combined with their overlapping substrate specificities, reveals great complexity in the way in which various zDHHC substrates are processed inside the cell to achieve certain subcellular localizations.
MATERIALS AND METHODS
Chemicals
Unless otherwise stated, all chemicals were purchased from Sigma (Dorset, United Kingdom).
Plasmids
EGFP-tagged SNAP25 and CSP were produced as previously described (Greaves and Chamberlain, 2006; Greaves et al., 2009). Plasmid DNA encoding mouse HA-tagged zDHHC3, zDHHC7, zDHHC13, and zDHHC17 was kindly provided by Masaki Fukata (National Institute of Physiological Sciences, Osaka, Japan; Fukata et al., 2004). Owing to a slightly different nomenclature used for the DHHC clones synthesized by the Fukata lab, zDHHC13 used in this study is the original DHHC22 clone (Fukata et al., 2004; Greaves et al., 2008, 2010); the National Center for Biotechnology Information (NCBI) reference for mouse zDHHC13 used here is NP_082307.1. The HA-zDHHC17 clone used here was that originally generated by the Fukata lab (Fukata et al., 2004) encompassing amino acids 11–632 according to NCBI reference NP_766142.2. ANK-domain swaps to generate 13-NAnk17 and 17-NAnk13 mutants were centered on amino acid N288 of zDHHC17 and N278 of zDHHC13. Briefly, restriction sites were introduced before the initiating methionine and after residue N288/N278 of the zDHHC enzymes by site-directed mutagenesis. After swapping of these regions into the corresponding zDHHC enzyme, the restriction sites were removed by site-directed mutagenesis. The GST-17NAnk construct was produced by subcloning the zDHHC17 cDNA corresponding to amino acids 11–305 into pGEX-KG vector. For the mating-based SUS, all cDNAs were cloned by Gateway technology (Life Technologies, Paisley, United Kingdom), first as entry clones in pDONR207 and then as destination clones in the corresponding destination vector; zDHHC enzyme cDNAs (without a STOP codon) were inserted in pMetYC-Dest (Grefen et al., 2001) for expression of zDHHC-ProteinA-LexA-VP16 protein, whereas CSP and SNAP25 cDNAs (having a STOP codon) were inserted into pNX35-Dest (Grefen and Blatt, 2012) for expression of NubG-HA-CSP and NubG-HA-SNAP25 proteins. His6-tagged CSP and SNAP25 were also produced by Gateway cloning, using the foregoing CSP and SNAP25 cDNAs and Gateway compatible pET-28B as destination vector. For mammalian cell expression of zDHHC17 truncation mutants, corresponding cDNAs were inserted by Gateway technology to Gateway-compatible HA-pEF-BOS, and STOP codons were inserted afterward, for expression of HA-zDHHC17 proteins.
Protein purification
Plasmids encoding the proteins to be purified were inserted into BL21(DE3)pLysS bacterial cells (Promega, Southampton, United Kingdom), and protein expression was induced by addition of 1 mM isopropyl-β-d-thiogalactoside in culture medium. For GST/GST-17NAnk purification, bacterial lysates were incubated with glutathione Sepharose 4B (GE Healthcare, Little Chalfont, United Kingdom), and after extensive washing with PBS, proteins were eluted by incubation in 50 mM Tris, pH 8, containing 10 mM reduced glutathione. For His6-CSP/SNAP25, bacterial lysates were incubated with Ni2+-NTA agarose (Qiagen, Manchester, United Kingdom), and after extensive washing with Washing buffer I (40 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid [HEPES], pH 8, 300 mM NaCl, 40 mM imidazole), proteins were eluted by incubation with Elution buffer (same as Washing buffer, but with 500 mM imidazole added). All eluted proteins were dialyzed overnight against 5 l of ice-cold phosphate-buffered saline (PBS) and subsequently kept in aliquots at −80°C. Protein purity and concentration of dialyzed samples were assessed, after SDS–PAGE, by Bio-safe Coomassie staining (Bio-Rad, Hemel Hempstead, United Kingdom) and quantification of bands against bovine serum albumin (BSA) standards (Fisher Scientific, Loughborough, United Kingdom) run in parallel.
Antibodies
Mouse GFP antibody (JL8) was from Clontech (California). Rat HA antibody (immunoblotting) was from Roche (Sussex, United Kingdom), and mouse HA antibody (immunofluorescence) was from Cambridge Bioscience (Cambridge, United Kingdom). Mouse SNAP25 antibody (SMI-81) was from Covance (New Jersey). Rabbit CSP antibody was from Enzo Life Sciences (Exeter, United Kingdom). Rabbit synuclein (ab52168), Myc (ab9106), and VP16 (ab4808) antibodies were from Abcam (Cambridge, United Kingdom). Alexa Fluor secondary antibodies used for immunofluorescence were from Life Technologies. IRDye mouse and rabbit secondary antibodies were from LI-COR (Cambridge, United Kingdom) and DyLight rat ones from Fisher Scientific.
Cell culture and transfection
HEK293T cells were maintained in DMEM (Life Technologies) supplemented with 10% fetal bovine serum in a humidified atmosphere containing 5% CO2. Cells were plated onto poly-d-lysine–coated 24-well plates for S-acylation assays or coverslips for immunofluorescence analysis. Transfections were performed using Lipofectamine 2000 according to the manufacturer's instructions (Life Technologies). Cotransfections with EGFP-substrate and HA-zDHHC plasmids for biochemical experiments used 0.8 and 1.6 μg respectively, unless otherwise stated. For immunofluorescence, 0.5 μg of plasmid DNA was used for transfections. Cells were analyzed ∼20 h posttransfection.
Immunofluorescence
HEK293T cells were transfected with 0.2 μg each of HA-DHHC17/13 and GRASP65-mCherry and incubated overnight at 37°C/5% CO2 in a humidified atmosphere. Cells were then fixed in 4% formaldehyde and permeabilized by incubating in PBS containing 0.3% BSA and 0.25% Triton X-100 for 6 min at room temperature. The permeabilized cells were washed and incubated for 60 min in PBS-BSA containing mouse HA antibody (1:50 dilution). The cells were washed again in PBS-BSA and incubated for 60 min in PBS-BSA containing anti-mouse antibody conjugated to Alexa Fluor 488 (1:400 dilution). The labeled cells were washed in PBS and briefly submerged in distilled H2O before being mounted onto glass slides using Mowiol. Cells were imaged using a Leica SP5 confocal microscope. Scale bars represent 5 μm.
S-acylation assays
For analysis of SNAP25 S-acylation, transfected cells on 24-well plates were incubated for 30 min in DMEM containing 1% defatted BSA and then incubated in the same medium containing 0.5 mCi/ml 3H-palmitic acid for 3–4 h at 37o C. The labeled cells were washed in PBS, lysed, and boiled in SDS-dissociation buffer (2% SDS, 25 mM dithiothreitol, 10% glycerol, 0.01% bromophenol blue, 50 mM Tris, pH 6.8), subjected to SDS–PAGE, and transferred onto nitrocellulose for immunoblotting analysis or 3H-palmitate detection, which relied on exposing the blots to light-sensitive film in the presence of a Kodak Biomax Transcreen LE (PerkinElmer, Seer Green, United Kingdom) for 4–18 d in the dark at −80°C. CSP S-acylation status was determined by a well-characterized change in migration pattern on SDS–PAGE gels (Chamberlain et al., 2001; Greaves and Chamberlain, 2006; Greaves et al., 2008, 2012), since this assay does not rely on radioactivity and can produce more accurate measurements of S-acylation due to quantification of both palmitoylated and total protein in the same gel. SDS–PAGE was performed using 12% polyacrylamide gels. After S-acylation of CSP, marked (∼7 kDa) reduction in migration was observed.
Mating-based split-ubiquitin system
The mating-based SUS in yeast is a very sensitive method for the detection of protein–protein interactions in vivo; this technique is specifically applicable for transmembrane proteins (Grefen et al., 2009) and hence can be used for the study of zDHHC enzyme interactions with other proteins. It relies on ubiquitin split into halves: the C-terminal half (Cub) is fused in between a transmembrane (bait) protein and a transcriptional reporter complex (PLV), whereas the N-terminal half (Nub) is fused to a prey protein. Bait and prey are initially expressed in different types of yeast; upon mating of these, bait and prey are coexpressed in the same cell, and their interaction leads to the reassembly of the ubiquitin molecule. The whole ubiquitin can be thus recognized by ubiquitin-specific proteases, which will cleave downstream of Cub, leading to the release of the PLV complex and subsequent activation of different reporter genes; of these, Ade2 and His3 encode for proteins required for synthesis of adenine and histidine, and thus their expression allows the yeast to grow on selective media lacking these two nutrients (growth assay). A point mutation (I13G) into the N-terminal half of ubiquitin (designated NubG) prevents spontaneous reconstruction of ubiquitin, which could occur in the absence of interaction if wild-type Nub (designated NubI) is used instead. A schematic of the SUS principle is shown in Figure 2A. Owing to the PLV being attached to the bait rather than the prey, the concentration of the latter has very little effect on reporter gene activation and hence on yeast growth; on the contrary, decreased concentration of bait can lead to reduced yeast growth, whereas its increased concentration can produce a strong reporter activation response even in limiting concentrations of the prey or in weak bait–prey interactions. Nevertheless, this can be overcome by addition of methionine in the yeast growth medium, which will restrict bait expression due to met25, a methionine repressible promoter in pMetYC-Dest (bait) plasmid (Grefen et al., 2001). Yeast transformation, mating, growth media preparation, and lysis of haploid yeast for Western blotting were done as described previously (Grefen et al., 2009). Briefly, overnight cultures of matings were resuspended in sterile water at indicated OD600, and 5 μl of each mating was dropped on synthetic defined (SD) medium (supplemented or not with methionine) for assessment of interactions and on SD media supplemented with adenine and histidine for verification of matings and yeast cell density. Plates were placed at 30°C and were scanned after incubation for the number of days indicated in the corresponding figure. When interactions of different baits were compared, all matings were grown for the same number of days; however, when bait expression, and thus corresponding NubI-mating growth, varied considerably, matings were incubated for an appropriate number of days to achieve similar (for all baits) growth with control prey (NubI).
Pull-down assays
For GST pull down of rat-brain proteins, rat brain was lysed in 1.5 ml of lysis buffer (20 mM HEPES, pH 7.4, 150 mM NaCl, 1% Triton X-100, and protease inhibitor cocktail) using a Dounce homogenizer, and the lysate was centrifuged (12,000 × g for 10 min at 4°C) to remove insoluble material. The supernatant was precleared by incubation with 250 μg of purified GST and 1 ml of glutathione-Sepharose beads for 2 h at 4°C. The cleared lysate was split into halves, and each half was incubated with 200 μl of glutathione beads and either 250 μg of purified GST or an equimolar quantity of purified GST-17NAnk (474 μg) overnight at 4°C. Unbound fractions were collected after centrifugation, and bound proteins were eluted, after extensive wash with lysis buffer, by boiling in sample buffer. A similar procedure was followed for the pull down of Myc-CSP from HEK293T cells. For His6-CSP and His6-SNAP25 pull down of HA-zDHHC enzymes, the procedure was as follows: HEK293T cells expressing the corresponding HA-zDHHC plasmids were lysed in 600 μl Binding buffer (20 mM Tris, pH 7.9, 150 mM NaCl, 1% Triton X-100, 20 mM imidazole, 20 mM 2-mercaptoethanol) and cleared by centrifugation (10,000 × g for 10 min at 4°C). A 240-μl amount of the supernatant was incubated for 2 h at 4°C with either 75 μg of purified His6-tagged SNAP25 (or equivalent amount of PBS) and 25 μl of Ni2+- nitriloacetic acid (NTA) agarose (Qiagen) or with 25 μl of Ni2+-NTA agarose (Qiagen) that had been preincubated with 75 μg of purified His6-tagged CSP for 30 min at 4°C. After extensive washing with Washing buffer II (same as Binding buffer but having 300 mM NaCl and 60 mM imidazole instead), bound proteins were recovered by boiling in sample buffer.
Quantification of blots and statistical analysis
Quantification of bands in 3H fluorographs and immunoblots was performed by densitometry, using ImageJ (National Institutes of Health, Bethesda, MD) software. All statistical tests were performed with Prism software (GraphPad, La Jolla, CA).
Acknowledgments
This work was funded by grants from the Medical Research Council (0601597/2) and the Wellcome Trust (WT094184MA).
Abbreviations used:
- ANK
ankyrin repeat
- CSP
cysteine-string protein
- SNAP25
synaptosomal-associated protein 25
- SUS
split-ubiquitin system.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E14-06-1169) on September 24, 2014.
REFERENCES
- Butland SL, Sanders SS, Schmidt ME, Riechers SP, Lin DT, Martin DD, Vaid K, Graham RK, Singaraja RR, Wanker EE, et al. The palmitoyl acyltransferase HIP14 shares a high proportion of interactors with huntingtin: implications for a role in the pathogenesis of Huntington's disease. Hum Mol Genet. 2014;23:4142–4160. doi: 10.1093/hmg/ddu137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain L, Graham M, Kane S, Jackson J, Maier V, Burgoyne R, Gould G. The synaptic vesicle protein, cysteine-string protein, is associated with the plasma membrane in 3T3-L1 adipocytes and interacts with syntaxin 4. J Cell Sci. 2001;114:445–455. doi: 10.1242/jcs.114.2.445. [DOI] [PubMed] [Google Scholar]
- Fukata Y, Fukata M. Protein palmitoylation in neuronal development and synaptic plasticity. Nat Rev Neurosci. 2010;11:161–175. doi: 10.1038/nrn2788. [DOI] [PubMed] [Google Scholar]
- Fukata M, Fukata Y, Adesnik H, Nicoll RA, Bredt DS. Identification of PSD-95 palmitoylating enzymes. Neuron. 2004;44:987–996. doi: 10.1016/j.neuron.2004.12.005. [DOI] [PubMed] [Google Scholar]
- Greaves J, Carmichael JA, Chamberlain LH. The palmitoyl transferase DHHC2 targets to a dynamic membrane cycling pathway: regulation by a C-terminal domain. Mol Biol Cell. 2011;22:1887–1895. doi: 10.1091/mbc.E10-11-0924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greaves J, Chamberlain LH. Dual role of the cysteine-string domain in membrane binding and palmitoylation-dependent sorting of the molecular chaperone cysteine-string protein. Mol Biol Cell. 2006;17:4748–4759. doi: 10.1091/mbc.E06-03-0183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greaves J, Chamberlain LH. DHHC palmitoyl transferases: substrate interactions and (patho)physiology. Trends Biochem Sci. 2011;36:245–253. doi: 10.1016/j.tibs.2011.01.003. [DOI] [PubMed] [Google Scholar]
- Greaves J, Gorleku OA, Salaun C, Chamberlain LH. Palmitoylation of the SNAP25 protein family: specificity and regulation by DHHC palmitoyl transferases. J Biol Chem. 2010;285:24629–24638. doi: 10.1074/jbc.M110.119289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greaves J, Lemonidis K, Gorleku OA, Cruchaga C, Grefen C, Chamberlain LH. Palmitoylation-induced aggregation of cysteine-string protein mutants that cause neuronal ceroid lipofuscinosis. J Biol Chem. 2012;287:37330–37339. doi: 10.1074/jbc.M112.389098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greaves J, Prescott GR, Fukata Y, Fukata M, Salaun C, Chamberlain LH. The hydrophobic cysteine-rich domain of SNAP25 couples with downstream residues to mediate membrane interactions and recognition by DHHC palmitoyl transferases. Mol Biol Cell. 2009;20:1845–1854. doi: 10.1091/mbc.E08-09-0944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greaves J, Salaun C, Fukata Y, Fukata M, Chamberlain LH. Palmitoylation and membrane interactions of the neuroprotective chaperone cysteine-string protein. J Biol Chem. 2008;283:25014–25026. doi: 10.1074/jbc.M802140200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grefen C, Blatt MR. Do calcineurin B-like proteins interact independently of the serine threonine kinase CIPK23 with the K+ channel AKT1? Lessons learned from a ménage à trois. Plant Physiol. 2012;159:915–919. doi: 10.1104/pp.112.198051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grefen C, Lalonde S, Obrdlik P. Curr Protoc Neurosci Chapter 5, Unit 5.27. 2001. Split-ubiquitin system for identifying protein-protein interactions in membrane and full-length proteins. [DOI] [PubMed] [Google Scholar]
- Grefen C, Obrdlik P, Harter K. The determination of protein-protein interactions by the mating-based split-ubiquitin system (mbSUS) Methods Mol Biol. 2009;2009:1–17. doi: 10.1007/978-1-59745-289-2_14. [DOI] [PubMed] [Google Scholar]
- Huang K, Sanders SS, Kang R, Carroll JB, Sutton L, Wan J, Singaraja R, Young FB, Liu L, El-Husseini A, et al. Wild-type HTT modulates the enzymatic activity of the neuronal palmitoyl transferase HIP14. Hum Mol Genet. 2011;20:3356–3365. doi: 10.1093/hmg/ddr242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang K, Sanders S, Singaraja R, Orban P, Cijsouw T, Arstikaitis P, Yanai A, Hayden MR, El-Husseini A. Neuronal palmitoyl acyl transferases exhibit distinct substrate specificity. FASEB J. 2009;23:2605–2615. doi: 10.1096/fj.08-127399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennings BC, Linder ME. DHHC protein S-acyltransferases use a similar ping-pong kinetic mechanism but display different acyl-coA specificities. J Biol Chem. 2012;287:7236–7245. doi: 10.1074/jbc.M111.337246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang R, Wan J, Arstikaitis P, Takahashi H, Huang K, Bailey AO, Thompson JX, Roth AF, Drisdel RC, Mastro R, et al. Neural palmitoyl-proteomics reveals dynamic synaptic palmitoylation. Nature. 2008;456:904–909. doi: 10.1038/nature07605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Mahajan A, Tsai M-D. Ankyrin repeat: a unique motif mediating protein−protein interactions. Biochem. 2006;45:15168–15178. doi: 10.1021/bi062188q. [DOI] [PubMed] [Google Scholar]
- Linder ME, Deschenes RJ. Palmitoylation: policing protein stability and traffic. Nat Rev Mol Cell Biol. 2007;8:74–84. doi: 10.1038/nrm2084. [DOI] [PubMed] [Google Scholar]
- Martin BR, Cravatt BF. Large-scale profiling of protein palmitoylation in mammalian cells. Nat Methods. 2009;6:135–138. doi: 10.1038/nmeth.1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell DA, Mitchell G, Ling Y, Budde C, Deschenes RJ. Mutational analysis of Saccharomyces cerevisiae Erf2 reveals a two-step reaction mechanism for protein palmitoylation by DHHC enzymes. J Biol Chem. 2010;285:38104–38114. doi: 10.1074/jbc.M110.169102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell DA, Vasudevan A, Linder ME, Deschenes RJ. Thematic review series: lipid posttranslational modifications. protein palmitoylation by a family of DHHC protein S-acyltransferases. J Lipid Res. 2006;47:1118–1127. doi: 10.1194/jlr.R600007-JLR200. [DOI] [PubMed] [Google Scholar]
- Muszbek L, Haramura G, Cluette-Brown J, Van Cott E, Laposata M. The pool of fatty acids covalently bound to platelet proteins by thioester linkages can be altered by exogenously supplied fatty acids. Lipids. 1999;34:S331–S337. doi: 10.1007/BF02562334. [DOI] [PubMed] [Google Scholar]
- Ohno Y, Kashio A, Ogata R, Ishitomi A, Yamazaki Y, Kihara A. Analysis of substrate specificity of human DHHC protein acyltransferases using a yeast expression system. Mol Biol Cell. 2012;23:4543–4551. doi: 10.1091/mbc.E12-05-0336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno Y, Kihara A, Sano T, Igarashi Y. Intracellular localization and tissue-specific distribution of human and yeast DHHC cysteine-rich domain-containing proteins. Biochim Biophys Acta. 2006;1761:474–483. doi: 10.1016/j.bbalip.2006.03.010. [DOI] [PubMed] [Google Scholar]
- Ren W, Sun Y, Du K. DHHC17 palmitoylates ClipR-59 and modulates ClipR-59 association with the plasma membrane. Mol Cell Biol. 2013;33:4255–4265. doi: 10.1128/MCB.00527-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocks O, Gerauer M, Vartak N, Koch S, Huang Z-P, Pechlivanis M, Kuhlmann J, Brunsveld L, Chandra A, Ellinger B, et al. The Palmitoylation machinery is a spatially organizing system for peripheral membrane proteins. Cell. 2010;141:458–471. doi: 10.1016/j.cell.2010.04.007. [DOI] [PubMed] [Google Scholar]
- Salaun C, Greaves J, Chamberlain LH. The intracellular dynamic of protein palmitoylation. J Cell Biol. 2010;191:1229–1238. doi: 10.1083/jcb.201008160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton LM, Sanders SS, Butland SL, Singaraja RR, Franciosi S, Southwell AL, Doty CN, Schmidt ME, Mui KKN, Kovalik V, et al. Hip14l-deficient mice develop neuropathological and behavioural features of Huntington disease. Hum Mol Genet. 2013;22:452–465. doi: 10.1093/hmg/dds441. [DOI] [PubMed] [Google Scholar]
- Thomas G M, Hayashi T, Chiu S-L, Chen C-M, Huganir Richard L. Palmitoylation by DHHC5/8 targets GRIP1 to dendritic endosomes to regulate AMPA-R trafficking. Neuron. 2012;73:482–496. doi: 10.1016/j.neuron.2011.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G, Cynader MS. Palmitoyl acyltransferase zD17 mediates neuronal responses in acute ischemic brain injury by regulating JNK activation in a signaling module. J Neurosci. 2011;31:11980–11991. doi: 10.1523/JNEUROSCI.2510-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Di Vizio DD, Kirchner M, Steen H, Freeman MR. Proteome scale characterization of human S-acylated proteins in lipid raft-enriched and non-raft membranes. Mol Cell Proteomics. 2010;9:54–70. doi: 10.1074/mcp.M800448-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yount JS, Moltedo B, Yang Y-Y, Charron G, Moran TM, Lopez CB, Hang HC. Palmitoylome profiling reveals S-palmitoylation-dependent antiviral activity of IFITM3. Nat Chem Biol. 2010;6:610–614. doi: 10.1038/nchembio.405. [DOI] [PMC free article] [PubMed] [Google Scholar]